

ABSTRACT
Glioblastoma (GBM) is resistant to most multimodal therapies. Clinical success of immune-checkpoint inhibitors (ICIs) has spurred interest in applying ICIs targeting CTLA4, PD1 or IDO1 against GBM. This amplifies the need to ascertain GBM's intrinsic susceptibility (or resistance) toward these ICIs, through clinical biomarkers that may also “guide and prioritize” preclinical testing. Here, we interrogated the TCGA and/or REMBRANDT human patient-cohorts to predict GBM's predisposition toward ICIs. We exploited various broad clinical biomarkers, including mutational or predicted-neoantigen burden, pre-existing or basal levels of tumor-infiltrating T lymphocytes (TILs), differential expression of immune-checkpoints within the tumor and their correlation with particular TILs/Treg-associated functional signature and prognostic impact of differential immune-checkpoint expression. Based on these analyses, we found that predictive biomarkers of ICI responsiveness exhibited inconsistent patterns in GBM patients, i.e., they either predicted ICI resistance (as compared with typical ICI-responsive cancer-types like melanoma, lung cancer or bladder cancer) or susceptibility to therapeutic targeting of CTLA4 or IDO1. On the other hand, our comprehensive literature meta-analysis and preclinical testing of ICIs using an orthotopic GL261-glioma mice model, indicated significant antitumor properties of anti-PD1 antibody, whereas blockade of IDO1 or CTLA4 either failed or provided very marginal advantage. These trends raise the need to better assess the applicability of ICIs and associated biomarkers for GBM.
KEYWORDS: Cancer immunotherapy, copy-number alterations (CNA), CTLA4, high-grade glioma, IDO1, immune-checkpoint blockers (ICBs), mutational burden, neoantigen, patient prognosis, PD1, T cells, Treg cells
Introduction
Glioblastoma (GBM), a sub-type of high-grade glioma, is an aggressive type of cancer afflicting the central nervous system (CNS) (associated with very low median survival of 12–15 mo).1,2 GBM is resistant to most existing multimodal therapies.3-5 Nearly 160 FDA-approved anti-GBM drugs have been applied, yet only a few are implemented as standard-of-care (e.g., temozolomide, since 2005).2 Current GBM treatment paradigm consists of maximal surgical resection followed by radiotherapy plus temozolomide.2 However, even this only marginally improves the prognosis of GBM patients.3-5 This disturbingly negative situation advocates application of novel anti-GBM therapies.
Anticancer immunotherapy especially immune-checkpoint inhibitors (ICIs), have shown great promise against aggressive cancers like melanoma and lung cancer that had otherwise failed to sufficiently respond to conventional therapies.2,6 Immune-checkpoints like Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA4), Programmed Cell Death 1 (PD1) and Indoleamine 2,3-Dioxygenase 1 (IDO1) primarily aim to avoid autoimmune reactions and hence typically function to inhibit T cell effector responses (including anticancer T cell immunity).7,8 Thus, ICIs help “revive” anticancer immunity by blocking these checkpoints.2,6 Of note, while CTLA4 and PD1 are mainly expressed on T cells, yet IDO1 can be derived from multiple sources, including cancer cells, innate immune cells and stromal cells.7
Past research has revealed GBM's relative susceptibility to highly efficacious immunotherapies like oncolytic viruses or dendritic cell (DC)-based vaccines5,9,10 – a major motivation behind using anti-CTLA4 antibodies (Abs), anti-PD1 Abs or IDO1 inhibitors against GBM.2,11 It has been reported that IDO1 can be upregulated during gliomagenesis thereby making it an attractive target for GBM immunotherapy.12 On the other hand, studies on neuronal autoimmune disorders have shown that the CNS particularly exploits the PD1-axis for maintaining immune-tolerance.2 Hence, anti-PD1 Ab is currently being prioritized for GBM immunotherapy.2 In fact the anti-PD1 Abs (e.g., NCT02085070, NCT02337491, NCT01952769) and anti-CTLA4 Abs (e.g., NCT01950195, NCT01703507, NCT02107755, NCT02097732, NCT02115139) are currently being tested, also as monotherapies, against GBM in various phase I/II/III clinical trials.2
Therapeutically challenging cancer-types like melanoma have responded to ICI monotherapy in a remarkable fashion, both in preclinical and clinical settings.13,14 To this end, it is necessary to identify whether (and to what extent) GBM responds to ICI monotherapy; and whether such responsiveness can be predicted by broad clinical biomarkers. This can help understand whether GBM exhibits pre-existing (intrinsic) susceptibility to ICIs like melanoma or lung cancer. These trends would not only help to delineate the most suitable subset of patients to be treated with ICIs, but also those that should be avoided (owing to the severity of possible autoimmune toxicities) or treated with additional therapies to augment ICI's impact. But, while the clinical results of ICIs against GBM are awaited yet some preclinical studies have presented contradictory results, reporting both success15 and complete failure16 of anti-CTLA4 or anti-PD1 mono-immunotherapies.
Recently, various predictive biomarkers of ICI responsiveness have been delineated that can help in broadly predicting whether GBM could be susceptible to ICI monotherapy.6 Such broad predictive biomarkers include (but are not limited to) the following:6,17,18 (1) overall mutational burden, which is a surrogate marker for neoantigen burden (neoantigen-specific T cells are particularly active in tumors responding to ICIs thereby making high mutational/neoantigen burden crucial for ICI responsiveness), (2) differential expression of immune checkpoints, (3) pre-existing or basal levels of tumor-infiltrating T lymphocytes (TILs), (4) correlation of immune-checkpoint expression with particular TILs/Treg-associated polarization or effector function markers and (5) prognostic impact of differential immune-checkpoint expression.2,6,17,18 In terms of consistency, current clinical data shows that high pre-existing/basal density of TILs and high mutational/neoantigen burden together predict positive responsiveness to ICIs.2,6 These biomarkers are considered to be resulting from prolonged carcinogenic insults and mutagenic clonal evolution.6,17,18 Carcinogenic insults, in particular, are considered to be the predominant source of high mutational burden (i.e., non-synonymous somatic single-nucleotide variations).19,20 In fact, some recent genetic analyses have shown that specific mutagens or carcinogens induce distinct mutational lesions in particular cancer types21 like mutational signatures 4 and 29 (e.g., C>A or CC>AA mutations induced by tobacco mutagens, hence showing high presence in smokers and/or those who chew tobacco), signature 7 (e.g., CC>TT mutations induced by ultraviolet radiation), signature 22 (e.g., T>A mutations induced by aristolochic acid) and signature 24 (e.g., C>A mutations induced by aflatoxin).21 In line with this, melanoma (SKCM), lung cancer (LUAD/LUSC) and bladder cancer (BLCA) represent one of the most ICI-responsive cancers; since a sizeable subset of patients of these cancer types possesses high mutational/neoantigen burdens and high pre-existing TILs.2,6
Of note, anti-PD1 Abs have shown promising results in patients with “hypermutant-GBM” (i.e., a pediatric-GBM “sub-type” with high mutational burden resulting from biallelic mismatch repair deficiency or bMMRD).22 However, it is necessary to consider that hypermutant GBM has the highest known mutational/neoantigens burden of all human cancers (even higher than melanoma and lung cancer). Hence, as such it represents an exception, because typical (adult) GBM exhibits much lower mutational burden.1 Also, a very limited number of hypermutant GBM patients were tested for the efficacy of anti-PD1 Abs.
Thus, for adult-GBM, it is necessary to ascertain whether the above-mentioned broad predictive biomarkers can estimate the responsiveness of GBM to single-agent ICIs. To this end, our primary aim was 2-fold, i.e., first to exploit these broadly applicable biomarkers to predict whether GBM has a predisposition for positive responses toward ICIs targeting CTLA4, PD1 or IDO1 in patients. This was tested by using two-independent (publicly-accessible) cohorts of GBM patients as applicable, i.e., The Cancer Genome Atlas (TCGA)23-25 and REpository of Molecular BRAin Neoplasia DaTa (REMBRANDT).26 And second, to use these biomarkers-based outcomes for “guiding and prioritizing” preclinical testing of corresponding ICI monotherapy in an orthotopic-murine glioma model.
Results
Glioblastoma exhibits relatively low mutational and predicted neoantigen burden
Considering the broad utility of high number of non-synonymous SNVs, further referred to as “mutational-burden,” as a positive predictive biomarker of ICI responsiveness;19,20 we first decided to ascertain the positioning of GBM, in this regard, relative to other cancer types. TCGA GBM data set was primarily used for these analyses since it provides systematic information on somatic mutational counts in different cancer types.
In line with various previous analyses,2,6 we observed that cancer types harboring carcinogen-induced mutational signatures25 had significantly higher mutational burden (Fig. 1A), e.g., melanoma (SKCM), lung cancer (LUAD/LUSC) or bladder cancer (BLCA). Instead, GBM, which does not usually possess a (non-therapeutic) carcinogen-induced mutational signature, clustered with other similar cancer types and exhibited relatively low mutational burden (Fig. 1A).2,6
Figure 1.

Glioblastoma (GBM) has one of the lowest overall mutational and predicted neoantigen burdens. (A) Presence or absence of carcinogen-induced mutational signatures (derived from COSMIC-database: http://cancer.sanger.ac.uk/cosmic/signatures) was used as a means to “classify” median (somatic) mutational burdens of 18 TCGA cancer-data sets (mean ± s.d., Mann–Whitney test; p-value as indicated); (B) Correlation between median mutational burdens of different TCGA cancer-data sets and median mutational counts of corresponding cell lines belonging to these cancer types from the CCLE-data sets. (C) Correlation of median mutational burdens and median-predicted neoantigen burden from the respective TCGA data sets derived from The Cancer Immunome Atlas at http://tcia.at. Abbreviations: BLCA, bladder urothelial carcinoma; BRCA, breast cancer; CESC, cervical squamous cell carcinoma; CRC, colorectal carcinoma; GBM, glioblastoma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe cancer; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian cancer; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; THCA, thyroid cancer; UCEC, uterine corpus endometrial carcinoma; Of note, mutational burden refers to nonsynonymous somatic single nucleotide variations.
Moreover, we observed that GBM not only exhibits low mutational burden on the level of the tumor, but also on the level of human GBM-derived cancer cell lines (Fig. 1B). In contrast, the representative cell lines from ICI-responsive cancer types like SKCM, LUAD or BLCA, also exhibited higher mutational burdens. Of note, we observed a significant positive correlation between mutational burdens in various tumor types and the mutational burdens in the corresponding human cancer cell lines (Fig. 1B). However, quantitatively speaking, the cancer cell lines had approximately five times less median mutational burden than the corresponding tumor types (Fig. 1B), possibly because some tumor cells with high mutational counts might be too genetically unstable for long-term persistence in in vitro culturing conditions.
Next, in an analysis depicting significant positive correlation between overall mutational burden and burden of predicted neoantigens (a phenomenon well established in the literature),19,20 GBM also displayed one of the lowest predicted-neoantigen burdens, when compared with the typical ICI-responsive cancer types like SKCM, LUAD/LUSC and BLCA (Fig. 1C).
In conclusion, our analysis shows that most GBM tumors are at the lower end of the mutational/neoantigen-burden's scale, compared with typical ICI-responsive cancer types.
Glioblastoma exhibits lower expression of CTLA4, PDCD1 and IDO1 compared with melanoma, lung cancer and bladder cancer
The observation that, relative to typical ICI-responsive cancer types, GBM has much lower mutational and/or neoantigen's burden made us curious about the overall expression of major immune-checkpoint coding genes, i.e., CTLA4, PDCD1 (codes for PD1) and IDO1, in GBM. To this end, we first decided to exploit the TCGA data set to analyze the differential expression of these immune checkpoints in GBM, as compared with typical ICI-responsive cancer types like SKCM, LUAD/LUSC and BLCA. Interestingly, SKCM, LUAD/LUSC and BLCA expressed CTLA4 (Fig. 2A), PDCD1 (Fig. 2B) and IDO1 (Fig. 2C) immune checkpoints at significantly higher levels than GBM. LUAD in general exhibited relatively high expression of all three immune checkpoints.
Figure 2.
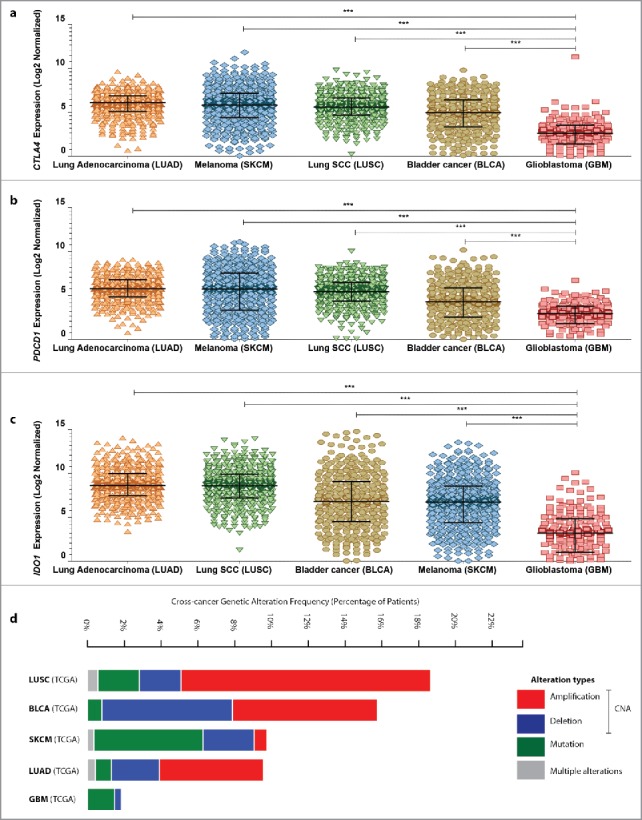
Melanoma, lung cancer and bladder cancer exhibit significantly higher expression of CTLA4, PDCD1 or IDO1 than glioblastoma in human patients. Analysis of differential expression of CTLA4 (A), PDCD1 (B) or IDO1 (C) between lung adenocarcinoma (LUAD; n = 517), melanoma (SKCM; n = 472), lung squamous cell carcinoma (SCC) (LUSC; n = 501), bladder cancer (BLCA; n = 408) and glioblastoma (GBM; n = 166) (from TCGA cohorts) (data are log2 normalized and presented as median with inter-quartile range; One-way ANOVA; significance set at p < 0.05; ***p < 0.001). Of note RNASeq, and not microarray, expression data was used for this analysis since comprehensive microarray data are not available for melanoma and bladder cancer within TCGA data set. (D) A combined cross-cancer genetic alteration frequency analysis (expressed as % of patients) for CTLA4, PDCD1 and IDO1 genes was performed using the TCGA cohorts of LUAD, LUSC, SKCM, BLCA and GBM. The graph indicates presence of either genetic mutations or specific copy-number alterations (CNA; like genetic deletion or amplification) or presence of multiple such alterations simultaneously (as indicated by the color-code in the legend within the figure).
Genomic DNA copy-number alterations (CNAs) are key genetic events in human cancer progression such that CNAs of particular genes may influence their subsequent expression,27,28 e.g., amplification-type CNAs of a gene have higher (but not absolute) probability of associating with upregulation of corresponding mRNA(s).27,29 Moreover, occurrence of CNAs, along with nucleotide mutations, is a good indication of genomic instability/variations associated with particular genes.30 Interestingly, in the TCGA cohorts, higher percentage of LUAD/LUSC, SKCM and BLCA patients exhibited CNAs (like deletions or amplifications) of CTLA4, PDCD1 and IDO1 genes, as compared with GBM (Fig. 2D). Importantly, no GBM patient in this cohort had amplification-type CNAs of CTLA4, PDCD1 and IDO1 as opposed to other cancer types analyzed here (Fig. 2D). This indicates that CTLA4, PDCD1 and IDO1 have higher chance of experiencing CNAs in ICI-responsive cancer types as compared with GBM.29 On the other hand, overall percentage of patients with mutations in these immune-checkpoint coding genes in GBM were to a certain extent comparable to BLCA, LUAD/LUSC (Fig. 2D).
In conclusion, GBM patients exhibit significantly lower mRNA expression of CTLA4, PDCD1 and IDO1 as well as lower tendency of displaying amplification-type CNAs associated with these genes, when compared with typical ICI-responsive cancer types like melanoma, lung cancer and bladder cancer.
Overall expression of CTLA4, PDCD1 and IDO1 is not drastically altered by gliomagenesis
Tumors tend to exploit immune checkpoints for immune tolerance and escape from anticancer T cells and hence frequently, tumor tissue has been reported to exhibit higher expression of immune checkpoints than corresponding normal tissue.7,18 To this end, we analyzed this in the GBM/normal brain samples from the TCGA and REMBRANDT data sets (Fig. 3). Interestingly, only in TCGA GBM cohort, CTLA4 showed significantly higher expression than corresponding normal brain samples (Fig. 3A). Although other analyses did not show significant upregulation of various checkpoints (Figs. 3B–F), it seemed that a small subset of patients with GBM tumors (5–10%) tend to show higher IDO1 expression than their normal counterparts (Figs. 3C and F). Although these trends were not significant, these were consistently observed in both TCGA and REMBRANDT data sets.
Figure 3.

Glioblastoma (GBM) tumor tissue does not show strong upregulation of CTLA4, PDCD1 and IDO1. Analysis of differential expression of CTLA4 (A, D), PDCD1 (B, E) and IDO1 (C, F) between normal brain tissue sample (for REMBRANDT, n = 21; for TCGA, n = 11) and GBM tissue sample (for REMBRANDT, n = 214; for TCGA, n = 202) (mean ± s.d., Mann–Whitney test; p-value as indicated). Of note, TCGA and REMBRANDT data sets were analyzed by different expression-analysis platforms and standardized by different post-processing and normalization analyses and hence their overall gene-expression counts have different (but proportional) scales.
Overall, it seems that the oncogenic process of gliomagenesis by itself does not cause significant upregulation of at least PDCD1 and IDO1 gene expression, with the exception of CTLA4 in the TCGA patient cohort.
Glioblastoma exhibits relatively low pre-existing presence of tumoral T cell biomarkers
Higher pre-existing or basal TILs positively predict responses to ICIs, e.g., melanoma tumors with high basal/pre-existing TILs tend to respond better to ICIs rather than tumors with low to negligible TILs.6,7,18 Thus, the amount of TILs is a suitable broad biomarker of ICI responsiveness.
Estimation of TILs can be achieved either via the “classical” immunophenotyping technique (immunohistochemistry or immunostaining-driven flow cytometry) or through the more recent technique of using (pre-established) T-cell-specific gene signatures.31-33 Since we did not have access to substantial number of GBM tumor tissues for the classical analyses, we decided to utilize the latter technique with the TCGA GBM cohort (as reported by us previously).5,33 TCGA GBM cohort was preferred since it is completely composed of primary-resected GBM tissues, thereby allowing for the best estimation of pre-existing/basal TILs.
Pan-cancer T cell subtype-specific gene signatures available from The Cancer Immunome Atlas34 were used to estimate the total TIL-fractions within 19 different TCGA tumor cohorts (Fig. 4A). Separate genetic signatures associated with different T cell sub-populations were layered together to estimate the overall TIL fractions for each cancer type. Interestingly, among these 19 different solid tumor types, GBM exhibited the least amount of TIL fractions (Fig. 4A). This was especially evident when compared with the higher amounts of TIL fractions in typically ICI responsive cancer types like SKCM, LUSC/LUAD and BLCA (Fig. 4A).
Figure 4.

Glioblastoma (GBM) exhibits sparse basal T cell-infiltrates and correlation of CTLA4 and IDO1 with T-cell-associated polarization biomarkers. (A) The overall (absolute) presence of different T-cells-associated genetic signatures or metagenes (indicated as color code) was estimated across 19 TCGA cancer-data sets using The Cancer Immunome Atlas. (B, C, D) Correlation of CTLA4, PDCD1, IDO1 expression with, GBM-specific Treg-metagene5 across TCGA (B) and REMBRADT data sets (C) (blue-box indicates co-clustering of immune checkpoint with Treg-metagene); or with, GZMA, PRF1 and IFNG across the same data sets (D). BLCA, bladder urothelial carcinoma; BRCA, breast cancer; CESC, cervical squamous cell carcinoma; CRC, colorectal carcinoma; GBM, glioblastoma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe cancer; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian cancer; PAAD, pancreatic adenocarcinoma; PRAD, prostate adenocarcinoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; THCA, thyroid cancer; UCEC, uterine corpus endometrial carcinoma.
In conclusion, GBM exhibits one of the lowest basal/pre-existing TIL-associated genetic signatures among various solid tumor types.
CTLA4 and IDO1 show association with Treg and effector T-cell-associated biomarkers
Low occurrence of basal TIL-associated genetic signatures in GBM raised the need to ascertain the exact association of above immune checkpoints with T cell polarization or activity-associated genetic signatures.2,6 We gave due consideration to two currently described scenarios in this regard, i.e., (1) the “canonical” association between immunosuppressive Treg cells and immune checkpoints, considering that the function of Tregs and CTLA4, PD1 and IDO1 in enforcing immunosuppression is overlapping2,6,17,34,35; and (2) the “paradoxical” association between immune checkpoints and T cell-effector markers (like IFNγ, Granzyme B-perforin), since activation of T cells eventually causes upregulation of immune checkpoints as an auto-regulatory loop in later stages of effector function (to avoid autoimmunity and resolve inflammation).2,6
To this end, we first analyzed the correlation between CTLA4, PDCD1 or IDO1 expression levels and a previously established Treg-specific genetic signature or metagene.5 Here, a Treg-specific metagene refers to a “multi-gene expression pattern”33 where aggregated patterns of various Treg-associated genes (experimentally validated by independent studies)31,32 are clustered to represent a correlated expression or co-expression matrix. This is because the tendency of these genes (i.e., CD247, CD2, CD3D, CD3G, GPR171, CD27, LCK, LTB, IL2RB and ICOS) to show co-expression has statistically higher (but not absolute) chance of indicating the presence of Tregs.31,33,34 The principle behind such immune cell-specific metagenes is discussed in details, elsewhere.31,33,34 In this study, the rational was, if an immune-checkpoint coding gene co-clusters with the Treg-metagene within human GBM tissue than the probability of that immune-checkpoint being expressed on Tregs is higher (but not absolute).
Interestingly, in a cohort-dependent fashion, either IDO1 (TCGA, Fig. 4B) or CTLA4 (REMBRANDT, Fig. 4C), but not PDCD1, showed considerable co-clustering with Treg metagene (as indicated by the tendency of IDO1 or CTLA4 to show nodal association with the “core” Treg-metagene). Of note, the “core” Treg metagene is indicated by red font coloring of the respective genes on the heatmaps (Figs. 4B and C). Noteworthy, this meta-analysis showed that the correlation between CTLA4 or IDO1 and Treg metagene was rather weak, as indicated by the lack of overlap between these immune checkpoints and the “core” of the metagene (Figs. 4B and C).
Next, we correlated CTLA4, PDCD1 and IDO1 expression levels with markers of T cell effector function, i.e., GZMA (coding granzyme B), PRF1 (coding perforin) and IFNG (coding IFNγ) which is displayed in a correlation heatmap. Interestingly, we observed a, cohort-specific, consistent positive correlation between these effector function-related factors (GZMA, PRF1, IFNG) and IDO1 (TCGA-cohort, Fig. 4D). On the other hand, in this cohort, CTLA4 achieved the next best correlation (with GZMA, IFNG), followed by PDCD1 (only with IFNG) (Fig. 4D).
In conclusion, mainly CTLA4 and IDO1 exhibited correlation with T cell polarization/function-related genetic markers, in a cohort-specific manner.
Differential CTLA4, PDCD1 and IDO1 expression fail to exhibit definitive prognostic impact in glioblastoma patients
In various publications it has been shown that high expression of immune checkpoints may associate with negative patient prognosis.17,34,35 To this end, we decided to ascertain the prognostic impact of immune-checkpoint gene expression in TCGA and REMBRANDT GBM cohorts. The differential expression of CTLA4, PDCD1 and IDO1 did not show strong association with poor or prolonged overall survival (OS) in GBM patients, neither in TCGA (Figs. 5A–C) nor in REMBRANDT (Figs. 5D–F) data sets. The hazard ratios (HRs) in all cases were neither less than 0.90 nor more than 1.10 thereby further substantiating the seemingly null prognostic impact of these immune-checkpoints' expression levels in GBM patients.
Figure 5.

CTLA4, PDCD1 and IDO1 differential expression does not exhibit significant prognostic impact in GBM patients. TCGA GBM-cohort (n = 540, A–C) and REMBRANDT GBM-cohort (n = 178, D–F) stratified (median) into “high-expression” (red; TCGA, n = 271; REMBRANDT, n = 89) or “low-expression” (black; TCGA, n = 269; REMBRANDT, n = 89) and represented as Kaplan–Meier plots (log-rank (Mantel-Cox) test; hazard ratios (HR)+95% confidence interval or CI).
Anti-PD1 therapy shows stronger preclinical efficacy than anti-CTLA4 or IDO1-targeted therapies
The clinical predictions on GBM responsiveness to ICIs on the basis of broad biomarkers were mixed. The predictive biomarkers of ICI responsiveness exhibited contradictory patterns in GBM patient cohorts, such that (i) four sets of biomarkers predicted ICI-resistance (i.e., low mutational/predicted-neoantigen burden, relatively low expression of immune-checkpoint coding genes, low basal/pre-existing TIL-associated signatures and no discernible prognostic impact of differential expression of immune-checkpoints), while (ii) two sets of biomarkers predicted susceptibility to at least anti-CTLA4 Abs and/or IDO1 inhibitors (i.e., high CTLA4 expression in GBM tissue compared with normal tissue and correlation of CTLA4/IDO1 with T cell polarization/function-related signatures).
These contradictory patterns on biomarker level made it challenging to prioritize, in an objective manner, the preclinical application of specific ICIs in murine settings. To this end, we decided to experimentally test the efficacy of ICIs targeting all three immune checkpoints, i.e., CTLA4, PD1 or IDO1 in orthotopic GL261 glioma-bearing mice. In one case, we administered pharmacological IDO1 inhibitors, i.e., 1-Methyl-DL-tryptophan (1-DL-MT, a mixture of levorotary/L and dextrorotary/D stereoisomers of 1-MT) or 1-Methyl-D-tryptophan (i.e., 1-D-MT)12 (Fig. 6A). In another case, we administered anti-CTLA4 or anti-PD1 Abs (Fig. 6B). Interestingly, both 1-DL-MT and 1-D-MT failed to prolong survival of GL261 glioma-bearing mice (Fig. 6C). This failure of IDO1 targeting was “phenocopied” on the level of Ido1−/− mice (Fig. 6D). Anti-CTLA4 therapy was able to only marginally (p = 0.05) extend the median survival of GL261 glioma-bearing mice (Fig. 6E). However, interestingly only anti-PD1 therapy significantly prolonged the median survival of GL261 glioma-bearing mice allowing some long-term survival (Fig. 6F).
Figure 6.

Anti-PD1 mono-immunotherapy exhibits the highest therapeutic efficacy against preclinical glioma. (A, B) Mice inoculated with GL261 cells intra-axially,5 were treated with IDO1-inhibitors (A) or anti-CTLA4, anti-PD1 antibodies (B). Kaplan–Meier survival curves for glioma-bearing mice treated with IDO1 inhibitors (CNTR, n = 13; +1-D-MT, n = 15; +1-DL-MT, n = 5) (C), Ido1+/+ (n = 14) vs. Ido1−/− (n = 17) mice (D) or treated with anti-CTLA4 (IgG Ab, n = 9; CTLA4 Ab, n = 10) (E) or anti-PD1 (IgG Ab, n = 8; PD1 Ab, n = 12) (F) antibodies (Log-rank-(Mantel-Cox)-test). (G–L) C57BL/6 mice were inoculated with live GL261 cells (Day 0), intra-axially and either injected with respective IgG antibodies (Ab), i.e., control mice or treated with anti-CTLA4 Ab (G–I) or anti-PD1 Ab (J–L). Thereafter the mice were killed at day 18–22 post-GL261 inoculation and the brains were isolated. Initially, total mononuclear immune cells were counted. Thereafter these were processed for FACS-based immunophenotyping for (G) CTLA4+CD4+T cells, (H) CTLA4+CD8+T cells, (I) CTLA4+Treg cells, (J) PD1+CD4+T cells, (K) PD1+CD8+T cells and (L) PD1+Treg cells. The histograms are representative of n = 3–4 mice/group (the percentage of CTLA4 or PD1 negative and positive T cells are indicated through the agency of histogram).
ICIs like anti-CTLA4 and anti-PD1 antibodies, exert their anticancer effects by effectively reducing the intra-tumoral persistence of CTLA4+ and PD1+ T cells (especially CTLA4+/PD1+ Treg cells), respectively.7,17,36 To this end, we interrogated whether brains inoculated with GL261-gliomas possessed CTLA4+/PD1+ T cell infiltrates; and whether ICIs targeting CTLA4/PD1 affected their persistence. GL261-glioma inoculated brains were indeed infiltrated by CTLA4+ or PD1+, CD4+ T cells, CD8+ T cells and Tregs (PD1+ T cells>CTLA4+ T cells) (Figs. 6G–L). Notably, more than half of Tregs infiltrating the GL261-glioma inoculated brains did not express CTLA4 or PD1 (Figs. 6I and L). However, in line with expectations, anti-CTLA4 or anti-PD1 antibodies were indeed able to effectively reduce the amount of CTLA4+ or PD1+ T cells infiltrating the GL261-glioma inoculated brains, respectively (Figs. 6G–L). Last but not least, we also confirmed that GL261 glioma cells tend to express surface PD-L1, one of the major ligands required for PD1 activation (Fig. S1).
Next, we decided to position our experimental results relative to the existing status quo in the field of ICI-based preclinical treatment of glioma, through a systematic literature meta-analysis. A survey of 15 relevant preclinical studies (Box S1) exploring the impact of ICIs targeting CTLA4, PD1 or IDO1 in preclinical (orthotopic) glioma murine models (most studies used either GL261 or SMA560 glioma), showed an interesting result (Fig. S2). ICIs targeting CTLA4 or IDO1 were reported, by most studies, to fail in significantly prolonging median survival of glioma-bearing mice (Fig. S2). In contrast, relatively more studies reported success of anti-PD1 therapy in prolonging median survival of glioma-bearing mice (Fig. S2).
Overall this shows that the propensity of anti-PD1 therapy to succeed in glioma-bearing mice is higher than IDO1-inhibitors or anti-CTLA4 therapy.
Discussion
Our observations reveal a discrepancy between broad predictive biomarkers of ICI responsiveness and preclinical efficacy of respective ICI monotherapy in GBM. The majority of broad biomarkers predicted ICI resistance in GBM. Few biomarkers, however, predicted some efficacy for ICIs targeting IDO1 or CTLA4 but not PD1. On the other hand, opposite to the biomarker-based trends, the preclinical efficacy of these ICIs in orthotopic GL261-glioma model revealed a significant susceptibility to anti-PD1 Abs, whereas the tumor-rejecting ability of IDO1 or CTLA4 targeting ICIs was null or poor, respectively.
The clinical biomarker analysis in GBM patients delineated low mutational/neoantigen burden, relatively lower tumoral expression of immune checkpoints and sparse pre-existing levels of TILs, features that do suggest that unlike melanoma or lung cancer, adult-GBM probably does not have intrinsic predisposition toward therapies targeting immune checkpoints.2,6,16 This does not mean that a subset-of-patients of GBM will not respond to ICI monotherapy (at least partially), however, these patients will have to be either delineated more stringently or alternative GBM-specific immune checkpoints will have to be characterized. Moreover, the biomarkers used in this study to estimate the ICI responsiveness of GBM are predictive of broad “resistant-phenotype,” and hence they may ignore particular GBM-specific features. Last but not least, our results are not representative of the ICI responsive, hypermutant pediatric-GBM since on one hand, hypermutant GBM patients are not well represented in TCGA/REMBRANDT cohorts and on the other hand, orthotopic GL261-glioma is not per se a good model of pediatric-GBM.
Interestingly, a recent study has shown that preclinical glioma models like GL261 do possess some neoantigens and may possess higher mutational burden than a typical primary human GBM tissue.1 Moreover, another recent study showed that murine cancer cell lines with higher mutational burden (e.g., cell lines derived from carcinogen-induced tumor) respond better to immune-checkpoint therapy than those with lower mutational burdens (e.g., cell lines derived from spontaneous or GEMM-derived tumors).37 Thus, it is tempting to speculate that GL261-based gliomas are more representative of medium-to-hyper mutant GBM rather than typical low mutational burden (adult) GBM, and hence are more responsive to ICIs as shown here and in previous studies.15 This is an interesting hypothesis that needs further validation. More research is required to understand the determinants of anti-glioma immunity and immunosurveillance in preclinical model and compare these with clinical determinants to reach better consensus on suitable biomarkers.38,39 Last but not least, beyond these glioma cell line-related differences there is also a distinct possibility that mice-to-human differences in CNS-associated immune responses and general biology of immune checkpoints may also be a source of the discrepancy between clinical biomarker-based predictions and preclinical therapeutic efficacies.39,40 For instance, we observed that more than half of the brain-infiltrating Tregs in GL261 glioma-bearing mice did not express considerable amounts of CTLA4 or PD1. This might indicate that alternative immune checkpoints (possibly specific to brain or CNS milieu) might be operating in a GBM micro-environment. Thus, in GBM contexts, CTLA4+/PD1+ Tregs could be either minor enforcers of immunosuppression41 or susceptible to being replaced by T cells expressing alternative immune checkpoints following ICIs treatment. For instance, a recent preclinical study found T cells exploiting T-cell immunoglobulin mucin-3 (TIM3) to drive lung cancer growth despite anti-PD1 therapy.42 Thus, future studies will have to concentrate on delineating such alternative GBM-associated immune checkpoints to design more tumor-specific ICIs.
Considering the relatively low intrinsic susceptibility of GBM to immunotherapy, ICIs may have a better chance of showing therapeutic efficacy if they are combined with other immunotherapies that can correct the low pre-existing TILs density within GBM tissue. Highly efficacious immunotherapies like next-generation DC vaccines (e.g., DC vaccines based on glioma cells that underwent immunogenic cell death or ICD),5,43,44 adoptive or chimeric antigen receptor (CAR) T cell immunotherapy (e.g., CAR T cells targeted against the glioma antigen IL13Rα2)45 or oncolytic viruses (e.g., Newcastle disease virus)9 can help in increasing intra-GBM TILs density thereby creating a conducive scenario for ICIs response. Such “smart” combinations will have to be tried pre-clinically and if found successful their translation toward the clinic should be expedited.
It is worth mentioning that a recent report estimating safety of anti-PD1 therapy (pembrolizumab) in combination with anti-angiogenic therapy (bevacizumab) in recurrent GBM, found this combination to be safe for patients, however, all enrolled patients experienced progressive disease despite therapy.46 As such it is too early to draw strong conclusions based on the above report, considering the very small number of patients, absence of proper control arms and follow-up criteria not tailored for long-term ICI response analysis. As the results of ongoing phase II/III ICI-clinical trials in GBM come out, the scenario would clear up, also with respect to the discrepancies we observe.2
Materials and methods
Cell culture, orthotopic GBM mice model and ICIs administration
GL261 murine glioma cells (received as a gift from Dr Eyupoglu, University of Erlangen, Germany) were cultured at 37°C under 5% CO2 in DMEM containing 4.5 g/L glucose and 0.11 g/L sodium pyruvate, supplemented with 2 mM glutamine, 100 units/mL penicillin, 100 µg/L streptomycin and 10% fetal calf serum. For orthotopic glioma murine model, female C57BL/6J mice (8–10 weeks old) were purchased from Harlan (Horst) or KU Leuven internal stock ad libitum. Animals were handled in accordance with the KU Leuven bioethics regulations. To generate intra-brain GBM, the mice were intra-axially injected with 5×105 GL261 cells as described previously.47 In brief, mice were anesthetized, set in a stereotactic frame (Kopf Instruments) and injected (in sterile circumstances) with the GL261 cells at 2 mm lateral and 2 mm posterior from the bregma, and at 3 mm underneath the dura mater. After intra-brain inoculation, the mice were monitored 2–3 times per week and clinical symptoms were noted with a neurologic-deficit grading scale modified from an experimental autoimmune encephalomyelitis model, described in the past.47 Mice were classified as long-term survivors if their survival reached beyond three times the median survival of the untreated control or CNTR mice. Also, as applicable, some animals received intra-peritoneal injections of anti-CTLA4 Abs (4F10) or anti-PD1 Abs (RMP1–14) (these antibodies were received from Louis Boon, Bioceros, Netherlands). IDO1 inhibitors (1-D-MT or 1-DL-MT, purchased from Sigma-Aldrich) were administered via oral gavage. The administration schedules and doses of these ICIs are described either in the figures as schemas or within the figure legends. As applicable, either representatives of two independent mice experiments (PD1, CTLA4 experiments) were shown or power analysis based on previous publications47-49 was used to reach necessary sample sizes in individual experiments (IDO1 experiments).
Literature meta-analysis for preclinical efficacy of ICIs in murine glioma model
PubMed was searched for applicable studies conducted in mouse, until 9th January, 2017. The following search keywords were used: (ido OR ctla-4 OR pd-1) AND (glioblastoma OR glioma) AND (murine OR mice OR mouse OR in vivo). To distinguish potentially relevant studies, the catalog of articles identified in the earlier search, were also scanned manually. Studies within the catalog were considered qualified if they met all of the subsequent criteria: (1) presented Kaplan–Meier plot based overall survival data and (2) data generated in syngeneic immunocompetent mice model. Studies were excluded because of following reasons: (1) not sufficient survival data reported, (2) letters, reviews, commentary, perspectives, case reports, conference abstracts, editorials or expert opinion, (3) studies reporting xenograft results in immunodeficient or humanized mice. Overall these search criteria helped short-list 15 research articles12,15,16,50-61 mentioned in Box S1.
Analysis of T cell biomarker-associated genetic signature and immune checkpoints
The metagene associated with Treg cells31,33 was derived from our previously published analysis, where GBM-tailored T cell-metagenes were established.5 The co-expression of CTLA4, PDCD1 and IDO1 was analyzed with respect to this Treg-metagene in TCGA and REMBRANDT GBM patient data sets to generate a (Pearson's) correlation submatrix. Data retrieved from cBioPortal62 or Project βstasis web-portals were used for the above matrix generation, as applicable. In another case, CTLA4, PDCD1 and IDO1 were correlated with expression of IFNG, GZMA, PRF1 in TCGA and REMBRANDT. These coefficient values were used for hierarchical clustering33 through the Cluster 3.0 software63 and the gene co-expression matrices were visualized as heatmaps through TreeView.64 Last but not least, to estimate the basal or pre-existing (total) T cell infiltrates-associated genetic signatures in different cancer types, we used the Cancer Immunome Atlas (https://tcia.at/home),34 to analyze the absolute amounts of tumor-associated T cell genetic signatures in 19 different TCGA tumor types (mentioned in the figure or figure legends). The “cell type fractions plot” was used to generate absolute fraction values with Cibersoft_LM22 deconvolution methodology.34
Analysis of CTLA4/PD1 expression on brain-infiltrating T cells
Brain-infiltrating mononuclear immune cells were isolated from GL261-inoculated mice (treated with IgG Ab or anti-CTLA4/PD1 Ab) as detailed previously.5,9,10 Surface staining of these mononuclear immune cells was performed with anti-CD4 PerCP-Cyanine5.5/APC-eF780, anti-CD3 FITC/PE/eFluor®450, anti-CD8 BV421/eFluor®605NC mAbs, anti-CTLA4 PE or anti-PD1 PE eBioscience or BD). Intracellular FoxP3 was detected using a FoxP3-PE staining kit (eBioscience) according to the manufacturer's instructions. Data acquisition was performed on LSRFortessa flow cytometer (BD Biosciences) and the FlowJo software was used for histogram analysis.
Prognostic impact and differential expression analysis of immune checkpoints
The differential expression levels of CTLA4, PDCD1 or IDO1 and associated clinical survival information (overall survival or OS) was retrieved and analyzed for the TCGA GBM patient data set (n = 540)23,24 and REMBRANDT GBM patients (n = 178)26 using the PROGgeneV2 web-platform65 and Project βstasis web-platform, respectively. These platforms stratified the respective patients on the basis of the median gene expression profile into two risk-groups, i.e., high risk or low risk.33 The respective patient risk groups were plotted with respect to OS to generate Kaplan–Meier curves using the Graphpad Prism software. HR (and its 95% confidence interval) and log-rank (Mantel-Cox) p values were calculated (statistical significance threshold set at p < 0.05).33 Patients surviving beyond the follow-up thresholds were censored. Last but not least, differences in expression of CTLA4, PDCD1 and IDO1 between normal brain tissue sample (for REMBRANDT, n = 21; for TCGA, n = 11) and GBM tissue sample (for REMBRANDT, n = 214; for TCGA, n = 202) were analyzed through the Project βstasis web-platform. Of note, only those GBM patients' data were analyzed within the TCGA data set, whose GBMs could be pathologically subdivided into classical, mesenchymal, neural or pro-neural phenotypes.
Cross-cancer analysis for differential expression and genetic alterations of immune checkpoints
Data for differential expression (RNASeq-based) analysis and genetic alterations (mutations or CNAs in respective genes) of CTLA4, PDCD1 or IDO1 between lung adenocarcinoma (LUAD; n = 517), melanoma (SKCM; n = 472), lung squamous cell carcinoma (SCC) (LUSC; n = 501), bladder cancer (BLCA; n = 408) and glioblastoma (GBM; n = 166) were retrieved from respective TCGA data sets available from the cBioPortal62 and further analyzed as detailed in the figure legends.
Analysis of mutational burden and predicted-neoantigens burden
The median somatic mutational burdens (i.e., non-synonymous somatic single nucleotide variations) for different TCGA cancer types were derived from Cancer Immunome Atlas (https://tcia.at/home),34 Major TCGA cancer types were classified to carry (validated) carcinogen-associated mutational signatures21 on the basis of the Catalog of Somatic Mutations in Cancer (COSMIC) database (http://cancer.sanger.ac.uk/cosmic/signatures) (see Results text for details). The mutational burden graph of these two groups was then plotted. In another case, the somatic mutational counts of various human cancer cell lines available in the Cancer Cell Line Encyclopedia (CCLE) database (https://portals.broadinstitute.org/ccle/), were retrieved from the cBioPortal62 database and correlated (Spearman's) with the (above) corresponding TCGA median mutational burdens. Last but not least, the overall median mutational burdens and predicted neoantigens burdens of various TCGA cancer types were derived from the Cancer Immunome Atlas (https://tcia.at/home),34 and plotted to derive overall correlation (Spearman's).
Statistical analysis
All statistical analyses were performed using either Prism software (GraphPad Software) or GraphPad QuickCalcs online software (http://www.graphpad.com/quickcalcs/index.cfm). Log-rank (Mantel-Cox) test or Mann–Whitney statistical test were used for statistical analysis, as applicable and unless otherwise mentioned (indicated in figure legends). The significance level was set at p <0.05 (*p <0.05, **p <0.01, ***p <0.001; values indicated in the figures).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
ADG was supported by FWO post-doctoral fellowship from FWO-Vlaanderen. LV and MVW were supported by Strategic Basic Research grant from IWT-Vlaanderen. This work is supported by FWO (G060713N, G076617N), Belgian State (IAP7/32) and KU Leuven (C16/15/073) grants to PA. We would like to acknowledge the strong scientific and financial support received from Prof. Stefaan Van Gool.
References
- 1.Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, Fu Y, Alexandrov A, Mardis ER, Artyomov MN et al.. Endogenous neoantigen-specific CD8 T cells identified in two glioblastoma models using a cancer immunogenomics approach. Cancer Immunol Res 2016; 4:1007-15; PMID:27799140; http://dx.doi.org/ 10.1158/2326-6066.CIR-16-0156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol 2015; 11:504-14; PMID:26260659; http://dx.doi.org/ 10.1038/nrneurol.2015.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polyzoidis S, Tuazon J, Brazil L, Beaney R, Al-Sarraj ST, Doey L, Logan J, Hurwitz V, Jarosz J, Bhangoo R et al.. Active dendritic cell immunotherapy for glioblastoma: Current status and challenges. Br J Neurosurg 2015; 29:197-205; PMID:25541743; http://dx.doi.org/ 10.3109/02688697.2014.994473 [DOI] [PubMed] [Google Scholar]
- 4.Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014; 15:e257-67; PMID:24872109; http://dx.doi.org/ 10.1016/S1470-2045(13)70585-0 [DOI] [PubMed] [Google Scholar]
- 5.Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, Agostinis P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med 2016; 8:328ra27; http://dx.doi.org/ 10.1126/scitranslmed.aae0105 [DOI] [PubMed] [Google Scholar]
- 6.Blank CU, Haanen JB, Ribas A, Schumacher TN. Cancer immunology. The “cancer immunogram.” Science 2016; 352:658-60 [DOI] [PubMed] [Google Scholar]
- 7.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/ 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garg AD, Romano E, Rufo N, Agostinis P. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: mechanisms and clinical translation. Cell Death Differ 2016; 23:938-51; PMID:26891691; http://dx.doi.org/ 10.1038/cdd.2016.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, De Vleeschouwer S, Agostinis P, Graf N, Van Gool SW. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer 2015; 136:E313-25; PMID:25208916; http://dx.doi.org/ 10.1002/ijc.29202 [DOI] [PubMed] [Google Scholar]
- 10.Vandenberk L, Garg AD, Verschuere T, Koks C, Belmans J, Beullens M, Agostinis P, De Vleeschouwer S, Van Gool SW. Irradiation of necrotic cancer cells, employed for pulsing dendritic cells (DCs), potentiates DC vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology 2016; 5:e1083669; PMID:27057467; http://dx.doi.org/ 10.1080/2162402X.2015.1083669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirzaei R, Sarkar S, Yong VW. T cell exhaustion in glioblastoma: Intricacies of immune checkpoints. Trends Immunol 2017; 38:104-15; PMID:27964820; http://dx.doi.org/ 10.1016/j.it.2016.11.005 [DOI] [PubMed] [Google Scholar]
- 12.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L et al.. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res 2014; 20:5290-301; PMID:24691018; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-0514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Eng J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M et al.. Pembrolizumab versus ipilimumab in advanced melanoma. N Eng J Med 2015; 372:2521-32; PMID:25891173; http://dx.doi.org/ 10.1056/NEJMoa1503093 [DOI] [PubMed] [Google Scholar]
- 15.Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J et al.. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res 2016; 4:124-35; PMID:26546453; http://dx.doi.org/ 10.1158/2326-6066.CIR-15-0151 [DOI] [PubMed] [Google Scholar]
- 16.Cockle JV, Rajani K, Zaidi S, Kottke T, Thompson J, Diaz RM, Shim K, Peterson T, Parney IF, Short S et al.. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro Oncol 2016; 18:518-27; PMID:26409567; http://dx.doi.org/ 10.1093/neuonc/nov173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chabanon RM, Pedrero M, Lefebvre C, Marabelle A, Soria JC, Postel-Vinay S. Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res 2016; 22:4309-21; PMID:27390348; http://dx.doi.org/ 10.1158/1078-0432.CCR-16-0903 [DOI] [PubMed] [Google Scholar]
- 18.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015; 348:56-61; PMID:25838373; http://dx.doi.org/ 10.1126/science.aaa8172 [DOI] [PubMed] [Google Scholar]
- 19.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/ 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 20.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM et al.. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015; 350:207-11; PMID:26359337; http://dx.doi.org/ 10.1126/science.aad0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL et al.. Signatures of mutational processes in human cancer. Nature 2013; 500:415-21; PMID:23945592; http://dx.doi.org/ 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V et al.. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 2016; 34:2206-11; PMID:27001570; http://dx.doi.org/ 10.1200/JCO.2016.66.6552 [DOI] [PubMed] [Google Scholar]
- 23.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH et al.. The somatic genomic landscape of glioblastoma. Cell 2013; 155:462-77; PMID:24120142; http://dx.doi.org/ 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061-8; PMID:18772890; http://dx.doi.org/ 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013; 45:1113-20; PMID:24071849; http://dx.doi.org/ 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madhavan S, Zenklusen JC, Kotliarov Y, Sahni H, Fine HA, Buetow K. Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol Cancer Res 2009; 7:157-67; PMID:19208739; http://dx.doi.org/ 10.1158/1541-7786.MCR-08-0435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Børresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci U S A 2002; 99:12963-8; PMID:12297621; http://dx.doi.org/ 10.1073/pnas.162471999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cava C, Bertoli G, Ripamonti M, Mauri G, Zoppis I, Della Rosa PA, Gilardi MC, Castiglioni I. Integration of mRNA expression profile, copy number alterations, and microRNA expression levels in breast cancer to improve grade definition. PLoS One 2014; 9:e97681; PMID:24866763; http://dx.doi.org/ 10.1371/journal.pone.0097681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017; 355:355; PMID:28126774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng C, Miao X, Li Y, Huang Y, Ruan J, Ma X, Wang L, Wu CI, Cai J. Determination of genomic copy number alteration emphasizing a restriction site-based strategy of genome re-sequencing. Bioinformatics 2013; 29:2813-21; PMID:23962614; http://dx.doi.org/ 10.1093/bioinformatics/btt481 [DOI] [PubMed] [Google Scholar]
- 31.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A et al.. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013; 39:782-95; PMID:24138885; http://dx.doi.org/ 10.1016/j.immuni.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 32.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD et al.. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015; 21:938-45; PMID:26193342; http://dx.doi.org/ 10.1038/nm.3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garg AD, De Ruysscher D, Agostinis P. Immunological metagene signatures derived from immunogenic cancer cell death associate with improved survival of patients with lung, breast or ovarian malignancies: A large-scale meta-analysis. Oncoimmunology 2016; 5:e1069938; PMID:27057433; http://dx.doi.org/ 10.1080/2162402X.2015.1069938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. bioRxiv 2016; 18(1):248-62 [DOI] [PubMed] [Google Scholar]
- 35.Kyi C, Postow MA. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett 2014; 588:368-76; PMID:24161671; http://dx.doi.org/ 10.1016/j.febslet.2013.10.015 [DOI] [PubMed] [Google Scholar]
- 36.Twyman-Saint\sVictor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM et al.. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015; 520:373-7; PMID:25754329; http://dx.doi.org/ 10.1038/nature14292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L et al.. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res 2016; 5(1):29-41; PMID:27923825 [DOI] [PubMed] [Google Scholar]
- 38.Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget 2015; 6:26841-60; PMID:26314964; http://dx.doi.org/ 10.18632/oncotarget.4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol 2017; 18:123-31; PMID:28092374; http://dx.doi.org/ 10.1038/ni.3666 [DOI] [PubMed] [Google Scholar]
- 40.Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ 2014; 21:26-38; PMID:23686135; http://dx.doi.org/ 10.1038/cdd.2013.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, Sedighim S, Treger J, Odesa S, Tucker A et al.. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro Oncol 2017; now287; PMID:28115578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H et al.. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nature communications 2016; 7:10501; PMID:26883990; http://dx.doi.org/ 10.1038/ncomms10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baert T, Garg AD, Vindevogel E, A VANH, Verbist G, Agostinis P, Vergote I, Coosemans AN. In vitro generation of murine dendritic cells for cancer immunotherapy: An optimized protocol. Anticancer Res 2016; 36:5793-801; PMID:27793901; http://dx.doi.org/ 10.21873/anticanres.11163 [DOI] [PubMed] [Google Scholar]
- 44.Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Breckpot K, Brough D, Chaurio R, Cirone M et al.. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol 2015; 6:588; PMID:26635802; http://dx.doi.org/ 10.3389/fimmu.2015.00588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J et al.. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Eng J Med 2016; 375:2561-9; PMID:28029927; http://dx.doi.org/ 10.1056/NEJMoa1610497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reardon David A., De Groot John Frederick, Howard Colman, Jordan Justin T, Daras Mariza, Jennifer Leigh Clarke et al.. Safety of pembrolizumab in combination with bevacizumab in recurrent glioblastoma (rGBM). J Clin Oncol 2016; 34:URL: http://meetinglibrary.asco.org/content/163977-176 [Google Scholar]
- 47.Maes W, Rosas GG, Verbinnen B, Boon L, De Vleeschouwer S, Ceuppens JL, Van Gool SW. DC vaccination with anti-CD25 treatment leads to long-term immunity against experimental glioma. Neuro Oncol 2009; 11:529-42; PMID:19336528; http://dx.doi.org/ 10.1215/15228517-2009-004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verschuere T, Toelen J, Maes W, Poirier F, Boon L, Tousseyn T, Mathivet T, Gerhardt H, Mathieu V, Kiss R et al.. Glioma-derived galectin-1 regulates innate and adaptive antitumor immunity. Int J Cancer 2014; 134:873-84; PMID:23929302; http://dx.doi.org/ 10.1002/ijc.28426 [DOI] [PubMed] [Google Scholar]
- 49.Vogel I, Verbinnen B, Maes W, Boon L, Van Gool SW, Ceuppens JL. Foxp3+ regulatory T cells are activated in spite of B7-CD28 and CD40-CD40L blockade. Euro J Immunol 2013; 43:1013-23; PMID:23348953; http://dx.doi.org/ 10.1002/eji.201242737 [DOI] [PubMed] [Google Scholar]
- 50.Hanihara M, Kawataki T, Oh-Oka K, Mitsuka K, Nakao A, Kinouchi H. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. J Neurosurg 2016; 124:1594-601; PMID:26636389; http://dx.doi.org/ 10.3171/2015.5.JNS141901 [DOI] [PubMed] [Google Scholar]
- 51.Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, Hoang K, Ashley C, McCall D, Rojiani AM et al.. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J Immunother Cancer 2014; 2:21; PMID:25054064; http://dx.doi.org/ 10.1186/2051-1426-2-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, Nicholas S, Kellett M, Ruzevick J, Jackson C et al.. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS One 2014; 9:e101764; PMID:25013914; http://dx.doi.org/ 10.1371/journal.pone.0101764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vom Berg J, Vrohlings M, Haller S, Haimovici A, Kulig P, Sledzinska A, Weller M, Becher B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J Exp Med 2013; 210:2803-11; PMID:24277150; http://dx.doi.org/ 10.1084/jem.20130678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT Jr.. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J Immunother 2012; 35:385-9; PMID:22576343; http://dx.doi.org/ 10.1097/CJI.0b013e3182562d59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, Cummings T, Allison JP, Bigner DD, Sampson JH. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res 2007; 13:2158-67; PMID:17404100; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-2070 [DOI] [PubMed] [Google Scholar]
- 56.Hardcastle J, Mills L, Malo CS, Jin F, Kurokawa C, Geekiyanage H, Schroeder M, Sarkaria J, Johnson AJ, Galanis E. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol 2016; now179; PMID:27663389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antonios JP, Soto H, Everson RG, Orpilla J, Moughon D, Shin N, Sedighim S, Yong WH, Li G, Cloughesy TF et al.. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016; 1:pii: e87059; PMID:27453950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, Liu A, Sankey EW, Tam A, Xu H et al.. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res 2016; 23(1):124-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mathios D, Park CK, Marcus WD, Alter S, Rhode PR, Jeng EK, Wong HC, Pardoll DM, Lim M. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int J Cancer 2016; 138:187-94; PMID:26174883; http://dx.doi.org/ 10.1002/ijc.29686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E et al.. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys 2013; 86:343-9; PMID:23462419; http://dx.doi.org/ 10.1016/j.ijrobp.2012.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, Garzon-Muvdi T, Kim E, Theodros D, Polanczyk M et al.. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med 2016; 8:370ra180; PMID:28003545; http://dx.doi.org/ 10.1126/scitranslmed.aag2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics 2004; 20:1453-4; PMID:14871861; http://dx.doi.org/ 10.1093/bioinformatics/bth078 [DOI] [PubMed] [Google Scholar]
- 64.Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics 2004; 20:3246-8; PMID:15180930; http://dx.doi.org/ 10.1093/bioinformatics/bth349 [DOI] [PubMed] [Google Scholar]
- 65.Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database. BMC Cancer 2014; 14:970; PMID:25518851; http://dx.doi.org/ 10.1186/1471-2407-14-970 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.