

Abstract
The heat-shock protein 90 (Hsp90) cochaperone FK506-binding protein 52 (FKBP52) upregulates, whereas FKBP51 inhibits, hormone binding and nuclear targeting of the glucocorticoid receptor (GR). Decreased cortisol sensitivity in the guinea pig is attributed to changes within the helix 1 to helix 3 (H1-H3) loop of the guinea pig GR (gpGR) ligand-binding domain. It has been proposed that this loop serves as a contact point for FKBP52 and/or FKBP51 with receptor. We examined the role of the H1-H3 loop in GR activation by FKBP52 using a Saccharomyces cerevisiae model. The activity of rat GR (rGR) containing the gpGR H1-H3 loop substitutions was still potentiated by FKBP52, confirming the loop is not involved in primary FKBP52 interactions. Additional assays also excluded a role for other intervening loops between ligand-binding domain helices in direct interactions with FKBP52 associated with enhanced receptor activity. Complementary studies in FKBP51-deficient mouse embryo fibroblasts and HEK293 cells demonstrated that substitution of the gpGR H1-H3 loop residues into rGR dramatically increased receptor repression by FKBP51 without enhancing receptor-FKBP51 interaction and did not alter recruitment of endogenous Hsp90 and the p23 cochaperone to receptor complexes. FKBP51 suppression of the mutated rGR did not require FKBP51 peptidylprolyl cis-trans isomerase activity and was not disrupted by mutation of the FK1 proline-rich loop thought to mediate reciprocal FKBP influences on receptor activity. We conclude that the gpGR-specific mutations within the H1-H3 loop confer global changes within the GR-Hsp90 complex that favor FKBP51 repression over FKBP52 potentiation, thus identifying the loop as an important target for GR regulation by the FKBP cochaperones.
The glucocorticoid receptor (GR), a member of the nuclear receptor superfamily of ligand-dependent transcription factors (1–3), mediates the physiological actions of glucocorticoid hormones essential in development, metabolism, immune and anti-inflammatory effects, and neuronal responses (4–11). GR is also an important therapeutic target, synthetic glucocorticoids being one of the most commonly prescribed pharmaceuticals. As for other nuclear receptor family members, GR shares a common structural organization consisting of a central DNA-binding domain, an N-terminal region containing activation function-1 (AF-1) and a C-terminal ligand-binding domain (LBD) that, in addition to binding corticosteroids, plays a role in receptor homodimerization (12, 13), contains an AF-2 that is tightly regulated by hormone binding and harbors the interaction site for heat-shock protein 90 (Hsp90) (1–3, 7). In the absence of ligand, GR localizes to the cytoplasm where it undergoes stepwise assembly with Hsp90/Hsp70 molecular chaperone machinery to a hormone-binding–competent form in which the mature apo-receptor complex consists of an Hsp90 dimer, p23, and one of the tetratricopeptide repeat (TPR)-containing cochaperones: the immunophilin FK506-binding proteins (FKBP) 51 and 52, cyclophilin 40 (CyP40), or protein phosphatase PP5 (7, 14–17). Through their TPR domains, the cochaperones compete for a common MEEVD peptide-binding site at the C terminus of Hsp90. Hormone binding promotes efficient translocation of GR to the nucleus where the activated receptor regulates target gene transcription (7, 18, 19).
Familial resistance to cortisol is a hypertensive and hyperandrogenic disorder characterized by elevated serum cortisol and ACTH concentrations, reflecting a generalized tissue resistance to the hormone and is associated with specific mutations in the GR LBD that result in reduced receptor ligand-binding affinity (20, 21). Studies of cortisol resistance observed as a normal condition in some New World species, including the guinea pig and certain primates, have revealed important insights into the molecular basis of end-organ resistance to cortisol arising from a lower GR-binding affinity for the hormone (22–25). In guinea pig GR (gpGR), decreased glucocorticoid sensitivity has been ascribed to the combined effect of 5 nonconservative residue changes within the unstructured loop separating helices 1 and 3 in the receptor LBD (12, 23, 26). On the other hand, resistance displayed by New World primates is caused not by amino acid sequence alteration, but primarily results from high-level expression of FKBP51 leading to the increased incorporation of the immunophilin into GR-Hsp90 molecular chaperone complexes and a reduced receptor hormone-binding affinity (25, 27–29). FKBP51 and its structurally similar cochaperone partner, FKBP52, contain two FKBP domains, the first of which, FK1, displays a peptidylprolyl cis-trans isomerase (PPIase) activity that may catalyze protein folding and is inhibited by FK506 (30), whereas the second, FK2, lacks catalytic function and drug-binding ability (14, 17, 31–33). Although the mechanism remains to be defined, inhibition of GR activity by FKBP51 requires both FK domains, as well as Hsp90 binding, but is independent of PPIase activity (27).
The dynamic assembly of immunophilin cochaperones into steroid receptor complexes is largely determined by their relative cellular expression and by receptor preferences for specific cochaperones, allowing selected cochaperones to differentially modulate receptor activity through unique receptor interactions (34–40). Although FKBP51 is the preferred cochaperone for mature GR-Hsp90 complexes (34–36) and inhibits GR function, FKBP52 has been shown to promote increased GR hormone-binding affinity and to potentiate the transcriptional activity of the receptor, both in yeast (41) and mammalian cells (35, 40). Studies of FKBP52-deficient mice have revealed a state of glucocorticoid resistance arising from loss of GR hepatic activity under metabolic stress (42) and have extended the critical physiological role of the immunophilin to cellular responses controlled by both the androgen receptor (AR) (43–45) and progesterone receptor (PR) (46, 47). The stimulatory effect of FKBP52 is directed through the GR LBD and requires FKBP52 binding to Hsp90, together with an intact FK1 domain, and although initially deemed to be essential (40, 41), FKBP52 catalytic (PPIase) function was subsequently shown not to be a requisite for potentiation (48). Rather, studies with AR and GR have led to proposals that a proline-rich loop overhanging the FK1 catalytic pocket in FKBP52 makes specific contact with a region of the receptor LBD, a structural feature also common to PR, stabilizing an LBD conformation optimal for high-affinity hormone binding and efficient transcriptional activation (17, 48). Indeed, there is recent evidence that, within the AR LBD, FKBP52 may have influence over some part of the binding function-3 (BF-3) regulatory surface known to allosterically modulate AR AF-2 activity (49, 50). BF-3 residues are conserved in GR and PR, as well as estrogen receptor-α (ERα) and the mineralocorticoid receptor (MR), remaining members of the steroid receptor subfamily (50). Although MR has a similar dependence to GR and PR on Hsp90 for hormone binding (51, 52), ERα is less reliant on Hsp90 for its hormone-binding ability, although it still assembles into ERα-Hsp90 complexes (37, 53, 54).
Although crystal structures of unliganded steroid receptor LBDs are unavailable, likely due to conformational instability of this domain in the absence of ligand and/or Hsp90 (14, 15, 17, 31, 55), structures of liganded AR, GR, and PR conform to a common fold for all nuclear receptors encompassing 12 α-helices (numbered H1 to H12 according to the apo-retinoic acid X receptor-α LBD structure) (12, 56–59). The short, barely discernible H2 in AR, GR, and PR forms part of an extended loop between H1-H3 (12, 56–58).
The counterinfluences of FKBP51 and FKBP52 on GR activity are compatible with a proposed model for GR hormonal activation in which a hormone-induced interchange of FKBP51 by FKBP52 in GR-Hsp90 complexes results in LBD conformational changes that promote increased hormone-binding affinity of the receptor and favor nuclear translocation of GR-Hsp90-FKBP52 complexes (26, 35, 40, 60–63). Thus, there is now a clear understanding that, for GR, steroid-binding affinity is determined in unliganded receptor by interactions between the receptor LBD and Hsp90, in concert with its cochaperones. Based on structural and functional studies of gpGR, which identified mutations within the H1-H3 loop to be responsible for lower cortisol-binding affinity, Fuller et al (26) suggested that the region might correspond to an interaction site for FKBP51 and FKBP52 with GR. They defined a hypothesis in which, mechanistically, the H1-H3 loop was deemed to serve as a contact point between receptor and either (or both) of the immunophilins. A favored interaction with FKBP51 over FKBP52, accompanied by changes in conformation of the ligand-binding pocket, would result in decreased receptor cortisol-binding affinity (26). In this work, we have assessed the role of the H1-H3 loop in the regulation of GR activity by the FKBP52 cochaperone in yeast and extended our study in this model system to conserved proline residues within other exposed interhelical regions of the GR LBD as potential contact sites for FKBP52 interaction. Additional studies demonstrated that substitution of the gpGR H1-H3 loop residues into rat GR (rGR) dramatically increased the potency of GR repression by FKBP51 without enhancing FKBP51 interaction. Because the gpGR-specific mutations within the H1-H3 loop result in reduced GR cortisol-binding affinity (23, 26), we conclude that the enhanced repression by FKBP51 may occur through global changes within the GR-Hsp90 complex that favor FKBP51 repression over FKBP52 potentiation, identifying the loop as an additional site for GR regulation by the FKBP cochaperones.
Materials and Methods
DNA constructs and mutagenesis
The yeast expression plasmids pG/N795 (expressing the full-length rGR), p423GPD/hFKBP52 and the lacZ reporter plasmid pUCΔSS-26x containing the glucocorticoid response element (41, 65) were provided by David Smith. The plasmid pRK5MCS/FKBP51-FLAG was provided by Theo Rein (38), pCMV6/FKBP51-FLAG and pCMV6/FKBP51 F130A-FLAG by Chad Dickey (66), pIRESpuro3/FKBP52 plasmid by John Price, pcDNA3/AR P892A by Mark Trifiro (67), and pPB3.luc by Robert Matusik (68).
The rGR DNA from the plasmid pG/N795 was used as a template to generate the rGR proline and gpGR mutant constructs. The rGR DNA was excised from pG/N795 with the restriction enzyme BamHI and ligated into pDrive cloning vector (QIAGEN, Hilden, Germany). Oligonucleotide primers were designed and synthesized (Sigma-Aldrich, St. Louis, Missouri) (Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) with mutagenesis performed according to the manufacturer's instructions (QuikChange site-directed mutagenesis kit; Stratagene, La Jolla, California). Once mutagenesis was confirmed, the mutated region of rGR was cassetted back into the parental pDrive/rGR construct using the restriction enzyme SphI. Correct orientation of the inserted fragments was checked with BamHI digests. The whole of the rGR DNA was then cloned into the pG plasmid using BamHI. The rGR combination mutant IHS/TS was created by performing site-directed mutagenesis on the already mutated rGR LYA562IHS. For mammalian transactivation studies, wild-type and mutated rGR were excised from their respective pG vector constructs with the restriction enzyme BamHI and ligated into pcDNA3.1. For coimmunoprecipitation studies, we used N-terminally hemagglutinin (HA)-tagged rGR (either wild-type or double IHS/TS mutant) cloned into pcDNA3.1 (pcDNA3.1/HA-rGR or pcDNA3.1/HA-rGR-IHS/TS). The HA-rGR constructs were generated by PCR from a pcDNA3.1/rGR template using a forward primer containing a BamHI recognition site, followed by the HA tag and a reverse primer containing the XhoI recognition site. The PCR amplicon was ligated into pDrive and the 5′-end of rGR sequenced to confirm the integrity of the BamHI site, HA tag, and rGR sequence up to the unique BglII site at position 708. The wild-type and IHS/TS-containing 3′ larger ends of the rGR sequence were then each cassetted into pDrive/HA-rGR from their respective untagged pcDNA3.1 constructs via BglII/XhoI digestion. The full-length HA-tagged wild-type and IHS/TS-mutant GRs were then subcloned from their respective pDrive constructs into pcDNA3.1 via BamHI/XhoI digestion.
The hFKBP52 DNA was excised from pIRESpuro3/hFKBP52 with the restriction enzymes NheI and NotI and ligated into pDrive cloning vector. Oligonucleotide primers were designed with a silent mutation within the sequence targeted by the small interfering RNA (siRNA) (Supplemental Table 1). After mutagenesis and sequence validation, the mutated hFKBP52 DNA was ligated into pcDNA3.1 using the above restriction enzymes to give pcDNA3.1/FKBP52 SM-1. Full-length hFKBP51 DNA was subcloned from pGEX4T.1/hFKBP51 (69) into pcDNA3.1 via EcoRI and XhoI restriction enzyme sites. To prepare the FKBP51 L119P mutant (primer details in Supplemental Table 1), FKBP51 DNA was ligated into the pDrive cloning vector and mutagenesis was performed as above.
Yeast rGR reporter assays
Yeast transformation and reporter assays were performed as described by Riggs et al (41), using Saccharomyces cerevisiae yeast strain W303a (kindly supplied by David Smith) with the exception that 25 nM deoxycorticosterone (DOC) was used to monitor wild-type receptor activity, whereas assessment of the activities of rGR proline mutants required 5 μM DOC, owing to reduced hormone responsiveness. β-Galactosidase activity was detected using a POLARstar OPTIMA microplate reader (BMG Labtech, Ortenberg, Germany). The β-galactosidase values, measured as relative light units, were plotted against the OD600 values to create a receptor transactivation curve standardized against culture growth.
Mammalian cell reporter assays
For these assays, we used HeLa cells, mouse embryo fibroblast (MEF) FKBP51 knockout (KO) cells (kindly provided by David Smith), or HEK293 cells. Cells were maintained in DMEM supplemented with 5% to 10% fetal bovine serum and 100 U/mL penicillin/100 μg/mL streptomycin. Forty-eight hours before each assay, cells were placed in phenol red-free DMEM supplemented with 5% to 10% charcoal-stripped fetal bovine serum and 100 U/mL penicillin/100 μg/mL streptomycin. All transfections were performed using Lipofectamine 2000 (Life Technologies, Invitrogen, Carlsbad, California). For the HeLa assays, cells were seeded at a density of 1.8 × 106 cells per 75-cm2 flask and the next day transfected with 5 nM siRNA targeting FKBP52 (Life Technologies, Ambion, Carlsbad, California; siRNA ID s50) or a negative control siRNA at 2 consecutive time points 24 hours apart to achieve optimal knockdown. Cells were then seeded into a 24-well plate at a density of 7 × 104 cells per well, and 24 hours after the final siRNA transfection, cells were transfected with 150 ng pcDNA3/AR P892A, 500 ng pPB3.luc, and 500 ng pcDNA3.1/FKBP52 SM-1 or empty pcDNA3.1 vector. Immediately after transfection, cells were treated with 10 nM dihydrotestosterone (DHT) or ethanol vehicle. For the MEF FKBP51-KO assays, cells were seeded into a 24-well plate at a density of 5.5 × 104 cells per well. After 24 hours, MEF FKBP51-KO cells were transfected with 100 ng pcDNA3.1/rGR (wild-type or mutant), 400 ng pGRE.luc, and 300 ng pcDNA3.1/hFKBP51 or empty pcDNA3.1. After another 24 hours, cells were treated with increasing concentrations of cortisol (Sigma-Aldrich) or ethanol vehicle. For the HEK293 assays, cells were seeded into a 24-well plate at a density of 1.05 × 105 cells per well. After 24 hours, cells were transfected with 100 ng pcDNA3.1/rGR (wild-type or mutant), 200 ng pGRE.luc, and 300 ng of the appropriate FKBP51 construct or empty pcDNA3.1. For the FKBP51 titration, increments of 50, 100, 200, 400, and 800 ng pRK5MCS/FKBP51-FLAG were transfected. After another 24 hours, cells were treated with 50 nM cortisol (Sigma-Aldrich) or ethanol vehicle.
For each assay, 20 hours after hormone treatment, cells were washed twice in PBS and harvested in 100 μL luciferase lysis buffer (30 mM Tris-HCl [pH 7.8], 2 mM EDTA, 10% [vol/vol] glycerol, 0.08% [vol/vol] Triton X-100, with 2 mM dithiothreitol added before use). Lysates were put through 1 freeze/thaw cycle and the supernatant collected after centrifugation at 19 000 g for 10 minutes at 4°C. Luciferase assays were performed according to the manufacturer's instructions (luciferase assay system; Promega, Madison, Wisconsin). Readings were taken using a POLARstar OPTIMA microplate reader and standardized against sample protein quantitated by Bradford analysis.
Western analysis
Yeast protein extraction was performed as described by Riggs et al (41). For the mammalian cell lines, protein was extracted as for the reporter assay. Typically, for each sample, 50 to 100 μg of whole-cell protein was electrophoresed through a 10% SDS-PAGE gel and transferred overnight to a Hybond-C Super nitrocellulose membrane (GE Healthcare, Buckinghamshire, United Kingdom) at 30 V at 4°C. The membrane was incubated in blocking buffer (1× Tris-buffered saline/0.02% Tween 20 [TBS-T] [pH 7.6], 3% skim milk) for 1 hour at room temperature and then incubated with primary antibody diluted in blocking buffer for 2 hours at room temperature. The following dilutions of primary antibodies were used: GR (H-300; Santa Cruz Biotechnology, Santa Cruz, California) at 1/300, AR (C-19; Santa Cruz Biotechnology) at 1/300, and FKBP52 (Hi52c, kindly provided by David Smith) at 1/1000. As an internal loading control for yeast protein, the ribosomal subunit L3 was probed using anti-L3 (kindly provided by Jonathan Warner) (70) at 1/2000. For HeLa and MEF cell lines, α-tubulin was probed using anti-α-tubulin (Sigma-Aldrich) at 1/10 000. The membrane was washed with blocking buffer (3 times for 10 minutes) and then incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (goat antimouse HRP or goat antirabbit HRP; Sigma-Aldrich) diluted 1/10 000 in blocking buffer for 1 hour at room temperature. Finally, the membrane was washed (3 times for 10 minutes) in 1× TBS-T and after incubation with Western Lightning Enhanced Chemiluminescence Reagent (ECL) Plus (PerkinElmer, Waltham, Massachusetts) the membrane was exposed to Amersham Hyperfilm ECL (GE Healthcare).
Coimmunoprecipitation assays
Examination of the comparative binding of FKBP51 to wild-type and mutant rGR
HEK293 cells were propagated in DMEM containing 10% charcoal-stripped fetal bovine serum to a confluency of approximately 60% in 25-cm2 flasks and transfected with pRK5MCS/FKBP51-FLAG (5 μg) and/or pcDNA3.1/HA-rGR or pcDNA3.1/HA-rGR-IHS/TS (4 μg) using Lipofectamine 2000. After 5.5 hours incubation, transfection medium was replaced with DMEM containing 10% charcoal-stripped fetal calf serum and the cells incubated for 60 hours, after which time monolayers were washed twice in PBS and the cells lysed in modified lysis buffer A′ (38) in which the NaCl concentration was reduced to 50 mM (50 mM NaCl, 20 mM Na2MoO4, 1 mM EDTA, 20 mM Tris-HCl [pH 7.5], 10% glycerol, and 0.5% Triton X-100) and protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). For each immunoprecipitation reaction, lysate protein (0.75–1.5 mg, depending on recovery for different experiments) was incubated overnight at 4°C in 25 μL (actual bead volume) of anti-HA agarose conjugate (Sigma-Aldrich) prepared according to the manufacturer. The next day, beads were washed 5 times in 1 mL of the above lysis buffer (without Triton-X-100) and the protein eluted from the beads in 32 μL of 2× sample buffer (0.12 M Tris-HCl [pH 6.8], 3.3% wt/vol SDS, and 10% glycerol) by gentle mixing and incubation at room temperature for 10 minutes. Bead suspension was then centrifuged and 30 μL supernatant withdrawn, to which was added 3 μL 2-mercaptoethanol. Samples were heated in boiling water for 5 minutes before separation by 10% SDS-PAGE and overnight transfer to a nitrocellulose membrane as described under “Western analysis.” Lysate proteins for each immunoprecipitation reaction (40 μg) were also separated by 10% SDS-PAGE and transferred to separate membranes. For immunodetection, membranes were cut in two for detection of HA-GR and FKBP51-FLAG and were incubated for 1 to 2 hours in blocking solution (TBS containing 3% skim milk powder) and then incubated (in fresh blocking solution) with either 1/5000 M2 monoclonal anti-FLAG (Sigma-Aldrich) for 2 hours or 1/5000 monoclonal anti-HA-HRP (Sigma-Aldrich) for 2.5 hours. Membranes for FLAG detection were washed and further processed with goat antimouse-HRP as described under “Western analysis.” Membranes for HA detection were washed in TBS-T (6 times for 5 minutes). Detection of immunoreactive bands by ECL was as described under “Western analysis.” The experiment was repeated 3 times, and FKBP51-FLAG immunoreactive bands (both in the coimmunoprecipitate and lysate) and HA-rGR wild-type and mutant immunoprecipitated bands were quantitated by densitometry using Scion Image software (Scion Corporation, Frederick, Maryland), correcting for background in each case. FKBP51-FLAG coimmunoprecipitating bands were normalized to both input lysate FKBP51-FLAG and HA-rGR bound to the beads, and the normalized FKBP51 bound to mutant was expressed as a percentage of that bound to wild-type GR. The mean and SE of these percentages were then calculated.
Examination of the comparative recruitment of endogenous Hsp90 and p23 to wild-type and mutant rGR complexes
pcDNA3.1/HA-rGR or pcDNA3.1/HA-rGR-IHS/TS were transfected into 25-cm2 flasks (in triplicate) and protein recovered from lysed cells as described above in the preceding paragraph. Lysate protein from pooled samples (3.7–5 mg) was precleared for approximately 2 hours at 4°C by gentle mixing with ∼45 μL GammaBind G Sepharose beads (GE Healthcare). Samples were then centrifuged, and approximately 12 μL of rabbit anti-HA antibody (Sigma-Aldrich; ∼0.65 mg/mL) was added to the supernatant and gently mixed overnight at 4°C, after which time the mixture was added to fresh GammaBind G Sepharose beads (∼40 μL) and incubated with gentle mixing for 4 hours at 4°C. Beads were then washed 7 times in chilled lysis buffer without Triton X-100 and immunocomplexes eluted from the beads as described above except that 2-mercaptoethanol was added directly to the sample buffer before boiling. Immunoprecipitates and lysate proteins (50–100 μg) were separated by SDS-PAGE and blotted onto nitrocellulose as described above. Immunodetection was similar to that described above with membranes cut in two for detection of Hsp90 using 1/5000 monoclonal anti-Hsp90 antibody H90-10 (Abcam, Cambridge, United Kingdom; 2 mg/mL), and p23 using 1/1000 monoclonal anti-p23 antibody JJ3 (Abcam), both with 2-hour incubations. The Hsp90 membranes were later stripped and examined for HA-GR as described above. The experiment was repeated 3 times with densitometric analysis of Hsp90, p23, and HA-GR mutant and wild-type bands (including correction for background) performed as described above. The amount of Hsp90 and p23 bound to wild-type was expressed as a percentage of that bound to mutant GR. The mean and SE of these percentages were then calculated.
Statistical analysis
The descriptive statistics were calculated and the graphs plotted using Microsoft Excel software. Statistical significance was determined by ANOVA, with the Dunnett correction for multiple comparisons, using SPSS software (version 18) or, in the case of Hsp90 and p23 recruitment to GR, a 2-sample t test (assuming equal variances).
Results
Five amino acid substitutions within the unstructured loop between H1 and H3 of the gpGR LBD differentiate gpGR from the human receptor, contributing to the low binding affinity phenotype (23, 26, 71, 72). It has been predicted that these crucial residues (Ile538, His539, Ser540, Thr545, and Ser546), lying on the surface of the gpGR LBD, might disrupt a contact domain for FKBP52 and at the same time result in conformational changes that compromise high-affinity cortisol binding (26). The amino acid sequence within the H1-H3 loop is fully conserved between human GR (hGR) and rGR, as well as the receptors of several other species, with the affected region of hGR reading 544LYAGYDSSV552 in comparison with IHSGYDSTS in the gpGR (see Figure 1A). We therefore used the previously developed S. cerevisiae model for GR potentiation by FKBP52 (41), with rGR altered in the H1-H3 loop with the gpGR-specific changes, in part (i.e., either IHS or TS) or in full (IHS/TS), to examine their effect on receptor potentiation by FKBP52. S. cerevisiae reporter strains were generated by stable transformation with a lacZ reporter plasmid derived from a hormone-inducible promoter, an expression plasmid for FKBP52 and an expression plasmid for wild-type or mutant rGR. Cultures were titrated with increasing concentrations of DOC, which functions as a potent GR agonist in yeast (73, 74). As expected, in the absence of FKBP52, rGR containing the guinea pig receptor mutations, either individually or in combination, displayed a decreased hormonal response with right-shifted dose-response curves relative to wild-type receptor, with the rGR IHS/TS mutant being least responsive (Figure 1B). Coexpression of FKBP52 increased the activity of wild-type rGR, as previously reported (41), as well as the activities of all mutated receptors, with a leftward shift in hormone-dose responses (Figure 1C). Interestingly, the activity of the rGR IHS mutant was restored to that of the wild-type receptor in the presence of FKBP52, whereas only a partial restoration was achieved for the rGR TS and IHS/TS mutants in yeast expressing the cochaperone (Figure 1C). These results demonstrated that the FKBP52 cochaperone can potentiate the transcriptional activity of the mutated GRs, albeit with varied efficiencies, thus discounting an essential role for this region in receptor-FKBP52 interaction. However, the inability of the rGR TS and IHS/TS mutants to fully replicate wild-type receptor activity under the stimulatory influence of FKBP52, especially at low hormone concentrations, suggests that the TS substitution might have some bearing on spatial relationships within the GR-Hsp90-FKBP52 complex important for receptor function.
Figure 1.

Influence of the nonconservative mutations within the H1-H3 loop in gpGR on FKBP52-mediated potentiation of rGR. A, The GR LBD sequences from helix 1 to helix 3 of the rat (CAA72938), human (NP_001018087), monkey (ABM91989), canine (XP_535225.2), bovine (NP_001193563.1), mouse (NP_032199.3), and guinea pig (NP_001166458) were aligned using the ClustalW program. The amino acids constituting the 2 helices are boxed, and the nonconservative residues in the gpGR are highlighted in black. B and C, The normalized rGR reporter expression rate in the absence (B) and presence (C) of FKBP52. Yeast was transformed with a β-galactosidase reporter pUCΔSS-26x, an expression plasmid containing the wild-type (WT) or mutated rGR, and p423GPD/hFKBP52 expression plasmid or empty vector control. Aliquots of yeast culture were dosed with increasing concentrations of deoxycorticosterone (DOC). The reporter expression rate of WT and mutated rGR were normalized and presented as a function of the log of DOC concentration. Graphs represent the mean normalized expression rate ± SEM of 3 independent experiments. Inset, activity levels in the presence of 5 nM DOC. *, P < .05; **, P ≤ .001; for comparison of normalized reporter expression rate of mutant rGR with that of wild-type rGR. Note, the titration curve for wild-type receptor in the absence of FKBP52 has been included in C to illustrate the level of FKBP52 potentiation.
Despite contrary evidence for the requirement of FKBP52 PPIase activity for potentiation, at least in the case of AR (48), we still considered the solvent-exposed loops within the GR LBD, incorporating conserved proline residues, as potential sites for FKBP52-receptor interaction. Alignment of AR, GR, and PR LBD sequences shows that 5 intervening loops separating LBD helices contain proline residues conserved in all of the receptors (Figure 2A). These prolines, identified by GR codons 541 and 553 in loop H1-H3, 582 (loop H3-H4), 625 (loop H5-H6), and 676 (loop H8-H9), are located in accessible positions in the GR LBD structure and in those of the other receptors, which generally resemble one another (compare AR, GR, and PR in Figure 2B). GR Pro750 in loop H11-H12 is identified closely with corresponding proline residues in AR and PR (Figure 2A). All of these prolines (AR Pro892, GR Pro750, and PR Pro906) are situated in a similar exposed and accessible location within the H11-H12 loop as shown in Figure 2B.
Figure 2.
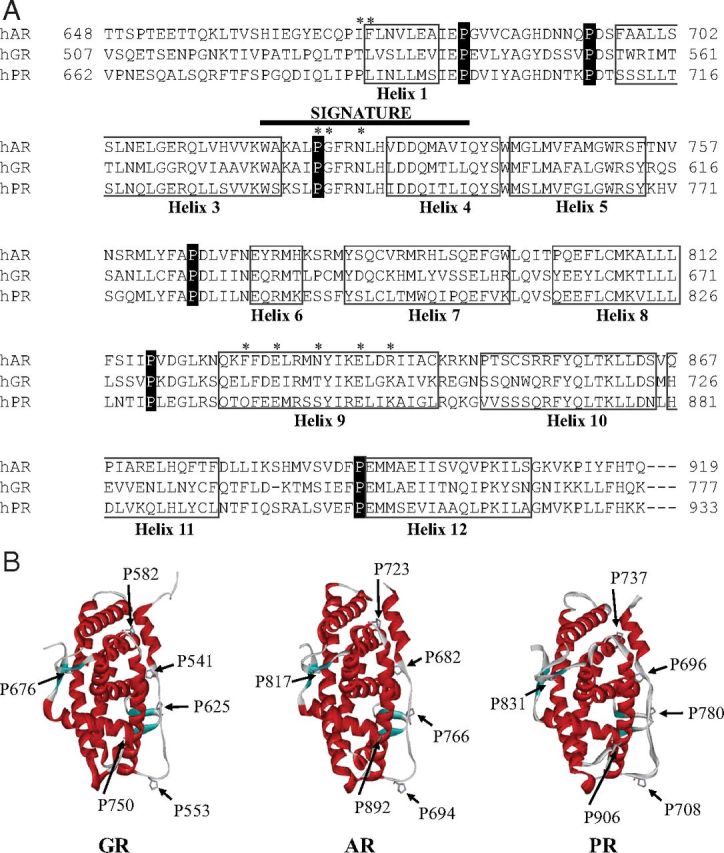
Alignment of steroid receptor LBDs to identify conserved proline residues. A, The LBD sequences of human AR (NP_000035), GR (NP_001018087), and PR (NP_000917) were aligned using the ClustalW program. The amino acids constituting the LBD helices are boxed, and the conserved proline residues are highlighted in black. The nuclear receptor signature sequence is indicated (thick black line). Residues that map to the BF-3 regulatory site defined for AR (50) are highlighted with an asterisk. B, Ribbon structures of the GR, AR, and PR were generated using the ViewerLite version 5.0 program and demonstrate the similar structural orientation of conserved proline residues (12, 56, 57). PDB ID codes are 3E7C, 1E3G, and 1A28, respectively.
We used the yeast-based assay to examine the interhelical loop prolines as candidate sites for GR-FKBP52 interaction. GR Pro582 was not included in the study because a serine-substituted mutant of the corresponding AR proline residue (AR P723S) was shown to display an enhanced activity similar to wild-type AR when coexpressed with FKBP52 in yeast (44). Remaining GR proline residues were substituted with alanine in an rGR background, and the mutated receptors were individually tested for potentiation by FKBP52. Yeast strains stably transformed with plasmids for proline-mutated rGR, a hormone-inducible β-galactosidase reporter gene, and FKBP52 were used in the assay. Our rationale was that a GR mutant demonstrating a basal response to hormone, but no further potentiation by FKBP52, would identify the corresponding loop as a possible target for FKBP52 interaction. In preliminary titration experiments with increasing DOC concentrations, we observed that all of the mutants had a markedly reduced basal activity compared with wild-type receptor and the assay DOC concentration was increased accordingly from 25 nM for wild-type rGR to 5 μM for the mutants. Figure 3, A–E, shows that the wild-type rGR and receptors with modified prolines at 541 (rGR P559A), 553 (rGR P571A), 625 (rGR P643A), and 676 (rGR P694A) were all potentiated by FKBP52. However, the receptor modified at Pro750 (rGR P768A) was found to be transcriptionally inactive in the absence and presence of FKBP52, even at high hormone concentrations, and the result was therefore uninformative (Figure 3F).
Figure 3.

FKBP52 potentiation of interhelical proline mutants within the GR LBD. A–F, Yeast were transformed with a β-galactosidase reporter pUCΔSS-26x, a receptor expression plasmid for either wild-type (wt) or mutated rGR and an FKBP52 expression plasmid or empty vector control. Aliquots of yeast culture were dosed with 25 nM (wt) or 5 μM (all mutants) of DOC. The reporter expression of each receptor in the presence or absence of FKBP52 is presented. Graphs are representative of 3 independent experiments. G, Levels of transformed protein were determined by running yeast protein extracts through a 10% SDS-PAGE gel and a Western analysis performed by transferring overnight to a nitrocellulose membrane and probing sequentially for GR (H-300 antibody), FKBP52 (Hi52c antibody), and the internal control L3 (α-L3 antibody). Vertical lines separate membranes derived from different gels. Abbreviation: RLU, relative light units.
Because H11-H12 loop residues in GR are involved in direct contact with ligand or reside close to amino acids crucial for interaction networks between ligand and receptor (12), mutations within this region may impact negatively on receptor hormone binding and transcriptional activity, resulting in low basal readout even in the presence of FKBP52. To further examine the H11-H12 loop as a possible contact domain for FKBP52, we turned our investigation to AR Pro892, a conserved proline within the equivalent region in AR (Figure 2A). A P892A substitution was previously identified by Trifiro and coworkers in individuals with complete androgen insensitivity syndrome (67). AR P892A displays significant, but reduced hormone-binding affinity, as well as a reduction by ∼80% in transcriptional activity, compared with wild-type AR. Because AR P892A retains some activity with respect to ligand-binding and transactivation properties, it was amenable for testing as a substrate for potentiation by FKBP52. HeLa cells treated with siRNA targeting FKBP52 or control siRNA were transfected with AR P892A plasmid, together with an androgen-responsive reporter gene, with or without an expression plasmid for silently mutated FKBP52. Figure 4 shows the mutant to have a reduced transcriptional activity in FKBP52 knockdown versus negative control siRNA-transfected cells treated with DHT and that this activity was dramatically increased with exogenously expressed FKBP52, thus confirming potentiation of the mutated receptor. Taken together, our results do not support a direct role for proline residues within the intervening loops between H1-H3, H5-H6, H8-H9, and H11-H12 in primary interactions with the FKBP52 cochaperone critical for increased receptor activity.
Figure 4.

FKBP52 enhances potentiation of the AR P892A mutant. A, The transcriptional activity of AR P892A in the presence or absence of overexpressed FKBP52 was determined with a luciferase reporter assay in the HeLa cell line. Cells were transfected with siRNA targeting FKBP52 or a negative control (Neg C) siRNA at 2 consecutive time points 24 hours apart. Cells were then seeded in triplicate into a 24-well plate, and 24 hours after the final siRNA transfection, cells were transfected with reporter plasmid pPB3.luc, receptor expression plasmid pcDNA3/AR P892A, and either an expression plasmid for FKBP52 or empty vector (EV). Cells were treated with 10 nM dihydrotestosterone (DHT) or ethanol (EtOH) vehicle for 20 hours before being harvested and the luciferase assay performed. Luciferase values were standardized against protein quantitated by Bradford analysis and plotted as a percentage of the maximum observed response to hormone. The graph represents the mean ± SEM of 3 independent experiments, *, P < .05; **, P < .001; for comparison of DHT-stimulated activity versus vehicle control in cells overexpressing FKBP52 in knockdown (KD) and control siRNA experiments, respectively. B, Protein extracts were run through a 10% SDS-PAGE gel and a western analysis performed by transferring overnight to a nitrocellulose membrane and probing for AR (C-19 antibody), FKBP52 (Hi52c antibody) and the internal control, α-tubulin (anti-α-tubulin antibody).
Although FKBP51 overexpression does not repress GR in yeast (41), the cochaperone has been shown to inhibit GR activity in mammalian cells (27, 28, 35, 38, 40). To provide further evidence that FKBP51 may attenuate gpGR responses to cortisol through the H1-H3 loop, we initially compared the hormone responsiveness of the wild-type and mutant rGR by determining the transcriptional activity of the receptors in FKBP51-KO MEFs exposed to cortisol concentrations ranging from 0 nM to 500 nM. Figure 5A shows that loss of FKBP51 maintains the dichotomy previously demonstrated for the activities of human and gpGRs (23), with rGR containing gpGR-specific mutations within the H1-H3 loop being much less responsive than wild-type receptor at low hormone levels. However, from a threshold level of approximately 50 nM cortisol, both wild-type and mutated rGR displayed similar activity with increasing hormone concentrations. The result suggests that, in the absence of FKBP51, wild-type rGR is more amenable than the mutated receptor to potentiation by endogenous FKBP52 (Figure 5A). We next used the FKBP51-KO MEFs to test the effect of FKBP51 overexpression on the transcriptional activity of wild-type rGR and receptor mutated within the loop, corresponding either partially (LYA562IHS or SV569TS) or fully (IHS/TS) to the gpGR H1-H3 loop sequence (see Figure 1A). Figure 5B shows that with 50 nM cortisol stimulation, FKBP51 overexpression did not repress wild-type rGR activity but significantly inhibited the responses of rGR containing the gpGR-specific SV569TS and IHS/TS mutations. The result complements our study of the influence of FKBP52 expression on rGR in yeast (Figure 1, B and C) and suggested that the H1-H3 loop gpGR-specific mutations may alter the interaction and perhaps the role of both FKBP cochaperones.
Figure 5.

GRs containing guinea pig H1-H3 loop mutations are more sensitive than wild-type (WT) receptor to repression by FKBP51 without an increased association with the cochaperone. A, MEF FKBP51-KO cells were transfected with a reporter plasmid pGRE.luc and a receptor expression plasmid for either rGR or mutated rGR. Cells were treated with increasing concentrations of cortisol or ethanol vehicle for 20 hours before being harvested. Luciferase values were standardized against protein and plotted as a percentage of the maximum observed response to hormone. Each graph represents the mean ± SEM of 3 independent experiments, *, P < .05 for comparison of transcriptional activity between wild-type and mutant rGR. B, MEF FKBP51-KO cells were transfected as above with the addition of pcDNA3.1/hFKBP51 expression plasmid or empty vector control. Cells were treated with 50 nM cortisol or ethanol vehicle for 20 hours before being harvested. Luciferase values were standardized against protein and plotted as the fold change of receptor activity in response to hormone treatment. *, P ≤ .002 for comparison of fold change in transcriptional activity for wild-type versus mutant rGR in cells overexpressing FKBP51. C, HEK293 cells were transfected with a reporter plasmid pGRE.luc, a receptor expression plasmid for either rGR or mutated rGR and pRK5MCS/FKBP51-FLAG expression plasmid or empty vector control. Cells were treated with 50 nM cortisol or ethanol vehicle for 20 hours before being harvested. *, P ≤ .035; **, P ≤ .001; for comparison of fold change in transcriptional activity for wild-type or mutant rGR in cells containing the empty vector control versus overexpression of FKBP51-FLAG. D, Representative blot of 3 independent experiments where HEK293 cells were transfected with plasmids expressing HA-rGR (wild-type or mutant [IHS/TS]) and either FKBP51-FLAG or empty vector control. After 60 hours incubation, the cells were lysed and protein extracts immunoprecipitated (IP) with anti-HA agarose conjugate. The comparative levels of FKBP51-FLAG that coimmunoprecipitated with wild-type and mutant HA-rGR, together with the levels of HA-rGR bound to the beads, were assessed by anti-FLAG and anti-HA Western blot analysis, respectively (upper panel). The relative levels of input protein in the lysates are shown in the lower panel.
The results of Figure 5B confirmed that FKBP51 add-back to FKBP51-deficient MEFs had a more profound inhibitory effect on the activity of mutated versus wild-type rGR. In another experiment, we used HEK293 cells treated with 50 nM cortisol to examine the effect of increasing exogenous FKBP51 expression levels on mutant and wild-type rGR activities. Although the relative basal responses for the 2 receptor forms, in the absence of exogenous FKBP51, differed between the 2 cell lines, likely owing to the different levels of endogenous FKBPs, Figure 5C reveals markedly different patterns of FKBP51-mediated suppression, with mutated rGR being much more sensitive to repression, the result thus reinforcing the data of Figure 5B.
To test whether gpGR recruits FKBP51 more efficiently through the H1-H3 loop, we performed coimmunoprecipitation experiments comparing the binding of FKBP51-FLAG to HA-tagged rGR or the rGR IHS/TS mutant. Figure 5D shows a representative blot of 3 independent experiments that, when normalized for immunoprecipitated HA-tagged receptors and for FKBP51-FLAG expression, all showed lower levels of FKBP51 bound to mutated rGR. The mean percentage of mutant versus wild-type rGR-bound FKBP51 was 65.3% ± 8.9%. In additional coimmunoprecipitation experiments, we examined the effect of 50 nM cortisol (administered to cell monolayers either 17 hours or 1 hour before cell lysis) on FKBP51 binding to wild-type and mutant HA-tagged rGR; however, neither hormone treatment appreciably changed the comparative binding of FKBP51 compared with the standard treatment without hormone (results not shown). Together the results presented in Figure 5 suggest that rather than enhancing FKBP51 interaction, the H1-H3 loop mutations may promote LBD conformational changes that not only confer a decrease in GR ligand-binding affinity (23, 26, 71) but also elicit consequences (such as a reduced association with FKBP52) that allow FKBP51 to profoundly inhibit receptor function.
Decreased expression of Hsp90 is associated with reduced responsiveness of GR to glucocorticoids (54, 75). The cochaperone p23 also serves as an essential component of mature GR-Hsp90 complexes where it binds directly to ATP-bound Hsp90, stabilizing GR-Hsp90 interaction and the ligand-activatable form of the receptor (76–80). We therefore performed additional coimmunoprecipitation reactions, comparing the recruitment of endogenous Hsp90 and p23 to wild-type and mutant HA-tagged GR constructs transfected into HEK293 cells. Surprisingly, there was a trend toward better recruitment of Hsp90 to mutant GR, whereas p23 binding was more variable (results not shown). The mean percentage of wild-type versus mutant Hsp90 binding was 73.4% ± 9.8%, whereas that for p23 was 96.9% ± 26.1% with neither significantly different at the P < .05 level.
Although Rein and coworkers (40) previously negated a role for FKBP51 PPIase function in the repression of GR signaling, it was possible that an altered LBD conformation induced by the gpGR mutations within the H1-H3 loop might expose LBD proline residues susceptible to FKBP51 catalytic activity. To examine this further, we used a reporter assay in HEK293 cells with the enzymatically deficient FKBP51 F130A mutant, characterized by Dickey and coworkers (66). This mutated residue is located within the PPIase pocket (32) and contrasts with the previously used FKBP51 F67D/D68V mutant (40) in which the modified residues, positioned near the entrance to the catalytic site, may potentially impact PPIase domain conformation and affect productive protein-protein interactions (48). Nevertheless, although there was some reduction in the level of wild-type rGR repression by FKBP51 F130A in comparison with the native immunophilin, significant inhibition of receptor activity was still observed, thus confirming that FKBP51 PPIase function is dispensable for GR suppression (Figure 6A). Similarly, there was no difference between the inhibitory capabilities of wild-type or mutant FKBP51 with rGR modified within the H1-H3 loop to the gpGR loop sequence (Figure 6A). Our results therefore confirm that FKBP51 prolyl isomerase activity is not essential for inhibition of GR signaling, generally.
Figure 6.

The enhanced FKBP51 repression of mutant rGR activity is not abrogated by mutation of FKBP51 PPIase function or by the activating Leu119Pro mutation. A, HEK293 cells were transfected with a reporter plasmid pGRE.luc, expression plasmids for either rGR or mutated rGR and either FKBP51-FLAG, FKBP51 F130A-FLAG expression plasmid, or empty vector control. Cells were treated with 50 nM cortisol or ethanol vehicle for 20 hours before being harvested. Luciferase values were standardized against protein and plotted as the fold change of receptor activity in response to hormone treatment. Each graph represents the mean ± SEM of 3 independent experiments. *, P = .004; **, P ≤ .0001; for comparison of fold change in transcriptional activity for wild-type (WT) or mutant rGR in cells containing the empty vector control versus overexpression of FKBP51-FLAG or FKBP51 F130A-FLAG. B, HEK293 cells were transfected as above, but with expression plasmids for either wild-type FKBP51, FKBP51 L119P mutant, or empty vector control. Cells were treated with 50 nM cortisol as above. **, P ≤ .001 for comparison of fold change in transcriptional activity for wild-type or mutant rGR in cells containing the empty vector control versus overexpression of FKBP51 or FKBP51 L119P.
The proline-rich loop within the FKBP52 PPIase domain (sequence 116AGS119PPKIPP124) is largely responsible for the functional difference between FKBP52 and FKBP51 relating to GR potentiation and repression of hormone binding, respectively (48). The corresponding FKBP51 sequence (116AGS119LPKIPS124) differs at residues 119 and 124. Proline substitution for Leu119 in FKBP51 converts the immunophilin to an intermediate potentiator of GR transcriptional activity, partially mimicking the role of FKBP52 (48) and strongly intimating that the respective proline-containing loops within the FK1 domains of FKBP51 and FKBP52 mediate the counterinfluences of these immunophilins on GR activity (17, 48). We used the FKBP51 L119P mutant in a HEK293 cell reporter assay to determine whether mutation of the key Leu119 residue affects FKBP51-mediated repression of GR. Figure 6B shows that this limited disruption of the FKBP51 proline-containing loop had no influence on FKBP51 suppression of activities for either wild-type rGR or receptor harboring gpGR-specific mutations within the H1-H3 loop.
Discussion
Glucocorticoid resistance may arise from disruptions in glucocorticoid signaling that normally target GR. As seen in humans and the guinea pig, mutations within the GR LBD causing a reduced cortisol-binding affinity illustrate one mechanism contributing to glucocorticoid resistance (20, 21, 23, 26). In New World primates, elevated FKBP51 expression resulting in GR-Hsp90 heterocomplexes containing a receptor with decreased hormone sensitivity provides another mechanistic explanation (25, 27–29). Furthermore, GR upregulation of FKBP51 expression in glucocorticoid target tissues provides an inhibitory feedback mechanism for decreasing glucocorticoid sensitivity (81, 82). This has been recently highlighted in humans, with increasing evidence of resistance to glucocorticoid therapies associated with high-level FKBP51 expression (83–85). In contrast to FKBP51, FKBP52 selectively enhances GR ligand-binding affinity and hormone-induced transcriptional activity, although FKBP51 can attenuate this FKBP52-mediated potentiation (35, 40, 41). At the cellular level, GR activity is therefore dependent on the relative expression levels of the 2 closely related FKBP cochaperones and their ability to compete for assembly into GR-Hsp90 complexes (17, 62, 86). Because FKBP52 potentiates GR through specific contacts with the LBD (17, 41, 48, 49, 62) and the differential sensitivity to cortisol between human and gpGR is conferred by the H1-H3 loop (23, 26), the unstructured loop was proposed as a candidate interaction site for FKBP52 (and/or FKBP51) (26). This study therefore focused on the mechanisms of decreased glucocorticoid sensitivity conferred by gpGR H1-H3 loop sequences within the receptor LBD. Here we show that by substituting the gpGR H1-H3 residues known to account for decreased hormone-binding affinity into the H1-H3 loop of rGR, a region fully conserved in hGR, the modified rGR (with sequence altered from LYAGYDSSV to IHSGYDSTS, see Figure 1A) was still potentiated in the presence of FKBP52, albeit less efficiently than wild-type rGR, allowing us to conclude that the H1-H3 loop is not a primary contact site for FKBP52. However, our observation that the rGR TS mutant failed to attain full wild-type receptor activity in the presence of FKBP52 suggests that this region of the loop, which resides closer to H3 of the receptor, might contribute to an interaction with FKBP52 not through active binding per se, but instead a spatial passive steric mode in the context of the entire chaperone complex. Several H3 residues comprise the GR ligand-binding pocket, accommodating the ligand through extensive hydrophobic and hydrophilic interactions (12). Additionally, it is worth noting that H1 has previously been implicated in interactions with Hsp90 (61), together with a region encompassing residues 626 to 653, involving H6 and H7, identified as a critical contact site (87). Another segment contributing to Hsp90 interaction is predicted between residues 568 to 626, which includes part of H3, the H3-H4 loop, H4 and H5, and much of the H5-H6 loop (87). This region also contains the highly conserved nuclear receptor signature motif (Figure 2A) as well as residues Y598, F602, and M604 recently identified to form part of an allosteric network governing GR dependence on Hsp90 and that help to alter receptor conformation in response to ligand binding, resulting in the formation of the AF-2 interaction site for transcriptional coregulators (88). Importantly, the M604 residue itself is directly involved in ligand binding (12, 89). It is of interest that MR, which displays high sequence identity with GR (90), contains a surface-exposed region, corresponding to the GR H5-H6 loop, which confers steroid-binding specificity, without directly contacting ligand (26, 91, 92). This region has similarly been implicated in ligand-binding specificity for the AR and PR (92). Hsp90 interaction with this region may impart distal conformational effects important for the shape of the ligand-binding pocket with consequences for ligand specificity (26, 93).
Our study also assessed the interhelical loops within the GR LBD as candidate sites for interaction with the FKBP52 cochaperone. Because AR, GR, and PR are each potentiated by FKBP52, most likely through a similar mechanism, we used the sequence alignments of the LBDs for these receptors to identify conserved residues within solvent-exposed, intervening loops between α-helices that might be targeted for mutation in functional studies. In our strategy, we focused on conserved prolines, substituting each with alanine within the rGR LBD, apart from rGR Pro600 (hGR Pro582), because the corresponding proline residue in AR, mutated to serine (i.e., AR P723S) had been shown by Smith and coworkers (44) to display enhanced activity in the presence of FKBP52. We found the transactivation properties of each of the mutants to be impaired compared with wild-type receptor, most likely owing to detrimental local or distal effects on residues directly involved in ligand binding (12, 93). For example, the H11-H12 loop GR P750A (rGR P768A) mutant, which was functionally inert, both in the presence and absence of FKBP52, is located close to LBD residues involved in direct contact with ligand or reside close to amino acids crucial for interaction networks between ligand and receptor (12). GR Pro750 is also functionally important for the structural integrity, mobility, and orientation of H12 (67, 94, 95), the molecular switch fundamental to recruitment of transcriptional coregulators in steroid receptor-mediated hormone action (96). GR Pro625, in the H5-H6 loop, is a key residue in the formation of a novel dimer interface in hormonally activated receptor (12) and appears central to the stability of GR-Hsp90 heterocomplexes (97), suggesting an overlap between the dimerization interface and an important contact domain for Hsp90 interaction. Additionally, the GR P625A (rGR P643A) mutant was shown previously to be transcriptionally impaired owing to a nuclear translocation defect, linking aberrant signaling to deficits in Hsp90 interaction (97). The GR P541A and P553A mutations, within the H1-H3 loop, are located either side of residues 547GYDSS551, which stabilize GR dimerization through an extensive hydrogen bond network between receptor LBDs (12). Pro541 mediates hydrophobic interactions within the receptor LBD, and P541A, a naturally occurring mutation, may therefore structurally destabilize this domain (12). With the exception of the uninformative GR P750A (rGR P768A) mutant in the H11-H12 loop, our functional analysis showed clearly that all of the GR proline mutants displayed FKBP52-mediated enhancement of transcriptional activity in yeast. Functional examination of the equivalent H11-H12 loop mutation in AR (AR P892A) confirmed potentiation of the mutated receptor with FKBP52 in a mammalian reporter assay. Our study, then, has excluded a role for proline residues within the intervening loops between H1-H3, H5-H6, H8-H9, and H11-H12 in direct interactions with FKBP52 associated with enhanced receptor activity. The H3-H4 loop, which was not examined in our study, contains residues that are located within the BF-3 regulatory surface recently identified in AR and proposed as the interaction site mediating FKBP52 potentiation of that receptor (49, 50). These AR residues, Pro723, Gly724, and Asn727, are conserved in GR and PR, whose activities are both enhanced by FKBP52. Cox and coworkers (49) have identified a small-molecule inhibitor of FKBP52-enhanced AR function that restricts the AR-Hsp90-FKBP52 complex to a cytoplasmic location at low hormone concentrations, suggesting that the inhibitor may interfere with a hormone-induced, FKBP52-dependent transitory change in AR conformation necessary for efficient nuclear translocation.
The processes of GR function in mammals are partly defined in terms of hormone-binding affinity and nuclear translocation, both being dependent on the competing influences of the FKBP51 and FKBP52 cochaperones in GR-Hsp90 heterocomplexes, with FKBP52 upregulating hormone-binding function and increasing GR targeting to the nucleus and FKBP51 favoring a lower hormone-binding affinity and locking the receptor in the cytoplasm, precluding nuclear transport (17, 26, 31, 35, 40, 41, 60–63, 98). In a model proposed for the regulation of receptor activity by the FK506-binding immunophilins, the functional differences between the two cochaperones have been attributed to differences in their FK1 domains and specifically the proline-rich loop. Incorporation of FKBP52 into GR-Hsp90 complexes promotes productive contacts of FK1 with the receptor LBD, stabilizing a high-affinity hormone-binding receptor conformation, whereas assembly of FKBP51 into receptor complexes results in a repressed hormone-binding conformation (62). The mechanism by which FKBP51 inhibits GR hormone-binding affinity is not well understood. A dose-response study we performed in FKBP51-deficient MEFs, with increasing cortisol concentrations, preserved the dichotomy for rGR containing the H1-H3 loop gpGR mutations against wild-type rGR, with a markedly reduced transcriptional activity being evident for the mutant receptor at low hormone levels and responses equivalent to those seen for wild-type receptor being observed with ≥50 nM cortisol (Figure 5A). The study also suggested that the remaining endogenous FKBP52 had a reduced ability to potentiate the activity of receptor containing the gpGR H1-H3 loop mutations, consistent with data revealed by our yeast assays (Figure 1, B and C). In a key experiment, examination of the direct influence of H1-H3 loop gpGR mutations on rGR responsiveness to 50 nM cortisol in FKBP51-deficient MEFs, with and without exogenous FKBP51 expression, provided compelling evidence that the loop changes increase the potency of FKBP51-mediated repression on GR function (Figure 5B). Titration of HEK293 cells exposed to 50 nM cortisol with increasing levels of FKBP51 revealed a more dramatic repression of mutant versus wild-type rGR (Figure 5C), reinforcing the results of Figure 5B and implying an altered interaction of mutated receptor with this immunophilin. Contrary to our hypothesis (26), coimmunoprecipitation experiments revealed that, compared with wild-type receptor, the loop mutations in rGR did not increase recruitment of FKBP51 (Figure 5D), even in the presence of 50 nM cortisol. Thus, rather than enhancing GR-FKBP51 interaction, the gpGR H1-H3 loop mutations promote changes in LBD conformation that result in decreased GR ligand-binding affinity and more efficient repression by FKBP51. Although, in comparison with FKBP52, FKBP51 slows the rate of nuclear translocation of hormone-activated GR (40), we considered that the physiological cortisol concentration (50 nM) used to generate the results of Figure 5, B and C, would minimally contribute to the observed inhibitory action of FKBP51 on GR signaling, when compared with the impact on receptor hormone binding (27). The mechanism, therefore, likely involves changes in the nature of the whole GR-Hsp90 complex that affect the ability of FKBP52 to potentiate GR function as well as its incorporation into the complex in the presence of other competing Hsp90-binding TPR cochaperones. Additional coimmunoprecipitation studies discounted the possibility that the mutated and wild-type receptors differ in their recruitment of Hsp90 and/or the p23 cochaperone, with resulting changes in the stability of GR complexes.
We extended our investigation of the mechanism of FKBP51-enhanced repression of rGR with gpGR-specific H1-H3 loop mutations over that of wild-type rGR to determine the role of functional regions within the FKBP51 FK1 domain mediating PPIase activity and a proline-rich loop thought to make detrimental contacts with receptor (48). Using an alternate PPIase mutant, FKBP51 F130A, we have provided direct evidence that FKBP51 PPIase function is not involved in the inhibitory activity of this immunophilin on either wild-type or mutant rGR, confirming the results of earlier studies with hGR (27, 40). Smith and coworkers (48) have previously demonstrated that proline substitution of Leu119 within the proline-rich loop of FKBP51 afforded the FKBP51 L119P mutant with FKBP52-like potentiating activities intermediate between FKBP51 and FKBP52 for GR in the yeast model. Overexpression of this mutant in HEK293 cells was shown not to diminish the level of FKBP51 repression, either of rGR with gpGR-specific H1-H3 loop mutations or that of wild-type rGR (Figure 6B), suggesting that, under our experimental conditions, FKBP51 L119P does not interfere sufficiently to disrupt the inhibitory actions of FKBP51. The functional differences between the 2 FKBPs may not just be limited to their FK1 domains, because sequences in the FK2 domains also contribute to the stimulatory and inhibitory properties of FKBP52 and FKBP51, respectively, suggesting the possibility of secondary contacts with the receptor LBD (27, 48).
Within the Hsp90 C-terminal domain, 2 flexible, solvent-exposed amphipathic helices have been identified with potential to serve as substrate-binding sites for this molecular chaperone (78, 99). Hsp90 helix 1 shares homology with the LXXLL recognition motif of transcriptional coactivators that interact with the agonist-induced AF-2 site of nuclear receptors (96) and Hsp90 helix 2 displays sequence similarity with the C-terminal H12 of steroid receptors (100). These helices are proposed to sequentially mimic interactions of the flexible receptor H12 within the GR LBD during hormone-induced conformational changes from apo-GR to hormone-bound GR (holo-GR) complexes (64, 99). Thus in apo-GR, Hsp90 helix 2 stabilizes unliganded GR by engaging the receptor in the position normally occupied by H12 in response to hormonal activation. This relocates H12 to a hydrophobic groove in the GR LBD, preventing transcriptional coregulator interaction (64). It is thought that structurally this corresponds to the native conformation of unliganded GR, with H12 oriented as in antagonist-bound receptor (64, 89). Agonist binding translates structural changes induced by hormone, from the ligand-binding pocket to the solvent-exposed hydrophobic groove, favoring a relocation of H12 and formation of the AF-2 contact domain for coactivator interaction. These changes in holo-GR LBD conformation are transmitted allosterically through residues that also contribute to Hsp90-dependent GR stabilization (64, 88) and might facilitate Hsp90 helix 1 binding to the hydrophobic groove as an alternative to recruitment of coactivators (64). It is conceivable that the hormone-induced interchange of FKBP51 by FKBP52 in GR-Hsp90 complexes, favoring high-affinity hormone binding and nuclear translocation of receptor complexes (60), results from changes in GR LBD conformation associated with the transfer of receptor-client protein interaction from Hsp90 helix 2 to helix 1 (64). The transition is likely to be regulated through stabilizing interactions between distinct exposed surfaces on the receptor LBD and contact sites on Hsp90 as well as FKBP51 and FKBP52. Our results suggest that the H1-H3 loop might provide one such modulatory surface that can dramatically affect GR regulation by the FKBP cochaperones. The H1-H3 loop may impact allosterically on proximally located LBD residues that contribute to GR Hsp90-dependent stability, nuclear translocation, and hormone-induced conformational changes, features that are linked to both FKBP51-mediated inhibition and FKBP52 potentiation of GR. Because, like GR, the activities of AR and PR are similarly subject to FKBP51- and FKBP52-mediated regulation, the corresponding H1-H3 loop in these receptors might also be relevant in the control of androgen and progestin signaling. Although the x-ray crystal structure of the apo-GR LBD is not yet available, the relative locations of GR LBD surfaces and specific residues, including the H1-H3 loop, that may contribute to or influence receptor interactions with Hsp90 and the FKBP cochaperones are illustrated in Figure 7, with the LBD structure characterized for the dexamethasone-liganded GR LBD (89).
Figure 7.

Relative location of the GR H1-H3 loop to AF-2 and BF-3 surfaces and selected residues important for GR function. A, Schematic of GR LBD showing location of dexamethasone ligand (blue), H1, and AF-2 helices H3, H4, and H12. B, Space-filling model showing GR H1-H3 loop residues (red); AF-2 (H3 and H4 residues, blue; H12 residues, light blue); BF-3 regulatory surface (including conserved, partially conserved, and retained polar and hydrophobic residues identified for AR BF-3 highlighted in Figure 2A, orange); Pro625, important for GR-Hsp90 stability and nuclear localization (green); and Met604, involved in GR dependence on Hsp90 and in transmitting hormone-induced conformational changes to the GR LBD surface (yellow). Structural data were obtained from the RCSB (Research Collaboratory for Structural Bioinformatics, www.pdb.org) protein data bank (PDB ID 1P93) (89). Images were generated using the ViewerLite version 5.0 program.
In conclusion, within GR-Hsp90 complexes, the GR H1-H3 loop acts as a regulatory surface that mediates the transmission of differential folding signals from Hsp90-associated FKBP51 and FKBP52 cochaperones to the unliganded receptor resulting in LBD conformational changes that determine both ligand-binding specificity and affinity and impact transcriptional activity by regulating nuclear translocation. Rather than through direct interaction with the FKBP cochaperones, the mechanism likely involves changes in the spatial orientation of specific, critical residues via allosteric networks.
Acknowledgments
We are grateful to David Smith for supplying pG/N795, p423GPD/hFKBP52, and pUCΔSS-26x plasmids, S. cerevisiae yeast strain W303a, the MEF cell line from the FKBP51-KO mouse strain, and Hi52c antibody; to Theo Rein for pRK5MCS/FKBP51-FLAG; to Chad Dickey for pCMV6/FKBP51-FLAG and pCMV6/FKBP51 F130A-FLAG; to Jonathan Warner for L3 antibody; to John Price for pIRESpuro3/FKBP52 plasmid; to Mark Trifiro for pcDNA3/AR P892A plasmid; and to Robert Matusik for pPB3.luc plasmid. We also thank Suzanne Brown for her assistance with the statistical analyses.
This work was supported by grants from the National Breast Cancer Foundation, the Cancer Council of Western Australia, the Sir Charles Gairdner Hospital Research Fund, and the National Health and Medical Research Council of Australia through a Project Grant (ID 458661) to T.R. and a Senior Principal Research Fellowship (ID 1002559) to P.J.F. Prince Henry's Institute is supported by the Victorian Government's Operational Infrastructure Support program.
Disclosure Summary: None of the authors have anything to disclose.
Footnotes
- AF-1
- activation function-1
- AF-2
- activation function-2
- AR
- androgen receptor
- BF-3
- binding function-3
- CyP40
- 40-kDa cyclophilin
- DHT
- dihydrotestosterone
- DOC
- deoxycorticosterone
- ECL
- enhanced chemiluminescence
- ERα
- estrogen receptor-α
- FKBP
- FK506-binding protein
- gpGR
- guinea pig GR
- GR
- glucocorticoid receptor
- HA
- hemagglutinin
- hGR
- human GR
- Hsp90
- heat-shock protein 90
- HRP
- horseradish peroxidase
- KO
- knockout
- LBD
- ligand-binding domain
- MEF
- mouse embryo fibroblast
- MR
- mineralocorticoid receptor
- PP5
- serine/threonine protein phosphatase type 5
- PPIase
- peptidylprolyl isomerase
- PR
- progesterone receptor
- rGR
- rat GR
- TPR
- tetratricopeptide repeat
- siRNA
- small interfering RNA
- TBS-T
- Tris-buffered saline/0.02% Tween 20.
References
- 1. Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fuller P. The steroid receptor superfamily: mechanisms of diversity. FASEB J. 1991;5:3092–3099. [DOI] [PubMed] [Google Scholar]
- 3. Mangelsdorf DJ , Thummel C , Beato M , et al. . The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cole TJ , Blendy JA , Monaghan AP , et al. . Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9:1608–1621. [DOI] [PubMed] [Google Scholar]
- 5. de Kloet ER , Joëls M , Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. [DOI] [PubMed] [Google Scholar]
- 6. Funder JW , Sheppard K. Adrenocortical steroids and the brain. Annu Rev Physiol. 1987;49:397–411. [DOI] [PubMed] [Google Scholar]
- 7. Heitzer MD , Wolf IM , Sanchez ER , Witchel SF , DeFranco DB. Glucocorticoid receptor physiology. Rev Endocr Metab Disord. 2007;8:321–330. [DOI] [PubMed] [Google Scholar]
- 8. McKay LI , Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-κB and steroid receptor-signaling pathways. Endocr Rev. 1999;20:435–459. [DOI] [PubMed] [Google Scholar]
- 9. Peeke PM , Chrousos GP. Hypercortisolism and obesity. Ann N Y Acad Sci. 1995;771:665–676. [DOI] [PubMed] [Google Scholar]
- 10. Reichardt HM , Kaestner KH , Tuckermann J , et al. . DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. [DOI] [PubMed] [Google Scholar]
- 11. Vegiopoulos A , Herzig S. Glucocorticoids, metabolism and metabolic diseases. Mol Cell Biol. 2007;275:43–61. [DOI] [PubMed] [Google Scholar]
- 12. Bledsoe RK , Montana VG , Stanley TB , et al. . Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. [DOI] [PubMed] [Google Scholar]
- 13. Savory JG , Préfontaine GG , Lamprecht C , et al. . Glucocorticoid receptor homodimers and glucocorticoid-mineralocorticoid receptor heterodimers form in the cytoplasm through alternative dimerization interfaces. Mol Cell Biol. 2001;21:781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pratt WB , Toft DO. Regulation of signaling protein function and trafficking by the Hsp90/Hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111–133. [DOI] [PubMed] [Google Scholar]
- 15. Ratajczak T , Ward BK , Minchin RF. Immunophilin chaperones in steroid receptor signalling. Curr Top Med Chem. 2003;3:1348–1357. [DOI] [PubMed] [Google Scholar]
- 16. Riggs DL , Cox MB , Cheung-Flynn J , Prapapanich V , Carrigan PE , Smith DF. Functional specificity of co-chaperone interactions with Hsp90 client proteins. Crit Rev Biochem Mol Biol. 2004;39:279–295. [DOI] [PubMed] [Google Scholar]
- 17. Smith DF , Toft DO. The intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol. 2008;22:2229–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beato M , Herrlich P , Schütz[b] G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–857. [DOI] [PubMed] [Google Scholar]
- 19. Karin M. New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell. 1998;93:487–490. [DOI] [PubMed] [Google Scholar]
- 20. Bamberger CM , Schulte HM , Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245–261. [DOI] [PubMed] [Google Scholar]
- 21. Kino T , Vottero A , Charmandari E , Chrousos GP. Familial/sporadic glucocorticoid resistance syndrome and hypertension. Ann N Y Acad Sci. 2002;970:101–111. [DOI] [PubMed] [Google Scholar]
- 22. Chrousos GP , Renquist D , Brandon D , et al. . Glucocorticoid hormone resistance during primate evolution: receptor-mediated mechanisms. Proc Natl Acad Sci U S A. 1982;79:2036–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keightley MC , Curtis AJ , Chu S , Fuller PJ. Structural determinants of cortisol resistance in the guinea pig glucocorticoid receptor. Endocrinology. 1998;139:2479–2485. [DOI] [PubMed] [Google Scholar]
- 24. Keightley MC , Fuller PJ. Anomalies in the endocrine axes of the guinea pig: relevance to human physiology and disease. Endocr Rev. 1996;17:30–44. [DOI] [PubMed] [Google Scholar]
- 25. Scammell JG , Denny WB , Valentine DL , Smith DF. Overexpression of the FK506-binding immunophilin FKBP51 is the common cause of glucocorticoid resistance in three New World primates. Gen Comp Endocrinol. 2001;124:152–165. [DOI] [PubMed] [Google Scholar]
- 26. Fuller PJ , Smith BJ , Rogerson FM. Cortisol resistance in the New World revisited. Trends Endocrinol Metab. 2004;15:296–299. [DOI] [PubMed] [Google Scholar]
- 27. Denny WB , Prapapanich V , Smith DF , Scammell JG. Structure-function analysis of Squirrel Monkey FK506-binding protein 51, a potent inhibitor of glucocorticoid receptor activity. Endocrinology. 2005;146:3194–3201. [DOI] [PubMed] [Google Scholar]
- 28. Denny WB , Valentine DL , Reynolds PD , Smith DF , Scammell JG. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology. 2000;141:4107–4113. [DOI] [PubMed] [Google Scholar]
- 29. Reynolds PD , Ruan Y , Smith DF , Scammell JG. Glucocorticoid resistance in the squirrel monkey is associated with overexpression of the immunophilin FKBP51. J Clin Endocrinol Metab. 1999;84:663–669. [DOI] [PubMed] [Google Scholar]
- 30. Galat A. Peptidylprolyl cis/trans isomerases (immunophilins): biological diversity–targets–functions. Curr Top Med Chem. 2003;3:1315–1347. [DOI] [PubMed] [Google Scholar]
- 31. Ratajczak T , Cluning C , Ward BK. Steroid receptor contact domains for functional interactions with Hsp90 and its cochaperones. Curr Top Steroid Res. 2009;6:21–31. [Google Scholar]
- 32. Sinars CR , Cheung-Flynn J , Rimerman RA , Scammell JG , Smith DF , Clardy J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc Natl Acad Sci U S A. 2003;100:868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu B , Li P , Liu Y , et al. . 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc Natl Acad Sci U S A. 2004;101:8348–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barent RL , Nair SC , Carr DC , et al. . Analysis of FKBP51/FKBP52 chimeras and mutants for Hsp90 binding and association with progesterone receptor complexes. Mol Endocrinol. 1998;12:342–354. [DOI] [PubMed] [Google Scholar]
- 35. Davies TH , Ning YM , Sánchez ER. Differential control of glucocorticoid receptor hormone-binding function by tetratricopeptide repeat (TPR) proteins and the immunosuppressive ligand FK506. Biochemistry. 2005;44:2030–2038. [DOI] [PubMed] [Google Scholar]
- 36. Nair SC , Rimerman RA , Toran EJ , et al. . Molecular cloning of human FKBP51 and comparisons of immunophilin interactions with Hsp90 and progesterone receptor. Mol Cell Biol. 1997;17:594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ratajczak T , Carrello A , Mark PJ , et al. . The cyclophilin component of the unactivated estrogen receptor contains a tetratricopeptide repeat domain and shares identity with p59 (FKBP59). J Biol Chem. 1993;268:13187–13192. [PubMed] [Google Scholar]
- 38. Schülke JP , Wochnik GM , Lang-Rollin I , et al. . Differential impact of tetratricopeptide repeat proteins on the steroid hormone receptors. PLoS ONE. 2010;5:e11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Silverstein AM , Galigniana MD , Chen MS , Owens-Grillo JK , Chinkers M , Pratt WB. Protein phosphatase 5 is a major component of glucocorticoid receptor·Hsp90 complexes with properties of an FK506-binding immunophilin. J Biol Chem. 1997;272:16224–16230. [DOI] [PubMed] [Google Scholar]
- 40. Wochnik GM , Rüegg J , Abel GA , Schmidt U , Holsboer F , Rein T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem. 2005;280:4609–4616. [DOI] [PubMed] [Google Scholar]
- 41. Riggs DL , Roberts PJ , Chirillo SC , et al. . The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003;22:1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Warrier M , Hinds TD , Ledford KJ , et al. . Susceptibility to diet-induced hepatic steatosis and glucocorticoid resistance in FK506-binding protein 52-deficient mice. Endocrinology. 2010;151:3225–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen H , Yong W , Hinds TD , et al. . Fkbp52 regulates androgen receptor transactivation activity and male urethra morphogenesis. J Biol Chem. 2010;285:27776–27784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheung-Flynn J , Prapapanich V , Cox MB , Riggs DL , Suarez-Quian C , Smith DF. Physiological role for the cochaperone FKBP52 in androgen receptor signaling. Mol Endocrinol. 2005;19:1654–1666. [DOI] [PubMed] [Google Scholar]
- 45. Yong W , Yang Z , Periyasamy S , et al. . Essential role for co-chaperone FKBP52 but not FKBP51 in androgen receptor-mediated signaling and physiology. J Biol Chem. 2007;282:5026–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tranguch S , Cheung-Flynn J , Daikoku T , et al. . Cochaperone immunophilin FKBP52 is critical to uterine receptivity for embryo implantation. Proc Natl Acad Sci U S A. 2005;102:14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang Z , Wolf IM , Chen H , et al. . FK506-binding protein 52 is essential to uterine reproductive physiology controlled by the progesterone receptor A isoform. Mol Endocrinol. 2006;20:2682–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riggs DL , Cox MB , Tardif HL , Hessling M , Buchner J , Smith DF. Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol Cell Biol. 2007;27:8658–8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Leon JT , Iwai A , Feau C , et al. . Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc Natl Acad Sci U S A. 2011;108:11878–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Estébanez-Perpiñá E , Arnold LA , Nguyen P , et al. . A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104:16074–16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alnemri ES , Litwack G. The steroid binding domain influences intracellular solubility of the baculovirus overexpressed glucocorticoid and mineralocorticoid receptors. Biochemistry. 1993;32:5387–5393. [DOI] [PubMed] [Google Scholar]
- 52. Nemoto T , Ohara-Nemoto Y , Sato N , Ota M. Dual roles of 90-kDa heat shock protein in the function of the mineralocorticoid receptor. J Biochem. 1993;113:769–775. [PubMed] [Google Scholar]
- 53. Aumais JP , Lee HS , Lin R , White JH. Selective interaction of Hsp90 with an estrogen receptor ligand-binding domain containing a point mutation. J Biol Chem. 1997;272:12229–12235. [DOI] [PubMed] [Google Scholar]
- 54. Picard D , Khursheed B , Garabedian MJ , Fortin MG , Lindquist S , Yamamoto KR. Reduced levels of Hsp90 compromise steroid receptor action in vivo. Nature. 1990;348:166–168. [DOI] [PubMed] [Google Scholar]
- 55. Picard D. Chaperoning steroid hormone action. Trends Endocrinol Metab. 2006;17:229–235. [DOI] [PubMed] [Google Scholar]
- 56. Matias PM , Donner P , Coelho R , et al. . Structural evidence for ligand specificity in the binding domain of the human androgen receptor: implications for pathogenic gene mutations. J Biol Chem. 2000;275:26164–26171. [DOI] [PubMed] [Google Scholar]
- 57. Williams SP , Sigler PB. Atomic structure of progesterone complexed with its receptor. Nature. 1998;393:392–396. [DOI] [PubMed] [Google Scholar]
- 58. Wurtz JM , Bourguet W , Renaud JP , et al. . A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3:87–94. [DOI] [PubMed] [Google Scholar]
- 59. Bourguet W , Ruff M , Chambon P , Gronemeyer H , Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-α. Nature. 1995;375:377–382. [DOI] [PubMed] [Google Scholar]
- 60. Davies TH , Ning YM , Sánchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem. 2002;277:4597–4600. [DOI] [PubMed] [Google Scholar]
- 61. Galigniana MD , Erlejman AG , Monte M , Gomez-Sanchez C , Piwien-Pilipuk G. The Hsp90-FKBP52 complex links the mineralocorticoid receptor to motor proteins and persists bound to the receptor in early nuclear events. Mol Cell Biol. 2010;30:1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Storer CL , Dickey CA , Galigniana MD , Rein T , Cox MB. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol Metab. 2011;22:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wolf IM , Periyasamy S , Hinds T , Yong W , Shou W , Sanchez ER. Targeted ablation reveals a novel role of FKBP52 in gene-specific regulation of glucocorticoid receptor transcriptional activity. J Steroid Biochem Mol Biol. 2009;113:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fang L , Ricketson D , Getubig L , Darimont B. Unliganded and hormone-bound glucocorticoid receptors interact with distinct hydrophobic sites in the Hsp90 C-terminal domain. Proc Natl Acad Sci U S A. 2006;103:18487–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schena M , Yamamoto KR. Mammalian glucocorticoid receptor derivatives enhance transcription in yeast. Science. 1988;241:965–967. [DOI] [PubMed] [Google Scholar]
- 66. Jinwal UK , Koren J , Borysov SI , et al. . The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J Neurosci. 2010;30:591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Elhaji YA , Stoica I , Dennis S , et al. . Impaired helix 12 dynamics due to proline 892 substitutions in the androgen receptor are associated with complete androgen insensitivity. Hum Mol Genet. 2006;15:921–931. [DOI] [PubMed] [Google Scholar]
- 68. Rennie PS , Bruchovsky N , Leco KJ , et al. . Characterization of two cis-acting DNA elements involved in the androgen regulation of the probasin gene. Mol Endocrinol. 1993;7:23–36. [DOI] [PubMed] [Google Scholar]
- 69. Allan RK , Mok D , Ward BK , Ratajczak T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: evidence that coumarin antibiotics disrupt Hsp90 dimerization. J Biol Chem. 2006;281:7161–7171. [DOI] [PubMed] [Google Scholar]
- 70. Vilardell J , Warner JR. Ribosomal protein L32 of Saccharomyces cerevisiae influences both the splicing of its own transcript and the processing of rRNA. Mol Cell Biol. 1997;17:1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Keightley MC , Fuller PJ. Unique sequences in the guinea pig glucocorticoid receptor induce constitutive transactivation and decrease steroid sensitivity. Mol Endocrinol. 1994;8:431–439. [DOI] [PubMed] [Google Scholar]
- 72. Keightley MC , Fuller PJ. Cortisol resistance and the guinea pig glucocorticoid receptor. Steroids. 1995;60:87–92. [DOI] [PubMed] [Google Scholar]
- 73. Garabedian MJ , Yamamoto KR. Genetic dissection of the signaling domain of a mammalian steroid receptor in yeast. Mol Biol Cell. 1992;3:1245–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kralli A , Bohen SP , Yamamoto KR. LEM1, an ATP-binding-cassette transporter, selectively modulates the biological potency of steroid hormones. Proc Natl Acad Sci U S A. 1995;92:4701–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bohen SP , Yamamoto KR. Isolation of Hsp90 mutants by screening for decreased steroid receptor function. Proc Natl Acad Sci U S A. 1993;90:11424–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dittmar KD , Demady DR , Stancato LF , Krishna P , Pratt WB. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery: the role of p23 is to stabilize receptor·Hsp90 heterocomplexes formed by Hsp90·p60·Hsp70. J Biol Chem. 1997;272:21213–21220. [DOI] [PubMed] [Google Scholar]
- 77. Morishima Y , Kanelakis KC , Murphy PJ , et al. . The hsp90 cochaperone p23 is the limiting component of the multiprotein hsp90/hsp70-based chaperone system in vivo where it acts to stabilize the client protein·hsp90 complex. J Biol Chem. 2003;278:48754–48763. [DOI] [PubMed] [Google Scholar]
- 78. Ali MM , Roe SM , Vaughan CK , et al. . Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McLaughlin SH , Sobott F , Yao ZP , et al. . The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J Mol Biol. 2006;356:746–758. [DOI] [PubMed] [Google Scholar]
- 80. Grad I , McKee TA , Ludwig SM , et al. . The Hsp90 cochaperone p23 is essential for perinatal survival. Mol Cell Biol. 2006;26:8976–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hubler TR , Scammell JG. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones. 2004;9:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Paakinaho V , Makkonen H , Jääskeläinen T , Palvimo JJ. Glucocorticoid receptor activates poised FKBP51 locus through long-distance interactions. Mol Endocrinol. 2010;24:511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34(Suppl 1):S186–S195. [DOI] [PubMed] [Google Scholar]
- 84. Holownia A , Mroz RM , Kolodziejczyk A , Chyczewska E , Braszko JJ. Increased FKBP51 in induced sputum cells of chronic obstructive pulmonary disease patients after therapy. Eur J Med Res. 2009;14(Suppl 4):108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Woodruff PG , Boushey HA , Dolganov GM , et al. . Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104:15858–15863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Stechschulte LA , Sanchez ER. FKBP51: a selective modulator of glucocorticoid and androgen sensitivity. Curr Opin Pharmacol. 2011;11:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dalman FC , Scherrer LC , Taylor LP , Akil H , Pratt WB. Localization of the 90-kDa heat shock protein-binding site within the hormone-binding domain of the glucocorticoid receptor by peptide competition. J Biol Chem. 1991;266:3482–3490. [PubMed] [Google Scholar]
- 88. Ricketson D , Hostick U , Fang L , Yamamoto KR , Darimont BD. A conformational switch in the ligand-binding domain regulates the dependence of the glucocorticoid receptor on Hsp90. J Mol Biol. 2007;368:729–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kauppi B , Jakob C , Färnegårdh M , et al. . The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem. 2003;278:22748–22754. [DOI] [PubMed] [Google Scholar]
- 90. Arriza JL , Weinberger C , Cerelli G , et al. . Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. [DOI] [PubMed] [Google Scholar]
- 91. Rogerson FM , Dimopoulos N , Sluka P , Chu S , Curtis AJ , Fuller PJ. Structural determinants of aldosterone binding selectivity in the mineralocorticoid receptor. J Biol Chem. 1999;274:36305–36311. [DOI] [PubMed] [Google Scholar]
- 92. Rogerson FM , Yao YZ , Elsass RE , Dimopoulos N , Smith BJ , Fuller PJ. A critical region in the mineralocorticoid receptor for aldosterone binding and activation by cortisol: evidence for a common mechanism governing ligand binding specificity in steroid hormone receptors. Mol Endocrinol. 2007;21:817–828. [DOI] [PubMed] [Google Scholar]
- 93. Nettles KW , Sun J , Radek JT , et al. . Allosteric control of ligand selectivity between estrogen receptors α and β: implications for other nuclear receptors. Mol Cell. 2004;13:317–327. [DOI] [PubMed] [Google Scholar]
- 94. Kallenberger BC , Love JD , Chatterjee VK , Schwabe JW. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat Struct Biol. 2003;10:136–140. [DOI] [PubMed] [Google Scholar]
- 95. Viguera AR , Serrano L. Stable proline box motif at the N-terminal end of α-helices. Protein Sci. 1999;8:1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Glass CK , Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 97. Caamaño CA , Morano MI , Dalman FC , Pratt WB , Akil H. A conserved proline in the Hsp90 binding region of the glucocorticoid receptor is required for Hsp90 heterocomplex stabilization and receptor signaling. J Biol Chem. 1998;273:20473–20480. [DOI] [PubMed] [Google Scholar]
- 98. Pratt WB , Galigniana MD , Harrell JM , DeFranco DB. Role of Hsp90 and the Hsp90-binding immunophilins in signalling protein movement. Cell Signal. 2004;16:857–872. [DOI] [PubMed] [Google Scholar]
- 99. Harris SF , Shiau AK , Agard DA. The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure. 2004;12:1087–1097. [DOI] [PubMed] [Google Scholar]
- 100. Jackson SE , Queitsch C , Toft D. Hsp90: from structure to phenotype. Nat Struct Mol Biol. 2004;11:1152–1155. [DOI] [PubMed] [Google Scholar]