

Abstract
Microglia play a pivotal role in innate immunity in the brain. During development, they mature from myeloerythroid progenitor cells in the yolk sac and colonize the brain to establish a resident population of tissue macrophages. In the postnatal brain, they exert phagocytosis and induce inflammatory response against invading pathogens. Microglia also act as guardians of brain homeostasis by surveying the microenvironment using motile processes. Cortical spreading depression (CSD) is a slowly propagating (2–5 mm/min) wave of rapid, near-complete depolarization of neurons and astrocytes followed by a period of electrical suppression of a distinct population of cortical neurons. Not only has CSD been implicated in brain migraine aura, but CSD-like events have also been detected in stroke and traumatic injury. CSD causes a considerable perturbation of the ionic environment in the brain, which may be readily detected by microglia. Although CSD is known to activate microglia, the role of microglial activation in CSD-related neurological disorders remains poorly understood. In this article, we first provide an overview of microglial development and the multiple functions of microglia. Then, we review existing data on the relationship between microglia and CSD and discuss the relevance of CSD-induced microglial activation in neurological disease.
Keywords: Microglia, cortical spreading depression, phagocytosis, high-mobility group box 1, synaptic pruning
Microglia: Multifunctional resident macrophages in the central nervous system
In the past, the central nervous system (CNS) was considered a site of immune privilege. Microglia, first described by Achúcarro and subsequently thoroughly characterized by his successor, Rio-Hortega,1 are now known to play a pivotal role in innate immunity within the CNS.2 In addition, emerging evidence demonstrates the presence of a conduit that enables communication between the brain parenchyma and cervical lymph nodes via the glymphatic system (an interstitial fluid (ISF)/cerebrospinal fluid (CSF) exchange system located in the perivascular space) (Figure 1).3–6 Along with brain microglia, this exchange system facilitates immune surveillance of brain ISF. These findings suggest that the CNS immune system is more constitutively active than previously envisioned. A currently supported view concerning the origin of microglia postulates that they are derived from erythromyeloid progenitors in yolk sac that enter the brain during different developmental stages in the brain.7 Yolk sac erythromyeloid progenitors express the chemokine receptor CX3CR1.8 To closely analyze the fate of erythromyeloid progenitors in the CNS during embryogenesis, Kierdorf et al.9 used Cx3cr1GFP/wt mice, which contain a GFP knock-in on one allele of the Cx3cr1 gene. They discovered that mouse microglia were derived from primitive c-kit+ erythromyeloid precursors present in the yolk sac of embryos at embryonic age of 7.5 to 8.0 days (E7.5–E8.0) (Figure 2(a)). These precursors develop into CD45+ c-kitlow CX3CR1− immature (A1) cells and mature into CD45+ c-kit− CX3CR1+ (A2) cells. Microglial maturation is accompanied by a reduction in CD31 expression and concomitant upregulation of the macrophage marker, F4/80, and macrophage colony stimulating factor receptor (MCSF-R). Proliferating A2 cells develop into microglia and invade the developing brain at E9.5, a process that requires activity of matrix metalloproteinase. After entering the CNS, microglia actively engulf synaptic materials and engage in synaptic pruning during postnatal development. Engulfed synaptic components have been identified in microglial lysosomes.10 Inhibition of microglia-mediated synaptic pruning results in aberrant brain circuitry with excessive dendritic spines and immature synapses.10 Within the postnatal brain, microglia are maintained through cellular division or expansion of progenitors that colonized the brain during CNS development. Microglia form an autonomous population of cells within the brain that demonstrate activities independent of circulating monocyte activity.
Figure 1.
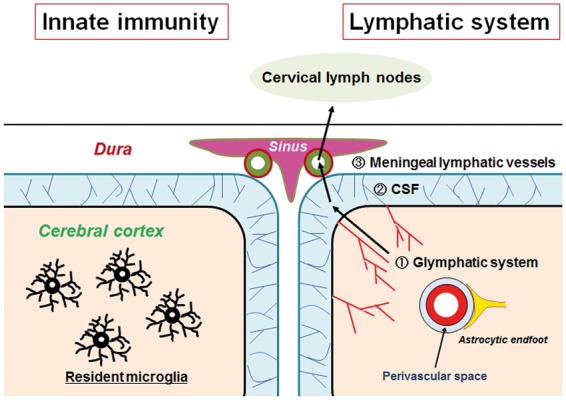
Immune surveillance in the CNS. Resident microglia are distributed in the brain parenchyma, and use motile processes to survey the microenvironment (left). They play a pivotal role in the CNS innate immunity. In parallel, the brain parenchyma is equipped with the glymphatic system, which clears interstitial solutes through the exchange of ISF and CSF along the perivascular space between the wall of the veins and astrocytic endfeet. Subsequently, CSF is absorbed into the meningeal lymphatic vessels, which are located in the vicinity of the venous sinus. Lymphatic fluid drains into the cervical lymph nodes (right). Both systems are considered to serve as major immune surveillance mechanisms.
Figure 2.

(a) Schematic representation of microglial development: Microglia are derived from c-kit-positive erythromyeloid precursors (EMPs) in the yolk sac at E7.5–E8.0. These precursors develop into CD45+ c-kitlow CX3CR1− immature (A1) cells and mature into CD45+ c-kit− CX3CR1+ (A2) cells. Proliferating A2 cells develop into embryonic microglia. Before completion of the blood–brain barrier (BBB), they invade the developing brain in a manner requiring matrix metalloproteinase (MMP) activity at E9.5. A2 cells colonize the brain tissue to establish a population of mature microglia. By contrast, blood monocytes are derived from hematopoietic stem cells (HSCs). Unless the BBB is disrupted, microglia and monocytes are independent of each other in the postnatal stage. (b) Multiple roles of microglia in the postnatal brain: Microglia in the postnatal brain are multifunctional. Primarily, they are involved in immune surveillance, and when necessary, they initiate an inflammatory response. Although this contributes to the elimination of pathogens, an excessive inflammatory response may result in bystander tissue damage. Microglia also phagocytose pathogens and apoptotic neurons and exert scavenging actions on tissue debris under pathological conditions. Microglia recognizes apoptotic cells by phosphatidylserine and other apoptotic signals. With regard to synaptic refinement, they carry out synaptic pruning in a manner requiring C3-C3R interactions. Microglia execute synaptic formation by secreting BDNF. In addition, microglia-derived TNFα can exert neuroprotective as well as neurodestructive action within different contexts.74 Microglia also play a role in angiogenesis through by synthesizing vascular endothelial growth factor (VEGF).75
In the postnatal brain, microglia have multiple functions (Figure 2(b)). As the powerhouse of local innate immunity, microglia protect the postnatal brain from infection.2 In response to pathogens invading the CNS, microglia act as a front-line defense system by phagocytosing pathogens, secreting humoral substances, such as proinflammatory cytokines, and producing reactive oxygen species (ROS) and reactive nitrogen species (RNS). Microglia-producing proinflammatory cytokines include interleukin-1β (IL-1β), IL-6, and tumor necrosis factor α (TNFα).11 These microglial actions collectively eliminate pathogens from the CNS. The activation pattern of microglia in response to invading pathogens is typical of the classical proinflammatory phenotype of macrophages (M1 polarization).12 This type of microglial activation is also involved in other disease processes. For instance, cerebral ischemia13 and amyloid β (Aβ)14 stimulate IL-1β synthesis in microglia through the activation of the inflammasome. Morphologically, activated microglia demonstrate enlarged cytoplasm and thickened processes. They tend to retract their processes and under extreme conditions, assume an amoeboid shape. Microglia also display an alternative activation pattern termed M2 polarization.12 M2-polarized microglia dampen inflammatory reactions and contribute to tissue remodeling and angiogenesis. Like other tissue macrophages, microglia engulf necrotic and apoptotic neurons. Dead neurons expose phosphatidylserine and other apoptotic “eat-me” signals on their cell surface, which prompts microglia to carry out phagocytosis.15 Such microglia-mediated clearance of unwanted neurons is an important process of proper brain tissue remodeling. With regard to microglia function under the physiological condition, it has been observed they are evenly distributed in the brain parenchyma.2 Recent in vivo imaging experiments have uncovered that these resident microglia constantly survey their surrounding microenvironment with their motile processes,16 which is likely relevant to immune surveillance. However, such motion has been observed even in the absence of offending microorganisms. Increasing evidence has revealed that microglia engulf presynaptic and postsynaptic elements by extending their processes to contact dendritic spines, axons, and synapses as observed in the developing brain.16,17 Microglia-mediated synaptic pruning is dependent on regional neuronal activity.18 In the visual cortex, visual experience causes microglial processes to change their morphology and motility; these processes alter distributions of extracellular space, display phagocytic structures, appose synaptic clefts more frequently, and envelope synapse-associated elements more extensively.19 Intriguingly, synaptic pruning occurs preferentially in response to less active inputs, which apparently contributes to activity-dependent synaptic plasticity and learning. Microglia express CR3, the high-affinity receptor for C3, which is required for the precise execution of synaptic pruning.20 Moreover, microglia are involved in synapse formation by secreting brain-derived neurotrophic factor (BDNF).21 Consistent with these data, genetic inhibition of microglia results in derangement of both synapse formation and elimination of dendritic spines.10
Clinical implications of cortical spreading depression
Cortical spreading depression (CSD) is defined as a slowly propagating (2–5 mm/min) wave of rapid, near-complete depolarization of neurons and astrocytes followed by a period of electrical suppression of a distinct population of cortical neurons (Figure 3). By a strict definition, CSD refers only to the electrical silence of brain electrical activity following spreading depolarizations.22 CSD is accompanied by secondary changes of cerebral blood flow (CBF), which comprised the following four distinct phases: (i) an initial, brief hypoperfusion, (ii) a marked, transient hyperemia, (iii) a later, smaller hyperemia, and (iv) a long-lasting oligemia. CSD was first observed in the cerebral cortex of healthy rabbits by Leao.23 Experimentally, CSD can be elicited by chemical stimulation (high potassium exposure, ATPase inhibitors, endothelin-1, N-methyl-D-aspartate (NMDA) receptor agonists), pinprick stimulation, and electrical stimulation of the intact cerebral cortex.27 CSD is considered the biological substrate of migraine aura.28 The most clinically convincing evidence for this came from an MRI study by Hadjikhani et al.,29 which demonstrated the clinico-radiological correlation of visual percept of aura symptoms and propagation of cortical blood oxygenation level-dependent (BOLD) signals in the visual cortex of a patient experiencing a migraine. In addition, CSD has been recorded by electrocorticography in patients with ischemic and hemorrhagic stroke and brain trauma.30,31 In most cases, however, electrical activity of brain tissue is already compromised before the development of spreading depolarizations. As a result, the characteristic temporal profile of CSD, in which rapidly evolving depolarizations of neural cells were followed by suppression of electrical activity, may not necessarily be observed. Because of this, “spreading depolarization” is now regarded as a more precise and preferred term to describe such conditions than CSD.24–26,32 Spreading depolarization can be elicited by cerebral ischemia or traumatic brain injury experimentally. Hereafter, we will use the term, CSD, in the broad sense. CSD can be induced in isolated brain slice cultures.33–38 Detailed electrophysiological analysis using hippocampal slices revealed that the apical dendrites of neurons were depolarized earlier than the somata during CSD.39,40 As compared to action potentials, CSD induces a greater magnitude of extracellular potential shift (typically 10–20 millivolts) which continues for much longer (at least several minutes). Correspondingly, CSD causes considerable alterations in the cerebral ionic environment. The initiation of CSD entails a rapid increase in [K+]e from 4 mM to 30–60 mM and a rapid decline in [Na+]e and [Cl−]e from 140 mM to 50–70 mM and [Ca2+]e from 1.5–2.0 mM to 0.2–0.8 mM.25,41,42 CSD also induces neurotransmitter release. For example, glutamate, the excitatory amino acid, is released into the extracellular space and binds to NMDA-type glutamate receptors to further promote the influx of Ca2+ and Na+.43,44 In response to these environmental changes, astrocytes take up excess potassium and glutamate released from neurons. Astroglial Na+, K+-ATPase activity is involved in these buffering actions. Consistent with these results, a loss-of-function mutation in the gene encoding the α2 isoform of the glial Na+, K+-ATPase (ATP1A2) causes familial hemiplegic migraine type 2 (FHM2), which is clinically characterized by prolonged aura episodes.45 Hence, clearance of potassium and glutamate from the extracellular space appears to be a critical event that determines predisposition to CSD inductions. Activation of the glial Na+, K+-ATPase is required to establish neuronal resting membrane potential and correct the CSD-induced derangement of the ionic environment. Since this process is accompanied by elevated ATP consumption. CSD imposes a considerable energetic burden on brain tissues. As expected, multiple consecutive CSD episodes robustly increase the cerebral metabolic rate of glucose (CMRGlu), as compared to a single CSD episode.46–48
Figure 3.
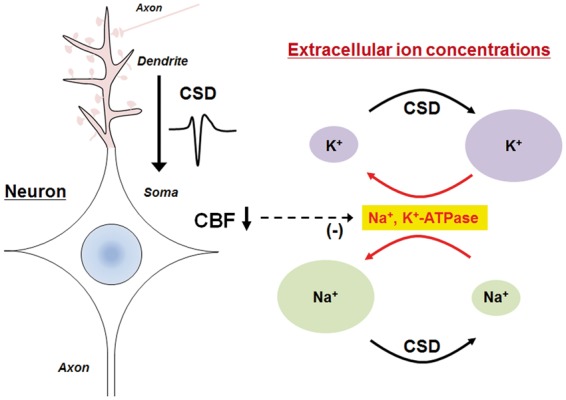
CSD-induced alterations in extracellular ion concentrations. CSD-associated depolarizations occur first in the dendrites and then propagate to the soma in cortical neurons. The initiation of CSD entails a rapid increase in [K+]e from 4 mM to 30–60mM and a rapid decline in [Na+]e from 140 mM to 50–70 mM (black arrows). Na+, K+-ATPase activity then rectifies the resultant abnormal ion distribution (red arrows). However, CSD-associated oligemia may have a negative impact on Na+, K+-ATPase activity.
Effects of CSD on microglial function
Microglia express voltage-sensitive ion channels, including Nav1.1, Kv1.3, and Kv1.5, and are thought to sense electrical activity pertaining to CSD.49–51 CSD initially elevates extracellular pH, which subsequently leads to a gradual decrease in tissue pH.52 Accumulation of protons may be detected by microglial transient receptor potential cation channel subfamily V, member 1 (TRPV1).53 A recent study revealed that microglia exhibit increased NMDA-dependent inward rectifying potassium conductance after CSD, which can be interpreted as a compensatory mechanism for elevated extracellular K+ concentration.54 However, little is known about microglia activity in response to CSD. Gehrmann et al.55 reported that the number of major histocompatibility complex (MHC) class II antigen-positive microglia significantly increased in the rat cerebral cortex between 16 and 24 h after CSD. This finding raised the possibility that CSD is able to elicit microglial immune reactions. Moreover, it has been shown that CSD can increase the proliferation and migration of microglia, both of which are well-recognized essential features of the immune response.36,56 Biochemically, there is evidence that CSD induces ROS production in microglia that are located in the affected brain tissues.37 In addition, CSD stimulates microglial secretion of IL-1β57 and TNFα35. Collectively, these data suggest that CSD can initiate inflammatory activation of microglia. Interestingly, ROS and TNFα have been shown to lower the threshold for CSD induction, thus forming a positive feedback mechanism that favors the perpetuation of CSD induction.35,37 On the other hand, TNFα has been shown to reduce CSD amplitudes in a dose-dependent manner.58 Hence, this proinflammarory cytokine seems to exert complex actions on CSD. In addition, IL-1β is also known to attenuate CSD amplitudes via the GABAA receptor activity at lower concentrations. However, at a high dose, IL-1β did not alter the magnitude of CSD amplitudes. Since these cytokines promote vascular permeability, as a whole, they are likely to have a deleterious effect on CNS tissue in CSD pathophysiology. Moreover, in hippocampal slice culture studies, selective depletion of microglial cells with clodronate conferred resistance to CSD induction.38 Conversely, restoration of microglial cells to previously depleted cultures restored the susceptibility to CSD.38 A similar phenomenon has been reported in an in vivo neuroimaging study.59 An in vivo calcium imaging technique demonstrated that microglia depletion led to a perturbation of neuronal calcium response.59 Hence, it is inferred that a certain microglia-regulated neuronal calcium response may be required for CSD occurrence. Furthermore, evidence shows that microglial CSD-generating activity can be inhibited by insulin-like growth factor-1 (IGF-1) treatment37 and environmental enrichment,38 the latter of which promotes M2-polarization of microglia (Figure 4). It has been demonstrated that IGF-1 antagonizes TNFα.37 These data were obtained in experimental settings where CSD was induced in cluster. Clinically, successive CSD/spreading depolarizations have been demonstrated in patients with stroke and traumatic brain injury.30,31 Experimental evidence demonstrates that CSD/spreading depolarizations contribute to the expansion of infarct volume by several mechanisms.60,61 First, a limited supply of ATP due to ischemia renders neurons unable to reestablish the resting membrane potential, which may lead up to the occurrence of terminal depolarization. Second, excess extracellular glutamate can induce excitotoxicity. IGF-1 treatment and other therapeutic interventions that promote M2-polarization of microglia may ameliorate secondary brain damage resulting from cerebral ischemia.
Figure 4.
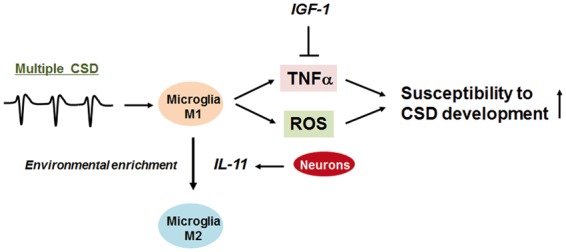
Microglia determine CSD susceptibility. Multiple CSD inductions cause M1-polarized activation of microglia. Resultant production of TNFα and ROS enhances the susceptibility of brain tissue to CSD development. The CSD-promoting action of TNFα can be inhibited by IGF-1. In addition, microglia polarized to the M2 activation mode by environment enrichment exhibit much less CSD susceptibility. The action of environmental enrichment can be mimicked by nasal administration of interleukin-11 (IL-11).
Microglial activation is dependent on the number of CSD inductions
As mentioned above, multiple CSD inductions are likely to activate microglia. Consequently, to determine if microglial activation occurs after a single CSD event, as seen in the usual migraine aura, we compared morphological changes of microglia in response to single and multiple CSD episodes. We found that a single CSD induction led to only subtle morphological changes in microglia, whereas multiple CSD inductions caused marked enlargement of microglia after 24 h.62 This morphological change normalized by 72 h after CSD inductions. However, there were no significant changes in the numbers of microglia. Our data suggest that a single episode of CSD is “innocuous” to the brain in terms of microglial activation. We did not detect activation of caspase-3 or DNA fragmentation detectable by the TUNEL assay (unpublished data). Nedergaard and Hansen63 reported that CSD was not associated with neuronal injury in the normal brain. This is also consistent with the traditionally held belief that a single attack of migraine with aura is a benign condition that does not cause any clinically relevant structural brain damage. We sought to clarify the events upstream of the multiple CSD-induced microglial enlargements. Karatas et al.64 reported that CSD causes the release of HMGB1 (high-mobility group box 1) from neurons via Pannexin 1 channels. Although HMGB1 is primarily located in the nucleus, upon injurious stimuli, it is released into the extracellular space, where it serves as a damage-associated molecular pattern (DAMP).65 We found that neuronal HMGB1 release was dependent on the number of CSD inductions such that only multiple CSD events were able to cause significant HMGB1 release from neurons (Figure 5).66 In addition, transcriptional activity of the HMGB1 gene in cortical neurons was enhanced in response to multiple CSD events, which may reflect an attempt to replenish the cellular pool of the HMGB1 protein.66 In general, DAMP molecules bind to their corresponding receptors, thus transmitting a danger signal to surrounding cells. Major HMGB1 receptors include toll-like receptor 2 (TLR2) and TLR4.65 The ligation of HMGB1 with these receptors initiates intracellular signaling cascades that involve MyD88 and IRAK4.67 We found that multiple CSD inductions enhanced the transcriptional activity of the TLR2, TLR4, MyD88, and IRAK4 genes in brain tissue. TLR2/4 is reportedly expressed in microglia. In our study, CSD inductions failed to cause morphological changes in microglia in TLR2/4 double knockout mice. Moreover, application of anti-HMGB1 antibody to the cortical surface attenuated the morphological alterations of microglia caused by multiple CSD inductions. These data indicate that the HMGB1–TLR2/4 axis plays a crucial role in the microglial activation caused by multiple CSD events. It is important to elucidate the functional significance of such activated microglia. We observed that the majority of hypertrophic microglia displayed prominent immunoreactivity for cathepsin D, a lysosomal acid hydrolase (Figure 6(a)).68 This finding was suggestive of an activated state of phagocytosis. Nevertheless, we did not observe any apoptotic changes in the brain tissue subjected to multiple CSD episodes, making it unlikely that activated microglia are involved in the execution of apoptosis or phagocytosis of dead neurons.68 Again, this was consistent with the previous report by Nedergaard and Hansen.63 An alternative concept is that activated microglial phagocytotic activity is involved in synaptic pruning (Figure 6(b)). As stated above, CSD elevates [Ca2+]i in dendritic spines of cortical neurons. A recent in vivo multiphoton microscopy study disclosed that CSD causes dramatic structural alterations of synapses between axons and the dendritic spines of cortical neurons.69 Several minutes after CSD, axonal bouton density increased by 20% and bouton size decreased by 25 to 40% compared to the resting state. Concomitantly, there was a morphological shift from predominantly stubby spines to thin or mushroom spines after CSD, which implies enhancement of synaptic excitability. Although single CSD events cause abnormalities in synaptic morphology and functionality, these alterations are short-lived and reversible. However, successive CSD episodes prevent the recovery of synaptic abnormalities, indicating that the accumulated stress of prior CSD events lead to irreversible dendritic injury.60 Thus, it is plausible that activated microglia engage in the repair of damaged synapses through synaptic pruning and formation after multiple CSD episodes.
Figure 5.

Microglial activation is influenced by the number of CSD episodes. A single CSD event, as seen in the usual aura of migraine patients, does not cause significant release of HMGB1 from cortical neurons. Conversely, clustering of CSD, as seen in stroke and brain trauma, induces a robust HMGB1 release from cortical neurons. Subsequently, HMGB1 acts on the TLR2/4 on the surface of microglia, which is followed by microglial activation, as demonstrated by their hypertrophic morphological alterations. The functional significance of microglial activation in this setting remains uncertain. They may contribute to neuroprotection, for example, through the synaptic repair on the dendritic spines of multiple CSD-affected cortical neurons. Alternatively, activated microglia may exert injurious actions.
Figure 6.

Upregulation of cathepsin D, a representative lysosomal acid hydrolase, in activated microglia subjected to multiple CSD episodes. (a) As compared to cortical microglia in untreated mice (upper row), those subjected to multiple (five times) CSD inductions (lower row) exhibit increased cathepsin D immunoreactivity (arrow). This finding raises the possibility that lysosomal activity is enhanced by multiple CSD events. The specimens were obtained from a cerebral cortex subjected to CSD. This is representative of our data obtained from six independent animals. Bar = 20 µm. (b) Microglia activated by multiple CSD events may be recruited to damaged dendritic spines for synaptic repair.
Concluding remarks: Unresolved issues concerning the role of microglia in CSD pathophysiology
Although aforementioned data suggest that microglia are required for the occurrence of CSD, the mechanisms by which microglia induce CSD remain unknown. Locally elevated extracellular K+ concentration is necessary for CSD induction.25,27 Provided that microglia raise extracellular K+ concentration before CSD occurrence, they would have to follow through in a voltage-independent manner. Microglia have been shown to express potassium intermediate/small conductance calcium-activated channels, subfamily N, member 4 (KCNN4/KCa3.1).70 This voltage-insensitive and calcium/calmodulin-regulated potassium channel has been implicated in lipopolysaccharide- and ischemia-induced brain damage.70 Activation of this channel can cause massive K+ efflux. Therefore, the involvement of KCNN4/KCa3.1 in the initiation of CSD may be worthy of investigation. Another unresolved issue is whether multiple CSD-induced microglial activation is an adaptive phenomenon or a harmful event to brain tissue. In-depth analysis of the molecules produced in these cells would be required to solve this problem. If microglia are indeed involved in synaptic pruning, the identification of synaptic components, like PSD95, within enlarged microglia should be an important confirmatory finding. Lastly, it remains uncertain whether immunological and inflammatory activation of multiple CSD-subjected microglia can influence systemic immunological activity. As mentioned above, the recently discovered lymphatic system makes possible immunological communication between the brain parenchyma and the peripheral lymphatic system. In this paradigm, the CSF space comprises part of the CNS lymphatic system. FHM is characterized by prolonged migraine aura, and neuroimaging data support repeated inductions of CSD.71 It has been reported that FHM attacks often cause high fever and in a fewer cases, CSF pleocytosis.71–73 It may, therefore, be interesting to explore whether a potential crosstalk between activated microglia and the CSF/lymphatic system is really relevant to CSD pathophysiology.
Acknowledgments
The authors are grateful to Drs. Toshihiko Shimizu and Tsubasa Takizawa for their insightful discussions.
Funding
The publication of this article was supported by a JSPS KAKENHI (Grant number 26460706 to MS) and a grant from Nippon Zoki Pharmaceutical Co., Ltd.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Mamoru Shibata was involved in concept, design, writing, and editing. Norihiro Suzuki was involved in writing and editing.
References
- 1.Tremblay ME, Lecours C, Samson L, et al. From the Cajal alumni Achucarro and Rio-Hortega to the rediscovery of never-resting microglia. Front Neuroanat 2015; 9: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casano AM, Peri F. Microglia: Multitasking specialists of the brain. Dev Cell 2015; 32: 469–77. [DOI] [PubMed] [Google Scholar]
- 3.Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 2012; 4: 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang L, Kress BT, Weber HJ, et al. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Transl Med 2013; 11: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015; 523: 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015; 212: 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat Rev Neurosci 2014; 15: 300–312. [DOI] [PubMed] [Google Scholar]
- 8.Bertrand JY, Jalil A, Klaine M, et al. Three pathways to mature macrophages in the early mouse yolk sac. Blood 2005; 106: 3004–3011. [DOI] [PubMed] [Google Scholar]
- 9.Kierdorf K, Erny D, Goldmann T, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 2013; 16: 273–280. [DOI] [PubMed] [Google Scholar]
- 10.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011; 333: 1456–1458. [DOI] [PubMed] [Google Scholar]
- 11.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci 2007; 8: 57–69. [DOI] [PubMed] [Google Scholar]
- 12.Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 2015; 7: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abulafia DP, de Rivero Vaccari JP, Lozano JD, et al. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab 2009; 29: 534–544. [DOI] [PubMed] [Google Scholar]
- 14.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008; 9: 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segawa K, Nagata S. An apoptotic ‘eat me’ signal: Phosphatidylserine exposure. Trends Cell Biol 2015; 25: 639–650. [DOI] [PubMed] [Google Scholar]
- 16.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005; 308: 1314–1318. [DOI] [PubMed] [Google Scholar]
- 17.Wake H, Moorhouse AJ, Jinno S, et al. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 2009; 29: 3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 2009; 10: 647–658. [DOI] [PubMed] [Google Scholar]
- 19.Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol 2010; 8: e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012; 74: 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parkhurst CN, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013; 155: 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011; 17: 439–447. [DOI] [PubMed] [Google Scholar]
- 23.Leao AAP. Spreading depression of activity in cerebral cortex. Electroencephalogr Clin Neurophysiol 1944; 7: 359–390.
- 24.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 2014; 15: 379–393. [DOI] [PubMed] [Google Scholar]
- 25.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015; 95: 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartings JA, Shuttleworth CW, Kirov SA, et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leao’s legacy. J Cereb Blood Flow Metab Epub ahead of print 21 June 2016. DOI: 10.1177/0271678X16654495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bures J, Buresova O, Krivanek J. The mechanism and applications of Leao’s spreading depression of electroencephalographic activity, Prague: ACADEMIA, 1974. [Google Scholar]
- 28.Milner PM. Note on a possible correspondence between the scotomas of migraine and spreading depression of Leao. Electroencephalogr Clin Neurophysiol 1958; 10: 705. [DOI] [PubMed] [Google Scholar]
- 29.Hadjikhani N, Sanchez Del Rio M, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A 2001; 98: 4687–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lauritzen M, Dreier JP, Fabricius M, et al. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 2011; 31: 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dreier JP, Reiffurth C. The stroke-migraine depolarization continuum. Neuron 2015; 86: 902–922. [DOI] [PubMed] [Google Scholar]
- 32.Hartings JA, Li C, Hinzman JM, et al. Direct current electrocorticography for clinical neuromonitoring of spreading depolarizations. J Cereb Blood Flow Metab Epub ahead of print 10 June 2016. DOI: 10.1177/0271678X16653135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snow RW, Taylor CP, Dudek FE. Electrophysiological and optical changes in slices of rat hippocampus during spreading depression. J Neurophysiol 1983; 50: 561–572. [DOI] [PubMed] [Google Scholar]
- 34.Kunkler PE, Kraig RP. Calcium waves precede electrophysiological changes of spreading depression in hippocampal organ cultures. J Neurosci 1998; 18: 3416–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grinberg YY, Dibbern ME, Levasseur VA, et al. Insulin-like growth factor-1 abrogates microglial oxidative stress and TNF-alpha responses to spreading depression. J Neurochem 2013; 126: 662–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grinberg YY, Milton JG, Kraig RP. Spreading depression sends microglia on Levy flights. PLoS One 2011; 6: e19294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grinberg YY, van Drongelen W, Kraig RP. Insulin-like growth factor-1 lowers spreading depression susceptibility and reduces oxidative stress. J Neurochem 2012; 122: 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pusic KM, Pusic AD, Kemme J, et al. Spreading depression requires microglia and is decreased by their M2a polarization from environmental enrichment. Glia 2014; 62: 1176–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herreras O, Somjen GG. Propagation of spreading depression among dendrites and somata of the same cell population. Brain Res 1993; 610: 276–282. [DOI] [PubMed] [Google Scholar]
- 40.Canals S, Makarova I, Lopez-Aguado L, et al. Longitudinal depolarization gradients along the somatodendritic axis of CA1 pyramidal cells: A novel feature of spreading depression. J Neurophysiol 2005; 94: 943–951. [DOI] [PubMed] [Google Scholar]
- 41.Phillips JM, Nicholson C. Anion permeability in spreading depression investigated with ion-sensitive microelectrodes. Brain Res 1979; 173: 567–571. [DOI] [PubMed] [Google Scholar]
- 42.Martins-Ferreira H, Nedergaard M, Nicholson C. Perspectives on spreading depression. Brain Res Brain Res Rev 2000; 32: 215–234. [DOI] [PubMed] [Google Scholar]
- 43.Mody I, Lambert JD, Heinemann U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol 1987; 57: 869–888. [DOI] [PubMed] [Google Scholar]
- 44.Marrannes R, Willems R, De Prins E, et al. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res 1988; 457: 226–240. [DOI] [PubMed] [Google Scholar]
- 45.De Fusco M, Marconi R, Silvestri L, et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 2003; 33: 192–196. [DOI] [PubMed] [Google Scholar]
- 46.Shinohara M, Dollinger B, Brown G, et al. Cerebral glucose utilization: Local changes during and after recovery from spreading cortical depression. Science 1979; 203: 188–190. [DOI] [PubMed] [Google Scholar]
- 47.Mies G, Niebuhr I, Hossmann KA. Simultaneous measurement of blood flow and glucose metabolism by autoradiographic techniques. Stroke 1981; 12: 581–588. [DOI] [PubMed] [Google Scholar]
- 48.Kocher M. Metabolic and hemodynamic activation of postischemic rat brain by cortical spreading depression. J Cereb Blood Flow Metab 1990; 10: 564–571. [DOI] [PubMed] [Google Scholar]
- 49.Schilling T, Quandt FN, Cherny VV, et al. Upregulation of Kv1.3 K(+) channels in microglia deactivated by TGF-beta. Am J Physiol Cell Physiol 2000; 279: C1123–C1134. [DOI] [PubMed] [Google Scholar]
- 50.Schilling T, Eder C. Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res 2007; 1186: 21–28. [DOI] [PubMed] [Google Scholar]
- 51.Black JA, Liu S, Waxman SG. Sodium channel activity modulates multiple functions in microglia. Glia 2009; 57: 1072–1081. [DOI] [PubMed] [Google Scholar]
- 52.Mutch WA, Hansen AJ. Extracellular pH changes during spreading depression and cerebral ischemia: Mechanisms of brain pH regulation. J Cereb Blood Flow Metab 1984; 4: 17–27. [DOI] [PubMed] [Google Scholar]
- 53.Schilling T, Eder C. Importance of the non-selective cation channel TRPV1 for microglial reactive oxygen species generation. J Neuroimmunol 2009; 216: 118–121. [DOI] [PubMed] [Google Scholar]
- 54.Wendt S, Wogram E, Korvers L, et al. Experimental cortical spreading depression induces NMDA receptor dependent potassium currents in microglia. J Neurosci 2016; 36: 6165–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gehrmann J, Mies G, Bonnekoh P, et al. Microglial reaction in the rat cerebral cortex induced by cortical spreading depression. Brain Pathol 1993; 3: 11–17. [DOI] [PubMed] [Google Scholar]
- 56.Urbach A, Brueckner J, Witte OW. Cortical spreading depolarization stimulates gliogenesis in the rat entorhinal cortex. J Cereb Blood Flow Metab 2015; 35: 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jander S, Schroeter M, Peters O, et al. Cortical spreading depression induces proinflammatory cytokine gene expression in the rat brain. J Cereb Blood Flow Metab 2001; 21: 218–225. [DOI] [PubMed] [Google Scholar]
- 58.Richter F, Lutz W, Eitner A, et al. Tumor necrosis factor reduces the amplitude of rat cortical spreading depression in vivo. Ann Neurol 2014; 76: 43–53. [DOI] [PubMed] [Google Scholar]
- 59.Szalay G, Martinecz B, Lenart N, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016; 7: 11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Risher WC, Ard D, Yuan J, et al. Recurrent spontaneous spreading depolarizations facilitate acute dendritic injury in the ischemic penumbra. J Neurosci 2010; 30: 9859–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hinzman JM, DiNapoli VA, Mahoney EJ, et al. Spreading depolarizations mediate excitotoxicity in the development of acute cortical lesions. Exp Neurol 2015; 267: 243–253. [DOI] [PubMed] [Google Scholar]
- 62.Takizawa T, Shibata M, Kayama Y, et al. High-mobility group box 1 is an important mediator of microglial activation induced by cortical spreading depression. J Cereb Blood Flow Metab Epub ahead of print 3 May 2016. DOI: 10.1177/0271678X16647398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nedergaard M, Hansen AJ. Spreading depression is not associated with neuronal injury in the normal brain. Brain Res 1988; 449: 395–398. [DOI] [PubMed] [Google Scholar]
- 64.Karatas H, Erdener SE, Gursoy-Ozdemir Y, et al. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013; 339: 1092–1095. [DOI] [PubMed] [Google Scholar]
- 65.Harris HE, Andersson U, Pisetsky DS. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol 2012; 8: 195–202. [DOI] [PubMed] [Google Scholar]
- 66.Takizawa T, Shibata M, Kayama Y, et al. Temporal profiles of high-mobility group box 1 expression levels after cortical spreading depression in mice. Cephalalgia 2016; 36: 44–52. [DOI] [PubMed] [Google Scholar]
- 67.Akashi-Takamura S, Miyake K. TLR accessory molecules. Curr Opin Immunol 2008; 20: 420–425. [DOI] [PubMed] [Google Scholar]
- 68.Zaidi N, Maurer A, Nieke S, et al. Cathepsin D: A cellular roadmap. Biochem Biophys Res Commun 2008; 376: 5–9. [DOI] [PubMed] [Google Scholar]
- 69.Eikermann-Haerter K, Arbel-Ornath M, Yalcin N, et al. Abnormal synaptic Ca(2+) homeostasis and morphology in cortical neurons of familial hemiplegic migraine type 1 mutant mice. Ann Neurol 2015; 78: 193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dale E, Staal RG, Eder C, et al. KCa 3.1-a microglial target ready for drug repurposing? Glia 2016; 64: 1733–1741. [DOI] [PubMed] [Google Scholar]
- 71.Iizuka T, Takahashi Y, Sato M, et al. Neurovascular changes in prolonged migraine aura in FHM with a novel ATP1A2 gene mutation. J Neurol Neurosurg Psychiatry 2012; 83: 205–212. [DOI] [PubMed] [Google Scholar]
- 72.Gekeler F, Holtmannspotter M, Straube A, et al. Diffusion-weighted magnetic resonance imaging during the aura of pseudomigraine with temporary neurologic symptoms and lymphocytic pleocytosis. Headache 2002; 42: 294–296. [DOI] [PubMed] [Google Scholar]
- 73.Pelzer N, Blom DE, Stam AH, et al. Recurrent coma and fever in familial hemiplegic migraine type 2. A prospective 15-year follow-up of a large family with a novel ATP1A2 mutation. Cephalalgia Epub ahead of print 24 May 2016. DOI: 10.1177/0333102416651284. [DOI] [PubMed] [Google Scholar]
- 74.Kraft AD, McPherson CA, Harry GJ. Heterogeneity of microglia and TNF signaling as determinants for neuronal death or survival. Neurotoxicology 2009; 30: 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brandenburg S, Muller A, Turkowski K, et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol 2016; 131: 365–378. [DOI] [PubMed] [Google Scholar]