

Abstract
Fancj, the gene associated with Fanconi anemia (FA) Complementation Group J, encodes a DNA helicase involved in homologous recombination repair and the cellular response to replication stress. FANCJ functions in part through its interaction with key DNA repair proteins, including MutL homolog-1 (MLH1), Breast Cancer Associated gene-1 (BRCA1), and Bloom syndrome helicase (BLM). All three of these proteins are involved in a variety of events that ensure genome stability, including the events of DNA double strand break (DSB) repair during prophase I of meiosis. Meiotic DSBs are repaired through homologous recombination resulting in non-crossovers (NCO) or crossovers (CO). The frequency and placement of COs are stringently regulated to ensure that each chromosome receives at least one CO event, and that longer chromosomes receive at least one additional CO, thus facilitating the accurate segregation of homologous chromosomes at the first meiotic division. In the present study, we investigated the role of Fancj during prophase I using a gene trap mutant allele. FancjGT/GT mutants are fertile, but their testes are very much smaller than wild-type litter-mates, predominantly as a result of impeded spermatogonial proliferation and mildly increased apoptosis during testis development in the fetus. This defect in spermatogonial proliferation is consistent with mutations in other FA genes. During prophase I, early events of synapsis and DSB induction/repair appear mostly normal in FancjGT/GT males, and the FANCJ-interacting protein BRCA1 assembles normally on meiotic chromosome cores. However, MLH1 focus frequency is increased in FancjGT/GT males, indicative of increased DSB repair via CO, and is concomitant with increased chiasmata at diakinesis. This increase in COs in the absence of FANCJ is associated with increased localization of BLM helicase protein, indicating that BLM may facilitate the increased rate of crossing over in FancjGT/GT males. Taken together, these results demonstrate a critical role for FANCJ in spermatogenesis at two stages: firstly in the proliferative activity that gives rise to the full complement of testicular spermatogonia and secondly in the establishment of appropriate CO numbers during prophase I.
Keywords: Gametogenesis, Fanconi anemia, MLH1, Meiotic prophase I, BRIP1, BACH1
Introduction
First described by Swiss pediatrician, Guido Fanconi, in 1927, Fanconi anemia (FA) is a rare autosomal or X-linked disorder characterized by a variety of congenital abnormalities, bone marrow failure, and increased cancer predisposition (Walden and Deans 2014). FA affects only 1:100,000 individuals, and yet involves no fewer than 16 distinct human complementation groups (Walden and Deans 2014). Cells from FA patients exhibit elevated sensitivity to DNA damaging agents, specifically those that induce interstrand DNA crosslinks (ICL) (Andreassen and Ren 2009; Muniandy et al. 2010; Rosselli et al. 2003). ICLs induce replicative stress and impair transcriptional activity by forming covalent attachments between the two strands of the DNA double helix, thereby preventing accessibility by the transcriptional and replicative machineries, leading to stalled replication forks. The FA network serves to alleviate this replicative stress by unhooking the ICL and repairing the resulting DNA double strand break (DSB) via homologous recombination (HR), while at the same time overcoming the stalled replication intermediate through the recruitment of translesion polymerases (Knipscheer et al. 2009).
Three major complexes characterize the FA pathway (reviewed by Walden and Deans 2014). The “anchor complex” consists of FANCM, FAAP24, and homodimers of the MHF1 and MHF2 proteins. The anchor complex recruits a second complex, the core complex, to the site of the DNA lesion. The role of the core complex, consisting of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FAAP20, and FAAP100, is to mono-ubiquitylate the FANCI and FANCD2 proteins, which together comprise the third complex, the “ID2 complex.” The ID2 complex is also the target of phosphorylation by the ATM/ATR kinases on several sites, and these post-translational modifications may be essential for stabilizing the complex and/or for modifying the activity of the complex. The ID2 complex, in turn, recruits a number of effector molecules, including structure-specific nucleases (FANCP/SLX4 and FANCQ/XPF), recombinases (FANCD1/BRCA2, FANCN/PALB2, and FANCO/RAD51C), and the helicase FANCJ/BRIP1 (Walden and Deans 2014).
The structural complexity of the ICL requires the action of multiple converging DNA repair pathways, and the FA network is presumed to form a signaling hub that regulates these repair events. Thus, the FA network of effector molecules interacts functionally with numerous other repair pathways, particularly those involving homologous recombination, nucleotide excision repair, and mismatch repair, enabling them to bridge and facilitate multiple repair activities for the successful resolution of the stalled replication fork. Along these lines, FA network members have been shown to interact with components of the DNA mismatch repair (MMR) pathway (notably the MutS homolog, MSH2, and the MutL homolog, MLH1) (Williams et al. 2011) and the homologous recombination (HR) repair pathway (notably Bloom syndrome mutated helicase, BLM, and breast cancer associated-1, BRCA1) (Walden and Deans 2014).
DNA repair activities are vital for all cell types, not least of which include the germ cells that are required for sexual reproduction. Indeed, the germ cell represents a unique case in which an extremely high level of proliferative activity during gonadal development results in the amplification of fewer than 150 germ cells at embryonic day (e) 8.5 into the final complement of >25,000 cells by e13.5 in the mouse (Tam and Snow 1981). This is followed during prophase I of meiosis by the onset of a genetically encoded genome-wide DNA damage/repair process that underlies the process of meiotic recombination, wherein the induction of DSBs throughout the genome triggers recombination events mediated by an array of key DNA repair and HR regulators (Baudat et al. 2013; Phadnis et al. 2011). Of the several hundreds of DSBs that form, only a small subset will be resolved as the crossovers (CO) that will ensure stable homolog interactions through until the first meiotic division (Bolcun-Filas and Schimenti 2012). These CO events are regulated through two pathways: the class I (ZMM) pathway that is dependent on the activity of members of the MMR family and which accounts for >90 % of all COs in the mouse (Kolas et al. 2005) and the minor class II pathway that is regulated by the MUS81-EME1 endonuclease (Holloway et al. 2008). The remaining DSBs are processed as non-crossovers (NCO) that, while promoting initial homolog interactions, do not contribute to the overall maintenance of these interactions through meiosis I. Thus, DSB repair events during prophase I must be strictly regulated through space and time to ensure that appropriate CO are established throughout the genome and that all DSBs are repaired prior to the first meiotic division.
Given the multiple steps at which DNA repair activities are critical for gametogenesis, it is not surprising that FA pathway components have been implicated in these events. Mouse mutants for a number of FA genes result in sub-fertility or infertility as a result of defects either in the proliferative events during gonadal development that give rise to the full cohort of germ cells or in defects associated with meiotic prophase I progression (Agoulnik et al. 2002; Houghtaling et al. 2003; Koomen et al. 2002; Lu and Bishop 2003; Noll et al. 2002; Whitney et al. 1996; Wong et al. 2003; Yang et al. 2001). For example, mutants in Fanca, a core complex component, show increased defects in meiotic chromosome pairing during prophase I, in addition to meiotic recombination repair defects (Wong et al. 2003), while Fancc mutants showed a reduced number of meiotic cells and impaired fertility (Whitney et al. 1996). Additionally, mice with Fancd2 mutation, a member of the ID complex, display morphological abnormalities in testis structure, resulting from fewer primordial germ cells, coupled with pairing defects during prophase I, which all culminate in a higher level of apoptosis during meiosis I (Houghtaling et al. 2003). Similarly, mice that are homozygous for the Chaos4 mutation in Fancm exhibit significantly reduced germ cell numbers at birth, resulting from decreased proliferation but without a concurrent increase in apoptosis (Luo et al. 2014). Taken together, these data suggest that FA genes are essential for the early proliferative events that give rise to the full population of primordial germ cells in the postnatal gonad, as well as for DSB repair events during prophase I.
Only one FA gene has yet to be examined via mutagenesis in the mouse: Fancj (also called Brip1 for BRCA1 interacting protein C-terminal helicase I) (Levitus et al. 2005; Litman et al. 2005). FancJ encodes a 5′ to 3′ DEAH helicase that was originally identified as a BRCA1-associated protein, hence its original name of BRCA1-associated c-terminal helicase I (BACH1) (Cantor et al. 2001). Similar to BRCA1, mutations in human FANCJ have been associated with breast cancer (Cantor et al. 2001; Seal et al. 2006), albeit at lower penetrance (Hiom 2010), leading to the suggestion that FANCJ functions with BRCA1 in HR events that underlie DSB repair. Indeed, FANCJ and BRCA1 co-localize at sites of DNA damage together with other mediators of DNA damage repair including TOPBP1, BLM, and components of the MMR pathway (including MLH1 and MSH2) (Cantor and Xie 2010; Gong et al. 2010; Leung et al. 2011; Peng et al. 2007; Peng et al. 2014; Suhasini and Brosh 2011; Suhasini et al. 2011; Zou et al. 2014). However, the phenotypes of human cells lacking either BRCA1 or FANCJ are different (Hiom 2010), suggesting that co-incident functions of BRCA1 and FANCJ make up only a subset of the overall roles of each protein.
FANCJ is thought to function downstream of FANCD2 monoubiquitylation and ICL unhooking to facilitate HR (Hiom 2010). In addition, human FANCJ is required for S phase progression (Kumaraswamy and Shiekhattar 2007) and can unwind G-quadruplex structures to maintain genome stability (Wu et al. 2008), similar to its Caenorhabditis elegans ortholog, DOG-1 (Kruisselbrink et al. 2008). These varied functions of FANCJ depend to differing extents on the interaction with MMR family members, with the BLM helicase, and with BRCA1, implicating FANCJ in a variety genome stabilizing processes.
Given these complex and varied functions of FANCJ, therefore, we asked what role this protein might play in germ cell proliferation and in the recombination events occurring during prophase I of meiosis. To study the role of FANCJ in gametogenesis, we generated a gene trap allele for Fancj using ES cells obtained from the NIH-sponsored Mouse Mutant Resource and Research Center (MMRRC) at the University of California at Davis. Homozygous gene trap mutant males showed complete loss of the FANCJ protein, were partially fertile, but had very small testes concurrent with significantly reduced numbers of spermatogonial stem cell precursors. During prophase I of meiosis, the events of synapsis and DSB-initiated recombination occur mostly normally in the FancjGT/GT males. However, there is a slight increase in aberrant synapsis events in some spermatocytes, coupled with a slight increase in the number of cells displaying delayed/aberrant DSB repair in pachytene. Moreover, we observe an overall significant increase in the number of DSB repair events that are repaired as crossovers. Interestingly, these changes in the progression of prophase I are not associated with changes in BRCA1 localization on asynapsed chromosomes, but are associated with upregulation of protein levels for members of the DNA MMR family, notably MLH1 and MLH3, which are fundamental components of the major crossover pathway in mammals. BLM helicase is also upregulated in the absence of FANCJ. Thus, we conclude that loss of FancJ alters the profile of DSB repair events such that there is an increase in BLM-related dissolution events and a concurrent increase in the repair of DSBs via the MLH1/3-dependent crossover pathway.
Materials and methods
Generation of the mice and genotyping
ES cells were obtained from The Mouse Biology Program at University of California at Davis, part of the NIH-sponsored MMRRC network. The ES cell clone number was BayGenomics RRI409 and was sequence verified by the MMRRC. Clone information is available on the MMRRC website at https://www.mmrrc.org. 5′ RACE revealed the appropriate gene modification at the Brip1 locus. The gene trap cassette is inserted after exon 5 of the gene, resulting in a disruption of the majority of the sequence that encodes the helicase domain, loss of the BRCA1 interaction domain, and partial loss of the nuclear localization sequence. The MLH1 interaction domain, encoded within exon 5, remains intact (Figure S1a). The parental ES cell line, E14Tg2a.4, was also obtained for use as a control in ES cell growth curve assays.
ES cells were injected into mouse blastocysts by the staff of the Cornell Transgenic Facility with 8 chimeric animals being obtained. Chimeric animals were screened by coat color and then by PCR genotyping of DNA extracted from tail snips. In all, we obtained seven chimeras, with between 10 and 70 % chimerism (assessed by coat color). The three mice with the highest degree of chimerism (between 60 and 70 %) were chosen for breeding and each achieved germline transmission of the altered Brip1 allele. This allele will be referred to herein as FancjGT.
To identify the mutant and wild-type alleles of Fancj, a genotyping strategy utilized one common forward primer and two reverse primers (Figure S1b). Their sequences are:
Exon 5 F2 GGTAGAGAAGAAAAGAATCCG
Exon 6 R1 GTGCATCTACATGGTGGACG
Construct R1 CACTCCAACCTCCGCAAACTC
The wild-type allele generates a 2.5-kb band, while the mutant allele generates a 900-bp band. PCR conditions included 54 °C annealing with 3 min extension for the wild-type allele, and 57 °C annealing for mutant with 2 min extension, for 35 cycles.
Sperm counts
The caudal epididymides were removed from adult mice and placed in prewarmed DMEM containing 4 % bovine serum albumin (Sigma-Aldrich). Each epididymis was squeezed with tweezers to extrude the sperm and then incubated at 32 °C/5 % CO2 for 20 min. A 20-μl aliquot of the sperm suspension was resuspended in 480 μl 10 % formalin, and the sperm cells counted.
Assessment of ES cell growth characteristics
E14 parental ES cells and Fancj mutant ES cells were plated out in triplicate at a density of 200,000 cells in each well of a 24-well plate in standard ES cell culture media plus leukemia inhibitory factor (LIF). Each day, the cells were counted and then re-plated in fresh medium for a total of 7 days.
Western blot
Whole testis protein was extracted from mice by sonication in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1 % SDS, 0.5 %, deoxycholate, 1 % NP40, supplemented with 100 μg/mL PMSF and 1× complete protease inhibitor (Roche). Samples were boiled for 5 min in sample buffer, electrophoresed on SDS-polyacrylamide gels (8–12 %), and transferred to nitrocellulose membranes. Primary-antibody incubation was performed for 12 h at 4 °C at 1:1000 dilution (antibodies used are: MUS81 (NBP1-32054 from Novus Biochemicals), MSH4 (ab95096 from Abcam), MSH5 (LS-C101911 from LsBio), MLH1 (550838 from BD pharmingen), β-Tubulin (T8328 from Sigma), β-Actin (A2228 from Sigma), BLM (a gift from Dr. Raimundo Freire), BRIP1/FANCJ (Novus, NBP1-31883), and BRCA1 (a gift from Dr. Satoshi Namekawa). Incubation with secondary antibodies was performed for 2 h (secondary HRP-conjugated antibodies were obtained from Pierce, Life Technologies). Signal detection was carried out using the superSignal substrate (Thermo Scientific). Images were captured with BIO RAD Image Lab 5.1 and analyzed by ImageJ (http://rsbweb.nih.gov/ij). Quantitation of Western blot signals was normalized to Actin or Tubulin signals, depending on which controls was used for a specific antibody.
Histology, TUNEL staining, and immunohistochemistry
Adult testes were fixed in Bouins fixative or in 10 % formalin for 6–12 h, depending on size. Fetal and neonatal gonads were fixed in the same fixatives, but for 4 h. All tissues were processed for histology by embedding in paraffin and sectioning at 5 μm. H&E staining, TUNEL (to detect apoptotic cells), and immunohistochemistry were performed as described previously (Holloway et al. 2011; Holloway et al. 2010; Holloway et al. 2014). Anti-TRA98 antibody (Abcam, #82527) was used at a concentration of 1:500.
Prophase I chromosome spreads and immunofluorescence staining
To obtain prophase I spermatocyte spreads, preparations were made according to methods used previously (Modzelewski et al. 2012). Briefly, testes from adult males were incubated in hypotonic extraction buffer (HEB; 30 mM Tris, pH 8.2, 50 mM sucrose, 17 mM trisodium citrate, 5 mM EDTA, 0.5 mM DTT, and 0.1 mM PMSF) on ice for 60 min. Small amounts of testicular tubule were transferred to 100 mM sucrose solution and further minced before spreading on slides coated in 1 % paraformaldehyde and 0.25 % Triton X-100. The slides were dried slowly in a humidified chamber for 3 h at room temperature and then washed in PBS containing 0.4 % Photoflo before staining. The primary antibodies used were either generated in our lab, or commercially available, or gifts from other labs (Kolas et al. 2005). The antibodies included: anti-RAD51 (Calbiochem, #PC130, 1:500), anti-γH2AX (Upstate Cell Signaling, NY, #05-636, 1:5000), anti-CREST (Fitzgerald Industries, MA, #90C-CS1058,1:10, 000), anti-SYCP3 (1:10,000), anti-SYCP1 (Abcam, MA, #15087, 1:500), anti-MLH1 (BD pharmingen, #550838, 1:200), anti-MLH3 (1:500) (Holloway et al. 2014), anti-BRCA1 (a gift from Dr. Satoshi Namekawa, 1:500), and anti-BLM (a gift from Dr. Raimundo Freire, 1:500). Secondary antibodies used were goat anti-mouse Alexa Fluor 488, goat anti-rabbit Alexa Fluor 555, goat anti-human Alexa Fluor 647, and goat anti-guinea pig Alexa Fluor 647 (all 1:1000, Invitrogen).
Diakinesis preparations to observe chiasmata
Diakinesis chromosome preparations were prepared as described previously (Evans et al. 1964; Uroz et al. 2008), with a slight modification (Holloway et al. 2010). In brief, testes were minced in hypotonic buffer (1 % sodium citrate) and incubated at room temperature for 20 min. The suspension was centrifuged, and the supernatant discarded. Cells were fixed twice: in methanol/glacial acetic acid/chloroform (30:10:0.15) and methanol/glacial acetic acid (3:1). Fixed cells were dropped onto slides and dried in the air. Slides were stained in Giemsa solution for 3 min, washed, and air dried, before being mounted under glass coverslips.
Image acquisition
All slides were visualized using the Zeiss Imager Z1 microscope (Carl Zeiss, Inc.). Images were captured using a high-resolution microscopy camera AxioCam MRm (Carl Zeiss, Inc.) and processed with AxioVision software (version 4.7.2; Carl Zeiss, Inc.).
Results
Growth rates of FancjGT/GT embryonic stem cells
ES cells harboring a gene trap insertion in the Fancj (Brip1) gene were obtained from the Knockout Mouse Program (KOMP) at the University of California at Davis. To assess the growth of FancjGT/GT embryonic stem (ES) cells, wild-type E14 parental ES cells were grown alongside targeted FancjGT/GT ES cells for a period of 7 days (Figure S2). The proliferative capacity of FancjGT/GT ES cells was significantly diminished compared to E14 parental ES cells when expressed as a percentage of initial seeding numbers of cells (Figure S2a; p<0.001 unpaired t test) or as an absolute cell number (Figure S2b; p<0.005 unpaired t test). While equivalent numbers of ES cells were seeded for each genotype, approximately 50 % fewer cells were observed in the dishes of FancjGT/GT ES cell clones as compared to those of E14 parental ES cells. This was not accompanied by increased apoptosis (not shown), but appeared instead to arise as a result of slower proliferative activity. Cell morphology and gross characteristics appeared normal in FancjGT/GT ES cells (not shown).
Reduced seminiferous epithelial cellularity, testis weights, and sperm counts in FancjGT/GT mice
Loss of Fancj expression in FancjGT/GT mice was confirmed by Western blotting of whole testis extracts with an anti-FANCJ antibody. The predicted protein size of 140 kDa is lost in the protein extracts from FancjGT/GT males (Figure S1c). FancjGT/GT mice were viable and fertile, with no overt changes in health status or body weight when compared to wild-type FanjGT/+ littermates (Fancj+/+: 25.2±1.3 g, Fancj+/GT: 33.2±4.3 g, FancjGT/GT: 27.7±4.2 g). Breedings of heterozygote pairs yielded homozyogous mutant, wild-type, and heterozygous pups at expected Mendelian frequencies (not shown).
Testes from FancjGT/GT male mice were compared to Fancj+/+ and FancjGT/+ littermates at 12–14 weeks of age. Testis weights of FancjGT/GT male mice were reduced by approximately 30 % compared to wild-type littermates (144.2±36.7 and 205.7±21.6 mg, respectively; p<0.05). Testis weights for FancjGT/+ littermates (199.0±21.5 mg) were not significantly different from that of wild-type males (Fig. 1a). This reduction in testis weight in FancjGT/GT male mice was accompanied by a 60 % reduction in epididymal spermatozoa compared to Fancj+/+ and FancjGT/+ littermates (Fig. 2b; p<0.005 and p<0.001, respectively).
Fig. 1.
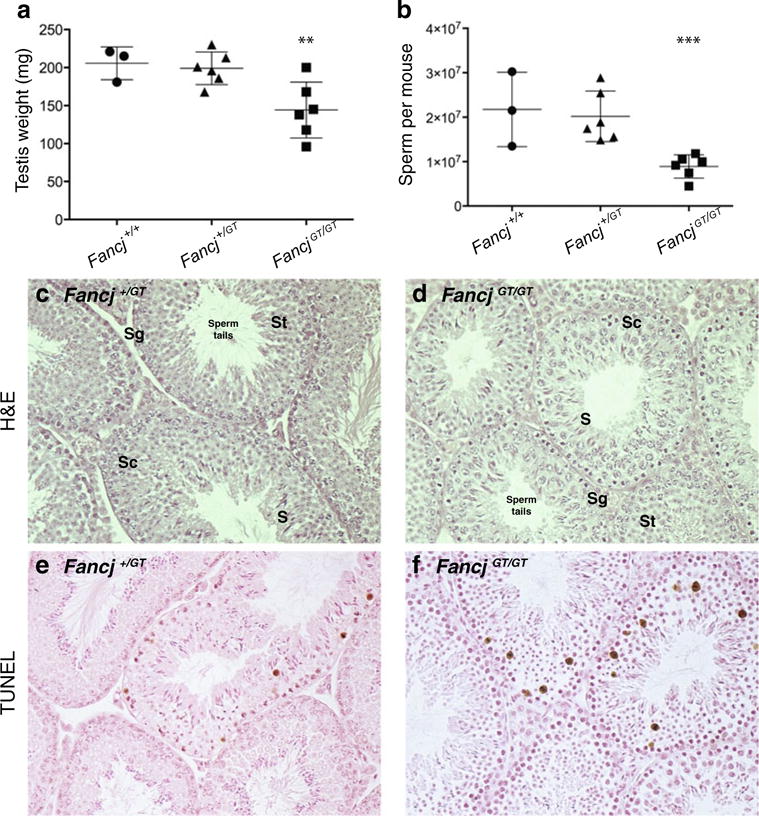
Testis weights and sperm numbers are significantly reduced in FancjGT/GT male mice compared to wildtype and heterozygous littermates. Testis weights (a) and epididymal spermatozoa counts (b) for Fancj+/+ (filled circles, n=3), FancjGT/+ (filled triangles, n=6), FancjGT/GT (filled squares, n=6) male mice. Testis weights are for pairs of testes and expressed in mg ± s.d. Sperm counts are presented as entire epididymal spermatozoa counts per mouse. Unpaired t tests were used to compare testis weights and sperm numbers between Fancj+/+ and FancjGT/GT: ***p<0.0001, **p<0.05. No statistically significant differences were found between Fancj+/+ and FancjGT/+ males for either testis weights or sperm counts. c–d Testicular histology for Fancj+/+ (c) and FancjGT/GT (d) males, as shown by H&E staining of paraffin-embedded testis sections. Sg, spermatogonia; Sc, spermatocytes; St, spermatids; S, spermatozoa. e, f Testicular TUNEL staining to reveal apoptotic cells in Fancj+/+ (e) and FancjGT/GT (f) males
Fig. 2.
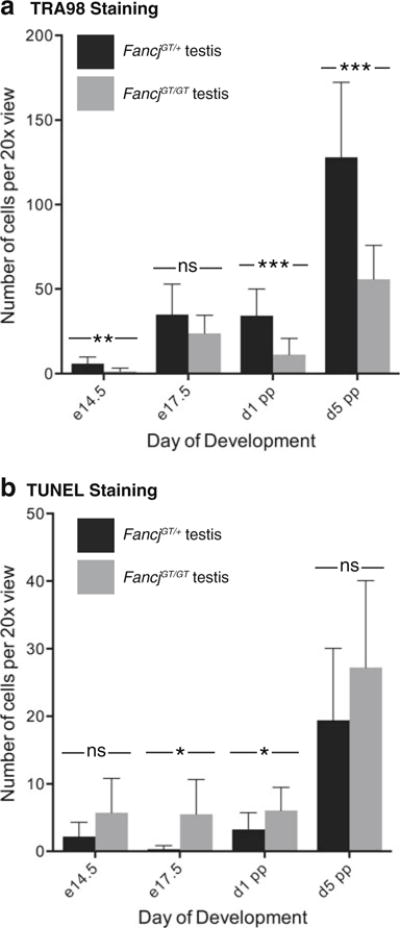
Impaired establishment of the primordial germ cell population in FancjGT/GT males during fetal development. Testis sections from FancjGT/+ (black bars) and FancjGT/GT (gray bars) males from e14.5 through until day 5 pp stained for the germ cell marker, TRA98 (a), and processed for apoptotic cell counts using the TUNEL assay (b). Values given are numbers of cells per ×20 view±standard deviation. Statistically significant differences are highlighted with asterisks: *p<0.05; **p<0.001; ***p<0.0001 (unpaired t test)
Histological analysis of FancjGT/+ and FancjGT/GT adult testis sections revealed a normal histological appearance of the seminiferous tubules and somatic cells of the testis, including Sertoli and Leydig cells (Fig. 1c, d). Germ cells at all stages of gametogenesis were apparent in the adult testis, including spermatogonial cells along the basement membrane of the seminiferous tubule, spermatocytes in the cell layers adjacent to the spermatogonia, and spermatids positioned closer to the lumen. Mature spermatozoa tails were apparent within the seminiferous tubular lumen (Fig. 1c, d), indicating full completion of all stages of spermatogenesis.
TUNEL labeling of apoptotic cells revealed similar numbers in FancjGT/+ and FancjGT/GT adult testis sections (6.2±4.3 and 4.6±3.8 TUNEL-positive cells per ×40 field, respectively), with no statistically significant difference between genotypes (p=0.3, unpaired t test). These data suggest that the reduced spermatozoa numbers and testis weights are due to reduced cellular proliferation in the germ cell pool during testicular development and/or slower progression through gametogenesis.
Reduced germ cell proliferation in fetal testis from FancjGT/GT males
To explore the basis for the reduced cellularity in the testes of FancjGT/GT adult males, we examined spermatogonial proliferation during early testicular development. During fetal life, primordial germ cells (PGC) originate as a small cluster of six cells in the proximal epiblast adjacent to the extraembryonic ectoderm at around e6.5 (Lawson and Hage 1994). By e8.5, these cells have proliferated to about 150, entering the embryonic tissues and migrate along the dorsal mesentery, arriving at the genital ridges by e10 (Ewen and Koopman 2010). Rampant proliferation of the PGCs occurs both during migration and upon arrival in the genital ridge such that, by e13.5, their numbers have increased to approximately 25,000 (Tam and Snow 1981). At this time, mitotic activity stops, and the now-female PGCs enter meiosis. In males, on the other hand, mitotic activity ceases and the PGCs enter G0 to become prospermatogonia. They resume mitotic activity only after birth, at approximately day 5 post-partum (Ewen and Koopman 2010). To examine PGC numbers in the absence of FANCJ, we quantitated total PGC numbers throughout early testis development. Figure 2 shows quantitation of germ cell numbers using TRA98 staining (Fig. 2a) and TUNEL staining (Fig. 2b) in embryonic and early postnatal testis sections from Fancj+/+ and FancjGT/GT males. The balance between cell proliferation and apoptosis can be observed via comparison of the two staining methods. One-way ANOVA revealed no significant difference between genotypes either for TRA98 or TUNEL across the range of ages studied, indicating no distinct difference in trend for either germ cell numbers or apoptosis. However, unpaired t tests comparing TRA98-positive germ cells at distinct ages revealed statistically significant decreases in germ cell numbers throughout gestation and into the early postpartum period in testes from FancjGT/GT males (Fig. 2a). This decreased germ cell number at each developmental time point in the FancjGT/GT males was associated with only mild increases in TUNEL-positive cells at these stages (Fig. 2b), suggesting that apoptotic cell death alone cannot account for the lower germ cell numbers in Fancj-deficient animals. For example, at e14.5, the earliest day studied and soon after male sex determination, very few PGCs are found in the FancjGT/GT males compared to wild-type animals, either because they are dying by apoptosis, or because the migration of PGCs to the developing gonad is delayed in the mutant animals, or because of reduced proliferation of PGCs upon their arrival in the testes of FancjGT/GT males. TUNEL labeling at this stage is only mildly increased at this timepoint, which suggests proliferative defects in the PGC population, either during migration or in the fetal testis. Similarly, between e17.5 and day 1 pp, there are increases in PGCs in both wildtype and mutant testes, but the proliferation is slower in the FancjGT/GT males. This reduced proliferation in the absence of FANCJ could be due to a lower starting population of PGCs or to increased apoptosis, which seems more rampant in later gestation. After day 1 pp, there is a rapid increase in PGCs in both mutant and wild-type testes, but this is offset by increased apoptosis in males from both genotypes. In particular, however, there is a dramatic loss of PGCs in the FancjGT/GT males, probably reflecting the increasing apoptosis in late gestation. Thus, the proliferation of PGCs is impaired in FancjGT/GT males, and this can only be partially explained by the increased apoptosis in the testes of these animals.
Early prophase I events are normal in FancjGT/GT males
To investigate whether FANCJ plays a role in meiotic prophase I progression, spermatocyte spreads were prepared from Fancj+/+ and FancjGT/GT adult males. These preparations were stained with antibodies directed against SYCP3, a key component of the axial element of the synaptonemal complex, and SYCP1, a component of the central element of the synaptonemal complex. Synapsis and prophase I progression were evident by the colocalization of these two immunofluorescent signals. Spermatocytes from FancjGT/GT males were evident throughout prophase I, with normal synapsis occurring in most cells at zygonema, and being complete by pachynema (Fig. 3).
Fig. 3.
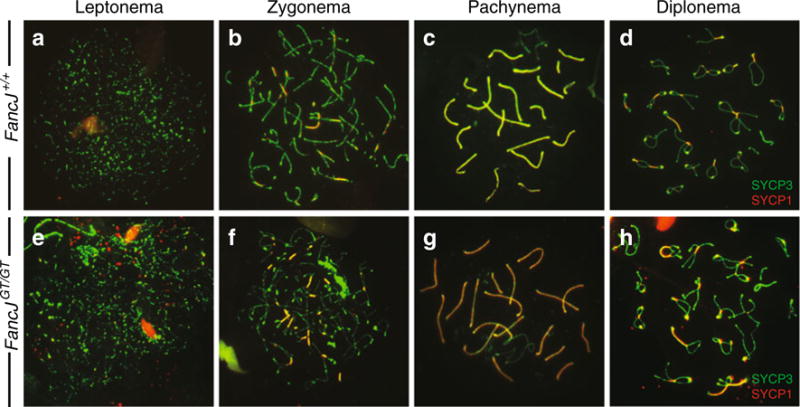
Normal chromosome synapsis during prophase I in FancjGT/GT males. Synapsis is observed by the localization of the synaptonemal complex proteins SYCP1 (red) and SYCP3 (green) on meiotic chromosome spread preparations from Fancj+/+ (a–d) and FancjGT/GT (e–f) males. Synapsed chromosome cores are indicated where red and green signals overlap to produce a yellow signal
While the majority of cells displayed normal synapsis and prophase I progression, we observed between 1 and 5 % of cells with abnormal chromosome configurations in each spread preparation from individual FancjGT/GT mice. Often, entire clusters of diplotene spermatocytes appear to show abnormal chromosome configurations, exemplified in Supplementary Figure S3a. In this example, showing three distinct cells, many abnormally asynapsed chromosomes are depicted (see white arrows for examples). The presence of >2 centromeres indicates mispairing between non-homologous chromosomes. Most usually, these abnormal configurations were only present in fewer than 1 % of cells from FancjGT/GT mice (meaning that 1 out of 100 cells from any one mouse showing abnormal pairing), but very occasionally the number of abnormally synapsing cells rose to as many as 5 % and higher. For example, the images in Figure S3a are taken from one FancjGT/GT mouse in which 5 cells out of 200 of the pachytene-like cells were found to display aberrant synapsis.
To investigate DSB repair events during prophase I in spermatocytes from FancjGT/GT males, chromosome spreads were stained with antibodies against specific markers of DSB induction and repair. The dynamics of DSB repair were similar in spermatocytes from Fancj+/+ males (Fig. 4a–d, f–j, and l) and FancjGT/GT littermates (Fig. 4m–x). The appearance of the phosphorylated form of histone H2AX, known as γH2AX, marks the first step of DSB repair and is used as a proxy for marking sites of DSB in leptonema (Fig. 4a, m) (Mahadevaiah et al. 2001). As DSBs are processed by the appropriate repair machineries, γH2AX signals decline, marking the progression from leptonema through zygonema (Fig. 4a, b, m, n). By pachynema, residual γH2AX is located throughout the chromatin of the sex chromosomes only, marking the domain of meiotic sex chromosome inactivation (MSCI; Fig. 4c, o). This MSCI-specific γH2AX staining diminishes through diplonema (Fig. 4d, p). The pattern of γH2AX localization through prophase I was largely similar between wild-type and FancjGT/GT spermatocytes; however, we observed delayed removal of γH2AX along the autosomes in a small subset of FancjGT/GT spermatocytes (Figure S3b, c). The frequency of this persistent γH2AX was in line with the cases of aberrant synapsis observed in the homozygous mutant animals. Interestingly, these aberrant synaptic events and delayed removal of γH2AX (presumably coupled with persistent DSBs) occur in small clusters of cells, usually consisting of 3–8 cells localized in close proximity on the slide, but represent fewer than 1 % of the cells imaged for each mouse, indicating that the cells likely arise from a similar location within the seminiferous epithelium, and that they may originate from the same spermatogonial precursor cell.
Fig. 4.
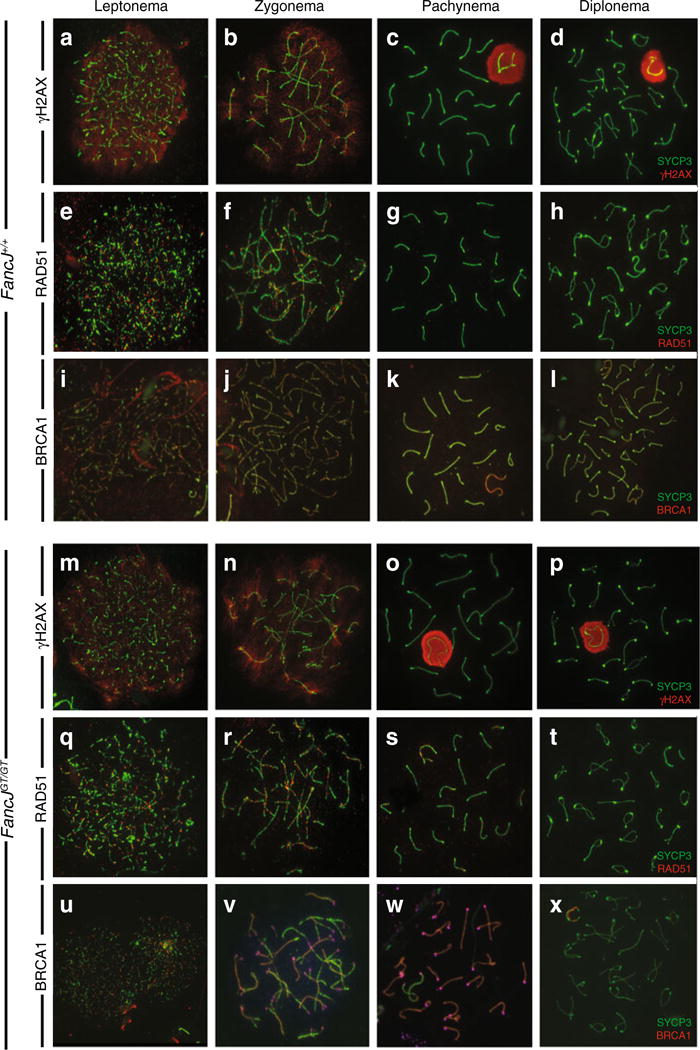
Prophase I progression in Fancj+/+ and FancjGT/GT males. Double strand break processing was monitored by staining for γH2AX to highlight the location and frequency of DSBs (a–d, m–p) and RAD51 (e–h, q–t) to highlight initial events of DSB processing. Additionally, BRCA1 is known to participate in various events during prophase I and localizes to chromosome cores (i–l, u–x). In addition, BRCA1 binds to FANCJ. However, loss of FANCJ in the FancjGT/GT males does not affect the normal localization of BRCA1 during prophase I
RAD51 is a single stranded binding protein of the RecA family. Together with DMC1, RAD51 facilitates early events of strand invasion following DSB induction, whereby a resected end of a DSB invades an opposing homologous chromosome to test for homology and to initiate a variety of DSB repair scenarios. As such, RAD51 marks all DSB events that are destined to be COs or NCOs. Similar to γH2AX, the accumulation of RAD51 on meiotic chromosome spreads from Fancj+/+ (Fig. 4e–h) and FancjGT/GT (Fig. 4q–t) males were mostly similar, showing a large number of foci at leptonema and zygonema that diminishes through late zygonema and is almost entirely absent at pachynema (Fig. 4g, s). RAD51 localization was completely gone from diplotene spermatocyte spreads (Fig. 4h, t). A small proportion of cells from FancjGT/GT males exhibited persistent localization of RAD51 into pachynema, but as with the γH2AX staining, this aberrant staining pattern was restricted to very few clusters of cells from homozygous mutant males. Given that fewer than 1 % of cells show this persistent RAD51, these cells were not always observed with each homozygous mutant animal since only a small proportion of the total spermatocyte pool is visualized with each antibody staining. Thus, with the exception of small clusters of cells, these observations would suggest that early DSB repair events are normal in the absence of FANCJ.
FANCJ was originally identified as BRIP1 through its interactions with BRCA1 (Cantor et al. 2001). Moreover, FANCJ and BRCA1 co-reside within a complex that involves other DNA damage response proteins, including TOPBP1 and MLH1 (Cantor and Xie 2010; Gong et al. 2010; Leung et al. 2011; Peng et al. 2007). However, BRCA1 and FANCJ are not entirely co-dependent since biallelic mutations in FANCJ cause Fanconi anemia, while mutations in BRCA1 are far more penetrant for breast cancer susceptibility (Hiom 2010; Seal et al. 2006). During prophase I of meiosis, BRCA1 is localized to early stretches of axial elements, forming a punctate staining pattern throughout the nucleus (Fig. 4i, u). As synapsis proceeds, BRCA1 is lost from synapsed bivalents, but remains on axial elements that have not yet undergone synapsis (Fig. 4j, v). By pachynema, BRCA1 no longer localizes to the autosomes, nor is it found on the paired region of the X and Y chromosomes (the pseudoautosomal region or PAR), but instead localizes intensely to the remaining asynapsed regions of the sex chromosomes where it persists into diplonema (Fig. 4k, l, w, and x). Importantly, this staining pattern was similar in Fancj+/+ (Fig. 4i–l) and FancjGT/GT (Fig. 4u, v, x) males, suggesting that FANCJ is not required for the localization and/or action of BRCA1 during prophase I of meiosis. Thus, the requirement for FANCJ in meiosis does not appear to involve its interaction with BRCA1.
Upregulation of BLM helicase localization during prophase I in FancjGT/GT males
Interactions between BLM helicase and FANCJ have been well characterized in different DNA damage scenarios and are thought to co-operate to maintain genomic stability downstream of HR (Suhasini and Brosh 2011). BLM and FANCJ interact both physically and functionally, and the level of cellular BLM protein is dependent on FANCJ (Suhasini et al. 2011). To explore the relationship between these two proteins during prophase I, therefore, we examined the localization of BLM protein on prophase I chromosomes from Fancj+/+ and FancjGT/GT males. As previously reported (Moens et al. 2000; Walpita et al. 1999), BLM localizes early in prophase I spermatocytes from wild-type males, appearing early in late leptonema, peaking in zygonema, and disappearing by mid-pachynema (Fig. 5a, b, s, d). In contrast, in FancjGT/GT males, BLM appears as discrete foci on chromosome cores from zygonema and persists in high numbers through pachynema and even as late as diplonema (Figs. 5e, 6l, and 7h). The intensity of BLM staining in Fancj mutant animals was higher throughout late prophase I compared to that seen in wild-type controls (Fig. 5a–h, lower panels). Thus, loss of FANCJ leads to persistent BLM localization on meiotic chromosome cores through prophase I.
Fig. 5.

Elevated and persistent BLM focus frequency at pachynema in FancjGT/GT males. BLM is localized in red and SYCP3 in green on meiotic chromosome spread preparations from Fancj+/+ (a–d) and FancjGT/GT (e–f) males through prophase I. Upper panels show the three-color images, and lower panels show just the BLM staining
Fig. 6.
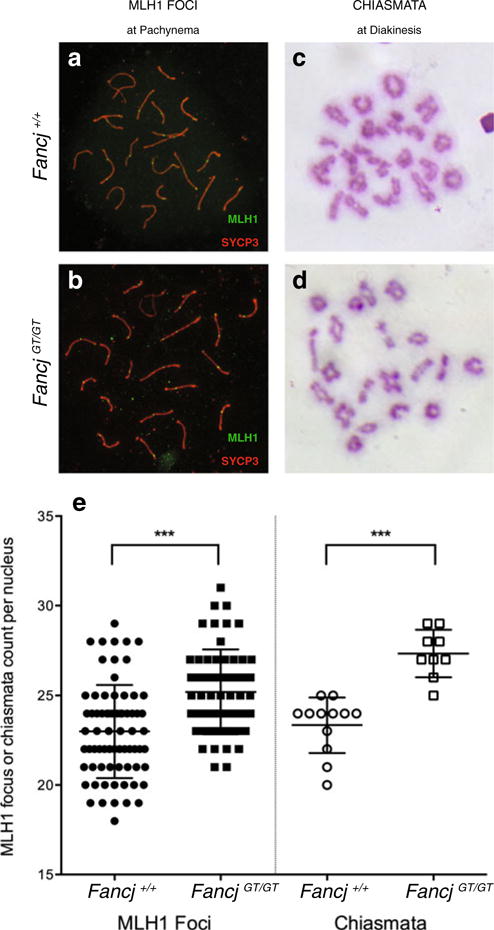
Elevated class I crossovers in FancjGT/GT males. a–b MLH1 localization (green) on meiotic chromosome cores stained with SYCP3 (red) in spermatocyte spreads from Fancj+/+ (a) and Fancj−/− (b) males. c–d Giemsa-stained diakinesis preps showing chiasmata configurations from Fancj+/+ (c) and Fancj−/− (d) males. e Quantitation and statistical comparison of MLH1 focus frequency and chiasmata counts from at least 3 mice for each genotype. Fancj+/+ shown with filled/open circles, Fancj−/− shown with filled/open squares. ***p<0.0001 by Mann–Whitney U test
Fig. 7.

Western blot analysis of whole testis lysates from Fancj+/+ and FancjGT/GT males reveals specific alterations in DNA MMR proteins and BLM. a–g Quantitation of protein levels for BRCA1, BLM, MLH1, MLH3, MSH4, MSH5, and MUS81 normalized to β-tubulin, showing significant increases in protein levels in FancjGT/GT (gray bars) compared to Fancj+/+ (black bars) testis extracts for all except BRCA1 and MUS81 (p<0.0001, unpaired t test). Representative Western blots are provided in panel h (at least 3 separate westerns were performed from two biological replicates) along with their respective controls (actin or tubulin, depending on the size of the protein of interest). Quantitation of each target protein is based on normalization to actin or tubulin
Class I crossover events are upregulated in FancjGT/GT males
DSB repair events are most frequently resolved as non-crossovers (NCO) through pathways that are not yet clearly defined in mammals. Approximately 10 % of DSB events are resolved as crossovers (CO), the majority of which arise through the class I (or ZMM) crossover pathway. This pathway is dependent on the ZMM proteins, which include MutS homologs of the DNA mismatch (MMR) family, MSH4 and MSH5 (de Vries et al. 1999; Edelmann et al. 1999; Hollingsworth et al. 1995; Kneitz et al. 2000; Paquis-Flucklinger et al. 1997; Ross-Macdonald and Roeder 1994). MSH4 and MSH5 form a heterodimer, MutSγ, which forms foci along meiotic chromosome cores from zygonema. Upon pachytene entry, a second MMR heterodimer associates with a subset of the MutSγ sites, and this heterodimer consists of MutL homologs, 1 and 3 (MLH1 and MLH3), collectively termed the MutLγ heterodimer (Baker et al. 1996; Edelmann et al. 1996; Kolas et al. 2005; Lipkin et al. 2002; Wang et al. 1999). MutLγ localization on DSB repair sites indicates the final designation marker for class I COs and thus can be used as a metric for the majority (90–95 %) of CO events. Interestingly, in spermatocytes from FancjGT/GT males, MLH1 focus frequency is elevated compared to that found in spermatocytes from Fancj+/+ males (Fig. 6a, b, e). Though this elevation is slight (25.2±1.6 foci per nucleus in FancjGT/GT compared to 22.9±2.6 in Fancj+/+), it is highly statistically significant given the usually tightly regulated MLH1 focus frequency observed in wild-type mice (p<0.0001 Mann–Whitney U test). Moreover, unlike the subtle changes in persistent DSBs described earlier, this phenotype is apparent amongst all pachytene spermatocytes from FancjGT/GT males. MLH3 focus frequency was similarly elevated (not shown).
To investigate whether the increased MLH1 foci contribute to an increased rate of crossing over, air-dried diakinesis spreads were prepared from testes of FancjGT/GT and Fancj+/+ littermates. Diakinesis spreads were stained with Giemsa to reveal the chromosome and chiasmata status. In line with the elevation in MLH1 foci, nuclei from FancjGT/GT spermatocytes showed an increase in chiasmata numbers compared to that seen in Fancj+/+ spermatocytes (Fig. 6c–e; 27.3±1.3 and 23.3±1.6 chiasmata per nucleus, respectively). This difference was highly significant when analyzed by Mann–Whitney U test (p<0.0001). The slightly higher chiasmata count relative to MLH1 focus count in both mutant and wild-type males reflects the additional chiasmata provided by other CO pathways that do not involve MutSγ and which most likely involve the Class II MUS81-regulated CO pathway (Holloway et al. 2008; Jessop and Lichten 2008). Thus, loss of FANCJ results in significantly up-regulated MLH1 focus frequency, leading to increased chiasmata counts.
Altered DNA damage repair protein levels in testis FancjGT/GT males
FANCJ has been shown to interact with numerous members of the DNA damage repair machinery, including BRCA1, BLM helicase, and MLH1 (Hiom 2010). Therefore, to investigate the impact of loss of FANCJ on the stability of these proteins, Western blots were performed using whole testis extracts from FancjGT/GT and Fancj+/+ littermates. BRCA1 protein levels were unaltered by the loss of FANCJ (Fig. 7a, h) in line with the unchanged BRCA1 localization observed in meiotic chromosome spreads (Fig. 4i–l). Similarly, MUS81 endonuclease, the major regulator of the class II CO pathway, was also unaffected by the loss of FANCJ (Fig. 7g, h). In contrast, however, BLM, MSH4, MSH5, MLH1, and MLH3 were all significantly elevated in testis extracts from FancjGT/GT males as compared to Fancj+/+ littermates (Fig. 7b, e, f, h), in agreement with the increased focus frequency for BLM, MLH1, and MLH3 in pachytene spermatocytes from the FancjGT/GT males.
Discussion
Results presented herein demonstrate multiple roles for FANCJ/BRIP1 in mammalian germ cells. Firstly, FANCJ is required for the normal proliferative activity that gives rise to the appropriate complement of germ cells that constitute the newly formed testis. Upon sexual differentiation, the PGC component of the FANCJ-deficient testis is already reduced relative to that found in wild-type littermates, and these numbers remain markedly reduced throughout late gestation. By the day of birth, the spermatogonial number is even further reduced in the FancjGT/GT males, with germ cells numbering fewer than 67 % of that seen in wild-type males. This greater drop in spermatogonial numbers through late gestation in FancjGT/GT males reflects mildly increased apoptotic activity, as measured by TUNEL labeling. Given that these cell losses occur towards the end of a phase of high proliferative activity in the developing testis, we infer that apoptosis is induced as a result of errors in cell proliferation that are likely to be caused during DNA replication. However, these losses are less severe than those observed in the germ cell pool in other FA mutants that constitute the core and ID2 complexes or in other downstream effectors of FA including FANCP/SLX4 (Holloway et al. 2011). These differences suggest that FANCJ represents one of several FA pathways that are collectively required to establish an adequate spermatogonial pool in the developing testis.
By day 5 post-partum, when proliferative activity resumes in the testis, the number of spermatogonial cells is reduced further relative to the wild-type complement, with fewer than 34 % normal spermatogonial numbers in the FancjGT/GT males. This further reduction coincides with a period of resumed proliferative activity in the testis and suggests that proliferation of the spermatogonial pool is impaired in the absence of FANCJ. Thus, we conclude that loss of FANCJ not only impedes proliferation of the PGC/spermatogonial pool in the testis, but also results in increased DNA damage as a result of errors in DNA replication, and this can lead to increased apoptosis within the pre- and neonatal testis. In support of this conclusion, other FA mutants show similarly reduced PGC proliferation and increased DNA damage in germ cells along with reduced testis weights in the adult males (Luo et al. 2014), pointing to an essential role for the FA pathway in protecting genome integrity in the proliferating germ lineage. In some cases, the loss of germ cells in FA mutants is more extreme, especially when the mutation involves a component of either the core complex or the ID2 complex. For example, in the absence of FANCC and/or FANCA, testis weights in mutant mice are reduced by >60 % on a C57Bl/6J strain background (Cheng et al. 2000; Noll et al. 2002; Wong et al. 2003). A similar degree of loss is observed in male mice bearing a homozygous mutation in another core complex component, Fancg (Koomen et al. 2002; Yang et al. 2001), and in an ID2 complex component, Fancd2 (Houghtaling et al. 2003). In all these cases, the mice were subfertile like the FancjGt/GT males, but their degree of germ cell loss and reduced testicular weight was more than double that observed in FancjGt/GT males (Fig. 1). We attribute these more severe testicular phenotypes to the fact that the loss of core and ID2 complex components is likely to impinge on several downstream pathways besides FANCJ, notably FANCP. In the case of FANCP (SLX4), our previous studies noted a 75 % loss of testis weight in adult Slx4GT/GT mutants (Holloway et al. 2011), suggesting that FANCP plays a more critical role in PGC proliferation events than does FANCJ.
Further defects associated with loss of FANCJ arise during prophase I of meiosis in males. The events of chromosome synapsis and early DSB induction/processing are largely normal in FancjGT/GT spermatocytes. A small proportion of the cells exhibit aberrant synapsis events and delayed DSB repair, but these cells tend to reside in small clumps of 3–8 cells, most likely reflecting a close proximity within their seminiferous tubular origins and/or shared clonal spermatogonial origins. This could suggest either that defects during spermatogonial proliferation may result in DNA damage that manifests as unrepaired DNA in the spermatocyte or that such mitotic DNA damage may affect the rate of DSB repair during meiosis. Alternatively, it is possible that spermatocytes progressing through prophase I in a syncytial formation may collectively undergo aberrant DSB repair, resulting in clusters of cells showing abnormal chromosome spread profiles. Whichever the case, the majority of spermatocytes from FancjGT/GT males show normal DSB repair progression, typified by progressive loss of γH2AX signal, acquisition and then loss of RAD51 signal, and accumulation of MutLγ in pachynema. Importantly, however, we find that the number of MLH1 and MLH3 foci on chromosome cores during pachynema is significantly elevated in spermatocytes from FancjGT/GT males compared to wild-type littermates. This increase in the loading of the MLH1/MLH heterodimer in prophase I, collectively termed MutLγ complex is indicative of increased utilization of the class I CO pathway. This is also associated with an increase in MLH1 and MLH3 protein in the testes of FancjGT/GT males. Similar increases in MutLγ focus frequency during prophase I are observed in mouse homozygous mutants for Mus81 and for Slx4 (Holloway et al. 2008; Holloway et al. 2011). However, loss of MUS81 or SLX4 results in normal rates of crossing over, as assessed by chiasmata counts, whereas loss of FANCJ results in increased chiasmata counts. The additional MutLγ foci observed in the absence of MUS81 or SLX4 appear to arise from a subset of DSB repair events that were already destined to become crossovers, most likely through the class II MUS81-regulated CO pathway (Berchowitz et al. 2007; Higgins et al. 2008; Holloway et al. 2008; Jessop and Lichten 2008; Oh et al. 2008). However, the additional MutLγ foci observed in FancJGT/GT spermatocytes appear to be additional events because the total number of chiasmata in these mutant animals still exceeds the number of MutLγ foci. These results suggest that a greater number of class I COs are derived from DSB events that would normally have become NCOs or from an increase in DSBs in general. We cannot differentiate with certainty between these two possibilities with the tools at hand currently, but we observe no difference in RAD51 focus frequency at zygonema (not shown), indicating a normal number of DSBs in FancJGT/GT spermatocytes.
Interestingly, the increased MutLγ focus frequency and protein level is associated with increased BLM helicase protein in whole testis extracts and on chromosome spreads, suggesting that BLM may participate in producing excess COs in the FancJGT/GT spermatocytes. Confounding this suggestion, however, is the observation that loss of BLM helicase in mouse meiosis is associated with increased COs (Holloway et al. 2010), while others have suggested that BLM and FANCJ may act coordinately in mitotic cells to unwind DNA at the end of a DSB and thereby to suppress repair by HR (Xu et al. 2015). The upregulated BLM localization through prophase I is similar to that seen in mutants lacking the class II crossover regulator, MUS81 (Holloway et al. 2008), suggesting that BLM participates in recruiting additional MLH1-MLH3-mediated crossover events in the absence of either MUS81 or FANCJ. However, this excess BLM is in contrast to events in somatic cells in culture, where loss of FANCJ results in >80 % reduction in the level of BLM protein (but not BLM transcript levels), suggesting that BLM stability is dependent on the presence of FANCJ (Suhasini et al. 2011). Taken together, therefore, it seems that the interaction between BLM and FANCJ may be context dependent and that, in prophase I spermatocytes at least, the interaction between FANCJ and MLH1 may be of greater significance for genome stability. Collectively, these observations point to a complex interplay between BLM and FANCJ that demands further investigation in a variety of cell types.
An interesting feature of our data was the observation that there was no impact on BRCA1 levels in spermatocytes from FancJGT/GT males, either by Western or on immunofluorescence chromosome spreads of mutant spermatocytes. However, there was an increase in MLH1 focus frequency and protein. Both MLH1 and BRCA1 interact with FANCJ, but each of these interactions with FANCJ are independent of the other protein (Wang et al. 2000). MLH1-FANCJ interaction is essential for some aspects of ICL repair (Cantor and Xie 2010), though the relationship is complex and poorly understood. For example, the sensitivity of FANCJ deficient cells to ICL inducing drugs can be rescued with a BRCA1-binding mutant of FANCJ, but not with a MLH1-binding mutant of FANCJ (Peng et al. 2007). Importantly, loss of MLH1 alone has no effect on ICL sensitivity, suggesting that the role of MLH1 in this context is dependent on its interaction with FANCJ (Peng et al. 2007). Taken together, these data suggest that the interaction between MLH1 and FANCJ can alter the spectrum of repair pathways utilized by the latter, and that the type of repair elicited may be context-dependent. In the context of our meiosis data, it appears that the loss of FANCJ can alter the utilization of MLH1 at sites of DSB repair, resulting in increased class I CO events. This could suggest that loss of FANCJ-MLH1 interactions can increase the accessibility of MLH1 to DSB repair intermediates, or that this increased MLH1 binding is conferred by an indirect action through BLM, as described above.
Recent studies have demonstrated that FANCJ interacts with the MutS homolog, MSH5 (Xu et al. 2015). MSH5 is a component of the MutSγ complex alongside MSH4, which functions as an essential regulator of the predominant crossover pathway during prophase I (Hollingsworth et al. 1995; Kneitz et al. 2000; Paquis-Flucklinger et al. 1997; Ross-Macdonald and Roeder 1994; Snowden et al. 2008). In the context of mammalian meiosis, the MutSγ complex is thought to facilitate DSB repair via crossing over through the predominant class I (or ZMM) pathway, in part by recruiting MutLγ (MLH1 and MLH3) (Franklin et al. 2006; Hoffmann and Borts 2004; Kolas et al. 2005; Santucci-Darmanin et al. 2002). The MutSγ complex also interacts with proteins that regulate the designation of DSB repair events to become COs, including the ring finger protein, RNF212 (Qiao et al. 2014). All these observations raise the question as to the functional importance of a potential FANCJ-MSH5 interaction in mammalian germ cells. It is possible that loss of FANCJ-MSH5 interactions could be responsible for increasing acquisition of MutLγ at DSB repair intermediate sites, leading to the increase in class I CO events.
Taken together, our data reveal an important role for FANCJ in germ cell proliferation during gonadal development and for modulation of CO rates through the regulation of the class I crossover pathway. We propose that several key interacting proteins could participate in this function, including MSH5, MLH1, and BLM, but that BRCA1 interactions with FANCJ are unlikely to be important in this context. Clearly, FANCJ represents a novel component of the class I regulatory machinery and understanding how the FA pathway mediates the activity of the FANCJ helicase, and how this function may be important for promoting or restricting class I CO events will be the subject of intense investigation in the coming years.
Supplementary Material
Acknowledgments
We thank Peter Borst for his assistance with mouse handling, genotyping, and maintenance. We are indebted to Dr. Satoshi Namekawa (Cincinnati Children’s Hospital, OH) and Dr. Raimundo Freire (University of Tenerife, Spain) for generously providing antibodies critical for these studies. We thank Simon Boulton (Francis Crick Institute, London) for providing advice and primer sequences for genotyping. We are grateful to Robert Munroe and Christian Abratte of the Cornell Stem Cell and Transgenic Core Facility for performing blastocyst injections. The Cornell Stem Cell and Transgenic Core is supported by NYSDOH contract C024174. This work was supported through funding from the NICHD to J.K.H. (HD065870) and from the NIGMS to P.E.C (GM097263). A.C. was supported by an award from the Dextra Undergraduate Research Endowment fund.
Footnotes
This article is part of a Special Issue on “Recent advances in meiotic chromosome structure, recombination and segregation” edited by Marco Barchi, Paula Cohen and Scott Keeney.
Electronic supplementary material The online version of this article (doi:10.1007/s00412-015-0549-2) contains supplementary material, which is available to authorized users.
Compliance with ethical standards Laboratory animals were maintained and utilized in accordance with Federal guidelines for the care and use of animals, and under a protocol approved by the Cornell Institutional Animal Care and Use Committee (IACUC).
All authors declare that they have no conflicts of interest relating to the work described herein.
References
- Agoulnik AI, et al. A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum Mol Genet. 2002;11:3047–3053. doi: 10.1093/hmg/11.24.3047. [DOI] [PubMed] [Google Scholar]
- Andreassen PR, Ren K. Fanconi anemia proteins, DNA interstrand crosslink repair pathways, and cancer therapy. Curr Cancer Drug Targets. 2009;9:101–117. doi: 10.2174/156800909787314011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SM, et al. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Baudat F, Imai Y, de Massy B. Meiotic recombination in mammals: localization and regulation. Nat Rev Genet. 2013;14:794–806. doi: 10.1038/nrg3573. [DOI] [PubMed] [Google Scholar]
- Berchowitz LE, Francis KE, Bey AL, Copenhaver GP. The role of AtMUS81 in interference-insensitive crossovers in A. thaliana. PLoS Genet. 2007;3:e132. doi: 10.1371/journal.pgen.0030132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolcun-Filas E, Schimenti JC. Genetics of meiosis and recombination in mice. Int Rev Cell Mol Biol. 2012;298:179–227. doi: 10.1016/B978-0-12-394309-5.00005-5. [DOI] [PubMed] [Google Scholar]
- Cantor SB, Xie J. Assessing the link between BACH1/FANCJ and MLH1 in DNA crosslink repair. Environ Mol Mutagen. 2010;51:500–507. doi: 10.1002/em.20568. [DOI] [PubMed] [Google Scholar]
- Cantor SB, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- Cheng NC, et al. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum Mol Genet. 2000;9:1805–1811. doi: 10.1093/hmg/9.12.1805. [DOI] [PubMed] [Google Scholar]
- de Vries SS, Baart EB, Dekker M, Siezen A, de Rooij DG, de Boer P, te Riele H. Mouse MutS-like protein Msh5 is required for proper chromosome synapsis in male and female meiosis. Genes Dev. 1999;13:523–531. doi: 10.1101/gad.13.5.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann W, et al. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85:1125–1134. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- Edelmann W, et al. Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nat Genet. 1999;21:123–127. doi: 10.1038/5075. [DOI] [PubMed] [Google Scholar]
- Evans EP, Breckon G, Ford CE. An air-drying method for meiotic preparations from mammalian testes. Cytogenet Cell Genet. 1964;3:289–294. doi: 10.1159/000129818. [DOI] [PubMed] [Google Scholar]
- Ewen KA, Koopman P. Mouse germ cell development: from specification to sex determination. Mol Cell Endocrinol. 2010;323:76–93. doi: 10.1016/j.mce.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Franklin FC, Higgins JD, Sanchez-Moran E, Armstrong SJ, Osman KE, Jackson N, Jones GH. Control of meiotic recombination in Arabidopsis: role of the MutL and MutS homologues. Biochem Soc Trans. 2006;34:542–544. doi: 10.1042/BST0340542. [DOI] [PubMed] [Google Scholar]
- Gong Z, Kim JE, Leung CC, Glover JN, Chen J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell. 2010;37:438–446. doi: 10.1016/j.molcel.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JD, Buckling EF, Franklin FC, Jones GH. Expression and functional analysis of AtMUS81 in Arabidopsis meiosis reveals a role in the second pathway of crossing-over. Plant J. 2008;54:152–162. doi: 10.1111/j.1365-313X.2008.03403.x. [DOI] [PubMed] [Google Scholar]
- Hiom K. FANCJ: solving problems in DNA replication. DNA Repair. 2010;9:250–256. doi: 10.1016/j.dnarep.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Hoffmann ER, Borts RH. Meiotic recombination intermediates and mismatch repair proteins. Cytogenet Genome Res. 2004;107:232–248. doi: 10.1159/000080601. [DOI] [PubMed] [Google Scholar]
- Hollingsworth NM, Ponte L, Halsey C. MSH5, a novel MutS homolog, facilitates meiotic reciprocal recombination between homologs in Saccharomyces cerevisiae but not mismatch repair. Genes Dev. 1995;9:1728–1739. doi: 10.1101/gad.9.14.1728. [DOI] [PubMed] [Google Scholar]
- Holloway JK, Booth J, Edelmann W, McGowan CH, Cohen PE. MUS81 generates a subset of MLH1-MLH3-independent crossovers in mammalian meiosis. PLoS Genet. 2008;4:e1000186. doi: 10.1371/journal.pgen.1000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway JK, Morelli MA, Borst PL, Cohen PE. Mammalian BLM helicase is critical for integrating multiple pathways of meiotic recombination. J Cell Biol. 2010;188:779–789. doi: 10.1083/jcb.200909048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway JK, et al. Mammalian BTBD12 (SLX4) protects against genomic instability during mammalian spermatogenesis. PLoS Genetics. 2011;7:e1002094. doi: 10.1371/journal.pgen.1002094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway JK, Sun X, Yokoo R, Villeneuve AM, Cohen PE. Mammalian CNTD1 is critical for meiotic crossover maturation and de-selection of excess pre-crossover sites. J Cell Biol. 2014;205:633–641. doi: 10.1083/jcb.201401122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17:2021–2035. doi: 10.1101/gad.11034031103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop L, Lichten M. Mus81/Mms4 endonuclease and Sgs1 helicase collaborate to ensure proper recombination intermediate metabolism during meiosis. Mol Cell. 2008;31:313–323. doi: 10.1016/j.molcel.2008.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneitz B, et al. MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 2000;14:1085–1097. [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolas NK, et al. Localization of MMR proteins on meiotic chromosomes in mice indicates distinct functions during prophase I. J Cell Biol. 2005;171:447–458. doi: 10.1083/jcb.200506170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koomen M, et al. Reduced fertility and hypersensitivity to mitomycin C characterize Fancg/Xrcc9 null mice. Hum Mol Genet. 2002;11:273–281. doi: 10.1093/hmg/11.3.273. [DOI] [PubMed] [Google Scholar]
- Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, Tijsterman M. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr Biol. 2008;18:900–905. doi: 10.1016/j.cub.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Kumaraswamy E, Shiekhattar R. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol. 2007;27:6733–6741. doi: 10.1128/MCB.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson KA, Hage WJ. Clonal analysis of the origin of primordial germ cells in the mouse. Ciba Found Symp. 1994;182:68–84. doi: 10.1002/9780470514573.ch5. discussion 84–91. [DOI] [PubMed] [Google Scholar]
- Leung CC, Gong Z, Chen J, Glover JN. Molecular basis of BACH1/FANCJ recognition by TopBP1 in DNA replication checkpoint control. J Biol Chem. 2011;286:4292–4301. doi: 10.1074/jbc.M110.189555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitus M, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nat Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- Lipkin SM, et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet. 2002;31:385–390. doi: 10.1038/ng931. [DOI] [PubMed] [Google Scholar]
- Litman R, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANC J. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Lu B, Bishop CE. Mouse GGN1 and GGN3, two germ cell-specific proteins from the single gene Ggn, interact with mouse POG and play a role in spermatogenesis. J Biol Chem. 2003;278:16289–16296. doi: 10.1074/jbc.M211023200. [DOI] [PubMed] [Google Scholar]
- Luo Y, Hartford SA, Zeng R, Southard TL, Shima N, Schimenti JC. Hypersensitivity of primordial germ cells to compromised replication-associated DNA repair involves ATM-p53-p21 signaling. PLoS Genet. 2014;10:e1004471. doi: 10.1371/journal.pgen.1004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevaiah SK, et al. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet. 2001;27:271–276. doi: 10.1038/85830. [DOI] [PubMed] [Google Scholar]
- Modzelewski AJ, Holmes RJ, Hilz S, Grimson A, Cohen PE. AGO4 regulates entry into meiosis and influences silencing of sex chromosomes in the male mouse germline. Dev Cell. 2012;23:251–264. doi: 10.1016/j.devcel.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens PB, Freire R, Tarsounas M, Spyropoulos B, Jackson SP. Expression and nuclear localization of BLM, a chromosome stability protein mutated in Bloom’s syndrome, suggest a role in recombination during meiotic prophase. J Cell Sci. 2000;113:663–672. doi: 10.1242/jcs.113.4.663. [DOI] [PubMed] [Google Scholar]
- Muniandy PA, Liu J, Majumdar A, Liu ST, Seidman MM. DNA interstrand crosslink repair in mammalian cells: step by step. Crit Rev Biochem Mol Biol. 2010;45:23–49. doi: 10.3109/10409230903501819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll M, et al. Fanconi anemia group A and C double-mutant mice: functional evidence for a multi-protein Fanconi anemia complex. Exp Hematol. 2002;30:679–688. doi: 10.1016/s0301-472x(02)00838-x. [DOI] [PubMed] [Google Scholar]
- Oh SD, Lao JP, Taylor AF, Smith GR, Hunter N. RecQ helicase, Sgs1, and XPF family endonuclease, Mus81-Mms4, resolve aberrant joint molecules during meiotic recombination. Mol Cell. 2008;31:324–336. doi: 10.1016/j.molcel.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquis-Flucklinger V, Santucci-Darmanin S, Paul R, Saunieres A, Turc-Carel C, Desnuelle C. Cloning and expression analysis of a meiosis-specific MutS homolog: the human MSH4 gene. Genomics. 1997;44:188–194. doi: 10.1006/geno.1997.4857. [DOI] [PubMed] [Google Scholar]
- Peng M, Litman R, Xie J, Sharma S, Brosh RM, Jr, Cantor SB. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Xie J, Ucher A, Stavnezer J, Cantor SB. Crosstalk between BRCA-Fanconi anemia and mismatch repair pathways prevents MSH2-dependent aberrant DNA damage responses. EMBO J. 2014;33:1698–1712. doi: 10.15252/embj.201387530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadnis N, Hyppa RW, Smith GR. New and old ways to control meiotic recombination. Trends Genet. 2011;27:411–421. doi: 10.1016/j.tig.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao H, et al. Antagonistic roles of ubiquitin ligase HEI10 and SUMO ligase RNF212 regulate meiotic recombination. Nat Genet. 2014;46:194–199. doi: 10.1038/ng.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosselli F, Briot D, Pichierri The Fanconi anemia pathway and the DNA interstrand cross-links repair. Biochimie. 2003;85:1175–1184. doi: 10.1016/j.biochi.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P, Roeder GS. Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Cell. 1994;79:1069–1080. doi: 10.1016/0092-8674(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Santucci-Darmanin S, Neyton S, Lespinasse F, Saunieres A, Gaudray P, Paquis-Flucklinger V. The DNA mismatch-repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum Mol Genet. 2002;11:1697–1706. doi: 10.1093/hmg/11.15.1697. [DOI] [PubMed] [Google Scholar]
- Seal S, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- Snowden T, Shim KS, Schmutte C, Acharya S, Fishel R. hMSH4-hMSH5 adenosine nucleotide processing and interactions with homologous recombination machinery. J Biol Chem. 2008;283:145–154. doi: 10.1074/jbc.M704060200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhasini AN, Brosh RM., Jr Fanconi anemia and Bloom’s syndrome crosstalk through FANCJ-BLM helicase interaction. Trend Genet. 2011 doi: 10.1016/j.tig.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhasini AN, et al. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011;30:692–705. doi: 10.1038/emboj.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PP, Snow MH. Proliferation and migration of primordial germ cells during compensatory growth in mouse embryos. J Embryol Exp Morphol. 1981;64:133–147. [PubMed] [Google Scholar]
- Uroz L, Rajmil O, Templado C. Premature separation of sister chromatids in human male meiosis. Hum Reprod. 2008;23:982–987. doi: 10.1093/humrep/dem427. [DOI] [PubMed] [Google Scholar]
- Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys. 2014;43:257–278. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- Walpita D, Plug AW, Neff NF, German J, Ashley T. Bloom’s syndrome protein, BLM, colocalizes with replication protein A in meiotic prophase nuclei of mammalian spermatocytes. Proc Natl Acad Sci. 1999;96:5622–5627. doi: 10.1073/pnas.96.10.5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TF, Kleckner N, Hunter N. Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci U S A. 1999;96:13914–13919. doi: 10.1073/pnas.96.24.13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. In Process Citation. [PMC free article] [PubMed] [Google Scholar]
- Whitney MA, et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88:49–58. [PubMed] [Google Scholar]
- Williams SA, et al. Functional and physical interaction between the mismatch repair and FA-BRCA pathways. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JC, Alon N, McKerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 2003;12:2063–2076. doi: 10.1093/hmg/ddg219. [DOI] [PubMed] [Google Scholar]
- Wu Y, Shin-ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wu X, Her C. hMSH5 facilitates the repair of camptothecin-induced double-strand breaks through an interaction with FANCJ. J Biol Chem. 2015 doi: 10.1074/jbc.M115.642884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Targeted disruption of the murine Fanconi anemia gene, Fancg/Xrcc9. Blood. 2001;98:3435–3440. doi: 10.1182/blood.v98.12.3435. [DOI] [PubMed] [Google Scholar]
- Zou J, Zhang D, Qin G, Chen X, Wang H, Zhang D. BRCA1 and FancJ cooperatively promote interstrand crosslinker induced centrosome amplification through the activation of polo-like kinase 1. Cell Cycle. 2014;13:3685–3697. doi: 10.4161/15384101.2014.964973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.