

Abstract
Physiologically based pharmacokinetic (PBPK) modeling can be used to predict drug pharmacokinetics in virtual populations using models that integrate understanding of physiological systems. PBPK models have been widely utilized for predicting pharmacokinetics in clinically untested scenarios during drug applications and regulatory reviews in recent years. Here, we provide a comprehensive review of the application of PBPK in new drug application (NDA) review documents from the US Food and Drug Administration (FDA) in the past 4 years.
A physiologically based pharmacokinetic (PBPK) model is a mathematical model that simulates pharmacokinetics of xenobiotics in the human or animal body by reflecting the current physiological understanding related to absorption, distribution, metabolism, and elimination. One salient feature is that PBPK models allow for the mechanistic and prospective prediction of a drug's pharmacokinetic profiles and aids in new drug development and regulatory decision‐making process.1, 2, 3, 4, 5 We surveyed the utilization of PBPK in the US Food and Drug Administration (FDA) approval, specifically focusing on their application and impact on labeling recommendations.
Overview of physiologically based pharmacokinetic appearance in the FDA new drug application review
Our survey covered all small molecule new molecular entity (NME) drugs that are intended for systemic use and were approved by the FDA between January 2013 and August 2016. The new drug application (NDA) review documents (Drugs@FDA, http://www.fda.gov/drugsatfda) and product labels were examined for PBPK‐related information for the 85 products that met the above criteria. There were a total of 18 products for which PBPK models were considered in the NDA review documents (Tables 1–4). In the majority of cases, PBPK models were used for the prediction of the effect of metabolic enzyme mediated drug‐drug interactions (DDIs; Figure 1a; Tables 1–4). Importantly, the frequency of model use is consistent with the perceived level of reliability.1, 3, 5
Table 1.
PBPK models in product labels or FDA review documents: New drug as a victim of DDIs or genetic variations
| ID | NME | Year of approval | Detail of predicted scenarios | Simulation results | Impact / outcome | Dataset or strategy for model development/validation | Referencesa |
|---|---|---|---|---|---|---|---|
| 1 | Aripiprazole lauroxil | 2015 | Combination of CYP3A/2D6 inhibitor and CYP2D6 pharmacogenetics | Clinically meaningful effects predicted | Predicted exposure change informed labeling recommendation (dose adjustments, avoid concomitant use, or no warnings) | DDI with ketoconazole and quinidine (aripiprazole) and CYP2D6 pharmacogenetics (aripiprazole and aripiprazole lauroxil) | |
| 2 | Ceritinib | 2014 | CYP3A moderate inhibitor and inducer, inhibitor effect on lower ceritinib dose | DDI with ketoconazole and rifampin | |||
| 3 | Cobimetinib | 2015 | CYP3A moderate inhibitor, CYP3A strong and moderate inducer | DDI with itraconazole | 27225997 | ||
| 4 | Eliglustat | 2014 | Combination of CYP3A/2D6 inhibitor and CYP2D6 pharmacogenetics | DDI with ketoconazole, paroxetine, metoprolol, or rifampin and CYP2D6 pharmacogenetics | |||
| 5 | Ibrutinib | 2013 | CYP3A moderate and weak inhibitor, CYP3A moderate inducer | DDI with ketoconazole and rifampin | 27367453 | ||
| 6 | Macitentan | 2013 | DDI with ritonavir, inhibitor effect on multiple dose macitentan | DDI with ketoconazole (single‐dose macitentan) | 26385839 | ||
| 7 | Naloxegol | 2014 | CYP3A moderate inducer | DDI with ketoconazole, diltiazem, quinidine, and rifampin | 27299937 | ||
| 8 | Olaparib | 2014 | CYP3A moderate inhibitor & inducer | DDI with ketoconazole and rifampin | |||
| 9 | Panobinostat | 2015 | CYP3A strong inducer | DDI with ketoconazole | |||
| 10 | Simeprevir | 2013 | CYP3A strong and weak inhibitor, rifampin (single dose) | DDI with ritonavir, darunavir/ritonavir, efavirenz, erythromycin, cyclosporine A, and rifampin (multiple dose) | 27896690 | ||
| 11 | Sonidegib | 2015 | CYP3A moderate inhibitor and inducer | DDI with erythromycin and rifampin | |||
| 12 | Belinostat | 2014 | UGT1A1 pharmacogenetics (*28 genotype) | (See text for details) | No clinical DDI/pharmacogenetic study available for validation | ||
| 13 | Osimertinib | 2015 | CYP3A inhibitor and inducer | Results not available | No impact on labeling recommendation, PMR/PMC to conduct DDI studies | No clinical DDI study available for validation |
CYP, cytochrome; DDI, drug‐drug interaction; ID, identification; NME, new molecular entity; PBPK, physiologically based pharmacokinetic; PMC, postmarketing commitment; PMR, postmarketing requirement; UGT1A1, UDP‐glucuronosyltransferase 1A1.
The numbers in the Reference column represent PubMed ID (if physiologically based pharmacokinetic models were published in scientific journals). New drug application review documents can be found at Drugs@FDA (http://www.fda.gov/drugsatfda). If not specified, Simcyp was used for PBPK simulations.
Table 4.
PBPK models in product labels or FDA review documents: Other areas of PBPK applications
| ID | NME | Year of approval | Detail of predicted scenarios | Simulation results | Impact / outcome | Referencesa |
|---|---|---|---|---|---|---|
| 27 | Ceritinib | 2014 | HI | Minimal effect predicted | No impact on labeling recommendation, PMR to determine HI effect | |
| 28 | Ibrutinib | 2013 | HI | Significant overestimation compared to interim clinical data | No impact on labeling recommendation, PMR to complete HI study | |
| 29 | Obeticholic acid | 2016 | HIb | Simulated plasma exposure matched observed parent and metabolite pharmacokinetic profile; predicted significantly smaller HI effect on hepatic exposures than plasma exposures | Helped regulatory recommendations of possible up‐titration for HI patients | |
| 30 | Simeprevir | 2013 | HI | Significant overestimation compared to interim clinical data | No impact on labeling recommendation, PMR to complete HI study | 27896690 |
| Mechanism of nonlinear pharmacokinetics | Saturation of OATP1B and CYP3A explained observed nonlinearity in exposure | No direct labeling impact, contributed to model development to inform DDI simulation | ||||
| Ethnic differences in exposure between whites and Asian | Observed plasma exposure difference reproduced with simulation; hepatic drug exposure simulated in different populations | No direct labeling impact |
CYP, cytochrome; DDI, drug‐drug interaction; HI, hepatic impairment; ID, identification; NME, new molecular entity; OATP, organic anion‐transporting polypeptide; PBPK, physiologically based pharmacokinetic; PMR, postmarketing requirement.
The numbers in the Reference column represent PubMed ID (if physiologically based pharmacokinetic [PBPK] models were published in scientific journals). NDA review documents can be found at Drugs@FDA (http://www.fda.gov/drugsatfda).
In‐house custom model built on Phoenix nonlinear mixed effects was used for PBPK simulation. If not specified, Simcyp was used for PBPK simulations.
Figure 1.
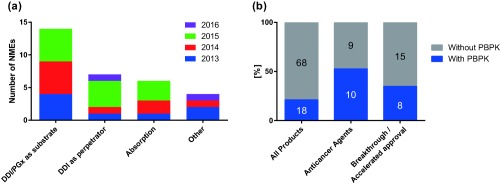
Overview of physiologically based pharmacokinetic (PBPK) information in product labels or US Food and Drug Administration (FDA) review documents for drugs approved by the FDA between January 2013 and August 2016. (a) Number of new molecular entities (NMEs) with information of PBPK for respective areas of applications. (b) Proportion of product labels/reviews containing PBPK information for drugs in all NMEs, anticancer agents, and NMEs with breakthrough therapy designation and/or accelerated approval status at the time of approval. The numbers on the bars represent the number of products in each category. Three of eight NMEs with PBPK in nononcology field were for rare diseases. Seven of eight NMEs with PBPK and breakthrough/accelerated approval status were anticancer agents. DDI, drug‐drug interaction; PGx, pharmacogenomics.
A key caveat of our survey was that it was limited to publicly available information. The utilization of PBPK models for sponsor's internal decision‐making and earlier stage and postapproval regulatory interactions, such as postmarketing requirement (PMR) or supplementary NDA, were not captured. PBPK models have great potential to influence decision‐making at different stages of drug development, such as initial dosing recommendations for pediatric clinical trials, design of DDI studies, etc.5 The current evaluation should be interpreted in the context of PBPK use during the final phase in the development of a new drug, because PBPK modeling strategies and the level of model validation can vary at different stages of drug development. This survey also did not capture the potential value of cost and speed savings by using PBPK vs. conducting clinical studies.
Utilization in the field of oncology
Interestingly, we observed wider acceptance of PBPK models in the field of oncology compared with other therapeutic areas (Figure 1b). One possible explanation is the difficulty of conducting clinical DDI studies in oncology, due to (1) shortage of appropriate patient populations or (2) ethical and safety concerns over exposing healthy volunteers to oncology medications. The first point is supported by the observation that three of eight NMEs with PBPK in the nononcology field were for rare diseases (such as eliglustat, macitentan, and obeticholic acid). Another possible explanation is that the higher levels of toxicity and narrower therapeutic windows for oncology drugs compared with drugs in other therapeutic areas warrant precise optimization of drug exposure. It is also noteworthy that 7 of 10 anticancer agents were given regulatory incentives to accelerate drug development, either with breakthrough therapy designation or accelerated approval, which may have contributed to a greater reliance on PBPK simulations.
New drug as a victim of drug‐drug interactions or genetic variations
PBPK models have been most extensively used for the prediction of the effect of DDI or pharmacogenetic effect on the pharmacokinetics of NME as a victim (Figure 1a). In particular, the extrapolation of the effect of strong inhibitors or inducers to less potent perpetrators constituted the majority of the applications (11 among 13 NMEs; Table 1, IDs 1–11), and all applications resulted in labeling recommendations (Table 1). In these cases, existing clinical data with strong perpetrator(s) were used to “anchor” the PBPK model performance, namely by accurately providing fraction metabolized by a particular enzyme, which provides a higher level of confidence in DDI prediction.1, 5 For example, a fourfold dose reduction of ibrutinib was recommended for patients taking moderate cytochrome P450 (CYP)3A inhibitors based on the PBPK model validated with clinical DDI data using strong perpetrators.
The elimination pathways for all the 11 NMEs involve CYP3A‐mediated metabolism, whereas two are metabolized by CYP2D6 in addition to CYP3A. This observation is not surprising, considering that CYP3A and CYP2D6 are involved in metabolism of a large proportion of marketed drugs, and well‐established probe perpetrators are available for these two enzymes. For two CYP3A and CYP2D6 dual substrates, aripiprazole lauroxil and eliglustat, complex interactions involving the inhibitors of these enzymes and CYP2D6 genotype have been predicted and utilized for dosing recommendations. Anchoring of model prediction with both a strong inhibitor and an inducer has been conducted in most cases, but there were two cases in which model validation with inhibitors was used for the prediction of inducer effect (cobimetinib, panobinostat), both of which resulted in a conservative labeling language of avoiding concomitant use with the strong inducers. This may suggest that the required level of PBPK model qualification depends on the context of PBPK application.
Because modification of recommended dose in the product label is considered a “high‐impact” application of PBPK,2 the use of a certain amount of clinical pharmacokinetic data as an external validation dataset has generally been required for including dosing recommendations in the product label. The case of belinostat is intriguing‐although there were no external data, the simulation performed by the FDA during the NDA review resulted in label language recommending a 25% reduction in the starting dose for homozygous carriers of a genetic polymorphism of its major metabolic enzyme, UDP‐glucuronosyltransferase 1A1. This is likely, in part, because the recommended starting dose is equal to the maximum tolerated dose. PMR to definitively examine the effect of pharmacogenetic alteration on belinostat pharmacokinetics and safety was included in the NDA approval. Therefore, even when the PBPK approach could not obviate the conduct of a dedicated clinical study, the model can inform the optimal use of medications.
New drug as a perpetrator of drug‐drug interactions
The second most frequent use of PBPK modeling is to predict the DDI potency of an NME as a perpetrator (inhibitor/inducer; Table 2). Four NMEs (IDs 14–17) have successfully utilized a PBPK modeling approach to demonstrate the lack of a clinically significant effect on the metabolic pathways of interest for the particular drug. Interestingly, strategies of model validation were different for these four NMEs. One method was to use a negative clinical DDI result with other CYP enzymes as an external validation (alectinib, panobinostat), whereas another was to use a sensitivity analysis on inhibition parameters (alectinib, canagliflozin). In the case of lenvatinib, an external model validation was seemingly not performed, presumably supported by general perception that PBPK‐based prediction of mechanism‐based inhibition leads to overestimation of DDIs.5 These observations based on limited cases suggest that required levels of model validation could be flexible, and that we may expect wider application of PBPK in this category.
Table 2.
PBPK models in product labels or FDA review documents: New drug as a perpetrator of drug‐drug interactions
| ID | NME | Year of approval | Detail of predicted scenarios | Simulation results | Impact / outcome | Dataset or strategy for model development/validation | Referencesa |
|---|---|---|---|---|---|---|---|
| 14 | Alectinib | 2015 | CYP2C8 inhibition | Clinically meaningful effect unlikely | Label states no clinically significant effect expected or label does not contain description on interaction potency | No effect on midazolam exposure in a clinical DDI study, sensitivity analysis of Ki,CYP2C8 | |
| 15 | Canagliflozin | 2013 | CYP2B6 inhibition | No effect on simvastatin and warfarin exposure in clinical DDI studies | 27862160 | ||
| 16 | Lenvatinib | 2015 | CYP2C8 and CYP3A inhibition | No external validation performed | |||
| 17 | Panobinostat | 2015 | CYP3A inhibition | Sensitivity analysis of CYP3A inactivation constant (kinact) | |||
| 18 | Brivaracetam | 2016 | CYP2C19 and OCT2/MATEs inhibition | Result not available | PBPK model not reviewed since basic DDI prediction model was sufficient | – | |
| 19 | Ceritinib | 2014 | CYP3A modulation | Clinically meaningful effect predicted | No impact on labeling recommendation, PMR/PMC to conduct DDI studies | No DDI data (as perpetrator) for validation | |
| 20 | Osimertinib | 2015 | CYP3A modulation | Result not available | No DDI data for validation |
CYP, cytochrome; DDI, drug‐drug interaction; ID, identification; MATE, multidrug and toxin extrusion transporter; NME, new molecular entity; OCT2, organic cation transporter 2; PBPK, physiologically based pharmacokinetic; PMC, postmarketing commitment; PMR, postmarketing requirement.
The numbers in the Reference column represent PubMed ID (if physiologically based pharmacokinetic models were published in scientific journals). NDA review documents can be found at Drugs@FDA (http://www.fda.gov/drugsatfda). If not specified, Simcyp was used for PBPK simulations.
There were two NMEs with PBPK models for which additional dedicated clinical DDI studies with CYP3A substrates were requested as PMR. For ceritinib, PBPK prediction resulted in a 5 to 10‐fold increase of midazolam exposure. In addition, neither of these two NMEs had supporting clinical data to verify model performance. Further examples will be needed to evaluate the ability of the PBPK approach to quantitatively predict positive DDI effects.1
Drug absorption and other areas of application
Most of the PBPK applications on absorption‐related pharmacokinetic changes were exploratory and did not impact the labeling recommendation (Table 3). One exception was panobinostat, for which the product label states “altered panobinostat absorption was not observed” with drugs that elevate the gastric pH in PBPK simulation.
Table 3.
PBPK models in product labels or FDA review documents: Drug absorption
| ID | NME | Year of approval | Detail of predicted scenarios | Simulation results | Impact / outcome | Referencesa |
|---|---|---|---|---|---|---|
| 21 | Alectinib | 2015 | Timing of food on alectinib exposureb | Less than 20% change predicted | PBPK model not reviewed because no obvious exposure‐response relationship identified | 27450228 |
| 22 | Ceritinib | 2014 | Food effect | Cmax change matched observation but AUC did not | No direct labeling impact, FDA exploratory simulations | |
| Effect of gastric pH change | Cmax and AUC decrease by 10% | |||||
| P‐gp contribution to intestinal absorption | Appeared to be minimal | |||||
| 23 | Ibrutinib | 2013 | Food effect to explain different exposure between healthy and oncology subject | Pharmacokinetic differences ascribed to type and timing of food on hepatic/intestinal blood flow rate | No direct labeling impact | |
| Intestinal exposure prediction | Dose staggering could lower the risk of P‐gp‐mediated DDI | |||||
| 24 | Naloxegol | 2014 | P‐gp contribution to intestinal absorption | Appeared to be minimal | No direct labeling impact | 27299937 |
| 25 | Panobinostat | 2015 | Effect of gastric pH changec | Minimal effect predicted (no change in the fraction of a drugs absorbed up to the gastric pH of 8.0) | Label states no ARA effect observed in simulation | |
| 26 | Sonidegib | 2015 | Food effect | Significant underestimation compared to clinical data | No direct labeling impact, FDA exploratory simulations |
ARA, acid reducing agents; AUC, area under the curve; Cmax, peak plasma concentration; FDA, US Food and Drug Administration; ID, identification; NME, new molecular entity; PBPK, physiologically based pharmacokinetic; P‐gp, P‐glycoprotein.
The numbers in the Reference column represent PubMed ID (if PBPK models were published in scientific journals). New drug application review documents can be found at Drugs@FDA (http://www.fda.gov/drugsatfda).
GastroPlus was used for PBPK simulation.
GastroPlus and Simcyp was used for PBPK simulation. If not specified, Simcyp was used for PBPK simulations.
Other PBPK applications observed in the NDA review documents include prediction of the effect of hepatic impairment (HI; Table 4), but PMRs were issued for three of four NMEs to conduct or complete dedicated clinical studies, presumably because of the low level of reliability.1 In the case of obeticholic acid, the effect of HI on hepatic exposure was explored with PBPK simulations, and this helped to inform the dosing recommendation in patients with HI.
Potential expansion of physiologically based pharmacokinetic model applications
The regulatory impacts of PBPK‐based predictions have been limited in areas other than metabolism‐based DDIs and pharmacogenetics, such as in transporter‐mediated DDIs or effect of acid reducing agents. This is largely attributable to lack of prediction performance verifications. Accumulated experience of PBPK application in these areas will help to establish the level of confidence necessary to inform regulatory decisions. Also, some degree of model validation with clinical data using the NME has generally been required for high‐impact regulatory decisions.2 Currently, validation of system components, such as hepatic metabolic activity in patients with renal impairment, based on other molecules that share same elimination pathways has not been considered sufficient. Such limitations have impeded the application of PBPK models to replace clinical studies for evaluating so‐called “intrinsic factors,” such as in pediatric populations, to evaluate ethnic differences in pharmacokinetics, or in patient populations with organ impairment. The authors believe that future verification efforts of PBPK‐based prediction performance with cross learning from other molecules, including both xenobiotics and endogenous biomarkers, will greatly expand the use of PBPK beyond DDI and pharmacogenetic applications and will contribute to acceleration of new drug development.
CONFLICT OF INTEREST
All authors are employees of Genentech, a member of the Roche Group. They have no other conflicts of interest to declare.
ACKNOWLEDGMENTS
The authors thank Drs. Amita Joshi, Leslie Chinn, and the Genentech Simcyp/GastroPlus Technical Working Group members for helpful discussions and feedback.
References
- 1. Wagner, C . et al Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst. Pharmacol. 4, 226–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. European Medicines Agency . Draft guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211315.pdf> (2016). Accessed 27 August 2016.
- 3. Jamei, M . Recent advances in development and application of physiologically‐based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr. Pharmacol. Rep. 2, 161–169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luzon, E. , Blake, K. , Cole, S. , Nordmark, A. , Versantvoort, C. & Berglund, E.G. Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin. Pharmacol. Ther. (2016). [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 5. Jones, H.M. et al Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin. Pharmacol. Ther. 97, 247–262 (2015). [DOI] [PubMed] [Google Scholar]