

Abstract
Loss-of-function mutations of the type 2 vasopressin receptor (V2R) in kidney can lead to nephrogenic diabetes insipidus (NDI). We studied a previously described, but uncharacterized, mutation of the V2R (N321K missense mutation) of a patient with NDI. The properties of the mutant receptor were evaluated. We constructed a highly sensitive Epac-based bioluminescence resonance energy transfer biosensor to perform real-time cAMP measurements after agonist stimulation of transiently transfected HEK293 cells with V2Rs. β-Arrestin binding of the activated receptors was examined with luciferase-tagged β-arrestin and mVenus-tagged V2Rs using the bioluminescence resonance energy transfer technique. Cell surface expression levels of hemagglutinin-tagged receptors were determined with flow cytometry using anti-hemagglutinin-Alexa 488 antibodies. Cellular localization examinations were implemented with fluorescent tagged receptors visualized with confocal laser scanning microscopy. The effect of various vasopressin analogs on the type 1 vasopressin receptor (V1R) was tested on mouse arteries by wire myography. The N321K mutant V2R showed normal cell surface expression, but the potency of arginine vasopressin for cAMP generation was low, whereas the clinically used desmopressin was not efficient. The β-arrestin binding and internalization properties of the mutant receptor were also different than those for the wild type. The function of the mutant receptor can be rescued with administration of the V2R agonist Val4-desmopressin, which had no detectable side effects on V1R in the effective cAMP generating concentrations. Based on these findings we propose a therapeutic strategy for patients with NDI carrying the N321K mutation, as our in vivo experiments suggest that Val4-desmopressin could rescue the function of the N321K-V2R without significant side effects on the V1R.
Members of the G protein–coupled receptor (GPCR) superfamily are a major group of cell surface receptors, which recognize hormones, neurotransmitters, and sensory information; thus, they play essential roles in physiological processes (1). In addition to their physiological importance, the pathological significance of GPCRs cannot be emphasized enough because at least 40% of modern therapeutic drugs target directly or indirectly these receptors and their signaling (2). Mutations of GPCRs are responsible for numerous human diseases and more than 600 loss-of-function mutations of GPCRs have been identified (3). Investigation of these mutations helps to reveal the functions and structures of the different GPCRs; moreover, it may provide clues to find drugs targeting receptors (4). One of the most extensively investigated receptors regarding inactivating mutations is the type 2 vasopressin receptor (V2R). Loss-of-function mutations of V2R can cause nephrogenic diabetes insipidus (NDI) with different mechanisms (5, 6).
V2Rs are localized to the basolateral plasma membrane of the principal cells in the kidney collecting ducts and have essential roles in mediating the water-conserving effect of arginine vasopressin (AVP). AVP is secreted from the neurohypophysis in response to increased plasma osmolality, and its effect on water reabsorption is mediated by V2R. Binding of AVP to the V2R, which is a Gs-coupled receptor, leads to cAMP-mediated translocation of aquaporin-2 to the apical plasma membrane. This regulation of aquaporin-2 water channel localization is crucial for increasing urine osmolality and reducing urine output in humans (7). Ligand binding of GPCRs also stimulates mechanisms that can lead to termination of signaling. Impairment of this process can cause diseases as well (8, 9). Desensitization and internalization of GPCRs are regulated by GPCR kinases and β-arrestins (10, 11). Binding of β-arrestin to the desensitized receptor is followed by the internalization, which decreases the amount of receptors in the plasma membrane. The balance between the internalization, degradation, synthesis, and recycling determinates essentially the hormone sensitivity of a tissue (12).
Diabetes insipidus (DI) is a syndrome characterized by polyuria, hyposthenuria, and polydipsia. NDI is caused by the impaired effect of AVP in the kidney, although the hormone secretion is normal. Almost 90% of cases of NDI are caused by loss-of-function mutations of the V2R. More than 200 mutations have been identified worldwide, many of them are missense mutations, which act by different mechanisms (13). Thus, mutations can be classified into several groups based on their consequences (14). Class I mutations of the AVPR2 gene lead to impaired transcription, mRNA processing, or translation of the receptor, resulting in truncated and rapidly degraded proteins. Class II mutations lead to the formation of misfolded full-length proteins, which are recognized by the quality control system of the endoplasmic reticulum (ER) and result in ER retention (15). Thus, class I and II mutations lead to hormone insensitivity because of the decreased number of cell surface receptors. Class III mutants are another group of missense mutations, which interfere with either G protein coupling or AVP binding, leading to inappropriate signal transduction without affecting the cell surface expression of the receptors. Class IV mutants have normal ligand binding, but their intracellular trafficking is altered, causing impaired cAMP signal production mostly due to constitutive β-arrestin–dependent internalization into endosomal vesicles (16).
Identification of the altered properties of mutated receptors could help to define therapeutic strategies for the treatment of patients with NDI. Possible therapeutic mechanisms include direct stimulation of signal generation, bypassing the receptor (14), and different strategies to rescue the receptor function. The most extensively investigated V2R mutants belong to the class II mutations, which cause ER retention. Pharmacological chaperones are chemical ligands, which facilitate the folding of receptors in the ER and rescue them from ER retention. This mechanism leads to increased plasma membrane expression of otherwise functional receptors. Pharmacological chaperones of the V2R can be antagonists (17–21) or agonists (22, 23).
In this study, we have identified the N321K mutation of the V2R in a patient with NDI by genomic DNA (gDNA) sequencing. Although this mutation was reported previously, the mechanism of its pathogenic effect has not been identified (6). Here we characterize the pharmacological and functional properties of this mutant receptor and based on these findings we propose a new therapeutic strategy for patients carrying this mutation.
Materials and Methods
Materials
Molecular biology enzymes were obtained from Fermentas, Stratagene, and Invitrogen. Cell culture dishes and plates for bioluminescence resonance energy transfer (BRET) measurements were purchased from Greiner. Lipofectamine 2000 and coelenterazine h were from Invitrogen. The anti-hemagglutinin (HA)-Alexa 488 mouse monoclonal antibody was purchased from Life Technologies. Unless otherwise stated, all other chemicals and reagents were purchased from Sigma-Aldrich. The human embryonic kidney (HEK293) cells were from ATCC.
Mutation analysis
Written informed consent was obtained from a male patient with NDI. gDNA was extracted from peripheral blood leukocytes using a DNA isolation kit (Boehringer Mannheim Corporation). The AVPR2 gene was amplified with PCR in fractions using the forward primer 5′-ATCACCTCCAGGCCCTCAGA-3′ and reverse primer 5′-ATGGGACGGCAGATGGCAC-3′, as well as the forward primer 5′-TGATCCTGGCCATGACGCTG-3′ and reverse primer 5′-AGAGGCAAGACACCCAACAGC-3′. The sizes of the PCR products were determined in agarose gel. The PCR products were purified for DNA sequencing and sequenced in both directions.
Molecular biology
The cDNA of the human arginine vasopressin receptor 2 (clone identification no.: AVR0200000; GenBank accession no: AY242131) was purchased from S&T cDNA Resource Center. The untagged and the HA-tagged V2Rs were subcloned into pcDNA3.1. For the construction of the super Renilla luciferase (Sluc)–tagged V2R, the receptor sequence was amplified from the cDNA clone and subcloned into a pEYFP-N1 vector (Clontech) containing the sequence of Sluc (24). To create the mVenus-tagged V2R, the amplified receptor was subcloned into a pEYFP-N1 vector containing the sequence of mVenus. Venus contained an A206K mutation holding the protein in monomeric form (25). β-Arrestin2-Renilla luciferase (Rluc) was constructed as described previously (26). The β-arrestin2 was subcloned into a pEYFP vector with replacement of enhanced yellow fluorescent protein (YFP) with humanized Renilla luciferase (Promega). The generation of the myristoylated and palmitoylated (MP)-YFP was described previously (27). Mutagenesis was performed using standard site-directed mutagenesis techniques to generate N321K receptor constructs. After verifying the mutations with dideoxy sequencing, the mutated fragment was exchanged between the wild-type (WT) and mutated portion with suitable restriction sites to avoid the generation of unwanted mutations outside the sequenced regions. The Epac-BRET sensor was based on the TEPACVV construct developed and kindly provided by Dr Kees Jalink (28). For the construction of the Epac-BRET sensor, the mTurquoise part of the TEPACVV was replaced with Sluc. The Sluc sequence was amplified with PCR and was subcloned into TEPACVV.
Cell culture and transfection
Cell culture and transfection protocols were described previously (27). The DNA amounts were 0.25 μg of receptor-containing construct/well and 0.25 μg of BRET partner–containing construct/well; the amount of Lipofectamine 2000 was 0.5 μl/well.
BRET measurements
BRET measurements were performed as described previously (27). We used either a Renilla luciferase-fused receptor as the energy donor and an enhanced YFP-tagged protein as the acceptor or we used an intramolecular BRET probe (cAMP-measuring Epac-BRET). Dose-response sigmoidal curves were generated using nonlinear regression. The statistical analysis was performed by two-way ANOVA and one-way ANOVA with a Tukey multiple comparison test.
Confocal microscopy
The cells plated on polylysine-pretreated glass coverslips (3 × 105 cells/35-mm dish) and were transiently transfected with the HA-tagged receptor constructs (2 μg of receptor/well; the amount of Lipofectamine 2000 was 4 μl/well.). After 24 hours, the cells were washed with PBS and were fixed with 4% paraformaldehyde solution for 10 minutes. Cells were washed 3 times with 10% fetal bovine serum–containing PBS solution. For immunostaining, the anti-HA-Alexa 488 mouse monoclonal antibodies were diluted to 1:250 in the presence or absence of 1% saponin, and the cells were incubated for 1 hour. After being washed with 10% fetal bovine serum–PBS, the coverslips were mounted on slides. The localization and distribution of the targeted probes were analyzed using a Zeiss LSM 510 confocal laser scanning microscope.
Flow cytometry
The cells plated on glass coverslips (3 × 105 cells/35-mm dish) were transiently transfected with the HA-tagged receptor constructs or pcDNA3.1 (2 μg of DNA/well; the amount of Lipofectamine 2000 was 4 μl/well). The cells were deattached by Versene reagent treatment and were centrifuged. The cells were suspended in ice-cold PBS and were centrifuged on 4°C. The cell pellets were suspended and incubated with diluted (1:100) anti-HA-Alexa 488 mouse monoclonal antibodies for 40 minutes on 4°C. After the labeling period, the cells were washed in ice-cold PBS. Flow cytometry measurements were performed with Beckman-Coulter SC. After the fluorescent intensity of the cells was measured, Gmean was calculated using WinMDI v2.9 (http://facs.scripps.edu/software.html). For the relative fluorescent intensity, the background (pcDNA3.1) was subtracted, and the data were normalized for the WT receptor. Statistical analysis was performed by two-way ANOVA.
Wire myography
Thoracic aortas from rats were removed and placed into cold Krebs' solution containing 119 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2 · 2H2O, 1.17 mM MgSO4 · 7H2O, 20 mM NaHCO3, 1.18 KH2PO4, 0.027 EDTA, and 10.5 glucose. Aortic rings were mounted onto a multichannel isometric myograph system (Danish Myo Technology). The thermostated (37°C) organ chambers of the myographs were filled with Krebs' solution, which was bubbled with carbogen gas (5% CO2 and 95% O2). The resting tension of aortic rings was set to 10 mN and allowed to equilibrate for 30 minutes. The integrity and functionality of the aortic rings were tested by124 mM K+-containing Krebs' solution (constriction) and after several washing cycles and a waiting period by 10 μM acetylcholine (vasodilation). Recording was performed with the PowerLab data acquisition system and the LabChart evaluation program (ADInstruments). Vasoconstrictor responses were calculated as percent values of the reference 1 μM phenylephrine-caused precontraction. Concentration-dependent vasoconstrictor response curves to agonists were obtained using parallel segments.
Results
The male patient was born in 1984 with polyuria and polydipsia, and NDI was diagnosed at the age of 18 months because desmopressin (dDAVP), a vasopressin analog, was ineffective during the early water deprivation test. Currently, his water consumption is approximately 12 L/d. Thiazide and amiloride diuretics were ineffective, and the water intake remained unchanged. Clinical laboratory tests of the patient revealed the following parameters (reference ranges shown in parentheses): serum sodium, 145 mmol/L (136–146 mmol/L); serum potassium, 4.3 mmol/L (3.5–5.0 mmol/L); and serum osmolality, 282 mOsm/kg (without any medication, at his usual daily water intake, 275–295 mOsm/kg). The urine specific gravity was 1003 g/cm3 (1002–1030 g/cm3), and the urine osmolality was 72 mOsm/kg (50–1200 mOsm/kg, depending on fluid intake).
The AVPR2 gene was amplified with PCR of the genomic DNA isolated from the peripheral blood of the patient (see details under Materials and Methods), and the mutation was identified by DNA sequencing. A missense mutation was found in the patient (Figure 1A), and this C → G substitution results in an asparagine lysine change (N321K) in the seventh transmembrane domain of the V2R (7). No other mutations in the AVPR2 gene were found. The family anamnesis of the patient suggests that the N321K substitution is not a de novo mutation, because symptoms of DI were present in at least 3 generations of his family (Figure 1B). However, it was not possible to achieve sequencing of the AVPR2 gene of other family members. According to the records, the grandmother's father had polydipsia and died at 82 years of age. The grandmother also had polydipsia and had a daily water consumption of 6 to 8 L. Her 11-month-old child died of exsiccosis. The mother and sister of the patient are healthy.
Figure 1.
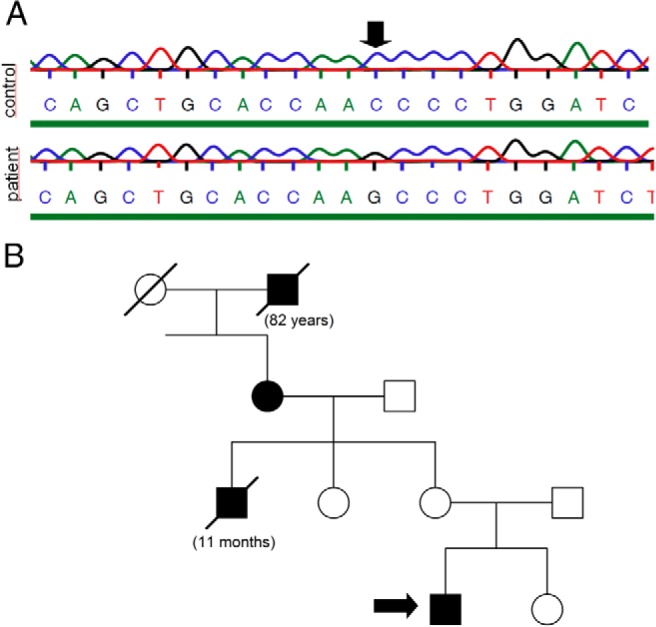
The results of DNA sequencing and the familiar anamnesis. A, gDNA was isolated from peripheral blood. After PCR amplification, the AVPR2 gene was sequenced. The chromatogram shows the results of the sequencing in the patient and in a healthy control subject. B, Familiar anamnesis of the patient. The filled mark indicates polydipsia-polyuria syndrome in the male (squares) and female (circles) members of the family. The scored marks indicate deceased family members.
We expressed HA-tagged WT or N321K mutant V2R transiently in HEK293 cells to examine the cellular localization of the receptors. Immunofluorescent staining of the receptor was performed using anti-HA antibodies tagged with Alexa 488 both in permeabilized and nonpermeabilized cells. Confocal microscopy revealed that the mutant N321K-V2R is localized in the plasma membrane of the transfected cells very similarly to that in the wt receptor (Figure 2, A and B). Immunostaining of permeabilized cells expressing either WT or mutant receptors showed marked intracellular fluorescence. The mock-transfected cells with empty pcDNA3.1 did not show any fluorescent staining (data not shown). Taken together, the mutant receptors showed cellular distribution that was very similar to that of the WT receptors. These data show that the N321K-V2R can reach the plasma membrane of the cells (Figure 2, A−D). Theoretically, the fusion of the mutant receptor with a tag (HA or fluorescent protein) could alter the trafficking; therefore, we used another approach to determine the localization of the receptors. To confirm the plasma membrane localization of the mutant receptor, we have also used HEK293 cells transiently expressing fluorescently tagged receptors. The mVenus tagged WT-V2R and N321K-V2R had similar cellular distribution in living cells as assessed by confocal microscopy (data not shown). We also compared the quantity of the expressed receptors on the surface of transfected cells performing flow cytometry measurements. HA-tagged WT-V2R and N321K-V2R were transiently transfected in HEK293 cells and labeled with anti-HA-Alexa 488 antibodies as described in the Materials and Methods. We did not detect any significant difference in the relative fluorescence intensities (RFIs) of the WT (RFI, 1.0) and N321K receptors (RFI, 0.954 ± 0.05; n = 3, P > .05) on the cell surface, indicating that the plasma membrane expression of the mutant receptor is similar to that of the WT receptor (data not shown).
Figure 2.

Examination of the cell surface expression of the N321K-V2R. Immunofluorescence microscopy analysis of HEK293 cells transiently expressing WT (A and C) or N321K (B and D) HA-tagged V2R. The samples were stained with anti-HA-Alexa 488 mouse monoclonal antibodies under permeabilized (C and D) and nonpermeabilized (A and B) conditions. Scale bars corresponds to 10 μm.
Because these data showed no evidence of the ER retention of transiently expressed N321K mutant receptors, the function of this mutant receptor was also evaluated. Because V2R is coupled to Gs, we used the BRET technique to monitor cAMP generation in living HEK293 cells upon agonist stimulation. The HEK293 cells were transiently transfected with the Epac-BRET sensor and with either WT-V2R or N321K-V2R constructs. The experiments were performed 24 hours after the transfection. The Epac-BRET probe reports when the Epac domain is loaded with cAMP, causing conformational changes that move the energy acceptor away from the donor, as was shown in a previous report that presented the corresponding fluorescence resonance energy transfer probes (28). Consequently, an increase in the intracellular cAMP level results in a decreased BRET ratio in our measurements. Figure 3A shows the real-time evaluation of cAMP levels in living cells expressing either WT-V2R or N321K-V2R. Cells were stimulated at the indicated time with 10 nM AVP (WT receptor, ■) or with 1 μM AVP (mutant receptor, ▴). It is noteworthy that the basal (before stimulus) BRET ratio is higher in the cells expressing the N321K-V2R than in the cells expressing the WT receptor, indicating that the basal cAMP concentration in the N321K-V2R–expressing cells is lower than that of the WT-V2R–expressing cells. Basal cAMP production of WT-V2Rs was already reported in COS7 cells, and it could be blocked with antagonists (29). In agreement with these data, the WT-V2R also possesses basal activity in HEK293 cells, whereas the N321K-V2R lacks constitutive activity in our expression system. Although the mutant receptor was able to stimulate cAMP production upon AVP stimulus with amplitude very similar to that of the WT receptor, the kinetics of the activation was different. The cAMP production was sustained in the cells expressing the WT-V2R, whereas it was apparently more transient in cells expressing the N321K-V2R mutant receptor (Figure 3A). We also determined the dose-response curve of the mutant and the WT receptor upon an AVP stimulus (Figure 3B). The effect of the hormone on the WT-V2R- and N321K-V2R-expressing HEK293 cells was calculated as the BRET ratio difference between the ligand and the vehicle-treated cells at the first time points after the treatment. The maximal BRET changes were similar for both receptors, but the potency of the N321K-V2R is dramatically decreased compared with that for the WT. The pEC50 of AVP for the WT receptor was 10.46 ± 0.04 M, whereas that for the mutant receptor was 6.49 ± 0.07 M.
Figure 3.

Measurement of the cAMP signal upon AVP and dDAVP stimuli. HEK293 cells were transiently transfected with the WT-V2R or the N321K-V2R and the Epac-BRET sensor. After 24 hours, the BRET measurements were implemented. A, Cells were stimulated with 10 nM AVP for the WT receptor (■) and with 1 μM AVP for the mutant receptor (▴) at the indicated time. B and C, Dose-response curve of AVP (B) and dDAVP (C). The effect of the hormone on the WT-V2R– and N321K-V2R–expressing HEK293 cells was calculated as the BRET ratio difference between the ligand (stim)–treated and the vehicle (nstim)–treated cells at the first time points after the treatment. Mean values ± SE are shown (n = 3).
The effect of the AVP analog dDAVP is fundamental in both the diagnosis and the treatment of DI. Therefore, we investigated the effect of dDAVP on cAMP production in HEK293 cells expressing WT-V2R and N321K-V2R (Figure 3C). According to our data, the dDAVP has a pEC50 of 9.23 ± 0.07 M for WT-V2R, whereas for the N321K mutant receptor, we could not measure detectable cAMP production upon dDAVP stimulation. The inefficiency of dDAVP on N321K-V2R cells is consistent with the clinical data of the patient who carries this mutation.
Next we examined the internalization properties of the mutant receptor compared with those of the WT receptor. First, we investigated the β-arrestin2 binding of the receptors using the BRET technique in living cells. Association of the receptor with β-arrestin2 was detected as the BRET signal elevation after the AVP stimulus indicating the interaction of mVenus-tagged WT-V2R with β-arrestin2-Rluc. Based on the kinetics of the binding, the WT-V2R is apparently a class B GPCR (Figure 4A) (30). Figure 4B shows the dose-response curves of β-arrestin2 binding to the receptors after 380 seconds of stimulation. The pEC50 of the β-arrestin2 binding dose-response curve to the WT receptor was 8.03 ± 0.005 M. Interestingly, we were not able to detect β-arrestin2 binding for the N321K-V2R even at high, supraphysiological levels of AVP (Figure 4B, ▴). These data prompted us to examine the internalization kinetics of the receptors. In this set of experiments, the receptors were tagged with a bioluminescence donor Renilla luciferase (V2R-Sluc), whereas the energy acceptor YFP was targeted to plasma membrane by fusing a small tag containing consensus sequences for myristoylation and palmitoylation (MP-YFP) (27, 31). The BRET ratio monitored the nonspecific resonance energy transfer, which is dependent on the distance between the donor and acceptor. As Figure 4C shows, stimulation of the WT V2R with 1 μM AVP (■) decreased the BRET ratio, reflecting the altered localization of the energy donor and the acceptor, which indicates the internalization of the cell surface–localized receptors into the endosomal compartments (31). The reduction in the BRET ratio between MP-YFP and N321K-V2R-Sluc upon stimulation with 1 μM AVP (▴) was smaller than that for WT-V2R-Sluc, suggesting that the internalization of N321K-V2R is reduced compared with that for the WT receptor.
Figure 4.

Examination of the internalization and β-arrestin-binding properties of N321K-V2R. HEK293 cells were transiently transfected with the plasmids of the indicated BRET partners and after 24 hours, the cells were exposed to AVP or vehicle (veh). A and B, β-Arrestin binding was measured with the transfection of WT- or N321K-V2R-mVenus and β-arrestin-Rluc plasmids. A, Cells were exposed either to 1 μM AVP (■) or vehicle (dashed line) at the indicated time points. B, Dose-response curve of β-arrestin-binding of AVP. The effect of the hormone on cells was calculated as the BRET ratio difference between the ligand (stim)–treated and the vehicle (nstim)–treated cells. C, Internalization kinetics was measured with the transfection of WT- or N321K-V2R-Sluc and MP-YFP plasmids. Cells were exposed to either 1 μM AVP or vehicle at the indicated time points. Mean values ± SE are shown (n = 3).
Theoretically, the functional impairment of the N321K-V2R mutant receptor can be repaired with an agonist that activates the receptor and has a proper potency in cAMP production. We tested several commercially available peptides, which are known ligands of the V2R. Our aim was to find a ligand that activates the mutant receptor–initiated cAMP generation and has a high V2R selectivity over that for the type 1 vasopressin receptor (V1R) to avoid the potential side effects in an in vivo system. Here, we present the results of cAMP measurements using a selective V2R agonist Val4-desmopressin (dVDAVP) (32), a V2R agonist but V1R antagonist deamino-Pen1,Val4-desmopressin (PVDAVP) (33), Lys8-vasopressin (LVP), and Asu1,6-arginine vasopressin (AsuAVP) (34). The dose-response curves were measured in transiently transfected HEK293 cells using our Epac-based cAMP-sensitive BRET probe (Figure 5). Table 1 shows the calculated EC50 values of the various peptides. The efficacy values of the dVDAVP, LVP, and AsuAVP peptides were similar to those of AVP (and dDAVP) after stimulation of the WT receptor expressed in HEK293 cells (Figure 5, A, C, and D, ■). As was expected, these peptides had dramatically decreased potency in N321K-V2R–expressing cells (Figure 5, A, B and D, ▴). For PVDAVP, the potency is slightly lower than the potencies of AVP, dVDAVP, LVP, and AsuAVP when the WT receptor was used (Figure 5B, ■); however, PVDAVP did not cause detectable cAMP production of the mutant receptor (Figure 5B, ▴). Of the peptides tested, the agonist dVDAVP had the highest potency (pEC50 = 6.3 ± 0.19 M) to activate the mutant receptor, which was comparable to the potency of AVP (6.492 ± 0.07 M) to stimulate the cAMP production of this receptor. This finding raised the possibility that dVDAVP can be used to rescue the function of N321K-V2R. Because the vasoconstrictor side effect of AVP analogs is a concern during the treatment of NDI, we have tested the effect of dVDAVP on V1R-initiated vasoconstriction of isolated mouse arteries by wire myography vessels. Figure 6 shows that increasing concentrations of AVP caused vasoconstriction through vascular smooth muscle cells, whereas even a 10−5 M concentration of dVDAVP was not able to evoke this effect.
Figure 5.

Measurement of the cAMP signal upon different agonist stimuli. HEK293 cells were transiently transfected with the WT-V2R or the N321K-V2R and the Epac-BRET sensor. After 24 hours, the BRET measurements were implemented. A–D, Dose-response curves of dVDAVP (A), PVDAVP (B), LVP (C), and AsuAVP (D). The effect of the hormones on the WT-V2R- and N321K-V2R-expressing HEK293 cells were calculated as the BRET ratio difference between the ligand (stim)-treated and the vehicle (nstim)-treated cells at the first time points after the treatment. Mean values ± SE are shown (n = 3).
Table 1.
Calculated pEC50 Values for the Various Peptides
| pEC50, M |
||
|---|---|---|
| WT | N321K | |
| AVP | 10.46 ± 0.04 | 6.492 ± 0.07 |
| dDAVP | 9.229 ± 0.07 | |
| dVDAVP | 10.02 ± 0.11 | 6.30 ± 0.05 |
| AsuAVP | 10.56 ± 0.04 | 5.94 ± 0.02 |
| LVP | 10.08 ± 0.02 | 5.52 ± 0.13 |
| PVDAVP | 8.75 ± 0.01 | |
Figure 6.

Effects of AVP and dVDAVP on vasoconstriction of mouse arterioles. Isolated mouse arterioles were exposed to increasing concentrations of AVP (■) or dVDAVP (▴). The values of the vasoconstrictor responses were calculated as percent values of the reference 1 μM phenylephrine-caused precontraction. The values are averages of 3 independent experiments. Mean values ± SEM are shown (n = 3).
Discussion
In this study we have characterized an N321K missense V2R mutation, which was identified from a Caucasian male patient with NDI. This mutation had been found previously in another patient, but the cellular consequences of the mutation were not examined before (6). The water deprivation test was carried out in childhood and clearly confirmed the diagnosis of DI; moreover, the administration of dDAVP did not have any effect on urine concentration. The familiar anamnesis strongly suggested a genetically inherited mutation, because the symptoms of DI were presented for at least 4 generations, which raised the possibility of an X-linked NDI. Sequencing of the gDNA demonstrated a C → G substitution in the AVPR2 gene, which results in an asparagine-lysine change at position 321 of the V2R. This asparagine is in the NPXXY motif, which is a conserved sequence in G protein–coupled receptors and is assumed to have a role in ligand binding, G protein–coupling, and internalization of the β-adrenergic receptor (35). Mutation of this asparagine residue in the type I angiotensin receptor (AT1aR) causes markedly reduced G protein interaction and generation of second messengers but has no effect on the internalization kinetics of the receptor (36). It was clearly shown that mutation of proline 322 in V2R leads to impaired coupling to Gs protein (4). According to our data, the plasma membrane expression of the N321K-V2R was similar to that of the WT receptor in transient expression systems (Figure 2). This result suggested that the mutant receptor is delivered to the cell surface, and an ER retention problem is not responsible for the phenotype of this patient. On the other hand, the stimulation of the mutant receptor with AVP revealed markedly decreased potency and unchanged efficacy in cAMP production compared with that for the WT receptor (Figure 3B). Interestingly, we have found that the basal activity of the N321K-V2R was also decreased compared with that of the WT-V2R (Figure 2A). However, the basal BRET ratio values (before stimulus) of the unstimulated N321K-V2R in cAMP measurements are identical to the BRET ratios of the WT receptor under maximal inhibition with high-dose antagonists (data not shown). Taken together, our results indicate that the mutant receptor has reduced second messenger formation capability in both the absence and presence of agonists, suggesting that the N321K mutation causes impaired G protein coupling. However, it is also important that the classification of a receptor mutation is not always unambiguous. NDI causing R137H-V2R was shown to belong to class IV because of the constitutive β-arrestin-dependent internalization of the receptors (16). Moreover, R137C-V2R and R137L-V2R mutations lead to nephrogenic syndrome of inappropriate dieresis due to the constitutive activity and β-arrestin-dependent internalization. The R137H-V2R also has impaired G protein coupling (37) and as was more recently shown, this mutant has altered trafficking to the plasma membrane as well (38). These results raise the possibility that one mutation can cause multiple effects on receptor function.
The hormone sensitivity of a tissue is also dependent on the internalization processes of receptors, which affect the receptor amount in the plasma membrane of the cells. We characterized and evaluated the internalization properties of the WT-V2R and N321K-V2R in HEK293 cells using the BRET technique to measure the β-arrestin2 binding of the stimulated receptors. The β-arrestin2 binding dose-response curve of the WT-V2R was right shifted compared with the cAMP dose-response curve, which reflects the presence of spare receptors in the plasma membrane and the enhancement of the generation of second messengers (39, 40). In contrary to the WT receptor, we could not detect β-arrestin2 binding of the N321K-V2R; therefore, we also examined the kinetics of the internalization properties of the receptors. We used the BRET-based approach, in which plasma membrane–targeted YFP served as an indicator of plasma membrane localization, and we measured the internalization of luciferase-tagged receptors from the cell surface upon stimulation (27). Although this method can also detect intramembrane movements of the receptor, we have used this method to monitor internalization of activated receptors, because internalization of the receptor leads to its divergence from the plasma membrane marker (31). The results showed that the N321K-V2R had markedly reduced internalization compared with that for the WT receptor. It is possible that the remaining internalization is the consequence of β-arrestin-independent processes. It is also interesting that, in contrast to the angiotensin receptor (35), a mutation in the NPXXY motif in the V2R leads to impaired β-arrestin binding and internalization. It is also notable that the reduced internalization does not necessarily mean continuous signaling of the receptor from the plasma membrane: the transient kinetics of the cAMP signal of the N321K-V2R suggests that this mutation has no major effects on the desensitization processes (Figure 2A). Because β-arrestin–mediated uncoupling of the GPCR from G proteins is not the only possible mechanism of desensitization, ie, phosphorylation of the receptor by messenger dependent kinases also can lead to termination of the receptor activation (41).
The clinical diagnosis of the NDI is also based on functional tests. The widely used tests are the water deprivation test and administration of dDAVP (42). Because dDAVP is an essential compound not only in the diagnostic procedures of NDI but also in the therapy of DI, we examined the effect of dDAVP on the mutant N321K receptor. As we mentioned before, we could not detect any cAMP signal upon dDAVP stimulus (Figure 3). These results were consistent with the clinical findings: administration of dDAVP in childhood did not produce any improvement in the observed parameters. The dose-response curve of the WT receptor showed that the potency of dDAVP was decreased compared with that of AVP. It was already known that although dDAVP is a V2R-specific agonist, it had a lower affinity to the receptor than AVP (43). Apparently, the N321K mutation induces a conformational change of the V2R, resulting in decreased affinity and/or G protein coupling, which led to lack of dDAVP effects on cAMP generation capability at the concentrations tested.
Theoretically, an altered receptor conformation can result in decreased potency for a certain agonist, but the potency of other agonists can be affected differently. An agonist, which could activate the mutant receptor despite the conformational change, can be the causal therapy in the case of a mutation such as N321K. To find such a compound, we tested several agonists with high affinity to the V2R (Figure 5 and Table 1). Statistical analysis showed that the effects of the agonists on the pEC50 values using a two-way ANOVA were significant (P < .0001). The WT receptor had similar potencies for LVP, dVDAVP, and AsuAVP. PVDAVP had the lowest EC50 for WT-V2R, and we could not detect any cAMP signal with the N321K-V2R, although this peptide was thought to be beneficial as a potential therapy because of its V1R antagonistic effect (33). dVDAVP had the highest potency on the N321K-V2R; moreover, the N321K mutation had a reduced effect for this compound: the difference between the mutant and the WT receptor in the potency was 1 order of magnitude less than that with other agonist (the ΔpEC50 value for dVDAVP was 3.71 M and values for AsuAVP and LVP were 4.62 and 4.56 M, respectively). With use of a one-way ANOVA statistical analysis with the Tukey multiple comparison test, the differences between the ΔpEC50 values of dVDAVP and the other agonists (AsuAVP and LVP) were significant (P < .005). The consequence of the N321K missense mutation is misfolding, resulting in a conformation with altered agonist sensitivity, which is more suitable for stimulation by dVDAVP than for other agonists. Fortunately, as was demonstrated earlier, the most effective AVP analog among the tested compounds, dVDAVP, is a selective V2R agonist (32). A potential high-dose agonist treatment could be limited because of the cross-reaction of the compound on the vascular V1Rs. The V1aR is expressed in various tissues such as in the walls of vascular vessels (44). Although the physiological concentration of AVP is lower than the concentration that exerts vasoconstriction, a high dose of vasopressin receptor agonist in the treatment of DI could provoke side effects (vasoconstriction and blood pressure elevation) through this system. The consequence of the agonist activity on V1R could be hypertension and according to studies in septic shock, decreased perfusion of the heart, kidney, and intestines (45). In this present study, we examined whether the high dose of dVDAVP, which is able to generate the cAMP signal through the activation of N321K-V2R could promote vasoconstriction in peripheral arterioles. As Figure 6 shows, we could not detect any constriction even at a 10 μM final dVDAVP concentration. This result suggests that an appropriate (moderately high) dose treatment could have beneficial effects on the NDI symptoms, including polyuria and polydipsia without the unsafe side effect of vasoconstriction.
Although dVDAVP did not become an alternative drug for dDAVP in the clinical therapy of NDI over the years, there are several studies in the literature about the clinical use of dVDAVP. Czakó et al (46) showed in a clinical trial not only that dVDAVP was more effective than dDAVP in patients with central DI but also that it had a short-term, moderate antidiuretic effect in patients with “ADH-resistant diabetes insipidus.” dVDAVP had a 3 times longer antidiuretic effect in central DI than dDAVP after IV injection. It has also been shown that intranasal use of dVDAVP was as effective as dDAVP in these patients.
In this study, we demonstrated that the disease-causing N321K mutation of the V2R does not lead to ER retention, and the N321K-V2R is present in the plasma membrane of HEK293 cells. The mutant receptor has unchanged efficacy but dramatically decreased potency for AVP. The N321K-V2R mutation leads to impaired internalization, most likely due to the lack of β-arrestin binding upon stimulation with agonist concentrations, which generate a maximal cAMP signal. The N321K-V2R is biased among different ligands, as the misfolding and the conformational change due to the mutation causes different sensitivities of agonists. According to our data, the function of the mutant receptor can be rescued with administration of the V2R receptor agonist dVDAVP, which had no detectable side effects on V1R in the effective cAMP signal causing concentration. Our in vivo experiments propose the possibility that the appropriate dosage of dVDAVP can rescue the function of the N321K-V2Rin a patient with NDI who has no significant side effects on the V1R. Based on these findings, a therapeutic strategy can be formed for patients with the N321K mutation in the V2R.
Acknowledgments
The excellent technical assistance of Ilona Oláh, Judit Rácz, and Mártonné Schulcz is greatly appreciated.
This work was supported by the Hungarian Ministry of National Resources (grant OTKA 100883) and the National Development Agency, Hungary (Grant TÁMOP 4.2.1.B-09/1/KMR-2010-0001).
Disclosure Summary: The authors have nothing to disclose.
Funding Statement
This work was supported by the Hungarian Ministry of National Resources (grant OTKA 100883) and the National Development Agency, Hungary (Grant TÁMOP 4.2.1.B-09/1/KMR-2010-0001).
Footnotes
- AsuAVP
- Asu1,6-arginine vasopressin
- AVP
- arginine vasopressin
- BRET
- bioluminescence resonance energy transfer
- dDAVP
- desmopressin
- dVDAVP
- Val4-desmopressin
- ER
- endoplasmic reticulum
- gDNA
- genomic DNA
- GPCR
- G protein–coupled receptor
- HA
- hemagglutinin
- LVP
- Lys8-vasopressin
- MP
- myristoylated and palmitoylated
- NDI
- nephrogenic diabetes insipidus
- PVDAVP
- deamino-Pen1,Val4-desmopressin
- RFI
- relative fluorescence intensity
- Rluc
- Renilla luciferase
- Sluc
- super Renilla luciferase
- V1R
- type 1 vasopressin receptor
- V2R
- type 2 vasopressin receptor
- WT
- wild-type
- YFP
- yellow fluorescent protein.
References
- 1. Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. [DOI] [PubMed] [Google Scholar]
- 2. Ma P, Zemmel R. Value of novelty? Nat Rev Drug Discov. 2002;1:571–572. [DOI] [PubMed] [Google Scholar]
- 3. Schöneberg T, Schulz A, Biebermann H, Hermsdorf T, Römpler H, Sangkuhl K. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol Ther. 2004;104:173–206. [DOI] [PubMed] [Google Scholar]
- 4. Ala Y, Morin D, Mouillac B, et al. . Functional studies of twelve mutant V2 vasopressin receptors related to nephrogenic diabetes insipidus: molecular basis of a mild clinical phenotype. J Am Soc Nephrol. 1998;9:1861–1872. [DOI] [PubMed] [Google Scholar]
- 5. Tao YX. Inactivating mutations of G protein-coupled receptors and diseases: structure-function insights and therapeutic implications. Pharmacol Ther. 2006;111:949–973. [DOI] [PubMed] [Google Scholar]
- 6. Arthus MF, Lonergan M, Crumley MJ, et al. . Report of 33 novel AVPR2 mutations and analysis of 117 families with X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2000;11:1044–1054. [DOI] [PubMed] [Google Scholar]
- 7. Moeller HB, Rittig S, Fenton RA. Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr Rev. 2013;34:278–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ligeti E, Csepanyi-Komi R, Hunyady L. Physiological mechanisms of signal termination in biological systems. Acta Physiol (Oxf). 2012;204:469–478. [DOI] [PubMed] [Google Scholar]
- 9. Rochdi MD, Vargas GA, Carpentier E, et al. . Functional characterization of vasopressin type 2 receptor substitutions (R137H/C/L) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: implications for treatments. Mol Pharmacol. 2010;77:836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Böhm SK, Grady EF, Bunnett NW. Regulatory mechanisms that modulate signalling by G-protein-coupled receptors. Biochem J. 1997;322(Pt 1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luttrell LM, Lefkowitz RJ. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115(Pt 3):455–465. [DOI] [PubMed] [Google Scholar]
- 12. Gáborik Z, Hunyady L. Intracellular trafficking of hormone receptors. Trends Endocrinol Metab. 2004;15:286–293. [DOI] [PubMed] [Google Scholar]
- 13. Spanakis E, Milord E, Gragnoli C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance. J Cell Physiol. 2008;217:605–617. [DOI] [PubMed] [Google Scholar]
- 14. Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27:2183–2204. [DOI] [PubMed] [Google Scholar]
- 15. Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–F270. [DOI] [PubMed] [Google Scholar]
- 16. Barak LS, Oakley RH, Laporte SA, Caron MG. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc Natl Acad Sci USA. 2001;98:93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morello JP, Salahpour A, Lapèrriere A, et al. . Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robben JH, Sze M, Knoers NV, Deen PM. Functional rescue of vasopressin V2 receptor mutants in MDCK cells by pharmacochaperones: relevance to therapy of nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2007;292:F253–F260. [DOI] [PubMed] [Google Scholar]
- 19. Wüller S, Wiesner B, Löffler A, et al. . Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J Biol Chem. 2004;279:47254–47263. [DOI] [PubMed] [Google Scholar]
- 20. Bernier V, Lagacé M, Lonergan M, Arthus MF, Bichet DG, Bouvier M. Functional rescue of the constitutively internalized V2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059. Mol Endocrinol. 2004;18:2074–2084. [DOI] [PubMed] [Google Scholar]
- 21. Ranadive SA, Ersoy B, Favre H, Cheung CC, Rosenthal SM, Miller WL, Vaisse C. Identification, characterization and rescue of a novel vasopressin-2 receptor mutation causing nephrogenic diabetes insipidus. Clin Endocrinol (Oxf). 2009;71:388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robben JH, Kortenoeven ML, Sze M, et al. . Intracellular activation of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus by nonpeptide agonists. Proc Natl Acad Sci USA. 2009;106:12195–12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jean-Alphonse F, Perkovska S, Frantz MC, et al. . Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J Am Soc Nephrol. 2009;20:2190–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woo J, von Arnim AG. Mutational optimization of the coelenterazine-dependent luciferase from Renilla. Plant Methods. 2008;4:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. [DOI] [PubMed] [Google Scholar]
- 26. Turu G, Szidonya L, Gáborik Z, et al. . Differential β-arrestin binding of AT1 and AT2 angiotensin receptors. FEBS Lett. 2006;580:41–45. [DOI] [PubMed] [Google Scholar]
- 27. Balla A, Tóth DJ, Soltész-Katona E, et al. . Mapping of the localization of type 1 angiotensin receptor in membrane microdomains using bioluminescence resonance energy transfer-based sensors. J Biol Chem. 2012;287:9090–9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klarenbeek JB, Goedhart J, Hink MA, Gadella TW, Jalink K. A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS One. 2011;6:e19170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takahashi K, Makita N, Manaka K, et al. . V2 vasopressin receptor (V2R) mutations in partial nephrogenic diabetes insipidus highlight protean agonism of V2R antagonists. J Biol Chem. 2012;287:2099–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shenoy SK, Lefkowitz RJ. Trafficking patterns of β-arrestin and G protein-coupled receptors determined by the kinetics of β-arrestin deubiquitination. J Biol Chem. 2003;278:14498–14506. [DOI] [PubMed] [Google Scholar]
- 31. Toth DJ, Toth JT, Gulyas G, et al. . Acute depletion of plasma membrane phosphatidylinositol 4,5-bisphosphate impairs specific steps in endocytosis of the G-protein-coupled receptor. J Cell Sci. 2012;125(Pt 9):2185–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sawyer WH, Acosta M, Balaspiri L, Judd J, Manning M. Structural changes in the arginine vasopressin molecule that enhance antidiuretic activity and specificity. Endocrinology. 1974;94:1106–1115. [DOI] [PubMed] [Google Scholar]
- 33. Manning M, Lowbridge J, Stier CT Jr, Haldar J, Sawyer WH. [1-Deaminopenicillamine,4-valine]-8-d-arginine-vasopressin, a highly potent inhibitor of the vasopressor response to arginine-vasopressin. J Med Chem. 1977;20:1228–1230. [DOI] [PubMed] [Google Scholar]
- 34. Hase S, Sakakibara S, Wahrenburg M, Kirchberger M, Schwartz IL, Walter R. 1,6-Aminosuberic acid analogs of lysine- and arginine-vasopressin and -vasotocin. Synthesis and biological properties. J Am Chem Soc. 1972;94:3590–3600. [DOI] [PubMed] [Google Scholar]
- 35. Barak LS, Ménard L, Ferguson SS, Colapietro AM, Caron MG. The conserved seven-transmembrane sequence NP(X)2,3Y of the G-protein-coupled receptor superfamily regulates multiple properties of the β2-adrenergic receptor. Biochemistry. 1995;34:15407–15414. [DOI] [PubMed] [Google Scholar]
- 36. Hunyady L, Bor M, Baukal AJ, Balla T, Catt KJ. A conserved NPLFY sequence contributes to agonist binding and signal transduction but is not an internalization signal for the type 1 angiotensin II receptor. J Biol Chem. 1995;270:16602–16609. [DOI] [PubMed] [Google Scholar]
- 37. Rosenthal W, Antaramian A, Gilbert S, Birnbaumer M. Nephrogenic diabetes insipidus. A V2 vasopressin receptor unable to stimulate adenylyl cyclase. J Biol Chem. 1993;268:13030–13033. [PubMed] [Google Scholar]
- 38. Kocan M, See HB, Sampaio NG, Eidne KA, Feldman BJ, Pfleger KD. Agonist-independent interactions between β-arrestins and mutant vasopressin type II receptors associated with nephrogenic syndrome of inappropriate antidiuresis. Mol Endocrinol. 2009;23:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol. 2006;20:953–970. [DOI] [PubMed] [Google Scholar]
- 40. Ehnis T, Hocher B, Abou-Rebyeh H, Oelkers W, Hensen J. Expression of vasopressin receptors (V2-subtype) on LLC-PK1 cells during cell culture. Eur J Clin Chem Clin Biochem. 1993;31:273–276. [DOI] [PubMed] [Google Scholar]
- 41. Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 42. Bichet DG. TMF: Nephrogenic Diabetes Insipidus. In Scriver's Online Metabolic and Molecular Bases of Inherited Disease; 2007. http://www.ommbid.com. Accessed 2010.
- 43. Manning M, Misicka A, Olma A, et al. . Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J Neuroendocrinol. 2012;24:609–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koshimizu TA, Nakamura K, Egashira N, Hiroyama M, Nonoguchi H, Tanoue A. Vasopressin V1a and V1b receptors: from molecules to physiological systems. Physiol Rev. 2012;92:1813–1864. [DOI] [PubMed] [Google Scholar]
- 45. Russell JA, Walley KR, Singer J, et al. . Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358:877–887. [DOI] [PubMed] [Google Scholar]
- 46. Czakó L, László F, Manning M. Treatment of diabetes insipidus with synthetic vasopressin derivatives (effect of 1-deamino-8-d-arginine vasopressin and 1-deamino-valine-8-d-arginine vasopressin) [in Hungarian]. Orv Hetil. 1980;121:773–778. [PubMed] [Google Scholar]