

ABSTRACT
Chronic inflammation can result from inadequate engagement of resolution mechanisms, mainly accomplished by specialized pro-resolving mediators (SPMs) arising from the metabolic activity of lipoxygenases (ALOX5/15) on ω-6 or ω-3 essential polyunsaturated fatty acids (PUFA). We previously demonstrated that formyl peptide receptor 1 (FPR1) suppresses gastric cancer (GC) by inhibiting its inflammatory/angiogenic potential. In this study, we asked whether FPR1 exploits inflammation resolution pathways to suppress GC angiogenesis and growth.
Here, we demonstrate that genetic or pharmacologic modulation of FPR1 in GC cells regulated ALOX5/15 expression and production of the SPMs Resolvin D1 (RvD1) and Lipoxin B4 (LXB4). SPM treatment of GC cells abated their angiogenic potential. Genetic deletion of ALOX15 or of the RvD1 receptor GPR32 increased the angiogenic and tumorigenic activity of GC cells thereby mimicking FPR1 loss. Deletion/inhibition of ALOX5/15 or GPR32 blocked FPR1-mediated anti-angiogenic activities, indicating that ALOX5/15 and GPR32 are required for FPR1's pro-resolving action. An ω-3- or ω-6-enriched diet enforced SPM endogenous production in mice and inhibited growth of shFPR1 GC xenografts by suppressing their angiogenic activity. These data implicate that FPR1 and/or pro-resolving pathway components might be used as risk/prognostic markers for GC; ω-6/3-enriched diets, and targeting FPR1 or SPM machinery may be exploited for GC management.
KEYWORDS: Angiogenesis, formyl peptide receptors, gastric cancer, lipoxin B4, pro-resolving pathways, resolvin D1
Introduction
Prolonged inflammation underlies the pathogenesis of various diseases including cancer.1 Recent years have seen a paradigm shift in our understanding of the etiopathogenesis of inflammation whereby chronic inflammation may not result only from persistence of inflammatory mechanisms, but may also arise from an inadequate engagement of the resolution mechanisms.2 Resolution is an active process brought about by a series of cellular mechanisms that are activated to restore tissue homeostasis.3 Lipid specialized pro-resolving mediators (SPMs), which are the main effectors of resolution, derive from the effects exerted by lipoxygenases (ALOX5 and 15) on ω-6 arachidonic acid (AA), or on ω-3 eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), which are essential polyunsaturated fatty acids (PUFA)3 (Fig. 1A). Recent evidence associates a deficit in pro-resolving pathways with the establishment of various inflammatory-related disorders (e.g., asthma, fibrosis, and autoimmune diseases).4 However, whether and how defects in resolution mechanisms affect cancer initiation and progression, and what signals control pro-resolving pathways in this context is unknown.
Figure 1.
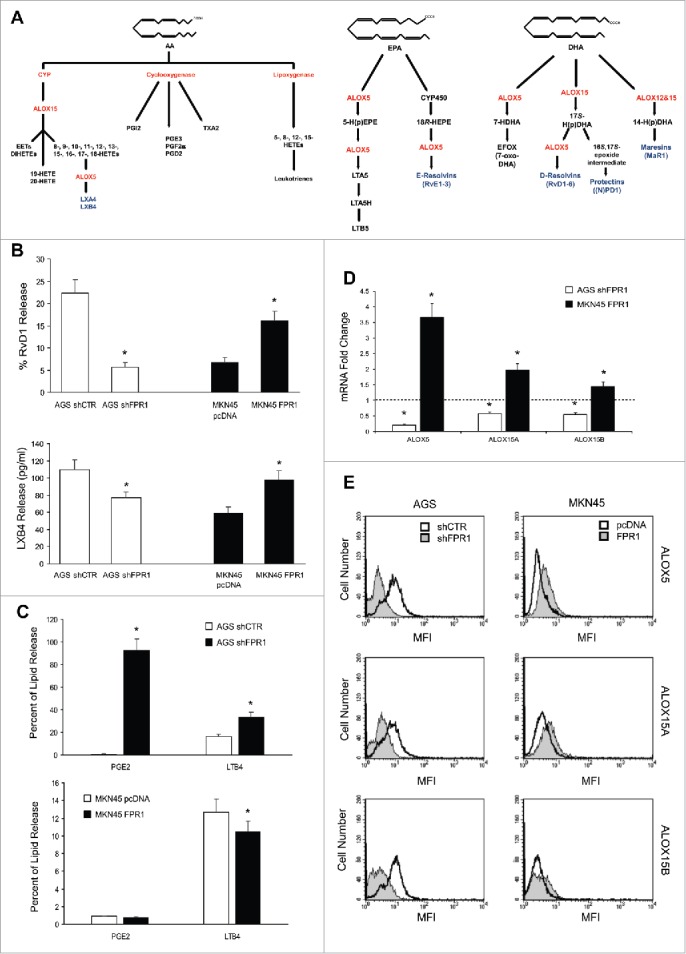
Relation between FPR1 expression and SPM biosynthesis machinery. (A) Schemes of arachidonic acid-AA, docosahexaenoic acid-DHA, and eicosapentaenoic acid-EPA metabolism. (B) AGS shFPR1 cl 15 cells produced significantly lower amounts of RvD1 and LXB4 compared to AGS shCTR cells, while MKN45 cells overexpressing FPR1 (MKN45 FPR1 cl 4) released higher amounts of RvD1 and LXB4 compared to empty vector transfected cells (MKN45 pcDNA), as evaluated by EIA assays. Data are represented as mean ± SD of five independent experiments. *p < .05 compared to the relative control. (C) Increased release of PGE2 and LTB4 from shFPR1 AGS cells and from MKN45 pcDNA cells compared to the relative control cells, assessed by EIA. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to the relative control. (D) AGS shFPR1 cl 15 expressed significantly lower levels and MKN45 FPR1 cl 4 significantly higher levels of ALOX5, ALOX15A, and ALOX15B mRNAs compared to relative controls (dotted line), as assessed by real-time PCR. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to the relative control. (E) ALOX5, ALOX15A, and ALOX15B protein levels were lower in AGS shFPR1 vs. AGS shCTR, and in MKN45 pcDNA vs. MKN45 FPR1 cells, as evaluated by cytofluorimetric analysis. One representative experiment out of three is shown.
Formyl peptide receptors (FPR1, 2, and 3) are pattern recognition receptors (PRR) of the G-protein-coupled (GPCR) family that recognize both exogenous and endogenous “danger” signals, and trigger inflammation and immune responses.5 FPRs can also trigger inflammation resolution, depending on the environmental context and on the specific ligand.6 We recently showed that genetic ablation of FPR1 in AGS gastric cancer (GC) cells, which constitutively express high levels of the receptor, increased their angiogenic and tumorigenic potential. Accordingly, enforced expression of FPR1 in MKN45 GC cells, which constitutively express low levels of the receptor, dramatically impaired GC cell xenograft angiogenesis and growth in immunodeficient mice.7
Since genetic deletion of FPR1 in GC cells increased angiogenesis and enhanced the response to pro-inflammatory cytokines,7 which is a phenotype suggestive of unresolved inflammation, we asked whether, at the gastric level, FPR1 might actively sustain pro-resolving pathways to inhibit GC angiogenesis and growth.
Here, we show that, in GC cells, FPR1 expression/activation levels directly correlate with ALOX expression, and with SPM Resolvin D1 (RvD1) and Lipoxin B4 (LXB4) (3) production (Fig. 1A). The increased angiogenic potential of GC cells lacking FPR1, previously reported,7 was reverted by RvD1 or LXB4 treatment. Genetic ablation of ALOX15 or of GPR32 (an RvD1 receptor)8 induced a pro-angiogenic phenotype in GC cells similar to that induced by FPR1 deletion. ALOXs and GPR32 are required for FPR1-mediated anti-angiogenic activity in GC cells. Consistently, administration of ω-3 or ω-6 PUFA-enriched diets, which enforces endogenous production of SPMs,9 inhibited xenograft growth of FPR1-silenced GC cells by ablating their angiogenic activity.
Our data indicate that FPR1 signaling activates a pro-resolving program in GC cells that inhibits angiogenesis and growth.
Results
FPR1 controls ALOX5 and ALOX15 expression and the production of SPMs in GC cells
To study whether pro-resolving pathway components are involved in the FPR1-mediated anti-angiogenic and tumor suppressor activity of GC, we used our previously generated GC cell lines, namely, FPR1-silenced AGS (AGS shFPR1) and MKN45 ectopically expressing FPR1 (MKN45 FPR1) or their relative controls (AGS shCTR and MKN45 pcDNA).7
RvD1 and LXB4 levels were significantly lower in AGS shFPR1 cells than in AGS shCTR cells. Consistently, MKN45 cells overexpressing FPR1 released higher amounts of RvD1 and LXB4 than did empty vector-transfected cells (Fig. 1B). RvD1 and LXB4 syntheses were reduced in shFPR1 but not in shFPR2 or shFPR3 AGS cells (not shown), which indicates that FPR1 plays a non-redundant role in controlling SPM production, as already observed for its tumor suppressor function.7 We evaluated the lipidomic profiles of controls and FPR1-depleted/overexpressing GC cell supernatants by liquid chromatography tandem-mass spectrometry (LC-MS/MS) (Fig. S1). Compared to controls, AGS shFPR1 cells produced significantly lower levels of SPMs, including 18-HEPE, a resolvin precursor, and LXB4 (Fig. S1A). MKN45 FPR1 cells released significantly higher levels of RvD3 and LXB4 than did control cells (Fig. S1A). LXA4 levels did not differ significantly among GC cells expressing different levels of FPR1 (Fig. S1B). SPM expression parallels the downregulation of pro-inflammatory eicosanoids.4 Consistently, AGS shFPR1 cells synthesized significantly higher amounts of LTB4 and PGE2 compared to shCTR cells, whereas MKN45 FPR1 cells released lower levels of PGE2 compared to empty vector transfected cells, as assessed by EIA (Fig. 1C).
The levels of the enzymes involved in SPM synthesis (ALOX5, ALOX15A, and ALOX15B)10,11 were significantly lower in FPR1-silenced GC cells than in shCTR cells, both at mRNA (Fig. 1D) and protein (Fig. 1E) level. The expression of SPM receptors GPR32, ChemR23, and BLT18 was lower in AGS shFPR1 cells than in controls (Fig. S1C and D). On the other hand, MKN45 FPR1 cells produced significantly higher levels of pro-resolving enzymes (Fig. 1D and E) and of SPM receptors (Fig. S1C and D). These data were reproduced on one more clone and a mass population of both AGS shFPR1 and MKN45 FPR1 GC cells (Fig. S2).
To approach the FPR1-SPMs association from a pharmacologic standpoint, we treated AGS and MKN45 cells with fMLF (10−9 M), which is an FPR1 agonist, or with cyclosporine H (CsH 800 nM), which is an FPR1 inverse agonist12 and verified their ALOX5/15 expression, SPM release, and synthesis of pro-angiogenic mediators (VEGF-A, -B, -C, -D, Ang1, and CXCL1). The release of RvD1 and LXB4 was significantly higher in AGS treated with fMLF (12 h) than in untreated cells. In contrast, RvD1 and LXB4 release was significantly lower in cells treated with CsH (12 h) than in controls (Fig. 2A). fMLF induced, whereas CsH reduced, ALOX5, ALOX15A, and ALOX15B mRNA expression in AGS and MKN45 cells (Fig. 2B). These observations were confirmed at the protein level (Fig. 2C). mRNA levels of pro-angiogenic molecules were significantly reduced by fMLF and induced by CsH treatment (Fig. 2D).
Figure 2.
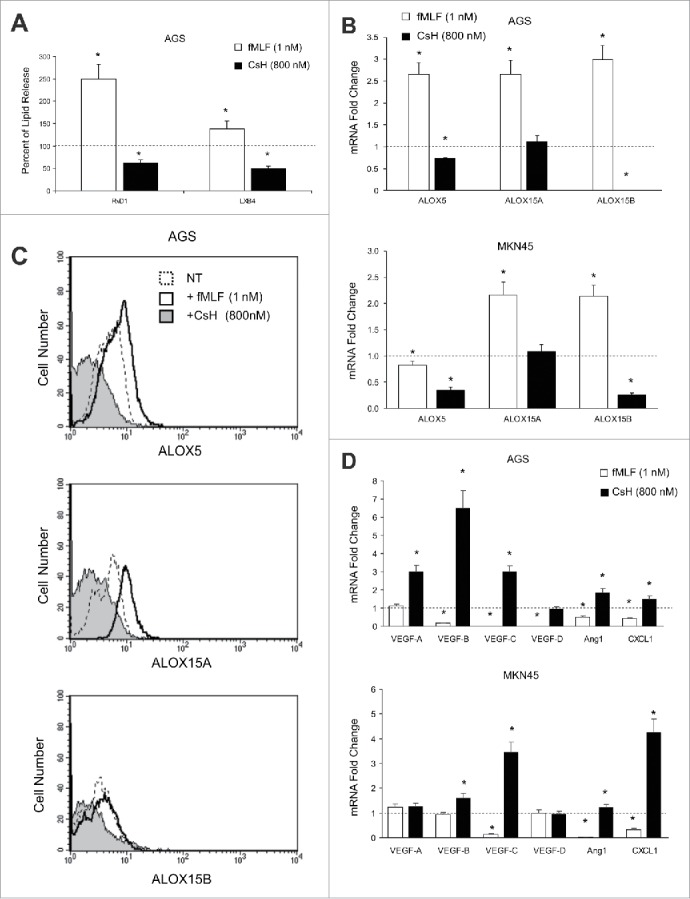
Effects of FPR1 pharmacologic modulation on SPM biosynthesis. (A) EIA assays showing that fMLF (agonist to FPR1-10−9 M) treatment of AGS significantly increases RvD1 and LXB4 release compared to untreated cells (dotted line). In contrast, CsH (inverse agonist to FPR1-800 nM) treatment significantly reduced RvD1 and LXB4 release compared to controls (dotted line). Data are represented as mean ± SD of three independent experiments. *p < .05 compared to untreated cells. (B) fMLF induced, whereas CsH inhibited, ALOX5, ALOX15A, and ALOX15B mRNA expression in AGS and MKN45 cells. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to untreated cells (dotted line). (C) fMLF induced, whereas CsH inhibited, ALOX5, ALOX15A, and ALOX15B protein expression in AGS cells, as evaluated by cytofluorimetric analysis. One representative experiment out of three is shown. (D) fMLF inhibited, whereas CsH induced, pro-angiogenic molecule mRNAs expression in AGS and MKN45 cells. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to untreated cells (dotted line).
SPMs inhibit the production of pro-angiogenic factors and restore the expression of ALOX5/15 and of SPM receptors in GC cells
To assess whether SPMs (RvD1 and LXB4) block the pro-angiogenic activity of GC cells, we treated or not AGS cells with RvD1 (1 nM) or LXB4 (1 nM), and evaluated the levels of pro-angiogenic factor mRNAs. RvD1 and LXB4 suppressed pro-angiogenic factor mRNA expression in GC cells (Fig. 3A). RvD1 treatment of GC cells significantly reduced VEGF-A release both in basal conditions and upon WKYMVm stimulation (pro-inflammatory FPR2/3 ligand) (Fig. 3B)7 Similar results were obtained with LXB4 (not shown). This effect was significantly more efficient in GC cells expressing low levels of FPR1 (AGS shFPR1 and MKN45 pcDNA) (Fig. 3A and B). Interestingly, RvD1 treatment restored, and in some cases increased above basal levels, the expression of ALOX5, ALOX15A, ALOX15B, and the SPM receptors (GPR32, ChemR23, and BLT1) at mRNA (Fig. 3C) and protein level (Fig. 3D and E) thereby triggering a positive feed-forward loop. These responses of AGS shCTR to RvD1 were significantly less efficient than that of AGSshFPR1 cells (Fig. 3C). Similar results were obtained with LXB4 (not shown).
Figure 3.
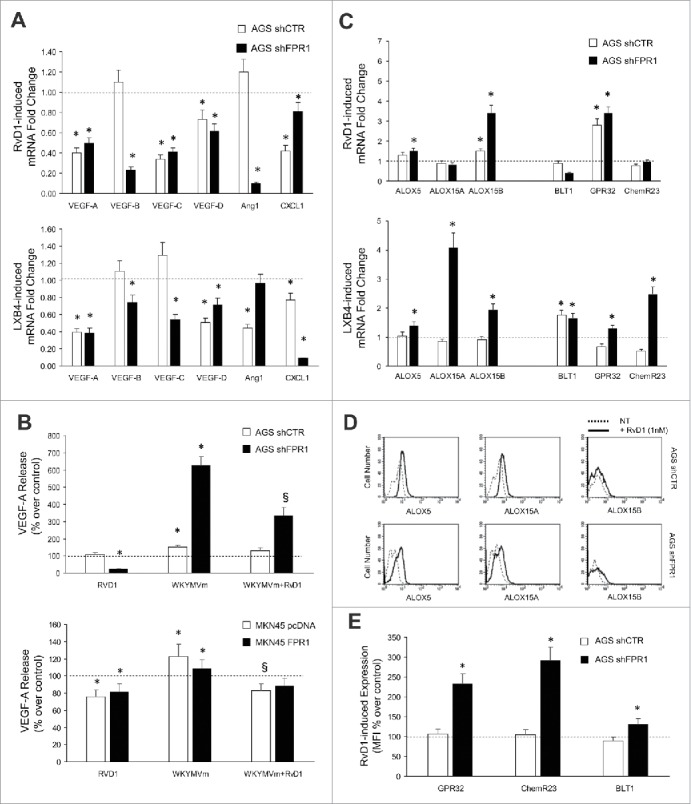
Anti-angiogenic effects of SPMs in GC. (A) RvD1 (1 nM) and LXB4 (1 nM) treatment significantly reduced mRNA expression of pro-angiogenic molecules (VEGF-A, -B, -C, -D, Ang1, and CXCL1) in AGS shCTR and shFPR1 cells. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to the relative untreated control (dotted line). (B) Reduction of spontaneous and WKYMVm-induced VEGF-A release upon RvD1 treatment of the indicated GC cells. Data are represented as mean ± SD of three independent experiments. *p < .05 vs. the relative untreated control (dotted line). (C) RvD1 and LXB4 treatment significantly induced the mRNA expression of the enzymes (ALOX5, ALOX15A, and ALOX15B) and receptors (BLT1, GPR32, and ChemR23) involved in pro-resolving pathways in AGS shCTR and shFPR1 cells. Data are represented as mean ± SD of three independent experiments. *p < .05 vs. the relative untreated control (dotted line). (D) RvD1 treatment significantly induced ALOX5, ALOX15A, and ALOX15B protein expression in AGS shCTR and shFPR1 cells, as evaluated by cytofluorimetric analysis. One representative experiment out of three is shown. (E) RvD1 treatment significantly induced GPR32, ChemR23, BLT1 protein expression in AGS shCTR and shFPR1 cells, as assessed by cytofluorimetric analysis. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to the relative untreated control (dotted line).
ALOX15- and GPR32-silencing increase angiogenesis and tumorigenesis of GC cells thereby mimicking the FPR1-silencing phenotype
To evaluate whether SPM pathway components are necessary to control the angiogenic activity of GC cells, we stably transfected AGS cells with vectors expressing shRNAs targeting ALOX5, ALOX15, GPR32, or control non-targeting shRNAs (shCTR). ALOX5-silencing induced a specific reduction of ALOX5 mRNA levels in AGS cells. However, AGS shALOX5 cells overexpressed ALOX15A and ALOX15B mRNAs (Fig. S3A). Transfection of shRNAs targeting ALOX15 significantly reduced ALOX15A and ALOX15B levels in AGS cells, but did not modify ALOX5 mRNA (Fig. S3A). The effects of shRNAs were confirmed at the protein level (Fig. S3B). shALOX15 and, to a lesser extent shALOX5, impaired AGS cell growth and survival, as did FPR1 silencing7 (not shown). shALOX5 and shALOX15 AGS cells (three clones/each) constitutively produced lower amounts of RvD1 and increased amounts of PGE2 and LTB4 versus controls, as did shFPR1 cells. LXB4 release was reduced in shALOX15, but not in shALOX5 cells (Fig. S3C). GPR32 and ChemR23 protein expressions were lower in AGS shALOX5 and shALOX15 cells than in controls (not shown). AGS shALOX5 and shALOX15 cells constitutively synthesized higher levels of VEGF-C and VEGF-D mRNAs and released increased amounts of VEGF-A than did controls (Fig. S3D).
To evaluate their tumorigenic potential, we injected AGS shCTR cells or ALOX-silenced clones subcutaneously in athymic mice. AGS shCTR and shALOX5 tumors reached a volume of about 1 mm3 at day 7, and regressed thereafter (Fig. 4A). In contrast, AGS shALOX15 cells formed larger non-regressing tumors, i.e., 2.8 mm3 (Fig. 4A), again replicating the phenotype of FPR1-silenced cells.7 The tumor growth advantage of shALOX15 cells was due to their higher vessel density compared to controls (Fig. 4B). No differences were detected in Ki-67+ and cleaved caspase 3+ cells, evaluated far from the necrotic areas, among shCTR, shALOX5, and shALOX15 cell xenografts (Fig. 4B). Our in vivo observations are corroborated by data, publicly available in the cBioPortal for Cancer Genomics database (http://www.cbioportal.org),13,14 reporting the RNAseq analysis of 295 human GC samples. These data show that enhanced mRNA expression of ALOX15, but not of ALOX5, was significantly associated with overall survival status and disease-free status15 in GC patients (Fig. 4C).
Figure 4.
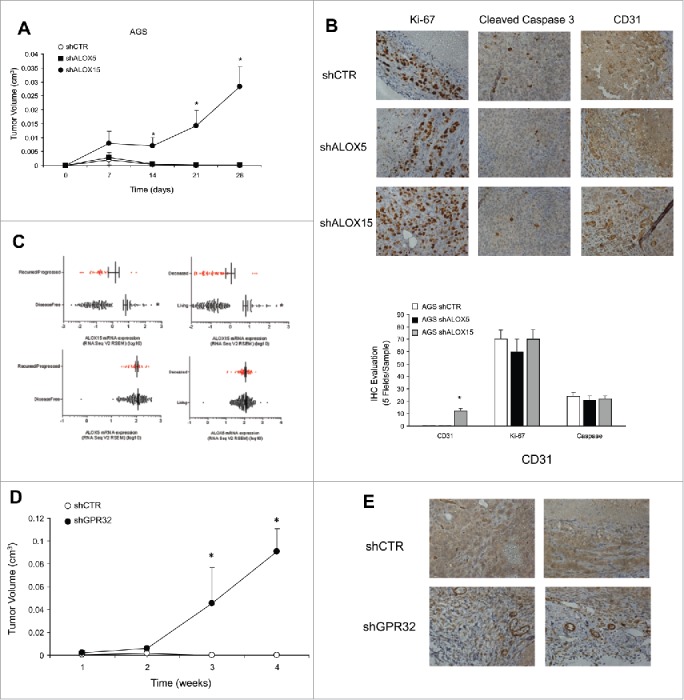
ALOXs and GPR32 involvement in GC angiogenic response. (A) Tumor growth curves of AGS shCTR, shALOX5 (three clones) and shALOX15 (three clones) xenografts in immunodeficient mice. *p < .05 compared to shCTR xenografts. (B) Representative images and quantification (five fields/sample) of the proliferation index (Ki-67), vessel density (CD31), and apoptotic rate (Cleaved Caspase 3), assessed by immunohistochemistry, of shCTR, shALOX5, and shALOX15 cell xenografts harvested 28 d post-inoculation. *p < .05 compared to shCTR xenografts. (C) ALOX15 and ALOX5 mRNA expression levels of 295 patients affected by gastric adenocarcinoma stratified for disease-free and overall survival status. *p < .05 between the two groups. (D) Tumor growth curves of AGS shCTR and shGPR32 xenografts (average of three clones) cells in immunodeficient mice *p < .05 compared to shCTR xenografts. (E) Representative images of the vessel density (CD31), assessed by immunohistochemistry, of shCTR and shGPR32 AGS cell xenografts harvested 28 d post-inoculation.
To analyze the effects of GPR32 silencing on the GC cell phenotype, we selected various AGS shGPR32 clones. The low levels of GPR32 were associated with a significant reduction of growth ability and with an increased apoptotic rate of GC cells in culture (not shown). The reduced level of GPR32 in AGS GC cells caused a significant reduction of ALOX5 and ALOX15A/B expression levels (Fig. S3E) and an increased expression of pro-angiogenic mediators at mRNA (Fig. S3E) and protein (Fig. S3F) level vs. shCTR cells. RvD1 did not reduce angiogenic factor mRNAs in shGPR32 cells (Fig. S3G). When xenotransplanted into nude mice, AGS shGPR32 formed significantly larger tumors than formed by shCTR cells (Fig. 4D). Consistently, the vessel density of shGPR32 xenografts was significantly higher than that of controls (Fig. 4E). No differences were detected in Ki-67+ and cleaved caspase 3+ cells in shCTR and shGPR32 xenografts (not shown).
FPR1-mediated suppression of angiogenesis in GC cells requires ALOX5, ALOX15, and GPR32
To assess whether FPR1-mediated suppression of angiogenesis requires ALOX5 and/or ALOX15, we stimulated with fMLF (10−9 M) GC cells in which ALOX expression/activity was abolished. As shown in Fig. 5A, fMLF significantly reduced the mRNA levels of pro-angiogenic factors (VEGF-B, VEGF-C, and CXCL1) in AGS shCTR, but not in shALOX5 and shALOX15 AGS cells. Similar results were obtained in AGS cells in which ALOXs were pharmacologically inhibited by 10 μM nordihydroguaiaretic acid-(NDGA) (Fig. 5B).16 These observations suggest that ALOX5 and ALOX15 are required to trigger the anti-angiogenic activity of FPR1 in GC cells.
Figure 5.
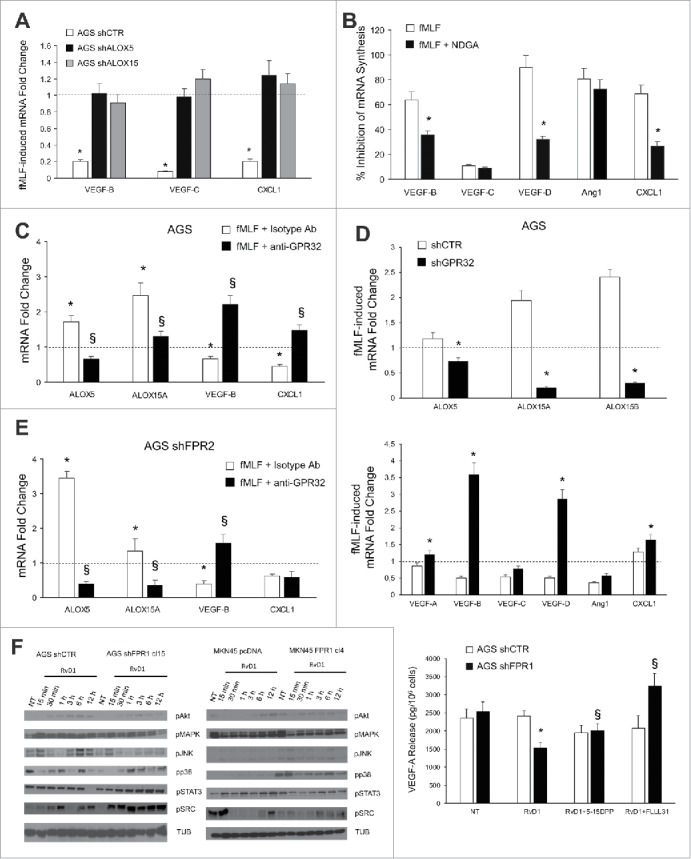
ALOXs and GPR32 are required for FPR1 tumor suppressor role. (A) VEGF-B, -C, and CXCL1 mRNA synthesis was inhibited in shCTR, but not in shALOX5 or shALOX15 AGS cells upon fMLF (10−9 M) treatment. Data are represented as mean ± SD of three independent experiments. *p < .05 vs. the relative untreated cells (dotted line). (B) Pre-treatment of AGS cells with NDGA (ALOX inhibitor) reverted the ability of fMLF to inhibit the mRNA synthesis of pro-angiogenic molecules. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to fMLF treated cells. (C) In AGS cells, fMLF induced ALOX5 and ALOX15A mRNA overexpression and concomitant VEGF-B and CXCL1 mRNA downregulation. These effects were reverted by a neutralizing GPR32 antibody. An isotype-matched antibody was used as a control. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to untreated cells (dotted line). §p < .05 compared to isotype-matched control. (D) fMLF significantly induced ALOX5, ALOX15A, and ALOX15B mRNA levels and significantly reduced the mRNA levels of pro-angiogenic mediators in AGS shCTR, but not in shGPR32 cells. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to shCTR cells. (E) A neutralizing GPR32 antibody inhibited fMLF-induced ALOX5 and ALOX15A mRNA overexpression and concomitant VEGF-B and CXCL1 mRNA downregulation in AGS shFPR2 cells. An isotype-matched antibody was used as a control. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to untreated cells (dotted line). §p < .05 compared to isotype-matched control. (F) Activation kinetics of AKT, MAPK, JNK, p38, STAT3, and SRC in AGS shCTR, AGS shFPR1, MKN45 pcDNA, MKN45 FPR1 cells assessed by western blot for their phosphorylated forms. Two STAT3 inhibitors (5-15 DPP and FLLL31) reverted the anti-angiogenic effects of RvD1 in AGS shFPR1 cells. Data are represented as mean ± SD of three independent experiments. *p < .05 compared to untreated cells (NT). §p < .05 compared to RvD1 treated cells.
To verify the requirement of the RvD1 pathway for FPR1-mediated anti-angiogenic activity, we stimulated parental GC cells with fMLF in the presence or absence of a GPR32-neutralizing antibody. fMLF (10−9 M) induced ALOX5 and ALOX15A mRNA expression and concomitant VEGF-B and CXCL1 mRNA downregulation in AGS. These effects were reverted by the GPR32-blocking antibody (5 μg/mL) (Fig. 5C). Similar results were obtained in MKN45 cells (Fig. S4A). Consistently, fMLF did not induce ALOXs or suppress pro-angiogenic mediators in AGS shGPR32 cells (Fig. 5D), although shCTR and shGPR32 cells released comparable amounts of RvD1 both in basal conditions and upon fMLF stimulation (not shown). We then assessed the involvement of FPR2, the other RvD1 receptor.8 fMLF stimulation of AGS shFPR2 cells induced ALOX5 and ALOX15A mRNA expression, and VEGF-B and CXCL1 downregulation, and these effects were reverted by the anti-GPR32 blocking antibody (Fig. 5E). Taken together, these results indicate that RvD1, by binding GPR32, but not FPR2, mediates FPR1's anti-angiogenic effects in GC cells.
We then asked which signaling pathways were involved in the anti-angiogenic response of GC cells to RvD1 and whether differences can be detected in FPR1-lacking cells compared to controls. We found that Akt, MAPK, JNK, and p38 signaling pathways were induced in GC cells treated with RvD1 (1 nM) for different times (15 min–12 h) without significant differences in the activation kinetic and strength between AGS shCTR and shFPR1 cells, or MKN45 pcDNA and FPR1 cells (Fig. 5F). In contrast, RvD1 treatment induced a significant STAT3 and SRC activation only in AGS shFPR1 and MKN45 pcDNA cells expressing lower levels of FPR1 compared to the relative control cells (Fig. 5F). RvD1-mediated reduction of VEGF-A release was reverted by pre-treatment of AGS shFPR1 cells with the two STAT3 inhibitors FLLL31 (10 μM) and 5–15DPP (15 μM)17,18 (Fig. 5F). Similar results were not detectable in AGS shCTR cells (Fig. 5F). STAT3 inhibition reverted RvD1 effects on CXCL1 and Ang1 mRNAs levels (Fig. S4B). Instead, when MAPK [U0 126 (25 μM)] or Akt [LY294002 (15 μM)] inhibitors7 were used, no effects on VEGF-A release were observed (Fig. S4C).
ω-6 and ω-3 diets reduce the tumorigenic potential of shFPR1 GC cells in vivo
Dietary interventions based on differential PUFA ω-3 or ω-6 intake affect AA-, EPA-, and DHA metabolism and the derived SPM endogenous production in mice (Fig. 1A).9 To assess the effects exerted by ω-6- or ω-3-enriched diets on GC cell growth, we xenotransplanted AGS shCTR or shFPR1 cells in immunocompromised mice randomly fed a classic, an ω-6/ω-3 balanced (grape seed-colza oils 50/50%, CTRL), an ω-6 enriched (grape seed oil 100%, ω-6), or an ω-3 enriched (colza/fish oils 80/20%, ω-3) diet, 2 weeks before and 4 weeks after xenotransplantation. No differences were detected between animals fed a classic diet and those fed a balanced diet (not shown) demonstrating that a 5% PUFA-enriched diet did not change the tumorigenic potential of GC cells. As shown previously,7 AGS shCTR cells formed small tumors that regressed in mice fed a balanced, ω-3-, or ω-6-enriched diet, without statistical differences among groups (Fig. 6A). In contrast, AGS shFPR1 cells formed tumors that progressively increased in size (Fig. 6A).7 Both ω-3 and ω-6-enriched diets significantly suppressed AGS shFPR1 xenograft growth. However, growth inhibition was significant at day 7 post-injection in ω-6-fed mice vs. day 14 in ω-3-fed mice (Fig. 6A). The administration of an ω-3 or ω-6 enriched diet to AGS shFPR1 xenotransplanted mice abolished statistical differences in tumor growth curves between shFPR1 and shCTR cells.
Figure 6.
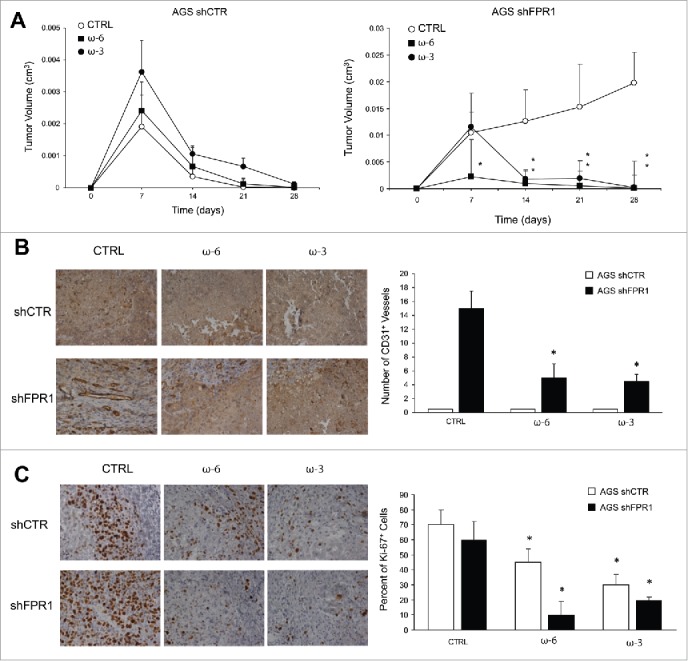
Effects of ω-6 and/or ω-3 increased consumption on GC growth/angiogenesis in a xenotransplantation mouse model. (A) Tumor growth of AGS shCTR and shFPR1 xenografts in CD1 nu/nu mice fed an ω-6/ω-3 balanced (grape seed/colza oils 50/50%, CTRL), an ω-6 enriched (grape seed oil 100%, ω-6), or an ω-3 enriched (colza/fish oils 80/20%, ω-3) diet. *p < .05 vs. CTRL diet. (B) Vessel density (CD31) assessed by immunohistochemistry of shCTR and shFPR1 cell xenografts harvested 28 d post-inoculation from mice fed the three diets as in panel A. Representative images and the relative quantifications (five fields/sample) are shown. *p < .05 vs. the relative CTRL diet. (C) Proliferation index (Ki-67) assessed by immunohistochemistry of shCTR and shFPR1 cell xenografts harvested 28 d post-inoculation from mice fed with the three diets. Representative images and the relative quantifications (five fields/sample) are shown.*p < .05 vs. the relative CTRL diet.
Vessel density was higher in shFPR1 xenografts than in shCTR xenografts (Fig. 6B); ω-6 and ω-3 diets reverted this difference (Fig. 6B). Vessel density was unchanged in shCTR xenografts irrespective of the diet administered (Fig. 6B). Both ω-6 and ω-3 diets significantly reduced Ki-67 staining of shFPR1, and, to a lesser extent, of shCTR excised tumors (Fig. 6C). Diet composition did not affect apoptotic rate as assessed by cleaved caspase-3 evaluation (not shown).
To investigate the effect of dietary intervention on mouse serum lipid composition, we profiled fatty acids by LC-MS/MS. Concentrations of AA were significantly higher in mice fed an ω-6 diet than in those fed a balanced or an ω-3 diet (Table 1). Consistently, EPA ad DHA concentrations were higher in ω-3-fed mice than in the other two groups of mice (Table 1). The concentrations of LXB4 were significantly higher in the sera of ω-6-fed mice than in controls. Concentrations of RvD1, RvD3, and RvE2 were significantly higher in the serum of mice fed an ω-3 diet than in controls. Interestingly, the ω-3 diet significantly reduced PGE2, LTB4, and TXB2 serum concentrations vs. controls (Table 1). Lipid serum concentrations did not differ among mice fed the same diet but xenotransplanted with shCTR or shFPR1 AGS cells (not shown).
Table 1.
Lipidomic profile of mice fed ω-6 and/or ω-3 enriched diets.
| DIET |
|||
|---|---|---|---|
| CTRL | ω-6 | ω-3 | |
| AA | 6353.6 ± 1444.1 | 16991 ± 925.8* | 5677.2 ± 1295 |
| PGD2 | 281 ± 43.9 | 237.3 ± 33.3 | 327 ± 75.1 |
| PGE2 | 183.5 ± 45.1 | 172 ± 66.7 | 92.7 ± 20* |
| PGF2α | 76 ± 33 | 57 ± 23 | 47.5 ± 14.7 |
| TXB2 | 740.8 ± 64.1 | 911.4 ± 147.1 | 466 ± 148.7* |
| LTB4 | 740.8 ± 141.2 | 775 ± 145.9 | 355.8 ± 109.8* |
| LXA4 | 5748.6 ± 258.5 | 5012.6 ± `185.1 | 5017 ± 230.5 |
| LXB4 | 134.3 ± 20.9 | 318.7 ± 33.4* | 183.4 ± 21.5 |
| EPA | 1476.2 ± 280.6 | 2167.8 ± 671.3 | 5028.1 ± 462.8* |
| RvE1 | 108.3 ± 29.9 | 32.6 ± 17.1 | 130.8 ± 26.4 |
| RvE2 | 3251.5 ± 532.2 | 3797 ± 474.2 | 8862.7 ± 576.1* |
| RvE3 | 213.4 ± 44.6 | 162.3 ± 42.5 | 249.7 ± 73.6 |
| DHA | 8886.4 ± 1586.2 | 10220.2 ± 1364.6 | 17447.5 ± 772* |
| RvD1 | 45.8 ± 19.5 | 52.3 ± 4.7 | 95.7 ± 12.5* |
| RvD2 | 46.6 ± 14.6 | 47.5 ± 4.9 | 86 ± 29.4 |
| RvD3 | 2664.7 ± 365.9 | 3252 ± 446.5 | 5101.1 ± 380.3* |
| RvD5 | 30.5 ± 5.9 | 20 ± 8.2 | 46.7 ± 18.2 |
| RvD6 | 56.7 ± 15.8 | 40.7 ± 17.4 | 98.2 ± 25.6 |
| PD1 | 56.6 ± 6.3 | 39.5 ± 6.3 | 56.5 ± 28 |
Lipid concentration in serum of mice fed the indicated diets (10 mice/group). Values are reported as nM.
p < .05 vs. the CTRL diet group.
To assess whether the effects exerted by an ω-3 or ω-6 diet on the proliferation rate of GC xenografts was a direct event or whether it was indirectly due to differences in tumor vascularization levels, we studied the effects of RvD1 (1 nM), LXB4 (1 nM), AA (20 μM), DHA (20 μM), and EPA (20 μM) on AGS shCTR and shFPR1 cell proliferation in vitro. We found no difference in BrdU incorporation between treated and untreated cells at the concentrations of lipids above mentioned (Fig. S5). AA, DHA, and EPA significantly reduced GC cell viability when used at higher concentrations (50 μM), as already reported by others.19-21 However, these concentrations were never reached in vivo in diet-fed mice.9,22 (Table 1).
Discussion
The role of FPR1 in cancer is complex. In certain cancer types, FPR1 exerts tumor-promoting activities by stimulating the motility and growth of cancer cells23-26 However, in mouse liver and colon, FPR1- or FPR2-genetic deletion abolishes epithelial homeostasis, increases inflammation after injury and, in colonic mucosae, increases tumorigenesis.27,28 Recent studies showed that decreased FPR1 concentrations in tumor-infiltrating antigen-presenting cells compromise chemotherapy-induced antitumor immunity.29,30 In human GC, high FPR1 expression, evaluated by immunohistochemistry, has been correlated with advanced tumor stage and poor overall survival.31 However, an FPR1 polymorphism linked to reduced FPR1 activity has been associated with an increased risk of stomach cancer in humans.32,33
Here, we provide evidence that FPR1, expressed by GC cells, plays a tumor suppressor role by constitutively activating an inflammation resolution program that includes the expression of ALOX5/15, SPMs (RvD1 and LXB4), and SPM receptors (BLT1, ChemR23, and GPR32). FPR1 loss/inhibition suppresses this program and induces an inflammatory/angiogenic phenotype in GC cells. Interestingly, the GC-associated FPR1 polymorphism was also associated with periodontitis,34 a disease whose pathogenesis has been linked to defective pro-resolving pathways and in which SPMs exert protective effects.35-38
We demonstrate that the anti-angiogenic potential of SPMs, already reported in other contexts,39 was more evident in cells expressing low levels of FPR1, despite their lower levels of SPM receptors (BLT1, GPR32, and ChemR23), vs. controls. This effect might be due to constitutively high levels of RvD1 and LXB4 produced by control cells, which could saturate/desensitize their receptors thereby rendering these cells less responsive to exogenous SPM stimulation.
Various studies have recognized that ALOXs exert tumor-promoting or tumor-suppressive activities.40 Our study introduces a new concept in this field, namely, that another component of pro-resolving pathways, i.e., the Resolvin D1 receptor GPR32,8 can exert tumor suppressor activity in GC. The LXB4 receptor remains unidentified; consequently, we were unable to investigate its role in GC. We demonstrate that the ALOX enzymes and GPR32 are induced by FPR1 and necessary for FPR1-mediated SPM production and anti-angiogenic activity in GC. In vivo, vessel density and tumor growth were higher in shALOX15 and shGPR32, but not in shALOX5, xenografts than in controls, despite their comparable angiogenic potential in culture. This discrepancy could be explained by the compensatory upregulation of ALOX15 expression in shALOX5 cells that enabled the production of other SPMs, including LXB4. Thus, qualitative or quantitative differences in SPM production between shALOX5 and shALOX15 cells and/or their interaction with tumor stroma might result in a different angiogenic potential in vivo. Be that as it may, in line with our data, an RNAseq analysis showed that higher ALOX15, but not ALOX5, mRNA expression was significantly associated with better overall survival of GC patients (TCGA, http://www.cbioportal.org).13-15
Several studies have demonstrated that ω-3 PUFA plays a direct anti-tumor or an adjuvant role in various cancers41,42 by modulating cell proliferation or apoptosis43-45 in the high micromolar range.13,19,20 We believe that, in our system, the effects of an ω-3 or ω-6 diet on xenograft growth were predominantly due to differences in vessel density rather than to a direct effect on cell proliferation. In fact, in the low micromolar range, which corresponds to the concentrations we found in vivo upon dietary modification,7,22 we found that PUFA suppressed the angiogenic response but did not directly affect GC cell proliferation in culture. In the present study, vessel density in shCTR xenografts did not differ among mice fed different diets, although cell proliferation was slightly, albeit significantly, reduced in mice fed an ω-3 or ω-6 diet. We ascribe this phenomenon to the very low in vivo angiogenic potential and to the massive presence of necrotic areas in these tumors.7 However, it should be noted that since PUFA administration induces the production of many intermediate metabolites that exert anti-angiogenic and anti-proliferative effects in vivo,46 we cannot exclude the possibility that in the xenograft microenvironment, these intermediates, together with PUFA, could reach a concentration sufficient to induce xenograft growth inhibition by directly modulating cell proliferation.
Pattern recognition receptors trigger inflammation,47 but can also promote its resolution.12,48,49 This is particularly important for the gastrointestinal tract, which, being continuously exposed to food- and microbioma-derived antigens, is subject to a chronic low-level of mucosal inflammation50 that must be counteracted to avoid tissue damage. In injured mouse colonic mucosa, FPR1 promotes tissue restitution by recognizing the endogenous N-terminal-derived annexin A1 (Ac2-26) peptides51,52 or specific commensal microbiota strains.53 Our data suggest that FPR1 is constitutively active in GC cells, which implies the presence of an endogenous ligand endowed with pro-resolving activity. A possible candidate ligand is peptide Ac2-26 that derives from AnxA1, which, as we previously showed, is constitutively expressed in GC cells.7
In conclusion, we describe a novel molecular mechanism in GC, actively controlled by FPR1, that links PUFA metabolism and SPM biosynthesis with angiogenesis. This raises the possibility of new prognostic tools and therapeutic interventions for GC. It is feasible that FPR1 and/or components of pro-resolving pathways may represent novel risk factors or prognostic markers of GC. Administration of SPMs or pharmacologic stimulation of FPR1, ALOX15, or GPR32 could suppress GC angiogenesis and growth. Lastly, as SPM production can be largely determined by diet, increasing ω-3 or ω-6 consumption might be a tool in the management of GC.
Materials and methods
Cell culture
The AGS and MKN45 cell lines derived from poorly differentiated human gastric adenocarcinoma were grown as previously described.7 To generate AGS cells stably expressing ALOX5, ALOX15, or GPR32 shRNA, we used pools of five constructs (Qiagen, Valencia, CA, USA) containing 21-mer short hairpin RNAs (shRNA) directed to various coding regions of each target gene. Transfectants were selected in medium with 500 ng/mL puromycin.
RNA isolation and real-time PCR
Total RNA was isolated and retrotranscribed according to the manufacturer's instructions (Promega, Madison, WI, USA). Real-time quantitative PCR was performed on the CFX96 system (Bio-Rad, Hercules, CA, USA) using the PE SYBR Green PCR kit (Applied Biosystems, Grand Island, NY, USA). The target-specific primers used for real-time PCR are listed in Table S1. No-reverse transcribed mRNA samples served as a negative control. The expression levels of each target were calculated relative to that of control cells, arbitrarily considered equal to 1. Results were normalized to β-actin mRNA levels.
Flow cytometric analysis
Cells were incubated (30 min at 4°C) with specific or isotype control antibodies (Abs). ALOX5, ALOX15A, and ALOX15B Abs were from Santa Cruz Biotechnology (Dallas, TX, USA), anti-GPR32 was from Acris (Herford, Germany), anti-BLT1 from LSBio (Seattle, WA, USA), and anti-ChemR23 from MyBiosource (San Diego, CA, USA). Cells were analyzed with an FACS Calibur cytofluorimeter using CellQuest software (BD Biosciences, Mississauga, ON, Canada). When necessary, we performed cell membrane permeabilization using the Cytofix/Cytoperm kit (BD Biosciences).
Protein studies
Protein extractions, immunoblotting, and immunoprecipitation were carried out according to standard procedures. Anti-phospho-MAPK, -Akt, -p38, -JNK, -STAT3, -SRC antibodies (Abs) were from Cell Signaling Technology (Danvers, MA, USA). Anti-tubulin was from Sigma-Aldrich (St. Louis, MO, USA), and secondary anti-mouse and anti-rabbit Abs coupled to HRP were from Bio-Rad.
Xenografts in mice
Each group of 10 mice (4-week-old female CD1 nu/nu mice, Charles River, Wilmington, MA) was inoculated subcutaneously with shCTR, shFPR1, shALOX5, shALOX15, shGPR32 AGS cells (1×107 cells/mouse). Tumor diameters were measured at regular intervals with a caliper. Tumor volumes (V) were calculated with the formula: V = A×B2/2 (A = axial diameter; B = rotational diameter). This study was conducted according to Italian regulations for experimentation on animals; the study protocol was approved by the Italian Ministry of Health. Paraffin-embedded tumors were analyzed by immunohistochemistry with anti-Ki-67 antibody from Biocare Medical (Concord, CA), anti-cleaved caspase 3, and anti-CD31 from R&D Systems (Minneapolis, MN).
Fatty composition of diets
The three isocaloric diets containing 5% fat (w/w) were designed as previously described.9,22 Pellets were prepared by Mucedola (Milan, Italy). We used grape seed and colza oils (50/50%) to prepare the pellets for the control diet (CTRL), grape seed oil (100%) for the ω-6 (ω-6), and colza oil/fish oils (80/20%) for an ω-3 fatty acid-enriched diet (ω-3). The oils were obtained from Sigma-Aldrich (St. Louis, MO). Pellets were stored under vacuum at −20°C. The diets were changed twice a week in each animal cage to avoid oxidative degradation of lipids.
ELISA and EIA assays
VEGF-A contents in culture supernatants were measured in duplicate determinations with a commercially available ELISA (R&D Systems). RvD1, LTB4, PGE2, and LXB4 contents in culture supernatants were measured in triplicate determinations with a commercially available EIA (Cayman Chemical, Ann Arbor, MI).
LC-MS/MS instrumentation and conditions
To extract the lipids, 120 μL samples (mice serum or cell culture supernatants), together with deuterated internal standards, were loaded onto solid phase extraction Waters Oasis HLB cartridges (60 mg sorbent, 30 μm particle size), and eluted with 2 mL methanol and 2 mL ethyl acetate into polypropylene tubes containing 6 μL of a glycerol solution (30% in methanol). The SPE eluates were dried and residues were then reconstituted in 100 μL methanol. 5 μL of methanol extract were analyzed by using a 4000QTrap mass spectrometer (Applied Biosystems) coupled to a 1,100 nanoHPLC system (Agilent Technologies, Waldbronn, Germany). The lipids were separated by using a micro-C18 column (10 cm x 1.0 mm, 5u). The mobile phase was generated by mixing eluent A (0.1% acetic acid) and eluent B (acetonitrile/isopropanol 50/50) and the flow rate was 30 nL/min. Starting condition was 20% to 95% B in 5 min. Tandem mass spectrometry was performed using a turbo ion spray source operated in a negative mode (curtain gas 20psi, GS 1/2 50/50psi, ion spray voltage −5,500 V, DP, −60 V; Dwell 25 ms and temperature of 550°C), and the multiple reaction monitoring mode was used to detect a unique product ion arising from collision-induced fragmentation of the protonated parent compound as reported in Table S2.
Statistical analysis
Values from groups were compared by using the paired Student t test or the Duncan test. A p value < 0.05 was considered statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
The authors whose names are listed above inform the Editors-in-Chief, Deputy Editors, Senior Editors, or Scientific Editors that they have NO relationships that they believe could be construed as resulting in an actual, potential, or perceived conflict of interest with regard to this manuscript.
Acknowledgments
We acknowledge Professor Gianni Marone for critical reading of the manuscript, Dr. Felice Rivellese who provided an insight that assisted the research and Jean Ann Gilder (Scientific Communication Srl, Naples, Italy) for writing assistance.
Funding
Grant Movie of the POR rete delle Biotecnologie in Campania, FIRB Merit grant of MIUR; Istituto Superiore di Oncologia grant (MIUR PON01_02782/12).
References
- 1.Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci 2012; 125(Pt 23):5591-6; PMID:23420197; http://dx.doi.org/ 10.1242/jcs.116392 [DOI] [PubMed] [Google Scholar]
- 2.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014; 510(7503):92-101; PMID:24899309; http://dx.doi.org/ 10.1038/nature13479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 2008; 8(5):349-61; PMID:18437155; http://dx.doi.org/ 10.1038/nri2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serhan CN, Chiang N, Dalli J. The resolution code of acute inflammation: novel pro-resolving lipid mediators in resolution. Semin Immunol 2015; 27(3):200-15; PMID:25857211; http://dx.doi.org/ 10.1016/j.smim.2015.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 2009; 61(2):119-61; PMID:19498085; http://dx.doi.org/25263443 10.1124/pr.109.001578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dufton N, Perretti M. Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacol Ther 2010; 127(2):175-88; PMID:20546777; http://dx.doi.org/25263443 10.1016/j.pharmthera.2010.04.010 [DOI] [PubMed] [Google Scholar]
- 7.Prevete N, Liotti F, Visciano C, Marone G, Melillo RM, de Paulis A. The formyl peptide receptor 1 exerts a tumor suppressor function in human gastric cancer by inhibiting angiogenesis. Oncogene 2015; 34(29):3826-38; PMID:25263443; http://dx.doi.org/ 10.1038/onc.2014.309 [DOI] [PubMed] [Google Scholar]
- 8.Cash JL, Norling LV, Perretti M. Resolution of inflammation: targeting GPCRs that interact with lipids and peptides. Drug Discov Today 2014; 19(8):1186-92; PMID:24993159; http://dx.doi.org/ 10.1016/j.drudis.2014.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gobbetti T, Ducheix S, le Faouder P, Perez T, Riols F, Boue J, Bertrand-Michel J, Dubourdeau M, Guillou H, Perretti M et al.. Protective effects of n-6 fatty acids-enriched diet on intestinal ischaemia/reperfusion injury involve lipoxin A4 and its receptor. Br J Pharmacol 2015; 172(3):910-23; PMID:25296998; http://dx.doi.org/ 10.1111/bph.12957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivanov I, Kuhn H, Heydeck D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 2015; 573(1):1-32; PMID:26216303; http://dx.doi.org/ 10.1016/j.gene.2015.07.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brash AR, Boeglin WE, Chang MS. Discovery of a second 15S-lipoxygenase in humans. Proc Natl Acad Sci U S A 1997; 94(12):6148-52; PMID:9177185; http://dx.doi.org/ 10.1073/pnas.94.12.6148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prevete N, Liotti F, Marone G, Melillo RM, de Paulis A. Formyl peptide receptors at the interface of inflammation, angiogenesis and tumor growth. Pharmacol Res 2015; 102:184-91; PMID:26466865; http://dx.doi.org/ 10.1016/j.phrs.2015.09.017 [DOI] [PubMed] [Google Scholar]
- 13.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6(269):pl1; PMID:23550210; http://dx.doi.org/ 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2(5):401-4; PMID:22588877; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research N Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513(7517):202-9; PMID:25079317; http://dx.doi.org/ 10.1038/nature13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu JM, Nurko J, Weakley SM, Jiang J, Kougias P, Lin PH, Yao Q, Chen C. Molecular mechanisms and clinical applications of nordihydroguaiaretic acid (NDGA) and its derivatives: an update. Med Sci Monit 2010; 16(5):RA93-100; PMID:20424564 [PMC free article] [PubMed] [Google Scholar]
- 17.Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E, Pandit B, Ihnat MA, Shenoy SS, Kulp S et al.. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res 2010; 70(6):2445-54; PMID:20215512; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uehara Y, Mochizuki M, Matsuno K, Haino T, Asai A. Novel high-throughput screening system for identifying STAT3-SH2 antagonists. Biochem Biophys Res Commun 2009; 380(3):627-31; PMID:19285012; http://dx.doi.org/ 10.1016/j.bbrc.2009.01.137 [DOI] [PubMed] [Google Scholar]
- 19.Finstad HS, Drevon CA, Kulseth MA, Synstad AV, Knudsen E, Kolset SO. Cell proliferation, apoptosis and accumulation of lipid droplets in U937-1 cells incubated with eicosapentaenoic acid. Biochem J 1998; 336 (Pt 2):451-9; PMID:9820824; http://dx.doi.org/ 10.1042/bj3360451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun SN, Jia WD, Chen H, Ma JL, Ge YS, Yu JH, Li JS. Docosahexaenoic acid (DHA) induces apoptosis in human hepatocellular carcinoma cells. Int J Clin Exp Pathol 2013; 6(2):281-9; PMID:23330014 [PMC free article] [PubMed] [Google Scholar]
- 21.Peng Y, Zheng Y, Zhang Y, Zhao J, Chang F, Lu T, Zhang R, Li Q, Hu X, Li N. Different effects of omega-3 fatty acids on the cell cycle in C2C12 myoblast proliferation. Mol Cell Biochem 2012; 367(1-2):165-73; PMID:22610825; http://dx.doi.org/ 10.1007/s11010-012-1329-4 [DOI] [PubMed] [Google Scholar]
- 22.Ducheix S, Montagner A, Polizzi A, Lasserre F, Marmugi A, Bertrand-Michel J, Podechard N, Al Saati T, Chetiveaux M, Baron S et al.. Essential fatty acids deficiency promotes lipogenic gene expression and hepatic steatosis through the liver X receptor. J Hepatol 2013; 58(5):984-92; PMID:23333450; http://dx.doi.org/ 10.1016/j.jhep.2013.01.006 [DOI] [PubMed] [Google Scholar]
- 23.Khau T, Langenbach SY, Schuliga M, Harris T, Johnstone CN, Anderson RL, Stewart AG. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J 2011; 25(2):483-96; PMID:20930115; http://dx.doi.org/ 10.1096/fj.09-154096 [DOI] [PubMed] [Google Scholar]
- 24.Yang Y, Liu Y, Yao X, Ping Y, Jiang T, Liu Q, Xu S, Huang J, Mou H, Gong W et al.. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am J Pathol 2011; 179(3):1504-12; PMID:21782780; http://dx.doi.org/ 10.1016/j.ajpath.2011.05.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belvedere R, Bizzarro V, Popolo A, Dal Piaz F, Vasaturo M, Picardi P, Parente L, Petrella A. Role of intracellular and extracellular annexin A1 in migration and invasion of human pancreatic carcinoma cells. BMC Cancer 2014; 14:961; PMID:25510623; http://dx.doi.org/ 10.1186/1471-2407-14-961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snapkov I, Oqvist CO, Figenschau Y, Kogner P, Johnsen JI, Sveinbjornsson B. The role of formyl peptide receptor 1 (FPR1) in neuroblastoma tumorigenesis. BMC Cancer 2016; 16:490; PMID:27432059; http://dx.doi.org/ 10.1186/s12885-016-2545-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen K, Liu M, Liu Y, Yoshimura T, Shen W, Le Y, Durum S, Gong W, Wang C, Gao JL et al.. Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J Clin Invest 2013; 123(4):1694-704; PMID:23454745; http://dx.doi.org/ 10.1172/JCI65569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giebeler A, Streetz KL, Soehnlein O, Neumann U, Wang JM, Brandenburg LO. Deficiency of formyl peptide receptor 1 and 2 is associated with increased inflammation and enhanced liver injury after LPS-stimulation. PloS One 2014; 9(6):e100522; PMID:24956481; http://dx.doi.org/ 10.1371/journal.pone.0100522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baracco EE, Pietrocola F, Buque A, Bloy N, Senovilla L, Zitvogel L, Vacchelli E, Kroemer G. Inhibition of formyl peptide receptor 1 reduces the efficacy of anticancer chemotherapy against carcinogen-induced breast cancer. Oncoimmunology 2016; 5(6):e1139275; PMID:27471610; http://dx.doi.org/ 10.1080/2162402X.2016.1139275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vacchelli E, Ma Y, Baracco EE, Zitvogel L, Kroemer G. Yet another pattern recognition receptor involved in the chemotherapy-induced anticancer immune response: formyl peptide receptor-1. Oncoimmunology 2016; 5(5):e1118600; PMID:27467929; http://dx.doi.org/ 10.1080/2162402X.2015.1118600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng TY, Wu MS, Lin JT, Lin MT, Shun CT, Hua KT, Kuo ML. Formyl Peptide receptor 1 expression is associated with tumor progression and survival in gastric cancer. Anticancer Res 2014; 34(5):2223-9; PMID:24778024 [PubMed] [Google Scholar]
- 32.Otani T, Ikeda S, Lwin H, Arai T, Muramatsu M, Sawabe M. Polymorphisms of the formylpeptide receptor gene (FPR1) and susceptibility to stomach cancer in 1531 consecutive autopsy cases. Biochem Biophys Res Commun 2011; 405(3):356-61; PMID:21216225; http://dx.doi.org/ 10.1016/j.bbrc.2010.12.136 [DOI] [PubMed] [Google Scholar]
- 33.Seifert R, Wenzel-Seifert K. Defective Gi protein coupling in two formyl peptide receptor mutants associated with localized juvenile periodontitis. J Biol Chem 2001; 276(45):42043-9; PMID:11559706; http://dx.doi.org/ 10.1074/jbc.M106621200 [DOI] [PubMed] [Google Scholar]
- 34.Maney P, Emecen P, Mills JS, Walters JD. Neutrophil formylpeptide receptor single nucleotide polymorphism 348T>C in aggressive periodontitis. J Periodontol 2009; 80(3):492-8; PMID:19254133; http://dx.doi.org/ 10.1902/jop.2009.080225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pouliot M, Clish CB, Petasis NA, Van Dyke TE, Serhan CN. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry 2000; 39(16):4761-8; PMID:10769133; http://dx.doi.org/ 10.1021/bi992551b [DOI] [PubMed] [Google Scholar]
- 36.Van Dyke TE, Hasturk H, Kantarci A, Freire MO, Nguyen D, Dalli J, Serhan CN. Proresolving nanomedicines activate bone regeneration in periodontitis. J Dent Res 2015; 94(1):148-56; PMID:25389003; http://dx.doi.org/ 10.1177/0022034514557331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, Van Dyke TE. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol 2007; 179(10):7021-9; PMID:17982093; http://dx.doi.org/16373400 10.4049/jimmunol.179.10.7021 [DOI] [PubMed] [Google Scholar]
- 38.Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, Petasis NA, Levy BD, Serhan CN, Van Dyke TE. RvE1 protects from local inflammation and osteoclast- mediated bone destruction in periodontitis. FASEB J 2006; 20(2):401-3; PMID:16373400; http://dx.doi.org/ 10.1096/fj.05-4724fje [DOI] [PubMed] [Google Scholar]
- 39.Jin Y, Arita M, Zhang Q, Saban DR, Chauhan SK, Chiang N, Serhan CN, Dana R. Anti-angiogenesis effect of the novel anti-inflammatory and pro-resolving lipid mediators. Invest Ophthalmol Vis Sci 2009; 50(10):4743-52; PMID:19407006; http://dx.doi.org/25458696 10.1167/iovs.08-2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klil-Drori AJ, Ariel A. 15-Lipoxygenases in cancer: a double-edged sword? Prostaglandins Other Lipid Mediat 2013; 106:16-22; PMID:23933488; http://dx.doi.org/25458696 10.1016/j.prostaglandins.2013.07.006 [DOI] [PubMed] [Google Scholar]
- 41.Dunbar BS, Bosire RV, Deckelbaum RJ. Omega 3 and omega 6 fatty acids in human and animal health: an African perspective. Mol Cell Endocrinol 2014; 398(1-2):69-77; PMID:25458696; http://dx.doi.org/ 10.1016/j.mce.2014.10.009 [DOI] [PubMed] [Google Scholar]
- 42.Gerber M. Omega-3 fatty acids and cancers: a systematic update review of epidemiological studies. Br J Nutr 2012; 107(Suppl 2):S228-39; PMID:22591896; http://dx.doi.org/ 10.1017/S0007114512001614 [DOI] [PubMed] [Google Scholar]
- 43.Song M, Nishihara R, Cao Y, Chun E, Qian ZR, Mima K, Inamura K, Masugi Y, Nowak JA, Nosho K et al.. Marine omega-3 polyunsaturated fatty acid intake and risk of colorectal cancer characterized by tumor-infiltrating T Cells. JAMA Oncol 2016; 2(9):1197-206; PMID:27148825; http://dx.doi.org/ 10.1001/jamaoncol.2016.0605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Black HS, Rhodes LE. Potential benefits of omega-3 fatty acids in non-melanoma skin cancer. J Clin Med 2016; 5(2):pii: E23; PMID:26861407; http://dx.doi.org/ 10.3390/jcm5020023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eltweri AM, Thomas AL, Metcalfe M, Calder PC, Dennison AR, Bowrey DJ. Potential applications of fish oils rich in omega-3 polyunsaturated fatty acids in the management of gastrointestinal cancer. Clin Nutr 2017; 36(1):65-78; PMID:26833289; http://dx.doi.org/21307302 10.1016/j.clnu.2016.01.007 [DOI] [PubMed] [Google Scholar]
- 46.Sapieha P, Stahl A, Chen J, Seaward MR, Willett KL, Krah NM, Dennison RJ, Connor KM, Aderman CM, Liclican E et al.. 5-Lipoxygenase metabolite 4-HDHA is a mediator of the antiangiogenic effect of omega-3 polyunsaturated fatty acids. Sci Transl Med 2011; 3(69):69ra12; PMID:21307302; http://dx.doi.org/ 10.1126/scitranslmed.3001571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140(6):805-20; PMID:20303872; http://dx.doi.org/ 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 48.Parlato M, Yeretssian G. NOD-like receptors in intestinal homeostasis and epithelial tissue repair. Int J Mol Sci 2014; 15(6):9594-627; PMID:24886810; http://dx.doi.org/ 10.3390/ijms15069594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer 2009; 9(1):57-63; PMID:19052556; http://dx.doi.org/ 10.1038/nrc2541 [DOI] [PubMed] [Google Scholar]
- 50.Fichtner-Feigl S, Kesselring R, Strober W. Chronic inflammation and the development of malignancy in the GI tract. Trends Immunol 2015; 36(8):451-9; PMID:26194796; http://dx.doi.org/ 10.1016/j.it.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Babbin BA, Laukoetter MG, Nava P, Koch S, Lee WY, Capaldo CT, Peatman E, Severson EA, Flower RJ, Perretti M et al.. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J Immunol 2008; 181(7):5035-44; PMID:18802107; http://dx.doi.org/23241962 10.4049/jimmunol.181.7.5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leoni G, Alam A, Neumann PA, Lambeth JD, Cheng G, McCoy J, Hilgarth RS, Kundu K, Murthy N, Kusters D et al.. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J Clin Invest 2013; 123(1):443-54; PMID:23241962; http://dx.doi.org/ 10.1172/JCI65831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alam A, Leoni G, Quiros M, Wu H, Desai C, Nishio H, Jones RM, Nusrat A, Neish AS. The microenvironment of injured murine gut elicits a local pro-restitutive microbiota. Nat Microbiol 2016; 1:15021; PMID:27571978; http://dx.doi.org/ 10.1038/nmicrobiol.2015.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.