

Abstract
Pancreatic β-cell death plays a role in both type 1 and type 2 diabetes, but clinical treatments that specifically target β-cell survival have not yet been developed. We have recently developed live-cell imaging-based, high-throughput screening methods capable of identifying factors that modulate pancreatic β-cell death, with the hope of finding drugs that can intervene in this process. In the present study, we used a high-content screen and the Prestwick Chemical Library of small molecules to identify drugs that block cell death resulting from exposure to a cocktail of cytotoxic cytokines (25 ng/mL TNF-α, 10 ng/mL IL-1β, and 10 ng/mL IFN-γ). Data analysis with self-organizing maps revealed that 19 drugs had profiles similar to that of the no cytokine condition, indicating protection. Carbamazepine, an antiepileptic Na+ channel inhibitor, was particularly interesting because Na+ channels are not generally considered targets for antiapoptotic therapy in diabetes and because the function of these channels in β-cells has not been well studied. We analyzed the expression and characteristics of Na+ currents in mature β-cells from MIP-GFP mice. We confirmed the dose-dependent protective effects of carbamazepine and another use-dependent Na+ channel blocker in cytokine-treated mouse islet cells. Carbamazepine down-regulated the proapoptotic and endoplasmic reticulum stress signaling induced by cytokines. Together, these studies point to Na+ channels as a novel therapeutic target in diabetes.
Pancreatic β-cell death is pathologically elevated in type 1 and type 2 diabetes, as well as in failing islet transplants. In type 1 diabetes, infiltrating autoreactive T cells secrete proinflammatory cytokines, such as TNF-α, IL-1β, and IFN-γ, which effectively coerce the vast majority of pancreatic β-cells into programmed cell death (1–4). Preventing β-cell death could reduce the burden on at-risk families, and protection of the few remaining β-cells in type 1 diabetic patients has the potential to delay disease progression (3). Furthermore, massive β-cell death before, during, and after clinical islet transplantation reduces the success of this promising therapy for type 1 diabetes (5–7). Type 2 diabetes results from the eventual loss of functional β-cell mass, which is in part due to increased β-cell death (2, 8). Thus, there is an urgent clinical need to develop or repurpose drugs that can enhance β-cell survival in both type 1 and type 2 diabetes.
Ion channels are a critical class of drug targets, and significant effort has been devoted to understanding the ionic basis of glucose-stimulated insulin secretion (9). Particular attention has been focused on K+ and Ca2+ channels, whereas Na+ channels are much less well understood despite their presence on β-cells (9). Moreover, the role of plasma membrane ion channels in cell fate decisions, such as apoptosis, remains to be fully elucidated. Unbiased screens are critical for the identification of new pathways involved in programmed β-cell death. We have recently developed rich multiparameter screening platforms with which multiple aspects of programmed cell death can be assessed simultaneously in a high-throughput manner (10).
Here, we report the results of a multiparameter, image-based high-throughput screen to identify drugs that prevent β-cell death in the context of cytotoxic cytokines, designed to mimic conditions that precipitate type 1 diabetes (11). We identified several novel antiapoptotic drugs, including carbamazepine, a use-dependent Na+ channel inhibitor. The confirmation of these effects with another use-dependent Na+ channel inhibitor strongly suggests a previously unappreciated role for Na+ channels in pancreatic β-cells. These data will support therapeutic efforts to inhibit β-cell death in diabetes.
Materials and Methods
Primary islet isolation, cell culture, and perifusion
Pancreatic islets were isolated from 12- to 20-week-old male or female C57BL/6J mice or MIP-GFP mice (The Jackson Laboratory) using collagenase and the filtration technique described previously (12, 13). Mice were housed in accordance with the University of British Columbia Animal Care Committee guidelines. The islets were further hand picked using a brightfield microscope. Islets were cultured overnight (37°C and 5% CO2) in RPMI 1640 medium (Invitrogen) with 9.9 mM glucose (Sigma-Aldrich), 100 U/mL penicillin, 100 μg/mL streptomycin (Invitrogen), and 10% vol/vol fetal bovine serum (FBS) (Invitrogen). Dynamic insulin secretion was measured by perifusion and RIA (14). MIN6 cells were cultured in DMEM (Invitrogen) containing 22.2 mM glucose, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% vol/vol FBS. Chemicals were from Sigma-Aldrich unless specified otherwise.
Multiparameter cell death screening platform
MIN6 cells expressing the eBFP2-DEVD-eGFP fluorescence resonance energy transfer (FRET) sensor for detection of caspase-3/7 activation were seeded into 96-well plates and cultured as described previously (13, 15). Cells were stained with 50 ng/mL Hoechst 33342 (Invitrogen), 0.5 μg/mL propidium iodide (PI) (Sigma-Aldrich), and a 3:500 dilution of annexin V–conjugated to Alexa Fluor 647 (Invitrogen). Cells were treated with test drugs in the presence of 22.2 mM glucose containing DMEM supplemented with 10% vol/vol FBS and a cytokine cocktail (25 ng/mL TNF-α, 10 ng/mL IL-1β, and 10 ng/mL IFN-γ; R&D Systems). After 30 hours of treatments, cells were imaged with ImageXpress Micro (Molecular Devices) at 37°C and 5% CO2. The fluorescent emission of Hoechst 33342 and enhanced blue fluorescent protein 2 (eBFP2), excited at 386/23 nm, was captured with a 445/20 nm filter. PI excitation and emission wavelengths passed through 562/40 and 624/40 nm filters, respectively. Alexa Fluor 647 excitation and emission wavelengths passed through 628/40 and 692/40 nm filters, respectively. Enhanced green fluorescent protein (eGFP) excitation and emission wavelengths passed through 472/30 and 520/35 nm filters, respectively. FRET between eBFP2 and eGFP decreases upon activation of caspase-3/7 and was measured using a 520/35 nm filter and normalized to eBFP2 emission intensity. Time-lapse images for the secondary screens were captured every 2 hours and analyzed using MetaXpress Software (Molecular Devices).
Chemical library
We used the Prestwick Chemical Library of 1120 drugs, which includes a diverse array of chemicals, including off-patent drugs (www.prestwickchemical.com). Compounds were pinned into the 96-well round bottom plates (Corning) at ∼15 μM using a 0.7-mm diameter 96-pin tool equipped onto a Biorobotics Biogrid II robot. Final compound concentrations of ∼8.5 μM were attained when treatments were added to the cells.
Electrophysiology
Ionic currents were recorded with standard whole-cell patch clamp methods. Patch pipettes were pulled from 1.5-mm OD thin-walled borosilicate glass (World Precision Instruments) to have tip resistances of 1 to 2 MΩ in standard recording solutions. The internal (pipette) solution contained 135 mM CsCl, 10 mM HEPES, and 10 mM EGTA, with pH adjusted to 7.2 using CsOH. The external solution contained 140 mM NaCl, 2 mM MgCl2, and 10 mM HEPES, with pH adjusted to 7.4 using NaOH. Currents were recorded with an Axopatch 200B patch clamp amplifier and Digidata 1322A analog-to-digital converter (Molecular Devices), filtered at 10 kHz, and digitized at 50 kHz. For collection of current-voltage relationships, P/4 leak subtraction was used, with a holding potential of −120 mV. For measurement of use-dependent inhibition, it was not possible to perform P/N leak subtraction, so only data from cells with small amounts of leak current were used. In these cases, we used offline linear leak subtraction, although this had very small effects on the appearance of the data.
RT-PCR and immunoblotting
Total RNA was isolated from fluorescence-activated cell sorting (FACS)–purified mouse primary islet cells using an RNeasy Mini kit (Qiagen). cDNA was generated using qScript cDNA SuperMix (Quanta Biosciences) and quantitative RT-PCR was conducted using TaqMan probes from Applied Biosystems and PerfeCTa qPCR SuperMix (Quanta) on a StepOnePlus device (Applied Biosystems). Relative gene expression changes were analyzed by the 2−ΔCt method, with Hprt1 or Ppia as reference genes. Immunoblotting was performed as described previously (14).
Calcium imaging
Mouse islet cells were transfected with the D3cpv, D1ER, or 4mtD3cpv FRET probe (16–18) for the detection of cytosolic, endoplasmic reticulum (ER), or mitochondrial Ca2+. In brief, dispersed mouse islet cells were transfected using the Neon system (Invitrogen) and imaged 48 hours later with ImageXpress Micro (Molecular Devices) at 20-minute intervals at 37°C and 5% CO2. Cells were imaged in RPMI 1640 medium with 20 mM glucose (14).
Statistical analysis
Data are expressed as means ± SEM unless otherwise indicated. Results were considered statistically significant at a value of P < .05 using the Student t test or ANOVA, where appropriate, with Prism software from GraphPad Software Inc.
Results
High-throughput screen for discovering drugs that protect β-cells
Image-based, high-throughput screening requires a high degree of reproducibility. For our screens, we chose the MIN6 β-cell line, which we and others have shown undergoes stereotypical apoptotic programmed cell death in response to proinflammatory cytokines (10). Apoptosis is the mode of cell death that is most commonly assessed in the fields of islet biology and diabetes research (1, 2), and guidelines on the measurement of apoptosis consistently state that multiple parameters should be assessed to distinguish it from other forms of cell death (19, 20). In the current study, we used a 4-parameter live-cell imaging approach to assess the effects of drugs on cytokine-induced β-cell apoptosis. To assess cell number, MIN6 cells were continuously cultured in a low concentration of Hoechst 33342, which we have previously shown does not have significant effects on cell survival (10). To assess phosphatidylserine translocation to the plasma membrane outer surface, an early event in the classic apoptotic cascade (21, 22), cells were cultured in a low concentration of Alexa Fluor 647–conjugated annexin V, which we have also shown to be nontoxic (10). Caspase-3 activation was assessed using a FRET sensor we recently modified to include eBFP2 and eGFP (10). The detection of late-phase cell death was monitored with incorporation of PI, which enters the cell only after the irreversible compromise of the plasma membrane. The combination of these fluorescent probes provided us with 4 distinct parameters for the analysis of β-cell death (Figure 1).
Figure 1.

Multiparameter, high-content screening for compounds that promote β-cell survival. MIN6 cells stably expressing the caspase-3 eBFP-DEVD-eGFP FRET sensor under the Ins1 promoter were seeded into 96-well plates and stained with Hoechst 33342, PI, and Alexa Fluor 647–conjugated annexin V. Cell death was induced with a cytokine cocktail of 25 ng/mL TNF-α, 10 ng/mL IL-1β, and 10 ng/mL IFN-γ, and cells were treated with an 8.5 μM concentration of each compound in the Prestwick Chemical Library, which includes US Food and Drug Administration–approved drugs. Cells were imaged after 30 hours of treatment with ImageXpress Micro, and images were analyzed using MetaXpress software. A–D, Z score values calculated from the numbers of total cells, PI-positive cells, annexin V–positive cells, and activated caspase-3–positive cells were determined and displayed as a heatmap where red and green represent high and low numbers, respectively. The top hits that promoted β-cell survival were selected from automated clustering into 20 different self-organizing maps (A). Scatter plots of PI (B), annexin V (C), and caspase-3 (D) vs total cell were generated. CBZ, carbamazepine.
Multiple antiapoptotic drugs identified with the high-throughput, high-content assay
We screened the Prestwick Chemical Library of 1120 drugs in an effort to identify chemicals and also molecular mechanisms that modulate apoptotic MIN6 cell death induced by a well-established toxic cocktail of cytokines. The results were automatically analyzed into self-organizing maps using the Acuity Express software program (Figure 1A). This approach groups the treatments according to similarity, taking into account the 4 measured cell death–associated parameters. Using this method, 19 distinct drugs showed a pattern that was similar to the “no cytokine” condition, meaning that these were the closest to completely blocking the apoptosis-inducing effects of the cytokines (Figure 1A). A variety of chemicals, from several classes, populated this small “hit” list (Table 1). Notably, we identified 5 vitamins, 5 antibiotics or antifungals, 6 drugs that modulate plasma membrane ion channels and/or membrane receptors, and 3 miscellaneous drugs that had significant protective effects on β-cell survival. The identification of direct antiapoptotic effects in a β-cell model was novel for most of the drugs. For 9 of these chemicals, we were unable to find evidence that they had been linked to apoptosis in any cell type. A literature search revealed that 4 of these chemicals had previously been shown to be antiapoptotic in another cell type, whereas 3 had been shown to be proapoptotic in other systems (Table 1). Two of these drugs, loperamide and carbamazepine, are known to either increase or decrease apoptosis, depending on the context. The possibility that these drugs might have specific effects on β-cells was intriguing.
Table 1.
Summary of “Hits” Identified as Similar to the “No Cytokine” Condition Using Self-Organizing Maps
| Hit Drug | Description | Known Effects on Apoptosis? | References |
|---|---|---|---|
| Pantothenic acid | Vitamin B5 | Antiapoptotic in some systems | 60 |
| Calciferol | VitaminD (D2, D3) | Proapoptotic in some systems | 61 |
| Cholecalciferol | VitaminD (D3) | Unknowa | |
| Tocopherol (R,S) | Vitamin E | Antiapoptotic in some systems | 62, 63 |
| Trolox | Vitamin E | Antiapoptotic in some systems | 64 |
| Amphotericin B | Pore-forming antibiotic, antifungal | Proapoptotic in some systems | 65–69 |
| Ceftazidime | Antibiotic | Proapoptotic in some systems | 70 |
| Cephalosporanic acid | Antibiotic | Unknowna | |
| Butoconazole | Antifungal | Unknowna | |
| Adamantanamine | Antiviral | Unknowna | |
| Alprenolol | Nonselective β-blocker, 5-HT1A antagonist | Unknowna | |
| Loperamide | μ-Opioid agonist, calcium channel inhibitor | Antiapoptotic or proapoptotic | 71–73 |
| Buflomedil | Vasodilator | Unknowna | |
| Tolazoline | Adrenergic blocking agent, peripheral vasodilator | Unknowna | |
| Carbamazepine | Na+ channel inhibitor, anticonvulsant, mood stabilizer | Antiapoptotic or Proapoptotic | 74–76 |
| Carcinine | β-Alanylhistamine, imidazole dipeptide | Antiapoptotic in photoreceptors | 77 |
| Meticrane | Diuretic | Unknowna | |
| Bisacodyl | Laxative | Unknowna | |
| Iopamidol | Contrast agent | Unknowna |
Listed as unknown as a search for the drug name and “apoptosis” yielded no results in PubMed.
The use of our multiparameter screening approach was validated when we identified mitoxantrone dihydrochloride as a drug that could completely prevent PI incorporation in the presence of toxic cytokines (Figure 1B). However, this was a false hit. In fact, there was a very high percentage of dying cells in the presence of this compound as shown by the dramatic cell loss, high annexin V fluorescence, and high caspase-3 activity (Figure 1, B–D), but this topoisomerase inhibitor had apparently prevented the PI from binding to the DNA. Had we not used a rich multiparameter approach in our initial screen, valuable time and resources could have been wasted following up this false-positive effect.
Carbamazepine blocks Na+ currents in primary mouse pancreatic β-cells
We chose a single drug for secondary validation. Of the 19 unique hits, we chose to focus on carbamazepine, a Na+ channel inhibitor, for 4 reasons. First, we were interested by the fact that carbamazepine has been found to have both proapoptotic and antiapoptotic effects in other systems. Second, ion channels are a highly druggable class of membrane proteins. Third, little was known about the role of voltage-gated Na+ channels in programmed cell death, in β-cells or in other cell types. Fourth, Na+ channels are not thought to play a major role in normal β-cell physiology, raising the possibility that targeting these channels might be specific to a state of β-cell stress and/or β-cell death.
Voltage-gated Na+ channels have not been extensively studied in β-cells, relative to other plasma membrane channels, so it was critical to evaluate the Na+ currents in our primary islet cell cultures and determine whether they were inhibited by carbamazepine. We used voltage clamp electrophysiology in the whole-cell configuration to measure Na+ in unambiguously identified β-cells from transgenic mice expressing green fluorescent protein (GFP) under the control of the Ins1 promoter (ie, MIP-GFP mice) (23). Indeed, many MIP-GFP β-cells in our cultures expressed Na+ currents with both a rapidly inactivating transient phase and an unusual sustained phase that was not sensitive to tetrodotoxin (Figure 2A and data not shown). Voltage-gated Na+ currents were most frequently measured from GFP-positive β-cells compared with β-cells identified simply by their larger morphology, suggesting that these currents may be more prominent in “mature” β-cells marked by activation of the mouse Ins1 promoter (24–26) than in immature β-cells or in other islet cell types.
Figure 2.

Use-dependent inhibition of voltage-gated Na+ currents in β-cells from MIP-GFP dispersed islets. A, GFP-positive cells were identified by epifluorescence, and whole-cell patch clamp recording was performed. Cells were held at −100 mV and pulsed in 10-mV steps between −90 mV and +50 mV. Rapidly decaying inward Na+ currents were apparent, along with a sustained inward current. The rapidly decaying current component was well inhibited by carbamazepine (CBZ). B, Peak current-voltage relationships for Na+ currents in the presence and absence of carbamazepine. C, Current traces depict pulses 1 and 25 in a repetitive train of 20-ms depolarizations from −100 mV to +10 mV (10-Hz frequency). In the presence of carbamazepine, there is a significant tonic block of the current starting at pulse 1 and further current reduction over the course of the pulse train. The sustained current component was not sensitive to use-dependent inhibition. D, Dispersed MIP-GFP islet cells were FACS sorted, and the expression of voltage-gated Na+ channels (Nav) and β-subunits (Navβ) was detected by quantitative RT-PCR (n = 3, mean ± SEM).
Importantly, we confirmed the molecular mechanism of carbamazepine. As expected, carbamazepine treatment resulted in the use-dependent inhibition of β-cell Na+ channels (Figure 2, A–C). The expression of voltage-gated Na+ channels and associated β-subunits was detected in FACS-purified β-cells (Figure 2D). Our results were consistent with those of other studies indicating the expression of Nav1.3 and Nav1.7 in mouse islet cells (27) and further indicated that Nav1.7 was predominantly expressed in β-cells (Figure 2D). These data collectively demonstrated that many mature β-cells express voltage-gated Na+ currents and validate the fact that carbamazepine can block these channels, but they do not necessarily show that the antiapoptotic effects of carbamazepine result from the use-dependent inhibition of Na+ channels.
Use-dependent Na+ channel blockers protect primary mouse β-cells from apoptosis
The use-dependent inhibition of Na+ currents by carbamazepine was reminiscent of the effects of lidocaine and other classic local anesthetics/anticonvulsants. To test the hypothesis that use-dependent inhibition of Na+ channels protects β-cells from cytokine-induced apoptosis, we chose to compare the effects of carbamazepine with those of lidocaine, which is also a use-dependent Na+ channel blocker. We compared these effects with those of tetrodotoxin, a Na+ channel blocker that is not use-dependent. Remarkably, both carbamazepine and lidocaine prevented cytokine-induced death of primary islet cells in a dose-dependent manner (Figure 3, A and B). On the other hand, tetrodotoxin was not protective (Figure 3C). We further tested whether these drugs might suppress programmed cell death induced by ER stress, which has been shown to share some mechanistic similarities with cytokine-induced apoptosis (28). Interestingly, all 3 drugs partially protected primary islet cells from death associated with the sarcoendoplasmic reticulum calcium ATPase inhibitor, thapsigargin (Figure 3, D–F). Studies with MIP-GFP islet cells further determined that the protection against cytokine-induced islet cell death was β-cell specific (Figure 4, A–C). The non–β-cell population, indicated by the lack of GFP expression, did not display any increase in cell death after cytokine treatment (Figure 4C). These data establish previously unappreciated roles for Na+ channels in β-cell death induced by cytokines and ER stress.
Figure 3.

Na+ channel inhibitors can reduce cytokine-induced primary islet cell death. Dispersed islet cells seeded into 96-well plate were stained with Hoechst 33342 and PI. Cells were treated with a cytokine cocktail (25 ng/mL TNF-α, 10 ng/mL IL-1β, and 100 ng/mL IFN-γ) (A–C) or 1 μM thapsigargin (D–F), in combination with 0.01 to 100 μM concentrations of Na+ channel inhibitors carbamazepine, lidocaine, and tetrodotoxin. Cells were imaged with ImageXpress Micro and the percentage of PI-positive (PI+) cells was calculated. Insets, area under the curve for the last 10 hours was calculated (n = 3–6, mean ± SEM; ✻, P < .05 compared with cytokine-treated cells).
Figure 4.

Na+ channel inhibitors can reduce cytokine-induced primary β-cell death. A–C, Dispersed MIP-GFP islet cells seeded into 96-well plates were stained with Hoechst 33342 and PI. Cells were treated with a cytokine cocktail (25 ng/mL TNF-α, 10 ng/mL IL-1β, and 100 ng/mL IFN-γ), in combination with 0.01 to 100 μM carbamazepine (CBZ). Cells were imaged with ImageXpress Micro, and the percentage of PI-positive cells in the islet cell (A), β-cell (B), and non–β-cell (C) populations was calculated. Insets, area under the curve for the last 10 hours was calculated (n = 4–6, mean ± SEM; ✻, P < .05 compared with cytokine-treated cells). D, Insulin level in medium collected 70 hours after treatment was measured (n = 5).
Carbamazepine does not affect insulin secretion
It was not clear what effects carbamazepine might have on insulin secretion. This is important because any β-cell–protecting drug should not have deleterious effects on insulin secretion. Moreover, it was important to test whether carbamazepine protected β-cells by modulating insulin secretion, which can have autocrine prosurvival effects (29, 30). Mouse islet cells treated with carbamazepine in the presence of cytokines did not display significant differences in insulin levels in the media collected 70 hours after treatment compared with that from islets treated with cytokines alone (Figure 4D). Islet perifusion studies further showed that carbamazepine does not acutely modulate insulin secretion under either basal or glucose-stimulated conditions (Figure 5). Thus, carbamazepine can protect β-cells from toxicity without significant effects on insulin secretion. These data also imply that the prosurvival effects were independent of autocrine insulin signaling.
Figure 5.

Insulin secretion is not modulated by acute carbamazepine (CBZ) treatment. Mouse islets were perifused with Krebs-Ringer bicarbonate buffer containing 3 or 15 mM glucose in combination with 100 μM carbamazepine (n = 3, mean ± SEM). DMSO, dimethyl sulfoxide.
Carbamazepine modulates cytosolic, ER, and mitochondrial Ca2+
We next assessed the molecular mechanisms associated with the protective effects of carbamazepine. Na+ channel modulation could influence intracellular Ca2+ levels by altering the electrical activity of β-cells (31, 32) and disruption of Ca2+ homeostasis can induce cell death (33). Long-term cytosolic Ca2+ imaging with genetically encoded Ca2+ biosensors revealed the elevation of intracellular Ca2+ in dying cells after treatment with a cocktail of proinflammatory cytokines and showed that this was reduced by carbamazepine (Figure 6A, right). The only cells that survived when treated with cytokines alone maintained a lower level of cytosolic Ca2+ (Figure 6A, left). These observations are consistent with the concept that carbamazepine protects β-cells by suppressing cytokine-induced Ca2+ excitotoxity via the use-dependent inhibition of Na+ channels.
Figure 6.
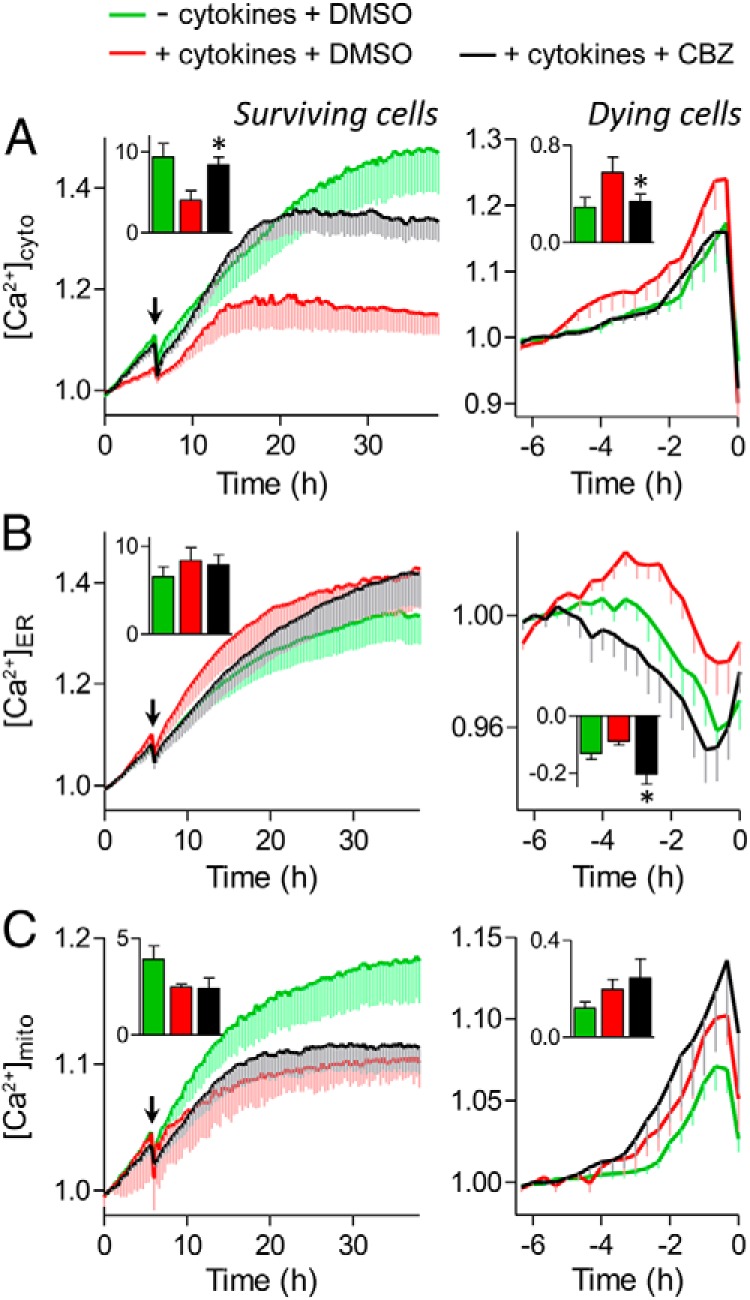
Carbamazepine (CBZ) modulates cytosolic, ER, and mitochondrial Ca2+ signaling. Dispersed mouse islet cells were transfected with D3cpv (A), D1ER (B), or 4mtD3cpv (C) genetically encoded Ca2+ biosensors to image cytosolic, ER, or mitochondrial Ca2+, respectively. Cells were incubated in 20 mM glucose, serum-free RPMI 1640 medium before the treatments with cytokines (red), cytokines and 100 μM carbamazepine (black), or no cytokines (green) at the indicated time (arrow). Left, Ca2+ levels of cells that remained alive during the 38 hours of imaging. Inset, area under the curve from the time of treatment to the end of the time course (n = 6–28 cells; ✻, P < .05 compared with cytokine treated). Right, Ca2+ levels of cells that died during the 38 hours of imaging. The FRET/cyan fluorescent protein ratios were normalized to the average reading from 5 to 6 hours before PI incorporation (0 hours). Inset, area under the curve from the time of Ca2+ influx or depletion to the time of PI incorporation (n = 11–26 cells; *, P < .05 compared with cytokine treated).
We also assessed Ca2+ levels under the same conditions within 2 key organelles, the ER and mitochondria. Interestingly, we also found that carbamazepine treatment could prevent the changes in the level of ER Ca2+ induced by cytokines and delay cell death after induction of Ca2+ depletion (Figure 6B, right). The prolonged depletion of ER Ca2+ was not associated with changes in mitochondrial Ca2+, suggesting that carbamazepine treatment may not alter the Ca2+-mediated crosstalk between ER and mitochondria (Figure 6C, right). Overall, these experiments demonstrate that inhibition of voltage-gated Na+ channel activity can modulate changes in Ca2+ disruptions induced by cytokines.
Carbamazepine decreases proapoptotic and ER stress signaling
Proinflammatory cytokines can induce apoptotic and ER stress signaling in β-cells (34, 35). Previous studies have also shown that proinflammatory cytokines can induce p38 MAPK activation and that TNF-α can enhance Na+ channel activity in mouse dorsal root ganglion neurons through a p38 MAPK-dependent mechanism (36). Thus, we considered the possibility that carbamazepine acts in part by blocking the activation of Na+ channels induced by p38 MAPK. Indeed, in islet cells treated with cytokines, we observed increases in cleaved caspase-3 and C/EBP homology protein (CHOP) levels and increases in activation of the p38 MAPK and c-Jun NH2-terminal kinase (JNK) pathways (Figure 7, A–D). Use-dependent blockage of voltage-gated Na+ channels by carbamazepine and lidocaine could decrease the elevation in cleaved caspase-3, CHOP, and phosphorylated JNK induced by cytokines (Figure 7, A, B, and D). Surprisingly, carbamazepine and lidocaine displayed differential effects on the phosphorylation of p38 MAPK (Figure 7C). The prosurvival effects of carbamazepine were associated with the down-regulation of proapoptotic and ER stress signaling but not with changes in BAD phosphorylation at serine-112 (Figure 7, E and F). Together, these experiments describe mechanisms involved in the protection of β-cells from cytokine- and ER stress–induced apoptosis by use-dependent Na+ channel inhibitors.
Figure 7.

Carbamazepine down-regulates cytokine-induced proapoptotic and ER stress signaling. A–E, Mouse islet cells were treated with 100 μM carbamazepine (CBZ), lidocaine (LIDO), or tetrodotoxin (TTX) under 20 mM glucose serum-free conditions for 24 hours. Immunoblotting of cleaved caspase-3, CHOP, phosphorylated and total p38 MAPK, JNK, and BAD is shown (n = 4–6; ✻, P < .05 compared with cytokine treated). F, Model of effects of carbamazepine on cell death signaling in β-cells.
Discussion
The goal of the present study was to identify small molecule drugs that protect pancreatic β-cells from programmed cell death induced by proinflammatory cytokines and to uncover the mechanisms of action. Our high-throughput screen identified a number of drugs as powerful prosurvival agents, including the use-dependent Na+ channel blocker carbamazepine. This observation prompted us to investigate the roles of Na+ channels in β-cells and pointed to an unexpected role of Na+ channels in β-cell death.
The physiological and pathophysiological roles of Na+ channels in β-cells remain poorly understood. Voltage-gated Na+ channels have been identified on β-cells from mice, rats, dogs, and humans (9, 37–42). It has been shown that Na+ channels are more active in canine and human β-cells than in rodent cells (40). Heterogeneity of Na+ currents and Na+ channel expression has been observed between subpopulations of β-cells (27). In our hands, the use of MIP-GFP cells permitted the recording of Na+ channels from many more cells, perhaps suggesting an enrichment of Na+ channels in mature β-cells. We have previously shown that β-cells with Ins1 promoter expression (ie, MIP-GFP cells) represent a subgroup of β-cells in a mature state (24–26). Na+ currents have been reported previously in MIP-GFP β-cells (23).
Insulin secretion from human β-cells is modulated by blockers of voltage-dependent Na+ channels (43). However, in general, studies with chemical inhibitors have suggested that Na+ channels have relatively modest effects on glucose-stimulated insulin secretion (9, 37, 39, 42). Our data suggest that use-dependent blockage of Na+ channels does not influence insulin secretion. Glucose induces cytoplasmic Na+ oscillations in pancreatic β-cells (44). Na+ channel activation modulates the frequency of Ca2+ oscillations in β-cells (32). In β-cells, increased ATP can shift the current-voltage and the voltage-dependent inactivation curves to the right (45). On the other hand, lifelong, complete knockout of the major regulatory Na+ channel β1-subunit (Scn1b, a component of Nav1.7) reduces insulin secretion (46). Tetrodotoxin blocks insulin secretion induced by carbachol (47). Na+ channels have also been shown to play a role in glucagon release from α-cells (48). Beyond hormone secretion, virtually nothing has been reported on roles for Na+ channels in β-cell fate decisions. To the best of our knowledge, our study is the first to report that Na+ channels participate in programmed cell death in β-cells or any endocrine cell type.
The role of Na+ channels in neuronal apoptosis has been the focus of several studies. Overall, these studies paint a complex picture. The potent Na+ channel blocker tetrodotoxin has been shown to protect neurons from hypoxia (49). Similarly, 4-[4-fluorophenyl]-2-methyl-6-[5-piperidinopntyloxy] pyrimidine hydrochloride, an inhibitor that blocks both Ca2+ and Na+ channels, protects neurons after ischemia (50). Moreover, Na+ channel mutations correlate with increased survival and disease severity in glioblastoma multiforme (51). On the other hand, mice lacking the Scn2a subunit gene, a component of the Nav1.2 channel, exhibit significant neuronal apoptosis (52). Neurons in culture and in vivo show activity-dependent survival, whereby inhibition of Na+ channels leads to apoptotic cell death (53–55) and selective activation of Na+ channels promotes survival (56). Other mutations in Na+ channels can have proapoptotic effects in the heart (57, 58). Together, these studies suggest that Na+ currents may play a deleterious role in neurons subjected to hypoxic or ischemic stresses, whereas a certain level of Na+ channel activity in basal conditions is essential for activity-dependent survival. This role is similar to the well established dual roles for Ca2+ channels in cellular survival. Collectively, these data support a model whereby plasma membrane excitability must be kept within a tight range to maintain optimal survival.
Before our study, very little was known about the effects of carbamazepine or other use-dependent Na+ channel inhibitors on pancreatic β-cells. Interestingly, it was recently shown that carbamazepine can rescue the trafficking defects of mutant KATP channels involved in congenital hyperinsulinism (59). Whether these effects of carbamazepine are involved in its ability to protect β-cells from apoptosis remains to be elucidated, but with the lack of changes in insulin secretion upon carbamazepine exposure, we do not favor a major role for this pathway in the effects observed in our study. In our view, it is more likely that carbamazepine and lidocaine protect β-cells via direct actions on Na+ channels and membrane excitability, similar to what may occur in neurons. Our studies suggest that inhibition of Na+ channels could reduce excessive membrane depolarization, thereby preventing the cytokine-stimulated influx of Ca2+ through voltage-gated Ca2+ channels. In addition, carbamazepine treatment extended the ER Ca2+ depletion time before cell death and improved the ER Ca2+ depletion threshold, which could have contributed to the observed down-regulation of ER stress signaling. Additional mechanistic studies into the prosurvival effects of carbamazepine are warranted.
In conclusion, we report the results of an unbiased high-throughput, high-content multiparameter screen that identified 19 drugs capable of protecting β-cells from cytokine-induced apoptosis. The unexpected observation that carbamazepine and lidocaine protected primary β-cells from apoptosis afforded new insight into the role of Na+ currents in apoptosis. Although, the current study focused on the validation of use-dependent Na+ channel inhibitors as antiapoptotic agents, additional insights are likely to come from the pursuit of other hits from our screen. The therapeutic impact for diabetes is intriguing especially because carbamazepine and many of the hits are already approved for the treatment of patients. Undoubtedly, these studies will continue to paint a complex picture of programmed cell death in the pancreatic β-cell.
Acknowledgments
This work was supported by grants from the JDRF and the Stem Cell Network (to J.D.J.). Y.H.C.Y. was supported by a scholarship from the Natural Sciences and Engineering Research Council of Canada.
Y.H.C.Y. designed and performed all nonelectrophysiology studies, analyzed data, and cowrote the manuscript. Y.Y.V. performed electrophysiology experiments. M.R. helped design the screen and provided access to critical reagents. H.T.K. designed and analyzed electrophysiology studies and wrote the relevant parts of the manuscript. J.D.J. conceived of the study, designed research, and cowrote the manuscript and takes full responsibility for all results.
Disclosure Summary: The authors have nothing to disclose.
Note Added in Proof
While our manuscript was under review, Cnop et al reported that carbamazepine protected the INS-1 beta-cell line from palmitate-induced death (Diabetes Epub Dec 30 2013).
Funding Statement
This work was supported by grants from the JDRF and the Stem Cell Network (to J.D.J.). Y.H.C.Y. was supported by a scholarship from the Natural Sciences and Engineering Research Council of Canada.
Footnotes
- CHOP
- C/EBP homology protein
- eBFP2
- enhanced blue fluorescent protein 2
- eGFP
- enhanced green fluorescent protein
- FACS
- fluorescence-activated cell sorting
- FBS
- fetal bovine serum
- FRET
- fluorescence resonance energy transfer
- GFP
- green fluorescent protein
- JNK
- c-Jun NH2-terminal kinase
- PI
- propidium iodide.
References
- 1. Mathis D, Vence L, Benoist C. β-Cell death during progression to diabetes. Nature. 2001;414:792–798. [DOI] [PubMed] [Google Scholar]
- 2. Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(suppl 2):S97-S-107. [DOI] [PubMed] [Google Scholar]
- 3. Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained β cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–2228. [DOI] [PubMed] [Google Scholar]
- 4. Nakanishi K, Watanabe C. Rate of β-cell destruction in type 1 diabetes influences the development of diabetic retinopathy: protective effect of residual β-cell function for more than 10 years. J Clin Endocrinol Metab. 2008;93:4759–4766. [DOI] [PubMed] [Google Scholar]
- 5. Robertson RP. Islet transplantation as a treatment for diabetes - a work in progress. N Engl J Med. 2004;350:694–705. [DOI] [PubMed] [Google Scholar]
- 6. Robertson RP. Islet transplantation a decade later and strategies for filling a half-full glass. Diabetes. 2010;59:1285–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shapiro AM, Lakey JR, Ryan EA, et al. . Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343:230–238. [DOI] [PubMed] [Google Scholar]
- 8. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. [DOI] [PubMed] [Google Scholar]
- 9. Misler S, Barnett DW, Gillis KD, Pressel DM. Electrophysiology of stimulus-secretion coupling in human β-cells. Diabetes. 1992;41:1221–1228. [DOI] [PubMed] [Google Scholar]
- 10. Yang YH, Johnson JD. Multi-parameter single-cell kinetic analysis reveals multiple modes of cell death in primary pancreatic β-cells. J Cell Sci. 2013;126(pt 18):4286–4295. [DOI] [PubMed] [Google Scholar]
- 11. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. [DOI] [PubMed] [Google Scholar]
- 12. Luciani DS, Ao P, Hu X, Warnock GL, Johnson JD. Voltage-gated Ca2+ influx and insulin secretion in human and mouse β-cells are impaired by the mitochondrial Na+/Ca2+ exchange inhibitor CGP-37157. Eur J Pharmacol. 2007;576:18–25. [DOI] [PubMed] [Google Scholar]
- 13. Yang YH, Szabat M, Bragagnini C, et al. . Paracrine signalling loops in adult human and mouse pancreatic islets: netrins modulate β cell apoptosis signalling via dependence receptors. Diabetologia. 2011;54:828–842. [DOI] [PubMed] [Google Scholar]
- 14. Yang YH, Manning Fox JE, Zhang KL, MacDonald PE, Johnson JD. Intraislet SLIT-ROBO signaling is required for β-cell survival and potentiates insulin secretion. Proc Natl Acad Sci USA. 2013;110:16480–16485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alejandro EU, Johnson JD. Inhibition of Raf-1 alters multiple downstream pathways to induce pancreatic β-cell apoptosis. J Biol Chem. 2008;283:2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gwiazda KS, Yang TL, Lin Y, Johnson JD. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in β-cells. Am J Physiol Endocrinol Metab. 2009;296:E690–E701. [DOI] [PubMed] [Google Scholar]
- 17. Luciani DS, Gwiazda KS, Yang TL, et al. . Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and β-cell death. Diabetes. 2009;58:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palmer AE, Giacomello M, Kortemme T, et al. . Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol. 2006;13:521–530. [DOI] [PubMed] [Google Scholar]
- 19. Kroemer G, Galluzzi L, Vandenabeele P, et al. . Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2009;16:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galluzzi L, Aaronson SA, Abrams J, et al. . Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- 22. Reutelingsperger CP, van Heerde WL. Annexin V, the regulator of phosphatidylserine-catalyzed inflammation and coagulation during apoptosis. Cell Mol Life Sci. 1997;53:527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leung YM, Ahmed I, Sheu L, et al. . Electrophysiological characterization of pancreatic islet cells in the mouse insulin promoter-green fluorescent protein mouse. Endocrinology. 2005;146:4766–4775. [DOI] [PubMed] [Google Scholar]
- 24. Szabat M, Lynn FC, Hoffman BG, Kieffer TJ, Allan DW, Johnson JD. Maintenance of β-cell maturity and plasticity in the adult pancreas: developmental biology concepts in adult physiology. Diabetes. 2012;61:1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szabat M, Pourghaderi P, Soukhatcheva G, Verchere CB, Warnock GL, Piret JM, Johnson JD. Kinetics and genomic profiling of adult human and mouse β-cell maturation. Islets. 2011;3:175–187. [DOI] [PubMed] [Google Scholar]
- 26. Szabat M, Luciani DS, Piret JM, Johnson JD. Maturation of adult β-cells revealed using a Pdx1/insulin dual-reporter lentivirus. Endocrinology. 2009;150:1627–1635. [DOI] [PubMed] [Google Scholar]
- 27. Vignali S, Leiss V, Karl R, Hofmann F, Welling A. Characterization of voltage-dependent sodium and calcium channels in mouse pancreatic A- and B-cells. J Physiol. 2006;572(pt 3):691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eizirik DL, Cnop M. ER stress in pancreatic β cells: the thin red line between adaptation and failure. Sci Signal. 2010;3:pe7. [DOI] [PubMed] [Google Scholar]
- 29. Mehran AE, Templeman NM, Brigidi GS, et al. . Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16:723–737. [DOI] [PubMed] [Google Scholar]
- 30. Johnson JD, Bernal-Mizrachi E, Alejandro EU, et al. . Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci USA. 2006;103:19575–19580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goncalves AA, Toyama MH, Carneiro EM, et al. . Participation of Na+ channels in the potentiation by Tityus serrulatus α-toxin TsTx-V of glucose-induced electrical activity and insulin secretion in rodent islet β-cells. Toxicon. 2003;41:1039–1045. [DOI] [PubMed] [Google Scholar]
- 32. Eberhardson M, Grapengiesser E. Role of voltage-dependent Na+ channels for rhythmic Ca2+ signalling in glucose-stimulated mouse pancreatic β-cells. Cell Signal. 1999;11:343–348. [DOI] [PubMed] [Google Scholar]
- 33. Harr MW, Distelhorst CW. Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harbor Perspect Biol. 2010;2:a005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cardozo AK, Ortis F, Storling J, et al. . Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005;54:452–461. [DOI] [PubMed] [Google Scholar]
- 35. Cardozo AK, Heimberg H, Heremans Y, et al. . A comprehensive analysis of cytokine-induced and nuclear factor-κB-dependent genes in primary rat pancreatic β-cells. J Biol Chem. 2001;276:48879–48886. [DOI] [PubMed] [Google Scholar]
- 36. Jin X, Gereau RW 4th. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J Neurosci. 2006;26:246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barnett DW, Pressel DM, Misler S. Voltage-dependent Na+ and Ca2+ currents in human pancreatic islet β-cells: evidence for roles in the generation of action potentials and insulin secretion. Pflugers Arch. 1995;431:272–282. [DOI] [PubMed] [Google Scholar]
- 38. Philipson LH, Kusnetsov A, Larson T, Zeng Y, Westermark G. Human, rodent, and canine pancreatic β-cells express a sodium channel α 1-subunit related to a fetal brain isoform. Diabetes. 1993;42:1372–1377. [DOI] [PubMed] [Google Scholar]
- 39. Pressel DM, Misler S. Role of voltage-dependent ionic currents in coupling glucose stimulation to insulin secretion in canine pancreatic islet B-cells. J Membr Biol. 1991;124:239–253. [DOI] [PubMed] [Google Scholar]
- 40. Pressel DM, Misler S. Sodium channels contribute to action potential generation in canine and human pancreatic islet B cells. J Membr Biol. 1990;116:273–280. [DOI] [PubMed] [Google Scholar]
- 41. Plant TD. Na+ currents in cultured mouse pancreatic B-cells. Pflugers Arch. 1988;411:429–435. [DOI] [PubMed] [Google Scholar]
- 42. Hiriart M, Matteson DR. Na channels and two types of Ca channels in rat pancreatic B cells identified with the reverse hemolytic plaque assay. J Gen Physiol. 1988;91:617–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Misler S, Dickey A, Barnett DW. Maintenance of stimulus-secretion coupling and single β-cell function in cryopreserved-thawed human islets of Langerhans. Pflugers Arch. 2005;450:395–404. [DOI] [PubMed] [Google Scholar]
- 44. Grapengiesser E. Glucose induces cytoplasmic Na+ oscillations in pancreatic β-cells. Biochem Biophys Res Commun. 1996;226:830–835. [DOI] [PubMed] [Google Scholar]
- 45. Zou N, Wu X, Jin YY, et al. . ATP regulates sodium channel kinetics in pancreatic islet β cells. J Membr Biol. 2013;246:101–107. [DOI] [PubMed] [Google Scholar]
- 46. Ernst SJ, Aguilar-Bryan L, Noebels JL. Sodium channel β1 regulatory subunit deficiency reduces pancreatic islet glucose-stimulated insulin and glucagon secretion. Endocrinology. 2009;150:1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hiriart M, Ramirez-Medeles MC. Muscarinic modulation of insulin secretion by single pancreatic β-cells. Mol Cell Endocrinol. 1993;93:63–69. [DOI] [PubMed] [Google Scholar]
- 48. Olsen HL, Theander S, Bokvist K, Buschard K, Wollheim CB, Gromada J. Glucose stimulates glucagon release in single rat α-cells by mechanisms that mirror the stimulus-secretion coupling in β-cells. Endocrinology. 2005;146:4861–4870. [DOI] [PubMed] [Google Scholar]
- 49. Banasiak KJ, Burenkova O, Haddad GG. Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience. 2004;126:31–44. [DOI] [PubMed] [Google Scholar]
- 50. Shi E, Kazui T, Jiang X, et al. . NS-7, a novel Na+/Ca2+ channel blocker, prevents neurologic injury after spinal cord ischemia in rabbits. J Thorac Cardiovasc Surg. 2005;129:364–371. [DOI] [PubMed] [Google Scholar]
- 51. Joshi AD, Parsons DW, Velculescu VE, Riggins GJ. Sodium ion channel mutations in glioblastoma patients correlate with shorter survival. Mol Cancer. 2011;10:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Planells-Cases R, Caprini M, Zhang J, et al. . Neuronal death and perinatal lethality in voltage-gated sodium channel alphaII-deficient mice. Biophys J. 2000;78:2878–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schonfeld-Dado E, Segal M. Activity-dependent survival of neurons in culture: a model of slow neurodegeneration. J Neural Transm. 2009;116:1363–1369. [DOI] [PubMed] [Google Scholar]
- 54. Schonfeld-Dado E, Fishbein I, Segal M. Degeneration of cultured cortical neurons following prolonged inactivation: molecular mechanisms. J Neurochem. 2009;110:1203–1213. [DOI] [PubMed] [Google Scholar]
- 55. Ikonomidou C. Triggers of apoptosis in the immature brain. Brain Dev. 2009;31:488–492. [DOI] [PubMed] [Google Scholar]
- 56. Salthun-Lassalle B, Hirsch EC, Wolfart J, Ruberg M, Michel PP. Rescue of mesencephalic dopaminergic neurons in culture by low-level stimulation of voltage-gated sodium channels. J Neurosci. 2004;24:5922–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang T, Yong SL, Drinko JK, et al. . LQTS mutation N1325S in cardiac sodium channel gene SCN5A causes cardiomyocyte apoptosis, cardiac fibrosis and contractile dysfunction in mice. Int J Cardiol. 2011;147:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Frustaci A, Priori SG, Pieroni M, et al. . Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation. 2005;112:3680–3687. [DOI] [PubMed] [Google Scholar]
- 59. Chen PC, Olson EM, Zhou Q, et al. . Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J Biol Chem. 2013;288:20942–20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Slyshenkov VS, Piwocka K, Sikora E, Wojtczak L. Pantothenic acid protects Jurkat cells against ultraviolet light-induced apoptosis. Free Radic Biol Med. 2001;30:1303–1310. [DOI] [PubMed] [Google Scholar]
- 61. Dunlap N, Schwartz GG, Eads D, et al. . 1α,25-Dihydroxyvitamin D3 (calcitriol) and its analogue, 19-nor-1α,25(OH)2D2, potentiate the effects of ionising radiation on human prostate cancer cells. Br J Cancer. 2003;89:746–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zakharova IO, Sokolova TV, Bayunova LV, Vlasova YA, Rychkova MP, Avrova NF. α-Tocopherol at nanomolar concentration protects PC12 cells from hydrogen peroxide-induced death and modulates protein kinase activities. Int J Mol Sci. 2012;13:11543–11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ahlemeyer B, Krieglstein J. Inhibition of glutathione depletion by retinoic acid and tocopherol protects cultured neurons from staurosporine-induced oxidative stress and apoptosis. Neurochem Int. 2000;36:1–5. [DOI] [PubMed] [Google Scholar]
- 64. Forrest VJ, Kang YH, McClain DE, Robinson DH, Ramakrishnan N. Oxidative stress-induced apoptosis prevented by Trolox. Free Radic Biol Med. 1994;16:675–684. [DOI] [PubMed] [Google Scholar]
- 65. Chen LY, Sheu MT, Liao CK, Tsai FC, Kao WY, Su CH. Taiwanofungus camphoratus (Syn Antrodia camphorata) extract and amphotericin B exert adjuvant effects via mitochondrial apoptotic pathway. Integr Cancer Ther. 2013;12:153–164. [DOI] [PubMed] [Google Scholar]
- 66. Cohen BE. Amphotericin B membrane action: role for two types of ion channels in eliciting cell survival and lethal effects. J Membr Biol. 2010;238:1–20. [DOI] [PubMed] [Google Scholar]
- 67. Mahmud H, Mauro D, Qadri SM, Foller M, Lang F. Triggering of suicidal erythrocyte death by amphotericin B. Cell Physiol Biochem. 2009;24:263–270. [DOI] [PubMed] [Google Scholar]
- 68. Odabasi Z, Karaalp A, Cermik H, et al. . Reduction of amphotericin B-induced renal tubular apoptosis by N-acetylcysteine. Antimicrob Agents Chemother. 2009;53:3100–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marklund L, Andersson B, Behnam-Motlagh P, Sandstrom PE, Henriksson R, Grankvist K. Cellular potassium ion deprivation enhances apoptosis induced by cisplatin. Basic Clin Pharmacol Toxicol. 2004;94:245–251. [DOI] [PubMed] [Google Scholar]
- 70. Kadota J, Mizunoe S, Kishi K, Tokimatsu I, Nagai H, Nasu M. Antibiotic-induced apoptosis in human activated peripheral lymphocytes. Int J Antimicrob Agents. 2005;25:216–220. [DOI] [PubMed] [Google Scholar]
- 71. Momeni HR, Jarahzadeh M. Effects of a voltage sensitive calcium channel blocker and a sodium-calcium exchanger inhibitor on apoptosis of motor neurons in adult spinal cord slices. Cell J. 2012;14:171–176. [PMC free article] [PubMed] [Google Scholar]
- 72. Gong XW, Xu YH, Chen XL, Wang YX. Loperamide, an antidiarrhea drug, has antitumor activity by inducing cell apoptosis. Pharmacol Res. 2012;65:372–378. [DOI] [PubMed] [Google Scholar]
- 73. Yoshida A, Tokuyama S, Iwamura T, Ueda H. Opioid analgesic-induced apoptosis and caspase-independent cell death in human lung carcinoma A549 cells. Int J Mol Med. 2000;6:329–335. [DOI] [PubMed] [Google Scholar]
- 74. Park HJ, Kim SK, Chung JH, Kim JW. Protective effect of carbamazepine on kainic acid-induced neuronal cell death through activation of signal transducer and activator of transcription-3. J Mol Neurosci. 2013;49:172–181. [DOI] [PubMed] [Google Scholar]
- 75. Gao XM, Margolis RL, Leeds P, Hough C, Post RM, Chuang DM. Carbamazepine induction of apoptosis in cultured cerebellar neurons: effects of N-methyl-d-aspartate, aurintricarboxylic acid and cycloheximide. Brain Res. 1995;703:63–71. [DOI] [PubMed] [Google Scholar]
- 76. Kim H, Bernard ME, Farkas A, et al. . Ionizing irradiation protection and mitigation of murine cells by carbamazepine is p53 and autophagy independent. In Vivo. 2012;26:341–354. [PMC free article] [PubMed] [Google Scholar]
- 77. Gavin BA, Arruda SE, Dolph PJ. The role of carcinine in signaling at the Drosophila photoreceptor synapse. PLoS Genet. 2007;3:e206. [DOI] [PMC free article] [PubMed] [Google Scholar]