

ABSTRACT
Purpose: Although local oncolytic viral therapy (OVT) may enhance tumor lysis, antigen release, and adaptive immune responses, systemic antitumor responses post-therapy are limited. Adoptive immunotherapy with autologous dendritic cells (DC) and cytokine-induced killer cells (DC–CIK) synergizes with systemic therapies. We hypothesized that OVT with Herpes Simplex Virus-granulocyte macrophage-colony-stimulating factor (HSV-GM-CSF) would induce adaptive T cell responses that could be expanded systemically with sequential DC–CIK therapy.
Patients and Methods: We performed a pilot study of intratumoral HSV-GM-CSF OVT followed by autologous DC–CIK cell therapy. In addition to safety and clinical endpoints, we monitored adaptive T cell responses by quantifying T cell receptor (TCR) populations in pre-oncolytic therapy, post-oncolytic therapy, and after DC–CIK therapy.
Results: Nine patients with advanced malignancy were treated with OVT (OrienX010), of whom seven experienced stable disease (SD). Five of the OVT treated patients underwent leukapheresis, generation, and delivery of DC–CIKs, and two had SD, whereas three progressed. T cell receptor sequencing of TCR β sequences one month after OVT therapy demonstrates a dynamic TCR repertoire in response to OVT therapy in the majority of patients with the systematic expansion of multiple T cell clone populations following DC–CIK therapy. This treatment was well tolerated and long-term event free and overall survival was observed in six of the nine patients.
Conclusions: Strategies inducing the local activation of tumor-specific immune responses can be combined with adoptive cellular therapies to expand the adaptive T cell responses systemically and further studies are warranted.
KEYWORDS: Dendritic cells, Herpes Simplex Virus, T cell receptor sequencing, oncolytic viral therapy
Introduction
Tumor cells demonstrate both impaired antiviral immune responses and higher permissiveness for viral replication.1,2 Oncolytic viral therapy (OVT) utilizes the selective replication of viruses in cancer cells to lyse tumor cells and release tumor antigens, while leaving normal tissue unaffected.3,4 OVT represents an emerging therapeutic modality that has achieved tumor regression in clinical trials.5 The US Food and Drug Administration (FDA) recently approved an OVT, Imlygic (talimogene laherparepvec or T-vec), for the treatment of advanced melanoma6 following the phase III OPTiM trial that demonstrated an improvement in durable response rates with a tolerable safety profile in patients receiving T-vec.7
OVT induces a local inflammatory response within the tumor and tumor antigen release primes tumor adaptive immunity, acting as an in situ vaccine. Although specific adaptive antitumor responses, often CD8+ T cell-mediated7,8 are induced, these responses have been limited compared with the immune responses within the injected tumor.9 In addition, analysis of non-injected tumors in patients who have undergone OVT injection of a target lesion has demonstrated a less robust immune response compared with the immune response found in the injected lesion.9 Methods to boost the systemic immune responses to OVT are being investigated, including combinations with other immune therapies, such as immune checkpoint inhibitors.10
Alternatives to systemic immune modulators may include adoptive transfer of autologous cellular therapies. In considering potential cellular therapies to modulate the systemic immune environment, it has been demonstrated that when OVTs expressing granulocyte macrophage-colony-stimulating factor (GM-CSF) are injected into tumors, such therapy both attracts and matures dendritic cells (DC).11 DC are potent antigen-presenting cells, promoting the generation of tumor antigen-specific helper and cytotoxic T cells, inducing antitumor immune responses.12-15 Ex vivo and in vivo experimental evidence has demonstrated that cytokine-induced killer (CIK) cells generated with a “cytokine cocktail”16,17 result in CIK cells with a high level of cytotoxic activity in a broad range of tumor cell lines, supporting their use for adoptive immunotherapy and have yielded encouraging results.18-20 It is hypothesized that the tumor-specific cellular components of CIK therapy may be responsible for the clinical benefits observed in some cancer patients who receive this therapy.21 Co-culture of CIK with cytokine producing DCs has elicited an increase in the antitumor activity of CIK cells.22 Thus, the combination of DCs and CIKs (DC–CIK) can lead to an increase in cytotoxic activity, more effective than either treatment alone,23 and have demonstrated encouraging clinical signals supporting investigation in a variety of malignancies.24,25
For this study, we used the oncolytic Herpes Simplex Virus (HSV) expressing GM-CSF (OrienX010), which was developed by OrienGene Biotechnology Ltd. The virus is based on the HSV-1 CL1 strain, which was isolated from a Chinese patient's mouth. For reference, we would point out that T-vec is based on the HSV-a strain JS-1. The genes encoding ICP34.5 (both copies) and ICP47 have been completely deleted, and the inactivated gene encoding ICP6 is inserted. The gene encoding human GM-CSF replaces the ICP34.5-encoding sequences.31
We hypothesized that DCs combined with cytokine-induced killer cells (DC–CIK) could expand the adaptive immune response induced by OVT. We, therefore, evaluated the antitumor and immunologic activity of a recombinant herpes simplex virus expressing human granulocyte macrophage colony stimulating factor (HSV-GM-CSF or OrienX010), given at three different dose levels in patients with metastatic cancer. In a subset of treated patients, OVT was followed by DC–CIK therapy. The clinical study was designed to assess the safety, clinical benefit, and immunologic activity of this sequential immunotherapy regimen in patients with measurable or evaluable metastatic cancer.
Results
Patient characteristics
From May 2013 to May 2014, nine total patients were enrolled in the OrienX010 Phase 1 dose escalation trial at the Capital Medical University Cancer Center, Beijing, China. Patients were required to provide informed written consent in order to participate in this trial. Patients who had residual tumor after receiving OrienX010 were then eligible to receive additional immunotherapy with DC–CIK, if they had either clinically progressed after receiving OrienX010, or had no further standard therapeutic options after receiving the OrienX010. The demographics for the enrolled patients are listed in Table 1. The patients enrolled in this trial had the following tumor types: breast carcinoma (2), head and neck squamous cell carcinoma (1), melanoma (2), GIST of the small intestine (1), rectal carcinoma (2), and malignant thyroid (1). Of the patients enrolled in this trial, only the patient with malignant thymoma had not received prior therapy. Eight of the patients had undergone prior systemic therapy for their malignancy, as reviewed in Table S2. All nine patients completed all planned treatment(s) with OrienX010.
Table 1.
Study of patient demographics in dose escalation of OrienX010.
| Demographic |
Number |
Percentage |
|---|---|---|
| Age (years) | ||
| Median | 56 | |
| Range | 25–70 | |
| Sex | ||
| Male | 5 | 56 |
| Female | 4 | 44 |
| Race | ||
| Asian | 9 | 100 |
| Diagnosis | ||
| Breast cancer | 2 | 22 |
| Malignant thymoma | 1 | 11 |
| Melanoma | 2 | 22 |
| Rectal cancer | 2 | 22 |
| GIST | 1 | 11 |
| Squamous cell carcinoma | 1 | 11 |
| Prior therapy lines of systemic therapy | ||
| Median | 2 | |
| Range | 0–10 | |
Five patients were consented for sequential DC–CIK therapy, one from the dose level 1 cohort, one from the dose level 2 cohort, and all three patients from the dose level 3 cohort. The time between the completion of OrienX010 and the initiation of DC–CIK therapy ranged from 1.5 to 11.6 mo. Patients who received sequential DC–CIK therapy received 1–3 cycles of DC–CIK therapy with doses ranging from 4.0 × 108 up to 2.6 × 109.
Gene copy and GM-CSF RNA analysis
In cohort 1 of OrienX010, post-injection serum from each patient was positive for OrienX010 gene copies. For the patients in the 2nd and 3rd cohorts who underwent multiple OrienX010 injections, post-injection serum from four of six patients were positive for the presence of OrienX010 gene copies. Quantification of GM-CSF RNA from fine needle aspirates (FNAs) of injected tumors demonstrated no difference in the presence of intratumoral GM-CSF RNA between pre- and post-OrienX010 treatment.
ELISA for anti-HSV-1 antibodies
In this study, a total of seven patients treated with the OrienX010 injections demonstrated an elevation in the amount of anti-HSV-1 antibody post-OrienX010 injection compared with pre-OrienX010 treatments. In cohort 1, all three patients treated in this cohort demonstrated an increase in anti-HSV-1 antibodies. In the 2nd and 3rd cohorts, two patients from each cohort demonstrated an increase in anti-HSV-1 antibodies.
Flow cytometry analysis
Flow cytometric analysis was performed on peripheral blood mononuclear cells (PBMCs) from peripheral blood obtained pre- and post-OrienX010 as well as pre-DC–CIK therapy and post each cycle of DC–CIK therapy. Flow analysis was done for the following cellular populations: CD3+, CD3+CD4+, CD3+CD8+, CD4+CD25+CD127low/−, CD8+CD28+, CD3-CD19+, CD3-CD16+CD56+, and the CD4+/CD8+ ratio. This analysis revealed no differences in the frequencies of the peripheral immune cell populations interrogated by flow at each of the above time points in this study.
Serology
Serum from six of the nine patients was antinuclear antibody (ANA) negative in both the pre- and post-OrienX010 injections. Serum from the three remaining patients (501, 603, and 604) demonstrated a positive ANA with a titer of >1:100 (positive ANA for this study was ≥1:100).
Toxicities OrienX010
The OrienX010 treatments of 4 × 108 (pfu) × 1 injection (dose level 1), 1 × 108 (pfu) × 3 injections (dose level 2), and 4 × 108 (pfu) × 3 injections (dose level 3) were all well tolerated with no dose limiting toxicity. The maximal tolerated dose was not reached in this trial. Table 2 illustrates the breakdown of adverse events (AEs) observed in the OrienX010 trial. There were no Grade 3 or greater AEs. Common AEs (occurring ≥20%) included fever (Grade 1–2), injection site pain (Grade 1–2), and fatigue (Grade 1–2). The remainder of the AEs were Grade 1 or 2, of these AEs, those felt related to the treatment were: fever and pain at injection site (definite), tachycardia, hematuria, AV block, GGT increase, insomnia, constipation, diabetes, ecchymosis, and leukopenia (possible).
Table 2.
Summary of adverse events and grade in OrienX010 vaccination dose escalation study.
| Single dose: 4 × 108 Pfu |
Multiple dose: 1 × 108 Pfu |
Multiple dose: 4 × 108 Pfu |
||||
|---|---|---|---|---|---|---|
| Body system/adverse events | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 |
| Blood and lymphatic system disorders | ||||||
| Leukopenia | 0 | 0 | 1 | 0 | 0 | 0 |
| Cardiovascular disorders | ||||||
| Tachycardia | 2 | 0 | 1 | 0 | 0 | 0 |
| Atrioventricular block | 1 | 0 | 0 | 0 | 0 | 0 |
| Endocrine disorders | ||||||
| Hyperglycemia | 0 | 0 | 1 | 0 | 0 | 0 |
| Gastrointestinal disorders | ||||||
| GGT increase | 1 | 0 | 0 | 0 | 0 | 0 |
| Constipation | 0 | 0 | 0 | 0 | 1 | 0 |
| Genitourinary disorders | ||||||
| Hematuria | 0 | 0 | 2 | 0 | 0 | 0 |
| General disorders and administration site conditions | ||||||
| Fever | 2 | 0 | 3 | 0 | 3 | 0 |
| Pain (at injection site) | 1 | 0 | 0 | 0 | 3 | 0 |
| Psychiatric disorders | ||||||
| Insomnia | 0 | 0 | 1 | 0 | 0 | 0 |
| Skin and subcutaneous tissue disorders | ||||||
| Purpura | 0 | 0 | 0 | 0 | 1 | 0 |
Toxicity—DC–CIK therapy
Overall, the DC–CIK therapy was well tolerated with only mild AEs. These AEs included mild transient fevers and chills. No Grade 3 or higher adverse events were experienced with the DC–CIK infusions and the observed AEs were similar to those we have previously published.20
Clinical outcomes: OrienX010 therapy
Overall, the clinical response to OrienX010 therapy across all dose levels included: seven patients with stable disease (SD) and two patients with progressive disease (PD). The Kaplan–Meier curves for progression free survival (PFS) and overall survival (OS) for the OrienX010 dose escalation trial are illustrated in Fig. 1A. The median PFS was 16.6 mo for all patients enrolled in the OrienX010 trial. The median OS has not yet been met in this trial after a median follow-up of 29.4 mo.
Figure 1.
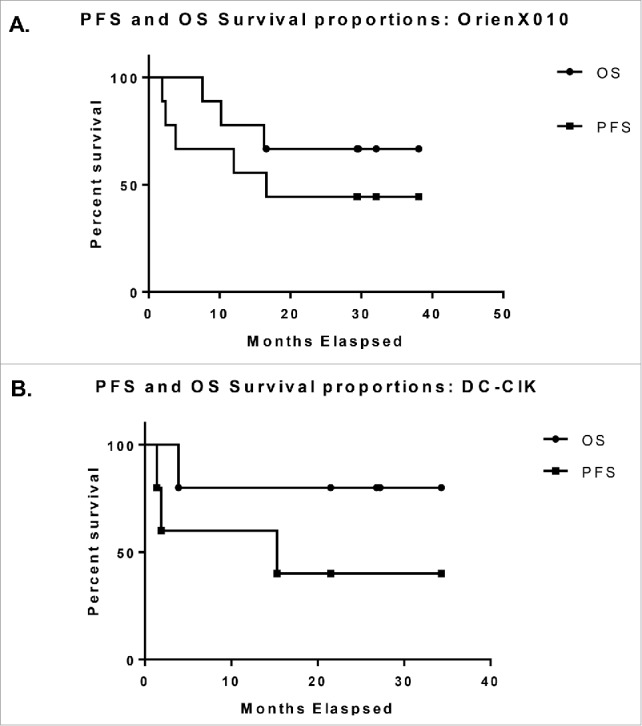
(A) Kaplan–Meier curves for progression free survival and overall survival for the OrienX010 dose escalation trial. The median PFS was 16.3 mo (497 d). Median OS has not been met. (B) Kaplan–Meier curves for progression free survival and overall survival for the DC–CIK therapy expansion trial. The median PFS was 9.9 mo (301 d). Median OS has not been met.
In the OrienX010 dose level 1 cohort, there were three patients with SD. Radiographically, there was demonstration of tumor necrosis in the injected tumor from one cohort 1 patient after OrienX010. In the dose level 2 cohort, there were two patients with SD and one patient with PD. In the dose level 3 cohort, there were two patients with SD and one patient with PD.
Patients who did not progress while on study were followed after completion of the study for evaluation of the duration of clinical response. No patients died while undergoing treatment on this study. During follow-up one patient in the dose level 1 cohort and two patients in the dose level 2 cohort have died.
Clinical outcomes: DC–CIK therapy
Five patients from the original nine patients enrolled in the OrienX010 trial went on to receive DC–CIK therapy. These patients included: one patient from OrienX010 dose level 1, one patient from dose level 2, and all three patients from dose level 3. The Kaplan–Meier curves for PFS and OS for the DC–CIK expansion cohort trial are illustrated in Fig. 1B. The median PFS from first dose of DC–CIK therapy was 15.3 mo for all patients enrolled in the DC–CIK therapy trial. The median OS has not yet been met in this trial after a median follow-up of 26.8 mo.
Clinical evaluation of the best response in the OrienX010 previously treated metastatic lesions in these five patients after treatment with DC–CIK demonstrated one partial response (PR), two SD, and two PD. The responses for each patient treated with the DC–CIK are now summarized. For the one patient from OrienX010 dose level 1, computed tomography (CT) imaging after OrienX010 treatment revealed overall SD (Fig. 2. A: submandibular mass pre-OrienX010: 2.5 cm × 2.0 cm and B: post-OrienX010: 2.1 cm × 2.1 cm). CT imaging after two cycles of DC–CIK therapy revealed overall SD with partial necrosis in the OrienX010 treated metastatic lesion, with no change in the size or diameter of this lesion. Repeat imaging for this patient after the final DC–CIK therapy revealed SD (Fig. 2C: post-DC–CIK therapy: 2.1 cm × 2.0 cm).
Figure 2.
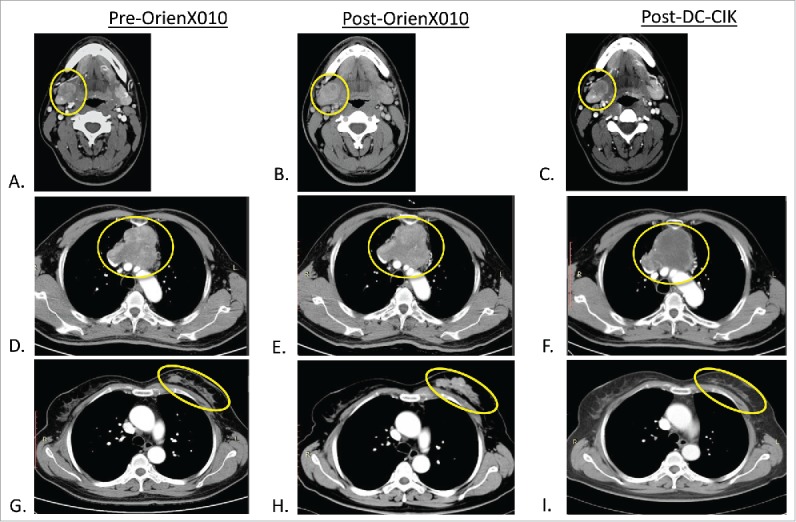
(A) Metastatic melanoma baseline CT of right submandibular (SM) met pre-OrienX010, (B) SM met post-OrienX010, and (C) SM met s/p after three cycles of DC -CIK therapy. (D) Malignant thymoma baseline CT of mediastinal mass (MM) pre-OrienX010, (E) MM after OrienX010, and (F) MM after two cycles of DC -CIK therapy. (G) Metastatic breast cancer baseline CT of left breast (LB) mass before OrienX010, (H) LB mass after OrienX010, and (I) LB mass after one cycle of DC -CIK therapy.
For the first patient from OrienX010 dose level 3, repeat staging after three doses of OrienX010 revealed SD (Fig. 2D: mediastinal mass pre-OrienX010: 8.3 cm × 6.7 cm and E: post-OrienX010: 8.6 cm × 7.5 cm). Repeat imaging after two cycles of DC–CIK therapy revealed overall SD in the OrienX010 treated mediastinal lesion by contrast CT scanning, with no change in the size or diameter but with apparent necrosis of this previously OrienX010 treated metastatic lesion (Fig. 2F: mediastinal mass post-DC–CIK therapy: 8.6 cm × 7.0 cm).
For the third patient from OrienX010 dose level 3, repeat staging after three doses of OrienX010 revealed SD (Fig. 2G: right breast lesion pre-OrienX010: 9.7 cm × 1.3 cm and H: post-OrienX010: 7.6 cm × 2.4 cm). CT imaging after one cycle of DC–CIK therapy revealed overall PR with evidence of necrosis and decrease in size in the previously OrienX010 treated lesion (Fig. 2I: right breast lesion post-DC–CIK therapy: 5.0 cm × 1.3 cm).
Evaluation of adaptive T cell responses: TCR sequencing analysis
To characterize response to therapy, we evaluated the clonal diversity of each patient's T-cell repertoire over time by serial T cell receptor (TCR) sequencing. As represented in Fig. 3A (Patient 401), colored bars reflect clone populations expanding and contracting over time; purple clones are present in the baseline sample and continue to be found in future samples, the orange fraction appears after OrienX010 injection, green clones first appear prior to DC–CIK therapy, and blue clones expand after DC–CIK treatment. For those patients who do not receive DC–CIK therapy, we cannot identify green and blue clone subsets. The gray subsets reflect clones that were sample specific.
Figure 3.
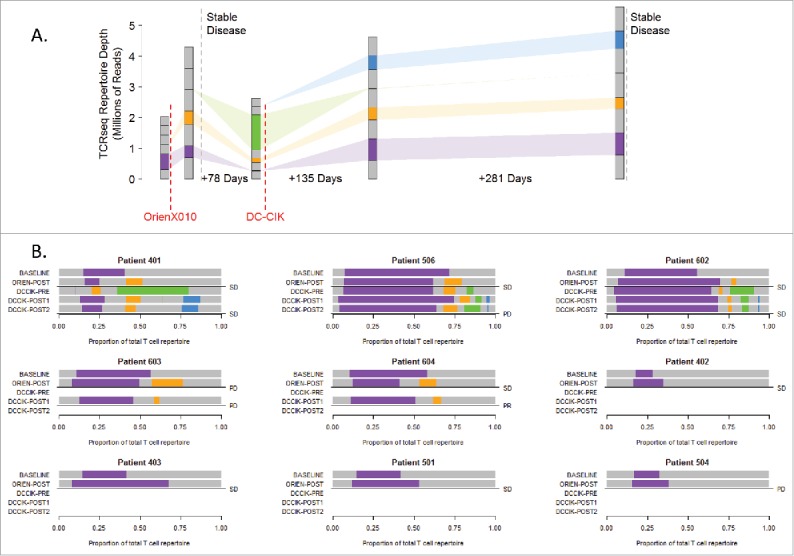
Clone populations in TCR β repertoires. (A) Schema for patient 401 tracks clone populations over time and b/w OrienX010 and DC–CIK treatments: purple clones first appear in the baseline sample, orange in the post-ORIEN sample, green in the pre-DC–CIK, and blue in the post-DC–CIK. (B) Patients 401, 506, 602, 603, and 604 received both OrienX010 and DC–CIK therapy, whereas patients 402, 403, 501, and 504 received OrienX010 only.
We represented serial repertoire sequencing for all patients in Fig. 3B where the bars reflect the relative frequency of patient-specific clone populations. In general, we note that the TCR β usage from peripheral blood samples from all patients characterizes a significant population of T cells pre-therapy as demonstrated by the purple region of the top bars. This baseline oligoclonal population is often expanded to represent a greater proportion of T cells following the OVT as shown by the expanded purple region in the second bars. This expansion either consists solely of the pre-existing clones (purple) or an expansion of a new set of clones as represented by the orange and green bars. These new clones may represent antigen spreading of the adaptive immune response in some patients.
Evaluation of clones immediately after the oncolytic therapy or immediately before DC-CIK therapy (the third bars) demonstrate that the DC–CIK therapies, as expected, tend to maintain expanded clonal populations. Of interest, we note that patients that had SD tended to have an increase in their pre-existing clonal population following OVT or the induction of new clones as demonstrated in patients 401, 506, 602, 604, 402, 403, and 501.
Patient 401 had SD, and the post-OrienX010 TCR usages represents a highly divergent sample compared with baseline in clonotypes with only 8.9% (732 of 23,906 clones) present in both samples as represented by the purple bars. Although the baseline AA TCR β sequencing demonstrated 13,521 unique clones, the post-OrienX010 TCR β AA sequencing demonstrated 23,906 unique clones, suggesting an expansion of different T cells post-therapy. Between these two samples there was conservation of ∼700 clones. In the post-OrienX010 sample a new TCR β population appears (orange bar). Interestingly, an increase in PBMC clonality is then seen in the follow-up sample, labeled pre-DC–CIK, with even fewer clones (58 clones), while a new population of TCR β clones appear at this time point as represented by a green bar. Of interest, is that following DC–CIK therapy, the original TCR clones represented by the purple and orange bars are present in the PBMC and maintained through post-DC–CIK therapy.
Analysis of the TCR usage from another patient with SD on OrienX010, Patient 506, demonstrates a population of T cells using TCR β sequences, represented by the purple bar, which is maintained as a dominant population throughout therapy. Nonetheless, expansion of an additional set of T cells using another set of TCR β sequences (orange bar) is noted after OrienX010 therapy, which then expanded to an additional set of TCR β sequences (green bar). All are maintained following DC–CIK therapy.
We note that in patient 504 (metastatic melanoma), there was only a minimal expansion post-OVT of their dominant clone and they had disease progression. In addition, patient 603 (rectal cancer) demonstrated contraction of their pre-existing dominant clone with limited antigen spreading and also had PD.
In evaluating the TCR β samples from the study, which includes significant heterogeneity of tumor types and a small number of patients with various HLA types, there were no unique consensus T cell populations identified from very high-level summaries of clone populations. In regards to specific clones elicited in the study, our analysis demonstrates that there are 12,244 unique clones across all nine patients (purple clones only—so they have to appear in at least two samples). Of these, 77 appear in more than one person (maximum one clone is found in six patients, the AA sequence for this clone is AWGQENTEAF).
Discussion
Approved therapies for metastatic disease remain non-curative for most solid tumors. Thus, new therapeutic approaches to elicit tumor cell death and/or induce protective host antitumor immunity are attractive alternatives. Recently, the HSV-1 OVT, T-vec, was FDA approved for unresectable melanoma, based on the results from the OPTiM trial demonstrating a higher durable response rate (CR+PR lasting >6 mo) and a trend to longer median OS.7 OVT takes advantage of the abnormal immune environment of the tumor leading to tumor lysis and release of tumor-specific antigens, which may elicit adaptive host immunity to these tumor-specific antigens.
Based on this concept, we performed a dose escalation study of the HSV1 based OVT, OrienX010 which also expresses GM-CSF to augment antigen presentation by the recruitment of DCs. OrienX010 was well tolerated with no dose limiting toxicities. Adverse events were Grades 1 and 2 only and maximum tolerated dose (MTD) was not reached with OrienX010 dose titration. In our exploratory analysis of the biologic assays in this study, serum analysis revealed that seven of the nine trial patients demonstrated an increase in anti-HSV antibodies after OrienX010 therapy, supporting a systemic immune response to OrienX010 (data not shown). In evaluating the OrienX010 gene copies in the post-injection blood samples, seven of the nine patients' post-OrienX010 blood samples demonstrated presence of these gene copies (data not shown). One curious finding was that there was no change in the GM-CSF RNA in either the injected tumors or the peripheral blood in the pre- and post-OrienX010 samples. We believe that this is due to the fact that the duration of the GM-CSF RNA is brief and checking RNA levels at earlier time points may have revealed its presence.
Systemic responses have previously been demonstrated following local oncolytic therapies, such as Tvec, but this has been in a minority of treated patients.26 Our exploratory analysis of phenotypic changes in circulating T-cell subpopulations did not demonstrate significant changes in Treg, activated CTLs, activated B cells, or NK cells in the peripheral blood after therapy with OrienX010 (data not shown). Ex vivo and in vivo experimental evidence has shown that CIK cells generated with a “cytokine cocktail”16,17 resulting in CIK cells with a high level of cytotoxic activity in a broad range of tumor cell lines18 and co-culture with DCs has elicited an increase in the antitumor activity of CIK cells.22 Thus, the combination of DCs and CIKs can lead to an increase in cytotoxic activity, more effective than either treatment alone,23 and have demonstrated encouraging clinical signals supporting investigation in a variety of malignancies.24,25 Therefore, in five patients with PD, residual tumor, or no therapeutic alternatives following OrienX010, we offered additional immunotherapy with autologous DC–CIK. Of the patients who went on to receive DC–CIK therapy, two patients had SD, whereas three progressed after receiving OrienX010. The adverse events noted in the patients who received the DC–CIK therapy were similar to what has been previously reported by our group and others.13,14
In this pilot study, we were encouraged by evidence that adaptive response to treatment were demonstrated locally as well as systemically. Based on TCR analysis of PBMC, we noted that the majority of patients expanded their pre-existing T cell clones, or develop a new clonal population in their post-OrienX010 samples, suggesting that the OVT resulted in the expansion of tumor-specific populations of activated T cells. Of interest, was these antigen-specific populations, quantified by the genetic signature of the TCR sequences, were maintained through the DC–CIK therapy.
Two patients, the single patient from OrienX010 dose level 1 and the first patient from OrienX010 dose level 3, developed a significant, but transient population of unique T-cell clones that expanded prior to DC–CIK therapy, and both of these patients had clinical benefit following both OVT and DC–CIK therapy, suggesting that the adaptive T cell responses to additional antigens were elicited, and expanded systemically to provide clinical benefit. Although the specific antigens recognized by these novel T cells populations have yet to be determined, it suggests that recognition of multiple antigens, and expansion of multiple T cells clones may be of benefit. Of note, it has been previously demonstrated that patients who developed an expansion of T cell clones in response to anti-PD-1 therapy, as determined by TCR sequencing, were the most likely to demonstrate a clinical response to treatment.27 Because of the small sample size, and the heterogeneity of tumor types, our current evaluation of the TCR usage from pre- and post- therapy PBMC samples has not demonstrated any common clonal populations.
In regards to specific clones elicited in the study, our analysis demonstrates that there were 12,244 new unique clones across all nine patients (purple clones only—so they have to appear in at least two samples). Of these, only 77 appear in more than one person and one clone found in six patients—(AWGQENTEAF). These new T cell clones may represent the activity of new clones responding to antigens released from lysed tumor cells following viral oncolytic therapy. We also expect that a proportion of these new clones may represent adaptive anti-viral T cell clones in response to the modified HSV utilized in the OrienX010 oncolytic therapy. Unfortunately, correlative studies (such as tetramer sorting or ELISPOT) to identify T cell populations specific for HSV antigens was unable to be performed as all of the peripheral blood samples had been depleted by the time TCR analysis of the peripheral blood T cell repertoire was completed. In future studies, we will plan to collect PMBC specifically for the purpose of HSV-specific ELISPOT assays. Of the seven patients with SD in the OrienX010 dose escalation trial, six demonstrated an increase in T cell clonality by TCR sequencing analysis. In the DC–CIK expansion cohort the one patient with a PR demonstrated an increase in TCR clonality during the DC–CIK therapy (Table S1).
Although a pilot study, we did measure clinical outcomes. We did find a median PFS of 16.6 mo from the first dose of OrienX010 therapy in all patients. At a median of 29.4 mo of follow-up, the median OS has not been met among all patients who received OrienX010. The median PFS from first dose of DC–CIK therapy was 15.3 mo for all patients enrolled in the sequential DC–CIK therapy trial. The median OS has also not yet been met in this portion of the trial after a median follow-up of 26.8 mo.
In this study, we have demonstrated that OVT can induce increased T cell clonality, likely through the resulting tumor antigen release during tumor lysis. In those patients who received additional adoptive immunotherapy, we also demonstrate that circulating T cell clones are maintained or expanded, suggesting this may be a marker of immune mediated systemic therapeutic response. Future studies are planned investigating the role of instituting check-point inhibitors to further augment the induced therapeutic clonal T cell populations and phenotypic analysis at later time points to identify T cell populations being expanded.
Methods
Patient selection
Patients were required to provide informed written consent in order to participate in this study under a protocol approved by the Beijing Shijitan Hospital Ethics Committee. Enrollment requirements were of age 18–70 y, advanced solid tumors having relapsed or failed standard therapy or advanced solid tumor type lacking effective first line therapy, ECOG status of 0–2, estimated survival of >3 mo, at least 4 weeks since exposure to chemotherapy or radiotherapy, and at least 6 weeks since exposure to nitrosoureas or mitomycin C. Exclusion criteria included untreated primary or metastatic CNS disease, adequate size of tumor allowing for injection of OrienX010, and serious concurrent illness. Disease lesions were considered candidate lesions if amenable to OrienX010 injection under direct visualization or ultrasound guidance with priority given to larger lesions. The volume of OrienX010 injected was based on the diameter of the target lesion (≤1.5 cm, up to 1 mL; >1.5 to ≤ 2.5 cm, up to 2 mL; >2.5 cm, up to 5 mL).
Study treatment and monitoring: OrienX010
In the dose escalation phase of testing OrienX010 in these advanced solid tumors, three dose levels of OrienX010 were selected, each dose level enrolled three patients. The starting dose of 4 × 108 (pfu) was chosen based on a previous study of OrienX010, with demonstrated safety and tolerability of single intratumoral doses from 1 × 106 to 1 × 108 (pfu). In our study, dose level 1, 4 × 108 (pfu) of OrienX010 was administered by one injection into the selected tumor. In dose levels 2 and 3, either 1 × 108 (pfu) or 4 × 108 (pfu), respectively, of OrienX010 was administered by intratumoral injection into the selected tumor every 2 weeks for three treatments (see Fig. S1).
Clinical tumor evaluations were conducted at baseline (days −7 to −1) in all cohorts and on day 28 in the dose level one cohort (e.g., week 4) and on day 57 in the dose level two and three cohorts. Tumor responses were evaluated by contrasted CT scan and/or magnetic resonance imaging (MRI) at baseline and after completion of treatment to evaluate for complete response (CR), PR, SD, or PD according to RECIST criteria version 1.1.
Study treatment and monitoring: DC–CIK therapy
Patients with residual tumor after completing the dose escalation trial of OrienX010 were eligible for sequential therapy with DC–CIK therapy, in addition they had to have either clinically progressed after receiving OrienX010 or had no further standard therapeutic options after receiving the OrienX010. Eligible patients for this sequential therapy underwent 1–3 cycles of DC–CIK immunotherapy as sequential immunotherapy with each cycle of DC–CIK therapy consisting of DC–CIK dosing on days 1, 3, and 5 (±1) of each cycle. Dosing of DC–CIK was patient dependent.
Clinical tumor evaluation was conducted at baseline prior to infusion of the DC–CIK therapy and after the first cycle of therapy or after completion of the DC–CIK therapy. Tumor responses were evaluated by CT and/or MRI for CR, PR, SD, or PD, according to the RECIST criteria version 1.1.
Generation and characterization of DC–CIK cells
Mononuclear cells were harvested from peripheral blood and expanded in vitro. For the induction of DC–CIKs, mobilization of PBMC was performed with SQ injection of GM-CSF 5 mcg/kg per day (Chugai Pharm Co. Ltd., Japan) to patients until the level of mononuclear cells reached 1.5 × 109/L. Then, PBMCs were separated by a COBE Spectra cell separator (COBE BCT, Lakewood, CO, USA) until CD34+ reaching ≥ 4.5 × 106/kg. Then, 40 mL of the apheresis product was co-cultured for 7 d with IL-4 (1,000 U/mL; R&D Systems, Inc., Minneapolis, MN), TNF-α (20 ng/mL; R&D Systems, Inc., Minneapolis, MN) and GM-CSF (800 U/mL; Amoytop Biotech Co., Ltd., Xiamen, China) in vitro to generate autologous DCs.
Mononuclear cells were separated by gradient centrifugation and activated in vitro with the recombinant cytokines IL-2 at 1,000 U/mL (Boehringer Mannheim, Germany), IFNγ at 1,000 U/mL (Boehringer Mannheim, Germany) and CD3 antibody at 1.7 μL/mL (Boehringer Mannheim, Germany) for 7–10 d.
The phenotypes of DCs (CD80, CD86, HLA-DR, CD1a, and CD11c) were detected by flow cytometry method, whereas CIKs were detected by CD3 and CD56. The proportion of CD80+ plus CD86+ cells reached greater than 80% among the cultured cells in the autologous DC-specific cultures. The cultured autologous DCs were then mixed with cultured CIKs at a proportion of 1:100, and then DC–CIK were harvested for intravenously administration to patients.
For the five patients who received DC–CIK therapy (401, 506, 602, 603, and 604), an average of 1.7 1± 0.66 × 109 of induced cells (combined preparation of DC and CIK cells) were infused each time.
Biological sample collection
In patients enrolled in the OrienX010 study, peripheral blood was collected at various time points pre- and post-therapy. In the single injection cohorts, peripheral blood was obtained at baseline and then post-OrienX010 injection at sequential time points. For multiple injections subjects, the blood sampling protocol were the same but with additional sampling at time points post-therapy. The presence of anti- HSV-1 antibodies pre- and post-OrienX010 therapy was evaluated using these samples. The plasma concentrations of these antibodies were determined using sandwich enzyme immunoassay kits from Boster Biological Technology (Wuhan, China) for HSV1 as previously described.28
In the OrienX010 single dose injection group: blood for OrienX010 gene copies was obtained at baseline, day 1, 1 h, 2 h, 4 h, 8 h, 24 h, 48 h, day 5, day 8, day 15, day, 22, and day 29 for 12 time points. For the subjects receiving multiple OrienX010 injections, the protocol was the same as the single injection with 10 additional time points gene copy analysis. For the multiple injection cohorts these time points were: baseline, day 1, 1 h, 2 h, 4 h, 8 h, 24 h, 48 h, day 5, day 8, day 14 (pre-2nd injection), day 28 (pre-3rd injection), day 29 1 h, 2 h, 4 h, 8 h, 24 h, 48 h, day 33, day 36, day 43, and day 57 for 22 time points.
GM-CSF Gene copy (by RT-PCR) was obtained by FNA from the injected tumors at baseline and at 48 h after OrienX010 treatment (including after each treatment for patients receiving multiple doses). Blood concentration of GM-CSF protein levels was evaluated from peripheral blood samples drawn day 8 and weekly through day 29 to compare the baseline gene copy and blood concentration of GM-CSF protein levels with later time points.
Serum ANA levels were detected by indirect fluorescent antibody (IFA) using HEp-2 cells as a substrate (Euroimmun, Germany), as per manufacturer's instructions. We defined positive ANA titer as titers of ≥1:100 by immunofluorescence (IF) such that one-part blood sample was mixed with 100 (or greater) parts of a diluting substance and ANA was still detectable.
The flow cytometric analysis of the frequency of lymphocyte subtypes (CD3+, CD3+CD4+, CD3+CD8+, CD4+/CD8+, CD4+CD25+CD127low/− CD8+CD28+, CD3−CD19+, and CD3−CD16+CD56+) was evaluated from peripheral blood. Blood samples for this analysis were obtained at baseline and weekly through day 29 in the single OrienX010 injection cohort and through day 56 in the multiple injection cohorts. In the patients who went on to receive DC–CIK therapy, additional flow was performed on blood samples obtained prior to initiation of DC–CIK therapy and after each cycle of DC–CIK therapy. Methods of flow cytometry and gating are previously described in Song et al.28,29
T cell receptor usage in peripheral blood
TCR usage was determined by DNA sequencing. PBMCs were isolated from whole blood using Ficoll-Paque plus (GE Healthcare). Total DNA was extracted from PBMCs/leukocytes using QIAamp DNA Blood Mini Kit (#51104, Germany) according to the manufacture's protocol, and evaluated using Qubit® 3.0 Fluorometer (Life Technologies, USA) and agarose gel electrophoresis (Biowest, Spain).
After quantification, multiplex PCR was performed using Multiplex PCR Kit (QIAGEN, Germany). 200 ng of total DNA was mixed with Vβ forward primers and Jβ reverse primers (0.2 μΜ each, designed), 25 μL 2× QIAGEN Multiplex PCR Master Mix, 5 μL Q-Solution, and DEPC-treated water added to make a total volume of 50 μL. The PCR program used was 1 cycle of 95°C for 15 min, and then 30 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 90 s, and extension for 30 s at 72°C, the last step was final extension for 5 min at 72°C and then down to 12°C. Size selection was also used for purification of 100–200 bp PCR productions by QIAquick Gel Extracton (QIAGEN, Germany).
DNA library preparation followed the manufacturer's instructions (Illumina) as described previously.30 We used the same workflow as described elsewhere to perform cluster generation, template hybridization, isothermal amplification, linearization, blocking and denaturization, and hybridization of the sequencing primers. Paired-end sequencing of samples was performed with a read length of 100 bp using the Illumina Hiseq2500 platform.
TCR analysis
For analysis of the TCR sequencing from this trial we evaluated only the AA sequences for the CDR3 regions of the TCR β chain. We selected to evaluate only AA sequences as our prior experience suggests that small DNA sequencing errors can lead to challenges in DNA data alignment. In this analysis, we evaluated all of the TCR sequencing data for each time point: baseline, post-ORIENX010 therapy, pre-DC–CIK therapy, post-DC–CIK therapy cycle 1, and post-DC–CIK therapy cycle 2.
Clinical evaluation of safety and clinical response
The clinical endpoints for safety in this study were MTD and dose limiting toxicity (DLT). For the OrienX010 portion of this study PFS was defined as the time from first OrienX010 tumor injection until disease progression or death whichever came first. OS was defined as the time from the first OrienX010 tumor injection until death due to any cause. For the DC–CIK therapy portion of this trial PFS was defined as the time from first treatment with DC–CIK therapy until disease progression or death whichever came first. OS was defined as the time from the first treatment with DC–CIK therapy until death due to any cause. PFS and OS were calculated using the Kaplan–Meier product limit method. Radiographic response was determined according to RECIST criteria 1.1.
Statistical analysis
Results from immunologic analyses were primarily descriptive. A Kaplan–Meier disease-free survival estimate and 95% confidence bounds was generated for the clinical data.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was a collaborative research project of Duke University Medical Center, Durham, NC, US and Capital Medical University Cancer Center, Beijing, China. This work was supported by Biocells (Beijing)-Capital Cancer Research Funding (2014 ZLZLXYM) and Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (XMLX201413).
References
- 1.Moehler M, Blechacz B, Weiskopf N, Zeidler M, Stremmel W, Rommelaere J, Galle PR, Cornelis JJ. Effective infection, apoptotic cell killing and gene transfer of human hepatoma cells but not primary hepatocytes by parvovirus H1 and derived vectors. Cancer Gene Ther 2001; 8:158-67; PMID:11332986; http://dx.doi.org/ 10.1038/sj.cgt.7700288 [DOI] [PubMed] [Google Scholar]
- 2.Donnelly O, Harrington K, Melcher A, Pandha H. Live viruses to treat cancer. J R Soc Med 2013; 106:310-4; PMID:23824333; http://dx.doi.org/ 10.1177/0141076813494196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol 2012; 30:658-70; PMID:22781695; http://dx.doi.org/ 10.1038/nbt.2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atherton MJ, Lichty BD. Evolution of oncolytic viruses: novel strategies for cancer treatment. Immunotherapy 2013; 5:1191-206; PMID:24188674; http://dx.doi.org/ 10.2217/imt.13.123 [DOI] [PubMed] [Google Scholar]
- 5.Carpenter SG, Carson J, Fong Y. Regional liver therapy using oncolytic virus to target hepatic colorectal metastases. Semin Oncol 2010; 37:160-69; PMID:20494708; http://dx.doi.org/ 10.1053/j.seminoncol.2010.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andtbacka RHI, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, Spitler LE, Puzanov I, Doleman S, Ye Y et al.. OPTiM: A randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol (Meeting Abstracts) 31, LBA9008- (2013). [Google Scholar]
- 7.Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther 1999; 10:385-93; PMID:10048391; http://dx.doi.org/ 10.1089/10430349950018832 [DOI] [PubMed] [Google Scholar]
- 8.Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, Guo ZS. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer 2013; 12:103; PMID:24020520; http://dx.doi.org/ 10.1186/1476-4598-12-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Sur Oncol 2010; 17:718-30; PMID:19915919; http://dx.doi.org/ 10.1245/s10434-009-0809-6 [DOI] [PubMed] [Google Scholar]
- 10.Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res 2016; 22:1048-54; PMID:26719429; http://dx.doi.org/ 10.1158/1078-0432.CCR-15-2667 [DOI] [PubMed] [Google Scholar]
- 11.Toda M, Martuza RL, Rabkin SD. Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte-macrophage colony-stimulating factor. Mol Ther 2000; 2:324-9; PMID:11020347; http://dx.doi.org/ 10.1006/mthe.2000.0130 [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Yu W, Li H, Yu J, Zhang X, Ren X, Cao S. Can the dual-functional capability of CIK cells be used to improve antitumor effects? Cell Immunol 2014; 287:18-22; PMID:24355711; http://dx.doi.org/ 10.1016/j.cellimm.2013.11.009 [DOI] [PubMed] [Google Scholar]
- 13.Wang QJ, Wang H, Pan K, Li YQ, Huang LX, Chen SP, He J, Ke ML, Zhao JJ, Li JJ et al.. Comparative study on anti-tumor immune response of autologous cytokine-induced killer (CIK) cells, dendritic cells-CIK (DC-CIK), and semi-allogeneic DC-CIK. Chin J Cancer 2010; 29:641-8; PMID:20591215; http://dx.doi.org/ 10.5732/cjc.009.10772 [DOI] [PubMed] [Google Scholar]
- 14.Zhong R, Han B, Zhong H. A prospective study of the efficacy of a combination of autologous dendritic cells, cytokine-induced killer cells, and chemotherapy in advanced non-small cell lung cancer patients. Tumour Biol 2014; 35:987-94; PMID:24006222; http://dx.doi.org/ 10.1007/s13277-013-1132-1 [DOI] [PubMed] [Google Scholar]
- 15.Holt GE, Podack ER, Raez LE. Immunotherapy as a strategy for the treatment of non-small-cell lung cancer. Therapy 2011; 8:43-54; PMID:21359153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kakimi K, Nakajima J, Wada H. Active specific immunotherapy and cell-transfer therapy for the treatment of non-small cell lung cancer. Lung Cancer 2009; 65:1-8; PMID:19062127; http://dx.doi.org/ 10.1016/j.lungcan.2008.10.018 [DOI] [PubMed] [Google Scholar]
- 17.Jakel CE, Schmidt-Wolf IG. An update on new adoptive immunotherapy strategies for solid tumors with cytokine-induced killer cells. Expert Opin Biol Ther 2014; 14:905-16; PMID:24673175; http://dx.doi.org/ 10.1517/14712598.2014.900537 [DOI] [PubMed] [Google Scholar]
- 18.Olioso P, Giancola R, Di Riti M, Contento A, Accorsi P, Iacone A. Immunotherapy with cytokine induced killer cells in solid and hematopoietic tumours: a pilot clinical trial. Hematol Oncol 2009; 27:130-9; PMID:19294626; http://dx.doi.org/ 10.1002/hon.886 [DOI] [PubMed] [Google Scholar]
- 19.Ma Y, Zhang Z, Tang L, Xu YC, Xie ZM, Gu XF, Wang HX. Cytokine-induced killer cells in the treatment of patients with solid carcinomas: a systematic review and pooled analysis. Cytotherapy 2012; 14:483-93; PMID:22277010; http://dx.doi.org/ 10.3109/14653249.2011.649185 [DOI] [PubMed] [Google Scholar]
- 20.Tao L, Huang G, Shi S, Chen L. Bevacizumab improves the antitumor efficacy of adoptive cytokine-induced killer cells therapy in non-small cell lung cancer models. Med Oncol 2014; 31:777; PMID:24271420; http://dx.doi.org/ 10.1007/s12032-013-0777-3 [DOI] [PubMed] [Google Scholar]
- 21.Gold JE, Ross SD, Krellenstein DJ, LaRosa F, Malamud SC, Osband ME. Adoptive transfer of ex vivo activated memory T-cells with or without cyclophosphamide for advanced metastatic melanoma: results in 36 patients. Eur J Cancer 1995; 31a: 698-708; PMID:7640041; http://dx.doi.org/ 10.1016/0959-8049(94)00523-8 [DOI] [PubMed] [Google Scholar]
- 22.Wongkajornsilp A, Wamanuttajinda V, Kasetsinsombat K, Duangsa-ard S, Sa-ngiamsuntorn K, Hongeng S, Maneechotesuwan K. Sunitinib indirectly enhanced anti-tumor cytotoxicity of cytokine-induced killer cells and CD3(+)CD56(+) subset through the co-culturing dendritic cells. PLoS One 2013; 8:e78980; PMID:24232460; http://dx.doi.org/ 10.1371/journal.pone.0078980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang X, Chen YT, Song HZ, Huang GC, Chen LB. Cisplatin pretreatment enhances anti-tumor activity of cytokine-induced killer cells. World J Gastroenterol 2011; 17:3002-11; PMID:21799646; http://dx.doi.org/ 10.3748/wjg.v17.i25.3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao B, Han M, Wang L, Gao X, Huang J, Huang M, Liu H, Wang J. Clinical outcomes of active specific immunotherapy in advanced colorectal cancer and suspected minimal residual colorectal cancer: a meta-analysis and system review. J Transl Med 2011; 9:17; PMID:21272332; http://dx.doi.org/ 10.1186/1479-5876-9-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L, Zhang W, Qi X, Li H, Yu J, Wei S, Hao X, Ren X. Randomized study of autologous cytokine-induced killer cell immunotherapy in metastatic renal carcinoma. Clin Cancer Res 2012; 18:1751-9; PMID:22275504; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-2442 [DOI] [PubMed] [Google Scholar]
- 26.Kaufman HL, Amatruda T, Reid T, Gonzalez R, Glaspy J, Whitman E, Harrington K, Nemunaitis J, Zloza A, Wolf M et al.. Systemic versus local responses in melanoma patients treated with talimogene laherparepvec from a multi-institutional phase II study. J Immunother Cancer 2016; 4:12; PMID:26981242; http://dx.doi.org/ 10.1186/s40425-016-0116-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-71; PMID:25428505; http://dx.doi.org/ 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song G, Wang X, Jia J, Yuan Y, Wan F, Zhou X, Yang H, Ren J, Gu J, Lyerly HK. Elevated level of peripheral CD8(+)CD28(−) T lymphocytes are an independent predictor of progression-free survival in patients with metastatic breast cancer during the course of chemotherapy. Cancer Immunol Immunother 2013; 62:1123-30; PMID:23604172; http://dx.doi.org/ 10.1007/s00262-013-1424-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song QK, Ren J, Zhou XN, Wang XL, Song GH, Di LJ, Yu J, Hobeika A, Morse MA, Yuan YH et al.. The prognostic value of peripheral CD4+CD25+ T lymphocytes among early stage and triple negative breast cancer patients receiving dendritic cells-cytokine induced killer cells infusion. Oncotarget 2015; 6:41350-9; PMID:26462021; https://dx.doi.org/ 10.18632/oncotarget.5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J et al.. The diploid genome sequence of an Asian individual. Nature 2008; 456:60-5; PMID:18987735; http://dx.doi.org/ 10.1038/nature07484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaufman HL, Kohlhapp FJ. Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015; 14:642-662; PMID:26323545; http://dx.doi.org/ 10.1038/nrd466 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.