

ABSTRACT
Combination of oncolytic virotherapy with immunomodulators is emerging as a promising therapeutic strategy for numerous tumor entities. In this study, we developed measles Schwarz vaccine strain vectors encoding immunomodulators to support different phases in the establishment of antitumor immune responses. Therapeutic efficacy of the novel vectors was evaluated in the immunocompetent MC38cea tumor model. We identified vectors encoding an IL-12 fusion protein (MeVac FmIL-12) and an antibody against PD-L1 (MeVac anti-PD-L1), respectively, as the most effective. Treatment of established tumors with MeVac FmIL-12 achieved 90% complete remissions. Profiling of the tumor immune microenvironment revealed activation of a type 1 T helper cell-directed response, with MeVac FmIL-12 ensuring potent early natural killer and effector T cell activation as well as upregulation of the effector cytokines IFN-γ and TNF-α. CD8+ T cells were found to be essential for the therapeutic efficacy of MeVac FmIL-12. Results of this study present MeVac FmIL-12 as a novel approach for targeted IL-12 delivery and elucidate mechanisms of successful immunovirotherapy.
KEYWORDS: Anti-PD-L1, cancer immunotherapy, interleukin-12, measles virus, Oncolytic viruses
Introduction
Intrinsic properties of cancer cells that are critical for continuous proliferation and tumor evolution likewise reduce protection against viral infections.1 This vulnerability is exploited therapeutically by application of oncolytic viruses (OV), which are currently gaining increasing attention as an attractive type of cancer treatment. Promising data for OV therapy are accumulating from numerous pre-clinical and clinical studies.2 Alongside the direct lysis of tumor cells determined by selective virus replication in malignant cells, activation of an antitumor immune response has been recognized as a critical determinant of OV efficacy.3 Thus, virotherapy can be considered a type of immunotherapy. Different OVs have been shown to induce immunogenic cell death,4-7 resulting in effective cross-presentation of tumor-associated antigens to T cells.8 Nevertheless, it is unlikely that a single OV or other immunotherapies administered as single agents will be sufficient to eradicate advanced malignancies. A variety of immunomodulation approaches have been explored to enhance the immunotherapeutic properties of different OVs.9 One of the best known immunomodulatory OV is Talimogene Laherparepvec (T-VEC), a herpes simplex virus encoding granulocyte macrophage colony-stimulating factor (GM-CSF)10 that was approved for treatment of advanced melanoma by the US Food and Drug Administration (FDA) in 2015.
Measles vaccine strain viruses (MeV) are a flexible platform to develop safe and effective oncolytic vectors.11 Clinical trials with MeV for treatment of different tumor entities, including multiple myeloma (NCT00450814), ovarian cancer (NCT02364713), hand and neck squamous cell carcinoma (NCT01846091) and glioblastoma multiforme (NCT00390299) are currently recruiting participants. In previous studies, we have demonstrated that immunomodulators, such as GM-CSF12 and antibodies against CTLA-4 and PD-L113 can improve MeV therapy in mouse tumor models. However, despite the promising pre-clinical data, understanding of the immunological determinants of efficacy and, therefore, the interplay of the oncolytic vector with a particular immunomodulator remains limited.
In this study, we developed measles Schwarz/Moraten vaccine strain (MeVac) viruses encoding immunomodulators to support different phases in the establishment of antitumor immune responses. We used MeVac vectors encoding GM-CSF14 to enhance antigen presenting cell maturation, the cytokine IL-1215 and the chemokine IP-10 (CXCL10)16 to activate and recruit effector cells, and antibodies against CTLA-4 and PD-L117 as well as a soluble form of the T cell costimulatory factor CD8018 to overcome immunosuppression within the tumor microenvironment. We identified MeVac encoding the IL-12 fusion protein (FmIL-12) and anti-PD-L1, respectively, as the most effective vectors in an immunocompetent murine colon adenocarcinoma model, MC38cea. We performed an analysis of therapy-induced immune effects to pinpoint determinants of successful therapy. We showed that MeVac FmIL-12 mediates potent early natural killer (NK) and T cell activation and production of IFN-γ and TNF-α. Depletion of immune cell subsets showed that CD8+ T cells are required for therapeutic efficacy of MeVac FmIL-12. Results of this study present MeVac encoding FmIL-12 as a potent immunomodulatory oncolytic MeV vector and give insights into activated antitumor immune effector mechanisms, creating a basis for further rational vector modifications.
Results
Targeted immunomodulatory oncolytic measles virus vectors
We constructed MeVac vectors encoding murine GM-CSF, IP-10, an IL-12 fusion protein (FmIL-12), antibodies against CTLA-4 and PD-L1, respectively, and a soluble form of murine CD80 (mCD80-Fc) in additional transcription units of the virus genome. Different positions were used for transgene insertion (Fig. 1A), avoiding insertion of large transgenes close to the MeV leader to limit virus attenuation. Vectors encoding enhanced green fluorescent protein (eGFP) in the leader position and an antibody constant region (IgG1-Fc) downstream of the MeV H open reading frame, respectively, were constructed as controls (Fig. 1A). Targeting of MC38cea cells19 was achieved using a single chain antibody (scAb) against CEA20 and the STAR system.21 The MeVac vector encoding eGFP and harboring the fully retargeted H protein (Hbl-αCEA) productively infected the producer Vero-αHis and target MC38cea cells, as indicated by syncytia formation, but not the parental Vero and MC38 cells, respectively (Fig. 1B), confirming specificity of the targeting.
Figure 1.

Cloning and characterization of recombinant measles virus vectors. (A) Schematic of recombinant measles Schwarz/Moraten vaccine strain (MeVac) genomes. T1—murine granulocyte macrophage colony-stimulating factor (mGM-CSF), murine IP-10 (mIP-10) or enhanced green fluorescent protein (eGFP); T2—murine IL-12 fusion protein (FmIL-12); T3—antibody against murine CTLA-4 or PD-L1 or a soluble form of murine CD80 (mCD80-Fc) or antibody constant region IgG1-Fc; Hbl-αCEA—MeVac H protein targeted to CEA; N, P, M, F, L—measles structural proteins; ld—measles leader; tr—measles trailer. (B) Targeted infection. Parental Vero and Vero-αHis expressing a single chain antibody against 6 histidine tag (His6) as well as parental MC38 and MC38cea cells expressing CEA were infected with MeVac encoding eGFP with H retargeted to CEA and including a C-terminal His6 tag (multiplicity of infection (MOI) = 1). Fluorescence microscopy pictures were taken 72 h post infection. Scale bars 100 µm. (C) Expression kinetics of MeVac-encoded FmIL-12. MC38cea cells were transduced with MeVac encoding FmIL-12 and eGFP as a control vector at MOI = 3. Supernatants were collected at the depicted time points and transgene expression was analyzed by ELISA. To control for unspecific binding values of MeVac eGFP supernatants were subtracted from the specific measurements. (D) Induction of IFN-γ production by MeVac-encoded FmIL-12. Murine splenocytes were stimulated with recombinant murine IL-2 and cultivated in the presence of medium from Vero-αHis cells infected with MeVac FmIL-12 or MeVac eGFP. After 48 h supernatants were collected and IFN-γ concentrations were measured by ELISA. Mean IFN-γ concentrations with standard errors of the mean of triplicate splenocyte cultures are shown for each FmIL-12 concentration. IFN-γ concentrations in the eGFP controls were close to background (data not shown). Representative data from one of two independent experiments are shown.
One-step growth curves were generated by transduction of MC38cea cells to characterize replication kinetics of the novel vectors. Titers for all vectors peaked between 36 h and 48 h post infection and declined afterwards (Fig. S1). It must be noted that MeV is adapted to primate cells.22 Accordingly, in one-step growth curves maximum titers in murine MC38cea cells were approximately one log10 lower than in primate Vero-αHis cells used for virus production (data not shown). For instance, MeVac encoding anti-CTLA-4 reached 4 × 105 ciu/mL in MC38cea and 2 × 106 ciu/mL in Vero-αHis cells in one-step growth curves. Of note, replication of MeVac GM-CSF was impaired, as it reached the lowest titers in the one-step growth curve and after several passages of propagation the concentration of virus particles in stocks never exceeded 5 × 106 ciu/mL. All vectors showed only mild cytotoxic effects in MC38cea cells, with the anti-PD-L1 encoding vector showing higher cytotoxicity than others. Cell viability started to increase 72 h after infection with all viruses (Fig. S2). These results reflect the limited replication and cytotoxicity of MeV in murine cells. The MC38cea model is, however, suited for studies of immunological aspects of MeV therapy.23 Expression of the immunomodulatory transgenes encoded by MeVac was assessed in supernatants of transduced MC38cea cells by ELISA at distinct time points after infection. Different patterns of expression kinetics were observed (Fig. 1C and Fig. S3a). Of note, different amounts of encoded immunomodulators were also present in virus suspensions (0 h). Expression of IgG1-Fc by the control vector was confirmed in supernatants from transduced cells by western blot (Fig. S3b). Notably, mIP-10 production was observed also in MC38cea transduced with MeVac eGFP (Fig. S3a) and untransduced MC38cea and MC38 (Fig. S4). Therefore, the MC38cea model was considered unsuitable for evaluation of mIP-10 in the context of MeV therapy. Further, functionality of MeVac-encoded immunomodulators was evaluated. Measles encoding GM-CSF has been studied previously.12 Functionality of MeVac-encoded mIP-10 was confirmed in a chemotaxis assay. Supernatants containing MeVac-encoded mIP-10 attracted more splenocytes than supernatants from cells infected with MeVac eGFP (data not shown). Functionality of MeVac-encoded anti-PD-L1, mCD80-Fc and anti-CTLA-4 was demonstrated by their ability to counteract MC38cea-mediated immunosuppression (Fig. S5). Functionality of MeVac-encoded FmIL-12 was assessed by induction of IFN-γ in murine splenocytes. At concentrations of 0.01–1 ng/mL, FmIL-12 led to a concentration-dependent increase in IFN-γ production, reflecting its immunostimulatory properties. In contrast, at high concentrations (10–1000 ng/mL) FmIL-12 led to a concentration-dependent decrease in IFN-γ production, which reflects overstimulation (Fig. 1D).
Immunomodulation enhances therapeutic efficacy of MeVac
All novel immunomodulatory MeVac constructs and selected combinations of the vectors were evaluated in the subcutaneous (s.c.) MC38cea model to identify the most effective therapeutic strategies as determined by tumor growth and survival (Fig. S6). These screening experiments identified MeVac anti-PD-L1 and MeVac FmIL-12 as the most promising vectors. Subsequently, therapeutic efficacy of MeVac FmIL-12 and MeVac anti-PD-L1 was directly compared in the MC38cea tumor model, including MeVac IgG1-Fc as a control. MeVac treatment led to a significant delay in tumor growth and prolonged survival in comparison to mock treatment, but no significant differences were observed between the virus treatment groups (Fig. 2A). However, the frequency of complete tumor remissions indicated differences in efficacy: A total of 4 out of 10 animals treated with MeVac IgG1-Fc experienced complete tumor remissions, in contrast to 6 and 9 out of 10 animals treated with MeVac anti-PD-L1 and MeVac FmIL-12, respectively (Fig. 2B). Efficacy of MeVac anti-PD-L1 was confirmed in two independent experiments. Therapeutic efficacy of MeVac FmIL-12 was validated in three independent experiments, reproducibly ensuring 88–90% complete tumor remissions. This suggests an added therapeutic benefit of anti-PD-L1 and especially FmIL-12 in the context of MeV therapy.
Figure 2.

Therapeutic efficacy of MeVac FmIL-12 and MeVac anti-PD-L1. (A and B) MC38cea cells were implanted subcutaneously (s.c.) into the right flank of C57BL/6J mice (10 animals per group). When tumors reached an average volume of 40 mm3 animals received intratumoral injections with 1 × 106 cell infectious units of MeVac encoding the respective transgenes in 100 µL or the respective amount of OptiMEM (mock) on four consecutive days. (A) Tumor volume distribution on day 16 post implantation. Dots representing individual mice and median values are shown. (B) Kaplan–Meier survival analysis. Complete tumor remission rates are shown for each group. (C–E) Systemic antitumor immunity in long-term survivors. Animals experiencing complete tumor remissions after MeVac treatment were rechallenged with s.c. MC38cea implantation 6 mo after initial tumor cell implantation. (C) Mice were monitored for tumor engraftment. Tumor rejection rates are shown. Splenocytes were collected from the rechallenged animals, stimulated with recombinant murine IL-2 and cocultivated with MC38cea cells (D) or with MC38 or B16 cells or DLD-1 cell lysate for one mouse from each group (E) at a ratio of 10:1. Supernatants were collected after 48 h and IFN-γ concentrations were measured by ELISA. Dots representing individual mice as well as medians are shown in d. Mean values with standard errors of the mean of two replicate measurements per sample are shown in e.
Protective antitumor immunity in long-term survivors of MeVac therapy
Animals experiencing complete tumor remissions were rechallenged with MC38cea cells 6 mo after the initial tumor cell implantation. All mice previously treated with MeVac anti-PD-L1 and MeVac FmIL-12 and 3 out of 4 of the animals previously treated with MeVac IgG1-Fc rejected secondary tumor engraftment, in contrast to naive control mice, where tumor engraftment was observed in all animals (Fig. 2C). This indicates establishment of a systemic antitumor immune response after MeVac treatment. Two weeks after tumor rechallenge, splenocytes from these animals were stimulated with MC38cea cells in vitro. A significantly higher IFN-γ production was observed in the MeVac FmIL-12 group compared to naive mice and the MeVac anti-PD-L1 group (Fig. 2D), suggesting a more potent cell-mediated immune memory in animals treated with MeVac FmIL-12. Stimulation with parental MC38 cells (Fig. 2E) induced a similar increase in IFN-γ concentration as stimulation with MC38cea (Fig. 2D), demonstrating that the recalled antitumor response is not CEA-restricted. Only a slight increase in IFN-γ concentration was detected after stimulation with unrelated murine B16 melanoma cells and IFN-γ concentration was close to background after stimulation with human DLD-1 cell lysate, showing that the observed response is specific for the MC38-derived tumor cells (Fig. 2E).
MeVac FmIL-12 induces a Th1-associated immune response
To identify immune effectors associated with the therapeutic efficacy of MeVac FmIL-12, we performed analyses of the immunological tumor milieu following virus treatment. Assuming that FmIL-12 exerts its specific effects rapidly after expression, tumor samples were collected 24 h after the last treatment. Reverse transcription quantitative PCR (RT-qPCR) analysis revealed an increase in mRNA for the transcription factor T-bet, which is associated with a T helper type 1 (Th1) response, in the MeVac FmIL-12 treatment group in comparison to controls (Fig. S7a). Levels of Foxp3 mRNA, associated with regulatory T cells, did not significantly differ between the treatment groups (Fig. S7b). This result suggests polarization toward a cell mediated antitumor immune response after treatment with MeVac FmIL-12.
MeVac FmIL-12 shapes intratumoral cytokine profiles
We also performed intratumoral cytokine profiling 24 h after the last treatment. MeVac FmIL-12 treatment led to a significant increase in IFN-γ and TNF-α concentration in comparison to both MeVac IgG1-Fc and mock treatment (Fig. 3A and B). Also a slight, but not significant increase in IL-6 concentration was observed in both the MeVac IgG1-Fc and MeVac FmIL-12 groups in comparison to mock (Fig. 3C). The cytokines IL-2, IL-4, IL-10 and IL-17 were close to background in all samples tested (data not shown).
Figure 3.
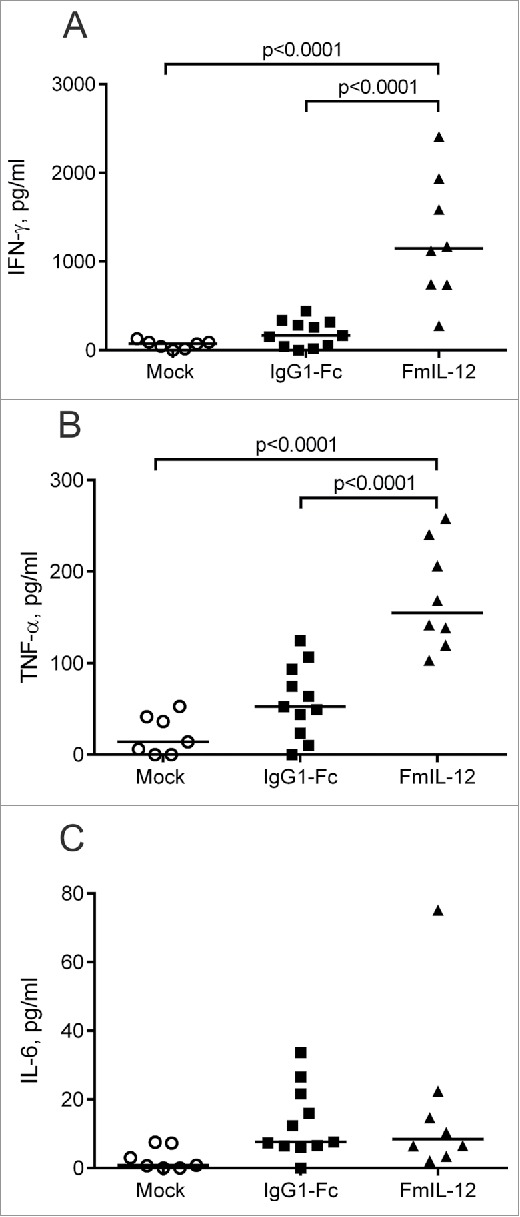
Intratumoral cytokine profiles after MeVac therapy. MC38cea tumor cells were implanted subcutaneously in C57BL/6J mice. When tumors reached an average volume of 120 mm3 animals received treatment with intratumoral injections of 1 × 106 cell infectious units of MeVac encoding FmIL-12 or IgG1-Fc in 100 µL or with the respective amount of OptiMEM (mock) on four consecutive days. Cytokine bead arrays were performed using protein extracts from tumors explanted one day after the last treatment. Dots representing tumor samples from individual mice and median values are shown. Representative results from one of two independent experiments are shown.
MeVac FmIL-12 induces activation and expansion of intratumoral immune effector cells
Tumor-infiltrating lymphocyte (TIL) subpopulations were analyzed 24 h after treatment with MeVac FmIL-12 in parallel to the cytokine profiling described above. Treatment with MeVac FmIL-12 led to a massive decrease of NK cell proportions (CD45+CD335+) in comparison to both MeVac IgG1-Fc and mock treatment (Fig. 4A). Further analyzing the NK cell population, a significant increase in the proportion of cells expressing the early activation marker CD69 could be observed after treatment with MeVac FmIL-12 in comparison to both control groups (Fig. 4B). The percentage of T cells (CD45+CD3+) increased significantly in the MeVac FmIL-12 group in comparison to both control groups (Fig. 4C). Furthermore, the activated cytotoxic T cell population (CD3+CD8+CD69+) was quantified. A slight, however not significant, increase of CD3+CD8+CD69+ percentages could be observed in the MeVac FmIL-12 group (Fig. 4D).
Figure 4.
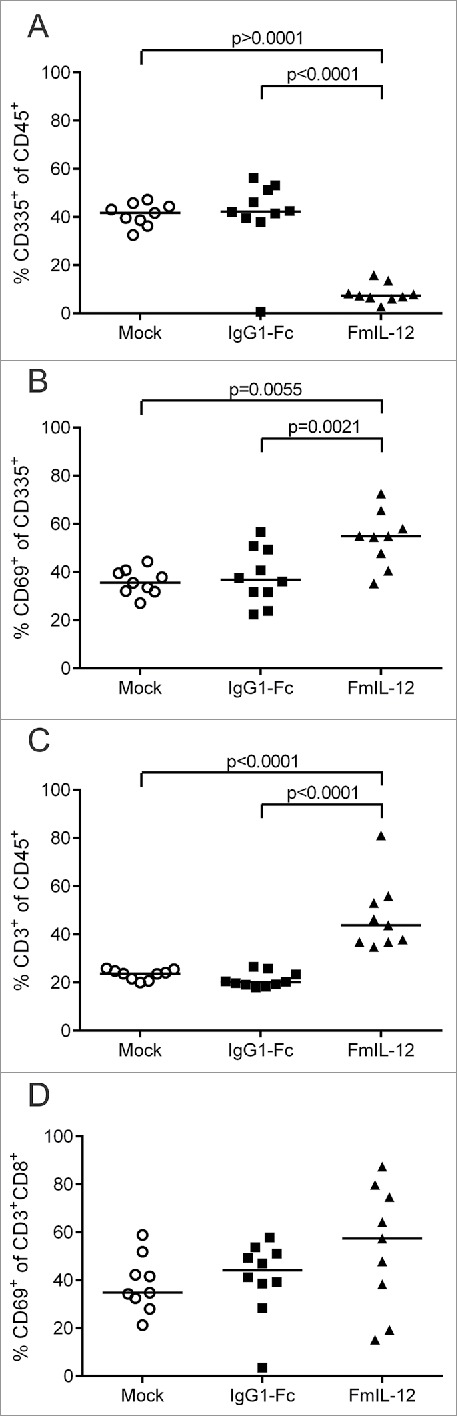
Tumor-infiltrating lymphocytes after MeVac therapy. MC38cea tumor cells were implanted subcutaneously in C57BL/6J mice and established tumors were treated with intratumoral injections of MeVac encoding FmIL-12, IgG1-Fc or carrier fluid (mock) on four consecutive days (the same experiment is described in Fig. 3). Single cell suspensions from the tumors were prepared and flow cytometry analysis was performed one day after the last treatment. Percentages of natural killer (NK) cells among all leukocytes (A), activated NK cells (B), T cells among all leukocytes (C) and activated cytotoxic T cells (D) are shown. Dots representing tumor samples from individual mice and median values are shown. Representative results from one of two independent experiments are shown.
Therapeutic efficacy of MeVac FmIL-12 is dependent on cytotoxic T cells
Depletion experiments were performed to assess the contribution of the main immune effector cell populations to therapeutic efficacy of MeVac FmIL-12 in the MC38cea tumor model. Depletion of the NK and CD4+ populations did not have a significant impact on survival of the animals after MeVac FmIL-12 treatment in comparison to animals with no immune cell depletions (data no shown). However, therapeutic effects were almost completely abrogated after depletion of the CD8+ T cell population (Fig. 5). These results show that while CD4+ and NK cells may contribute to the effects observed after MeVac FmIL-12 treatment, CD8+ cells are essential for therapeutic efficacy.
Figure 5.
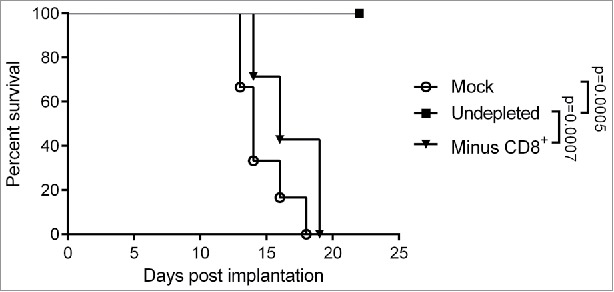
Impact of CD8+ T cell depletion on the therapeutic efficacy of MeVac FmIL-12. C57BL/6J mice were depleted of CD8+ T cells (Minus CD8+) by intraperitoneal antibody injections or left undepleted (mock, undepleted). MC38cea tumor cells were implanted subcutaneously and established tumors were treated with intratumoral (i.t.) injections of 1 × 106 cell infectious units of MeVac encoding FmIL-12 (Minus CD8+, undepleted) in 100 µL on four consecutive days. Mock treated animals received i.t. OptiMEM injections. Kaplan–Meier survival analysis is shown.
Discussion
In this study, we establish MeVac FmIL-12 as an effective immunovirotherapeutic. Furthermore, we identify immune signatures associated with successful immunomodulation in the context of MeV therapy.
While previous MeV oncolysis studies used Edmonston B strain derivatives,12,13,23 this study introduces Schwarz/Moraten (MeVac) measles vaccine strain vectors. MeVac vectors are more attenuated24 and more immunogenic25 than the Edmonston B derivatives, making them superior in terms of safety and immunomodulatory potential, respectively. MeVac is also the vaccine strain used for measles immunization in Europe, and therefore, a candidate for further clinical development. Thus, we chose MeVac for this study to deepen understanding of its immunomodulatory properties.
MeVac FmIL-12 and MeVac anti-PD-L1 were selected as the most promising therapeutics from a panel of vectors encoding immunomodulators targeting different phases in the “cancer immunity cycle”.26 We have studied measles virus encoding anti-PD-L1 previously.13 Other oncolytic vectors encoding IL-12, such as vesicular stomatitis virus,27 adenovirus28 and herpes virus29 have been developed, but to our knowledge this is the first report of MeV encoding IL-12 for cancer therapy. We demonstrated that MeVac-encoded immunomodulators are expressed and functional. However, expression kinetics and replication kinetics differed between individual vectors. Thus, the observed therapeutic outcomes are a result of a complex interplay of the viral vector, the expression kinetics of the encoded immunomodulator, the infected tumor cells and the tumor microenvironment. Treatment with MeVac FmIL-12 and MeVac anti-PD-L1 increased complete tumor remissions in the fully immunocompetent murine MC38cea tumor model in comparison to the control vector encoding the antibody constant region IgG1-Fc. Notably, treatment with the FmIL-12 encoding vector led to complete tumor remissions in 90% of animals with established tumors. This is an unprecedented rate of complete tumor remissions in pre-clinical studies of MeV efficacy in immunocompetent tumor models.12,13,23 IL-12 has long been known for its potent antitumor properties in different animal tumor models, but modest efficacy and severe toxicities have hampered clinical translation.30 Recent clinical trials investigating IL-12 for use in cancer treatment focus on local delivery using non-viral (NCT01440816, NCT01502293) and viral vectors (NCT01397708) to minimize systemic toxicities. The challenge of IL-12 delivery lies in its heterodimeric structure, as equal levels of both the p40 and the p35 subunit are required to generate the biologically active IL-12p70.15 To avoid separate expression of both IL-12 subunits, we generated a MeVac vector encoding an IL-12 fusion protein.31 With the MeVac FmIL-12 vector, we therefore present a novel approach for targeted delivery of this potent cytokine.
Immune profiling early after treatment gave insights into mechanisms of action of immunomodulatory MeVac. The effects observed in these experiments seem mainly attributable to vector-mediated FmIL-12 expression, as changes in T-bet mRNA levels, cytokine profile and TIL subpopulations were unique to the MeVac FmIL-12 group and not observed in both control groups. The only exception was a slight increase of IL-6 in both virus treatment groups, representing acute inflammation induced by the MeVac vector. The most significant change in cytokine profiles was an increase of IFN-γ and TNF-α after MeVac FmIL-12 treatment. IL-12 signaling is known to induce expression of both IFN-γ and TNF-α.15 The observed IFN-γ and TNF-α upregulation is consistent with the observed increased levels of T-bet in the MeVac FmIL-12 group, collectively indicating polarization toward a Th1-associated immune response, which is a well characterized property of IL-12.15
IL-12 is a well-known activator of NK and T cells, which augments their cytotoxic potential through upregulation of Granzyme B and Perforin as well as induction of IFN-γ.15 The analysis of TILs revealed a decrease of NK cells and an increase of the early activation marker CD69 on the NK cell population after MeVac FmIL-12 treatment. Most probably, powerful direct activation of NK cells through IL-12 signaling leads to activation induced death of the effector NK cells, which may or may not be associated with target tumor cell killing. Additionally, an increase in intratumoral T cells was observed after MeVac FmIL-12 therapy, consistent with the known proliferative effect of IL-12 on activated T cells.32 As activation was observed in both NK and T cell populations, both could contribute to the therapeutic efficacy of MeVac FmIL-12. Depletion experiments, however, revealed that the CD4+ and NK cell populations do not significantly influence therapeutic efficacy of MeVac FmIL-12, while the CD8+ T cells are essential. Previous studies suggest that the relative importance of different immune cell populations for the antitumor properties of IL-12 is dependent on the dose, treatment schedule and the tumor model.33–35 It has also been suggested that the importance of an immune cell population to the therapeutic efficacy of IL-12 is determined by its overall contribution to tumor control in a given model.35 The critical role of the CD8+ T cell population for MeVac FmIL-12 efficacy could therefore be determined by the central role of the T cell response in the control of MC38 tumors.36 Furthermore, the tumor rechallenge experiment demonstrated that MeVac therapy induces a robust memory response. Interestingly, a higher IFN-γ production upon in vitro restimulation suggested that animals treated with MeVac FmIL-12 may mount a larger or stronger memory response than animals treated with the control virus. As CD8+ T cells were crucial for the efficacy of MeVac FmIL-12, most probably the T cell memory response also mainly determined the restimulation experiment results. This would be in line with prolonged survival of memory T cells primed in presence of IL-12, as reported previously.37
Also after mock treatment, MC38cea tumors harbored high NK and T cell infiltrates. Nevertheless, tumors progressed rapidly, indicating a state of immunosuppression which is a known characteristic for MC38 tumors.38 Our results indicate that MeVac FmIL-12 can overcome suppressive signaling and, through activation of the abundant immune effectors, initiate rapid killing of the surrounding tumor cells. Results of this study present MeVac FmIL-12 as a potent therapeutic for activation of a Th1 effector response in tumors with high immune cell infiltration under immunosuppression. Tumors with different immune signatures might, however, benefit from different types of immunomodulation. To this end, MeV can be envisioned as a flexible platform for targeted oncolysis and delivery of immunomodulators tailored to the individual tumor immune environment. To approach this goal, future clinical trials of immunovirotherapy must incorporate a translational research program analogous to the immune profiling performed in this study. Surely, differences in interplay between immune effectors and virus can be expected in the context of a human immune system. Nevertheless, this pre-clinical study encourages translation of oncolytic MeV encoding IL-12 into clinical application.
Materials and methods
Construction of recombinant measles viruses
All measles virus constructs used in this study were based on Schwarz/Moraten vaccine strain (MeVac) genome (obtained from S. Bossow). Cassettes encoding murine GM-CSF (426 bp) and eGFP (720 bp) described previously12 were excised as MluI-AscI fragments from pCG vectors and inserted into an additional transcriptional unit (ATU) in the MeVac genome upstream of the N open reading frame (ORF) via the unique AscI restriction site. Cassettes encoding antibodies against murine CTLA-4 (1584 bp) and PD-L1 (1596 bp), and the antibody constant region IgG1-Fc (864 bp) described previously13 were excised from pCG vectors as MluI-PauI fragments and inserted into the MeVac genome in an ATU downstream the H ORF via the unique MauBI restriction site. The cassette encoding the murine IL-12 fusion protein (FmIL-12) consisting of murine IL-12 p40 and p35 subunits linked by a (Gly3Ser)4 was generated based on results obtained by Lieschke et al.31 Murine IL-12 p40 and p35 subunits were amplified by PCR using pUMVC3-mIL-12 (provided by V. Teichgräber) as a template. By additional PCRs p40 was flanked with an MluI site at the 5′ end and with a 45 bp linker fragment at the 3′ end using primers 5′–ACTAGTACGCGTGGCCACCATGTGTCCTCAGAAGCTAACCATCTCCTGGT–3′and 5′–CGACCCACCACCGCCCGAGCCACCGCCACCGGATCGGACCCTGCAGGGAACACATGCCCACTT–3′. The signal peptide at the 5′ end of p35 was deleted and p35 was flanked with the (Gly3Ser)4 linker at the 5′ end, and a PauI restriction site at the 3′ end by amplification with primers 5′–GGCGGTGGTGGGTCGGGTGGCGGCGGATCCAGGGTCATTCCAGTCTCTGGACCTGCCAGGTGTCTT and 5′–AAAGTCGACTGCGCGCTATTATCAGGCGGAGCTCAGATAGCCCATCACCCT–3′. The modified p35 and p40 constructs were fused with primers flanking the 5′ end of p40 and the 3′ end of p35 in an overlap PCR to obtain the final FmIL-12 construct, which was cloned into pCG. FmIL-12 was excised as an MluI-PauI fragment (1650 bp) from the pCG vector and inserted into the MeVac genome in an ATU downstream of the P ORF via the unique MauBI site. The cassette encoding murine Ip-10 (Cxcl10) was designed as an MluI-AscI fragment containing the 295 bp ORF of mIP-10 preceded by a Kozak sequence and followed by a two nucleotide spacer (TA). The mIp-10 was amplified with primers 5′–CCCTTTACGCGTGCCACCATGAACCCAAGTGCTGCCGTC–3′ and 5′–CCCTTTGGCGCGCCTATTAAGGAGCCCTTTTAGACC – 3′ using cDNA from splenocytes of a C57BL/6J mouse as a template and cloned into the pJET 1.2/blunt cloning vector (Thermo Scientific, K1231). The mIP-10 cassette was inserted into the MeVac genome in an ATU upstream of the measles N ORF as an MluI-AscI fragment via the unique AscI restriction site. Murine Cd80 was inserted into the MeVac genome as a soluble form of the protein (CD80-Fc).18 CD80-Fc was constructed by fusing the extracellular part of murine CD80 with the human IgG1-Fc region used in the anti-CTLA-4 and anti-PD-L1 constructs via fusion PCR. The first PCR fragment consisting of an MluI restriction site followed by a Kozak sequence, the murine CD80 signal peptide, the extracellular domain of murine CD80 (up to the asparagine in position 246) and the first 26 nucleotides of the hinge of IgG1-Fc was amplified with primers 5′–TTTACGCGTGCCACCATGGCTTGCAATTGTCAGTTG–3′ and 5′–TCAGAAGAAATGAGGCAAGCAGAGAAGTCGACGAGGCCAAATCTTGTGACAA–3′ using cDNA from C57BL/6J mouse splenocytes as a template. The second PCR fragment consisting of the human IgG1-Fc region followed by a myc tag, stop codon and AscI restriction site was amplified with primers 5′–GTCGACGAGGCCAAATCTTGTGACAA–3′ and 5′–CTCATCTCAGAAAAAGATCTGAATTAGGCGCGCCCCCTTT–3′ using human IgG1-Fc as a template. The resulting PCR products were fused with primers flanking the 5′ end of the first PCR product and the 3′ end of the second PCR product in an overlap PCR to obtain the mCD80-Fc construct, which was cloned into pCG. After excision as an MluI-AscI fragment mCD80-Fc (1614 bp) was inserted into the MeVac genome in an ATU downstream of the H ORF via the unique MauBI site. In all constructs, the MeVac H protein was fully retargeted to human CEA by fusion of a scAb to the mutated H protein analogously to Edmonston B strain constructs, as described previously.23 All MeVac genomic constructs were designed to comply with the “rule of six” in the measles genome to ensure successful rescue of recombinant virus particles.39,40
Cell culture
Vero African green monkey kidney cells (CCL-81) and B16 murine melanoma cells (CRL-6322) were obtained from the American Type Culture Collection. The Vero-αHis cell line stably expressing a scAb against His6 tag21 was a gift from S. J. Russell (Mayo Clinic, Rochester, MN). Murine colon adenocarcinoma cells MC38cea19 (transduced for stable expression of human CEA) and the parental MC38 cell line were a gift of R. Cattaneo (Mayo Clinic, Rochester, MN). DLD-1 cells were obtained from S. Fröhling (DKFZ, Heidelberg, Germany). Vero and MC38 cell lines were cultivated in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, 31966047) supplemented with 10% Fetal Calf Serum (FCS) (Biosera, FB-1000/500). B16 and DLD-1 cells were cultivated in Roswell Park Memorial Institute (RPMI) 1640 medium (Life Technologies, 61870044) with 10% FCS. All cell lines were maintained at 37°C in a humidified atmosphere with 5% CO2 and routinely tested for mycoplasma contamination.
Virus propagation and titration
Recombinant MeVac particles were obtained from cDNA constructs,40,41 and propagated on Vero-αHis cells.21 For propagation, Vero-αHis cells were infected at a multiplicity of infection (MOI) of 0.03 and cultivated at 37°C 5% CO2 until syncytia had spread across the whole cell layer (36–48 h post infection). Subsequently, culture medium was completely removed, cells were scraped and collected and viral particles released by one freeze-thaw cycle. Cell debris was removed by centrifugation (5 min, 6000×g at 4°C) and aliquots of the supernatant were stored at −80°C. The amount of viral particles was determined by 1:10 serial dilution titrations in octuplicates on 1.5 × 104 Vero-αHis cells per well in 96-well cell culture plates. Individual syncytia were counted 72 h post infection and titers calculated as cell infectious units (ciu) per mL.
Virus replication kinetics
MC38cea cells were seeded in 12-well plates (1 × 105 cells per well). After 12 h the cell culture medium was removed and cells were infected with the respective viruses at MOI = 3 in 300 µL OptiMEM (Life Technologies, 31985070) in triplicates for each time point. After adsorption for 2 h the inoculum was removed and substituted with 1 mL DMEM+10% FCS per well. Cells were scraped in the culture medium at the designated time points, collected and snap-frozen in liquid nitrogen. The amount of viral particles was determined by serial dilution titration assay in quadruplicates as described above.
Cell viability assay
MC38cea cells were seeded in 6-well plates (2 × 105 cells per well). After 12 h the cell culture medium was removed and cells were infected with the respective viruses at MOI = 5 in 800 µL OptiMEM in triplicates. After adsorption for 2 h the inoculum was removed and substituted with DMEM+10% FCS. At the designated time points cell viability was determined using the Colorimetric Cell Viability Kit III (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide, XTT) (PromoKine, PK-CA20–300–1000) according to the manufacturer's instructions.
Transgene expression
MC38cea cells were seeded in 12-well plates (1 × 105 cells per well). After 12 h the cell culture medium was removed and cells were infected with the respective viruses at MOI = 3 in 300 µL OptiMEM in triplicates. After adsorption for 2 h, 700 µL DMEM+10% FCS were added. Supernatants were collected at the designated time points. “0h” represents the time point of inoculation. Expression of the respective immunomodulators was detected by ELISA. Commercially available ELISA kits were used for detection of mGM-CSF, FmIL-12, mIP-10 (R&D Systems, MGM00, M1270, MCX100) and CD80-Fc (Boster Biological Technology, EK0708) according to the manufacturers' instructions. Anti-CTLA-4 and anti-PD-L1 were detected by binding to the respective murine proteins by ELISA as described previously.13 Western blot analysis was performed as described before.13
Isolation of murine splenocytes
Spleens were isolated aseptically and maintained in RPMI 1640 at 4°C until further processing. Spleens were passed through 100 µm nylon cell strainers (BD Biosciences, 352360) into 10 mL RPMI 1640 and pelleted. The pellets were resuspended in 1 mL ACK Lysing solution (Life Technologies, A1049201) for red blood cell lysis, incubated 10 min at room temperature and pelleted. Cells were resuspended in DPBS (Life Technologies, 14190169) and kept on ice until further use.
Production of MeVac-encoded immunomodulators for functional assays
Vero-αHis cells were seeded in 15 cm cell culture dishes and infected with MeVac encoding the respective transgenes at MOI = 0.03. Supernatants were collected (15 mL per plate) when syncytia had spread across the whole cell layer (ca. 36–44 h post infection). Supernatants were snap-frozen in liquid nitrogen, stored at −80°C and sterile filtered before use. Expression of the immunomodulators was confirmed by ELISA as described above.
Functional assays
MC38cea cells (2 × 105) were incubated with 15 mL medium containing MeVac-encoded anti-PD-L1, anti-CTLA-4, mCD80-Fc or IgG1-Fc. The treated MC38cea cells were resuspended in 100 µL activation medium consisting of RPMI 1640 with 5% FCS, 1% Penicillin–Streptomycin (P/S) (Life Technologies, 15070063), 500 nM ionomycin (Cayman Chemical Company, Cay-11932) and 5 nM phorbol 12-myristate 13-acetate (PMA) (Cayman Chemical Company, Cay-10008014) and seeded in a 96-well plate. 2 × 105 freshly isolated splenocytes from a C57BL/6J mouse in 100 µL activation medium were added per well. After 24 h coculture of splenocytes with the treated MC38cea cells, supernatants were collected and IFN-γ concentration was determined using mouse IFN-γ ELISA Ready-SET-Go!® (eBioscience, 88–7314–22) according to the manufacturer's instructions. A chemotaxis assay was performed to assess functionality of MeVac-expressed mIP-10. Freshly isolated C57BL/6J mouse splenocytes were cultivated for 48 h in a medium consisting of RPMI 1640, 10% FCS, 1% P/S, 50 U/mL IL-2. The activated splenocytes were added to the upper part and supernatants from cells infected with MeVac mIP-10 or MeVac eGFP to the lower part of 24-well cell culture plate with transwell migration chambers (6.5 mm, pore size 5 µm) (Corning, 29442–118). Splenocyte migration was allowed for 3 h at 37°C. Supernatants from the lower part of the chamber were collected and cells were counted using trypan blue staining (Sigma-Aldrich, T8154) for dead cell exclusion and a Neubauer hemocytometer. To assess FmIL-12 functionality 2 × 106 freshly isolated splenocytes from a C57BL/6J mouse were resuspended in RPMI 1640 supplemented with 10% FCS, 1% P/S and 50 U/mL recombinant murine IL-2 (Miltenyi, 130–094–054) and varying concentrations of MeVac-encoded FmIL-12 or respective amounts of supernatant from cells infected with MeVac encoding eGFP. Splenocytes were seeded in 12-well plates and incubated for 48 h at 37°C 5% CO2. Supernatants were collected and IFN-γ concentration was determined by ELISA as described above.
In vivo experiments
All animal experimental procedures were approved by the Animal Protection Officer at the German Cancer Research Center (Heidelberg, Germany) and by the regional council according to the German Animal Protection Law. MC38cea cells were implanted subcutaneously (s.c.) into the right flank of six to eight weeks old C57BL/6J mice (Harlan Laboratories, Rossdorf, Germany or bred in the Central Animal Laboratory of the German Cancer Research Center) in 100 µL DPBS with 1 × 106 cells per mouse. Treatment was initiated when the average tumor volume reached 40–70 mm3 (efficacy experiments) or 120 mm3 (cytokine profiling and TIL analysis). Mice received intratumoral (i.t.) injections with 100 µL of the respective virus suspensions diluted in OptiMEM to doses of 5 × 105 or 1 × 106 cell infectious units on four or five consecutive days (see figure legends for treatment schedules). Mice in the mock group received treatment with 100 µL OptiMEM. Tumor volumes were determined every third day by measuring the largest and smallest diameter with a caliper and calculating the volume using the formula: largest diameter × (smallest diameter)2 × 0.5. Mice were killed when tumor volumes exceeded 1500 mm3, ulceration occurred or animals were moribund. For immune cell depletion experiments mice received intraperitoneal injections with antibodies against CD4, clone GK1.5 (Biolegend, 100435), CD8, clone 2.43 (BioXCell, BE0061) or NK1.1, clone PK136 (Biolegend, 108712). The CD4 and CD8 depletion groups received injections of 100 µg antibody in 100 µL 3 d before s.c. implantation of MC38cea cells, on the day of tumor cell implantation, 3 d post tumor cell implanatation and followingly once a week. The NK1.1 depletion group received injections with 200 µg of the antibody in 200 µL 3 and 2 d before tumor cell implantation, on the day of tumor cell implantation, one and 3 d post tumor cell implantation and followingly once a week. The depletion efficiency in all groups was more than 95% as controlled by flow cytometry in peripheral blood of selected animals once a week.
Tumor-specific IFN-γ memory recall with murine splenocytes
MC38cea, MC38 and B16 cells were treated with 20 µg/mL mitomycin-C (Sigma-Aldrich, M4287) for 2 h with shaking at 37°C. Subsequently, cells were washed three times with DPBS and resuspended in activation medium consisting of RPMI 1640 with 10% FCS, 1% P/S and 50 U/mL recombinant murine IL-2. Freshly isolated murine splenocytes were resuspended in activation medium. Cocultures were prepared in 24-well plates by seeding 1 × 105 tumor cells with 1 × 106 splenocytes per well in 0.5 mL total volume of activation medium. As controls 1 × 106 splenocytes were cocultivated with DLD-1 cell lysates prepared by lysis of 1 × 106 cells per mL with one freeze-thaw cycle. Cells were cocultivated for 48 h, supernatants were collected and IFN-γ concentration was assessed by ELISA as described above.
Quantification of T-bet and Foxp3 mRNA by RT-qPCR
Pieces of freshly explanted MC38cea tumors were immersed in RNAlater (Qiagen, 76104) and stored at −20°C until further processing. Total RNA was extracted using the RNeasy Protect Mini Kit (Qiagen, 74124) according to the instructions of the manufacturer. Integrity of the extracted RNA was evaluated in an agarose gel electrophoresis. cDNA was synthesized using the Maxima H Minus First Strand cDNA synthesis kit (Thermo Scientific, K1682) with 1 μg RNA per sample and the oligo (dT) primers according to the instructions of the manufacturer. Primers were designed to span exon–exon junctions for amplification of specific products on T-bet (158 bp), Foxp3 (112 bp) and the reference gene L13a (131 bp) – T-bet Fw 5′–TAAGCAAGGACGGCGAATGTT–3′, T-bet Rev 5′–TGCCTTCTGCCTTTCCACAC–3′, FoxP3 Fw 5′–GGCCCTTCTCCAGGACAGA–3′, FoxP3 Rev 5′–GCTGATCATGGCTGGGTTGT–3′, L13A Fw 5′–GGCTGCCGAAGATGGCGGAG–3′, L13A Rev 5′–GCCTTCACAGCGTACGACCACC–3′. RT-qPCR was prepared with 1 μL cDNA, 200 nM respective forward and reverse primers, 10 μL Power SYBR® Green PCR Master mix (Thermo Scientific, 4309155) and water up to 20 μL total reaction volume. The reaction conditions were as follows—10 min initial denaturation at 95°C, followed by 40 cycles of 15 s denaturation at 95°C, 60 s annealing and extension at 55°C (T-bet) or 60°C (Foxp3 and L13a), and 5 s fluorescence detection at 72°C on a Roche LightCycler® 480 System. Melting curve analysis was performed to identify specific amplification. Minus reverse transcriptase controls and no template controls were run in parallel with the cDNA samples. Stable expression of the reference gene among treatment groups was validated using the Normfinder Software.42
Intratumoral cytokine profiling
Protein extraction from freshly harvested MC38cea tumors was performed as described in.43 In brief, freshly isolated MC38cea tumors were snap-frozen in liquid nitrogen and stored at −80°C until processing. Samples were thawed on ice, minced in small pieces and homogenized with a pestle in lysis buffer with 10 mM Tris–HCl (pH 8.0) (Sigma-Aldrich, T2694), 150 mM NaCl (Sigma-Aldrich, S5150), 10% Glycerol (Sigma-Aldrich, G7757), 5 mM EDTA (Sigma-Aldrich, G7757), cOmplete Mini, EDTA-free Protease Inhibitor Cocktail Tablet (Sigma-Aldrich, 05892791001), 1% NP-40 (Abcam, ab142227). Lysates were incubated for 1 h at 4°C with rotation and sonicated afterwards with an intermittent (0.5 min) on and off sonication regimen for 7 min (high intensity) using Diagenode Bioruptor® Standard with a cooling water pump (Diagenode). Lysates were cleared by centrifugation at 13000×g for 15 min and aliquots of supernatants were stored at −80°C until analysis. Cytokine concentrations were measured with the Mouse Th1/Th2/Th17 Cytokine Bead Array Kit (BD Biosciences, 560485) according to the manufacturer's instructions.
Flow cytometry of tumor-infiltrating lymphocytes
Single cell suspensions were prepared from freshly isolated MC38cea tumors. Tumors were minced in small pieces in RPMI + 10% FCS + 200 U/mL Collagenase Type I (Life Technologies, 17018029) and incubated for 30 min at 37°C with gentle vortexing every 10 min. The resulting cell suspensions were passed through 100 µm nylon cell strainers and cells were spun down and resuspended in DPBS. Subsequently 2 × 106 cells per sample were resuspended in 100 µL DPBS and stained with the antibodies CD45.2-PerCP-Cy™5.5 (clone 104) (BD Biosciences, 552950), CD3 Molecular Complex-Alexa Fluor® 700 (clone 17A2) (BD Biosciences, 561388), CD4-APC-Cy™7 (clone GK1.5) (BD Biosciences, 552051), CD8a-APC (clone 53–6.7) (BD Biosciences, 561093), CD69-PE (clone H1.2F3) (Biolegend, 104507), CD335-FITC (clone 29A1.4) (Biolegend, 137605). For dead cell staining samples were resuspended in DPBS with 1 µg/mL DAPI (4′,6-diamidino-2-phenylindole) (Sigma-Aldrich, D8417) before acquisition. Data were acquired on a BD FACS AriaII instrument with FACS Diva software version 8.0.1 and analyzed with FlowJo V10 (Tree Star Inc.). Only samples with at least 3000 events were included in the analysis.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software (version 6.01; GraphPad Software). Tumor volume differences in in vivo experiments, ELISA results in restimulation experiments, RT-qPCR, flow cytometry and cytokine profiling results were analyzed by one-way ANOVA with Tukey's multiple comparison post-hoc-test. Multiplicity adjusted p values are reported for data analyzed with ANOVA. Results were considered statistically significant if p values were lower than 0.05. Tumor volume distributions were analyzed for the last day when all animals where alive. Survival curves were analyzed by log-rank (Mantel–Cox) test with Bonferroni correction for multiple comparisons. Results were considered statistically significant if the p value was lower than the corrected threshold after Bonferroni correction.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank J. Albert and B. Hoyler for their excellent technical assistance. We acknowledge S. J. Russell, R. Cattaneo, V. Teichgräber, S. Bossow and S. Fröhling for providing cell lines and plasmids.
Funding
This study was supported by the German National Science Foundation (DFG, grant EN 1119/2–1 to C.E.E.). R.V. was supported by a fellowship from Melanie and Eduard zur Hausen Foundation. M-C.B-D. was supported by a fellowship from the Canadian Institutes of Health Research.
References
- 1.Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe 2014; 15:260-5; PMID:24629333; http://dx.doi.org/ 10.1016/j.chom.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pol J, Buqué A, Aranda F, Bloy N, Cremer I, Eggermont A, Erbs P, Fucikova J, Galon J, Limacher JM et al.. Trial watch-oncolytic viruses and cancer therapy. Oncoimmunology 2016; 5:e1117740; PMID:27057469; http://dx.doi.org/ 10.1080/2162402X.2015.1117740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015; 14:642-62; PMID:26323545; http://dx.doi.org/ 10.1038/nrd4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, Peach H, Phillips RM, Bond J, Pandha H et al.. Measles virus causes immunogenic cell death in human melanoma. Gene Ther 2013; 20:7-15; PMID:22170342; http://dx.doi.org/ 10.1038/gt.2011.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, Okazaki T, Marumoto T, Takahashi A, Takayama K et al.. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res 2012; 72:2609-21; PMID:22461509; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3185 [DOI] [PubMed] [Google Scholar]
- 6.Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, Bramante S, Parviainen S, Kanerva A, Loskog AS et al.. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res 2012; 72:2327-38; PMID:22396493; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2975 [DOI] [PubMed] [Google Scholar]
- 7.Angelova AL, Grekova SP, Heller A, Kuhlmann O, Soyka E, Giese T, Aprahamian M, Bour G, Rüffer S, Cziepluch C et al.. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J Virol 2014; 88:5263-76; PMID:24574398; http://dx.doi.org/ 10.1128/JVI.03688-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guillerme JB, Boisgerault N, Roulois D, Ménager J, Combredet C, Tangy F, Fonteneau JF, Gregoire M. Measles virus vaccine-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells. Clin Cancer Res 2013; 19:1147-58; PMID:23339127; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2733 [DOI] [PubMed] [Google Scholar]
- 9.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer 2014; 14:559-67; PMID:24990523; http://dx.doi.org/ 10.1038/nrc3770 [DOI] [PubMed] [Google Scholar]
- 10.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS et al.. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015; 33:2780-88; PMID:26014293; http://dx.doi.org/ 10.1200/JCO.2014.58.3377 [DOI] [PubMed] [Google Scholar]
- 11.Russell SJ, Peng KW. Measles virus for cancer therapy. Curr Top Microbiol Immunol 2009; 330:213-41; PMID:19203112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grossardt C, Engeland CE, Bossow S, Halama N, Zaoui K, Leber MF, Springfeld C, Jaeger D, von Kalle C, Ungerechts G. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum Gene Ther 2013; 24:644-54; PMID:23642239; http://dx.doi.org/ 10.1089/hum.2012.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, Shevchenko I, Umansky V, Nettelbeck DM, Weichert W et al.. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther 2014; 22:1949-59; PMID:25156126; http://dx.doi.org/ 10.1038/mt.2014.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood 2012; 119:3383-93; PMID:22323450; http://dx.doi.org/ 10.1182/blood-2011-11-370130 [DOI] [PubMed] [Google Scholar]
- 15.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003; 3:133-46; PMID:12563297; http://dx.doi.org/ 10.1038/nri1001 [DOI] [PubMed] [Google Scholar]
- 16.Homey B, Muller A, Zlotnik A. Chemokines: agents for the immunotherapy of cancer? Nat Rev Immunol 2002; 2:175-84; PMID:11913068; http://dx.doi.org/ 10.1038/nri748 [DOI] [PubMed] [Google Scholar]
- 17.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haile ST, Dalal SP, Clements V, Tamada K, Ostrand-Rosenberg S. Soluble CD80 restores T cell activation and overcomes tumor cell programmed death ligand 1-mediated immune suppression. J Immunol 2013; 191:2829-36; PMID:23918985; http://dx.doi.org/ 10.4049/jimmunol.1202777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbins PF, Kantor JA, Salgaller M, Hand PH, Fernsten PD, Schlom J. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res 1991; 51:3657-62; PMID:1712245 [PubMed] [Google Scholar]
- 20.Hammond AL, Plemper RK, Zhang J, Schneider U, Russell SJ, Cattaneo R. Single-chain antibody displayed on a recombinant measles virus confers entry through the tumor-associated carcinoembryonic antigen. J Virol 2001; 75:2087-96; PMID:11160713; http://dx.doi.org/ 10.1128/JVI.75.5.2087-2096.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura T. Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, Russell SJ. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol 2005; 23:209-14; PMID:15685166; http://dx.doi.org/ 10.1038/nbt1060 [DOI] [PubMed] [Google Scholar]
- 22.Vincent S, Tigaud I, Schneider H, Buchholz CJ, Yanagi Y, Gerlier D. Restriction of measles virus RNA synthesis by a mouse host cell line: trans-complementation by polymerase components or a human cellular factor(s). J Virol 2002; 76:6121-30; PMID:12021345; http://dx.doi.org/ 10.1128/JVI.76.12.6121-6130.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Parker WB, Sorscher EJ, Cattaneo R. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol Ther 2007; 15:1991-97; PMID:17712331; http://dx.doi.org/ 10.1038/sj.mt.6300291 [DOI] [PubMed] [Google Scholar]
- 24.Schwarz AJ. Preliminary tests of a highly attenuated measles vaccine. Am J Dis Child 1962; 103:386-9; PMID:13909753 [DOI] [PubMed] [Google Scholar]
- 25.Combredet C, Labrousse V, Mollet L, Lorin C, Delebecque F, Hurtrel B, McClure H, Feinberg MB, Brahic M, Tangy F. A molecularly cloned Schwarz strain of measles virus vaccine induces strong immune responses in macaques and transgenic mice. J Virol 2003; 77:11546-554; PMID:14557640; http://dx.doi.org/ 10.1128/JVI.77.21.11546-11554.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39:1-10; PMID:23890059; http://dx.doi.org/ 10.1016/j.immuni.2013.07.012 [DOI] [PubMed] [Google Scholar]
- 27.Shin EJ, Wanna GB, Choi B, Aguila D 3rd, Ebert O, Genden EM, Woo SL. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope 2007; 117:210-4; PMID:17204993; http://dx.doi.org/ 10.1097/01.mlg.0000246194.66295.d8 [DOI] [PubMed] [Google Scholar]
- 28.Bortolanza S, Bunuales M, Otano I, Gonzalez-Aseguinolaza G, Ortiz-de-Solorzano C, Perez D, Prieto J, Hernandez-Alcoceba R. Treatment of pancreatic cancer with an oncolytic adenovirus expressing interleukin-12 in Syrian hamsters. Mol Ther 2009; 17:614-22; PMID:19223865; http://dx.doi.org/ 10.1038/mt.2009.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Derubertis BG, Stiles BM, Bhargava A, Gusani NJ, Hezel M, D'Angelica M, Fong Y. Cytokine-secreting herpes viral mutants effectively treat tumor in a murine metastatic colorectal liver model by oncolytic and T-cell-dependent mechanisms. Cancer Gene Ther 2007; 14:590-97; PMID:17431402; http://dx.doi.org/ 10.1038/sj.cgt.7701053 [DOI] [PubMed] [Google Scholar]
- 30.Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002; 13:155-68; PMID:11900991; http://dx.doi.org/ 10.1016/S1359-6101(01)00032-6 [DOI] [PubMed] [Google Scholar]
- 31.Lieschke GJ, Rao PK, Gately MK, Mulligan RC. Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat Biotechnol 1997; 15:35-40; PMID:9035103; http://dx.doi.org/ 10.1038/nbt0197-35 [DOI] [PubMed] [Google Scholar]
- 32.Gately MK, Desai BB, Wolitzky AG, Quinn PM, Dwyer CM, Podlaski FJ, Familletti PC, Sinigaglia F, Chizonnite R, Gubler U et al.. Regulation of human lymphocyte proliferation by a heterodimeric cytokine, IL-12 (cytotoxic lymphocyte maturation factor). J Immunol 1991; 147:874-82; PMID:1713608 [PubMed] [Google Scholar]
- 33.Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, Gately MK. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 1993; 178:1223-30; PMID:8104230; http://dx.doi.org/ 10.1084/jem.178.4.1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kodama T, Takeda K, Shimozato O, Hayakawa Y, Atsuta M, Kobayashi K, Ito M, Yagita H, Okumura K. Perforin-dependent NK cell cytotoxicity is sufficient for anti-metastatic effect of IL-12. Eur J Immunol 1999; 29:1390-96; PMID:10229107; http://dx.doi.org/ 10.1002/(SICI)1521-4141(199904)29:04%3c1390::AID-IMMU1390%3e3.0.CO;2-C [DOI] [PubMed] [Google Scholar]
- 35.Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol 2000; 165:2665-70; PMID:10946296; http://dx.doi.org/ 10.4049/jimmunol.165.5.2665 [DOI] [PubMed] [Google Scholar]
- 36.Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, Church SE, Maby P, Vasaturo A, Angelova M et al.. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med 2016; 8:327ra326; PMID:26912905; http://dx.doi.org/14978082 10.1126/scitranslmed.aad6352 [DOI] [PubMed] [Google Scholar]
- 37.Chang J, Cho JH, Lee SW, Choi SY, Ha SJ, Sung YC. IL-12 priming during in vitro antigenic stimulation changes properties of CD8 T cells and increases generation of effector and memory cells. J Immunol 2004; 172:2818-26; PMID:14978082; http://dx.doi.org/ 10.4049/jimmunol.172.5.2818 [DOI] [PubMed] [Google Scholar]
- 38.Medina-Echeverz J, Fioravanti J, Zabala M, Ardaiz N, Prieto J, Berraondo P. Successful colon cancer eradication after chemoimmunotherapy is associated with profound phenotypic change of intratumoral myeloid cells. J Immunol 2011; 186:807-15; PMID:21148040; http://dx.doi.org/ 10.4049/jimmunol.1001483 [DOI] [PubMed] [Google Scholar]
- 39.Calain P, Roux L. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J Virol 1993; 67:4822-30; PMID:8392616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dötsch C, Christiansen G, Billeter MA. Rescue of measles viruses from cloned DNA. Embo J 1995; 14:5773-84; PMID:8846771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin A, Staeheli P, Schneider U. RNA polymerase II-controlled expression of antigenomic RNA enhances the rescue efficacies of two different members of the Mononegavirales independently of the site of viral genome replication. J Virol 2006; 80:5708-15; PMID:16731909; http://dx.doi.org/ 10.1128/JVI.02389-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004; 64:5245-50; PMID:15289330; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-0496 [DOI] [PubMed] [Google Scholar]
- 43.Amsen D, de Visser KE, Town T. Approaches to determine expression of inflammatory cytokines. Methods Mol Biol 2009; 511:107-42; PMID:19347295; http://dx.doi.org/ 10.1007/978-1-59745-447-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.