

Abstract
Inactivating mutations of KISS-1 receptor (KISS1R) have been recently described as a rare cause of isolated hypogonadotropic hypogonadism transmitted as a recessive trait. Few mutations have been described, and the structure-function relationship of KISS1R remains poorly understood. Here, we have taken advantage of the discovery of a novel mutation of KISS1R to characterize the structure and function of an uncommon protein motif composed of 3 proline-arginine-arginine (PRR) repeats located within the intracellular domain. A heterozygous insertion of 1 PRR repeat in-frame with 3 PRR repeats leading to synthesis of a receptor bearing 4 PRR repeats (PRR-KISS1R) was found in the index case. Functional analysis of PRR-KISS1R showed a decrease of the maximal response to kisspeptin stimulation, associated to a lower cell surface expression without modification of total expression. PRR-KISS1R exerts a dominant negative effect on the synthesis of the wild-type (WT)-KISS1R. This effect was due to the nature of inserted residues but also to the difference of the length of the intracellular domain between PRR-KISS1R and WT-KISS1R. A molecular dynamic analysis showed that the additional PRR constrained this arginine-rich region into a polyproline type II helix. Altogether, this study shows that a heterozygous insertion in KISS1R may lead to hypogonadotropic hypogonadism by a dominant negative effect on the WT receptor. An additional PRR repeat into a proline-arginine-rich motif can dramatically changed the conformation of the intracellular domain of KISS1R and its probable interaction with partner proteins.
Isolated hypogonadotropic hypogonadism (IHH) is defined by low sex steroid hormone synthesis with low LH and FSH levels. The congenital form of this condition is a rare disease. Several genes have now been linked to IHH. Some were natural candidate genes, such as those encoding for the GnRH receptor (1) or its ligand, GnRH (2). Others were characterized by genome mapping and encoded for kisspeptins (Kps) (3) and their receptor (KISS1R) (4, 5) or neurokinin B and its receptor (Tachykinin receptor 3) (6). Kp and neurokinin B are both neuropeptides expressed in hypothalamic neurons as well as their receptors. They belong to a complex neuroendocrine network aiming to control synthesis and secretion of GnRH. They are major determinants of the gonadotropic axis reactivation leading to pubertal onset (7). They also relay the estradiol positive and negative feedback on the gonadotropic axis leading to the LH ovulatory peak (8, 9).
KISS1R is a G protein-coupled receptor (GPCR) expressed in GnRH neurons (9). Kp activates phospholipase Cβ and MAPK signaling pathways and depolarize GnRH neurons by activating nonselective transient receptor potential cation channels and by inhibiting inwardly rectifying potassium channels (9).
To date, 11 mutations in KISS1R have been identified in IHH (4, 5, 10–15). IHH due to KISS1R mutations is transmitted as a recessive trait, although late puberty has been reported in heterozygous parents of at least 1 index case (4). Compound heterozygous mutations (5, 12) or homozygous mutations have been reported (4, 5, 10, 11, 14, 15). However, there are no striking genotype-phenotype correlations for KISS1R mutations. Complete loss-of-function mutations do not necessarily cause complete gonadotropic deficiency, and variable phenotypes could be observed in patients carrying the same KISS1R mutation (4, 14). Similar reproductive phenotype variability was also reported in Kiss1-deleted mouse mode (16, 17). In very few cases, 1 heterozygous mutation in KISS1R was shown to be associated with a mutation in other IHH genes (18).
In the present study, we identified a heterozygous variant in KISS1R in 1 patient harboring IHH. This variant is characterized by the addition of one proline and two arginines in the intracellular domain of the receptor. The absence of a second event on the other allele, the normal coding sequence in other IHH genes, and the uncommon location of the insertion in the intracellular domain of KISS1R led us to suspect that the mutated receptor may have a dominant negative effect on the wild-type (WT)-KISS1R. Moreover, this proline-arginine-arginine (PRR) repeat motif seemed unique in mammalians, and its biochemical function, if any, was unknown. We have therefore analyzed the functional effects of the PRR insertion in the intracellular domain of KISS1R and modeled the structural consequence of this insertion. Functional results were then correlated with the phenotype.
Materials and Methods
Patient
The proband was a 16-year-old boy who was referred for impuberism and obesity. He was born at term, and there was no history of cryptorchidism. At the initial presentation, he was Tanner stage 1. The body mass index was 31 kg/m2. His penis length was 5 cm, and right and left testicular volumes were 3 mL. There was no anosmia. The born age was 13 years. He reported episodic nocturnal enuresis. Laboratory investigations revealed low plasma testosterone levels (1 nmol/L) with low LH (2.9 IU/L) and low FSH (5.3 IU/L) levels. The other endocrine axes were normal. The family history revealed obesity in the mother and the sister and a fertility problem in the father, but it was not possible to obtain further details.
Mutation analyses
The sequencing of KISS1R exons have been performed from DNA extracted from blood lymphocytes as previously described (4). Briefly, the 5 exons of KISS1R were amplified by PCR, and the PCR products were sequenced with BigDye dideoxyterminator cycle sequencing kit and the 3100 sequencer (Applied Biosystems, Courtaboeuf, France) with the same primers. Both parents gave informed consent for genetic analysis of the propositus DNA, but after the characterization of the mutation in the proband, they did not agree to determine whether the mutation was de novo or was transmitted by one of them or to the proband sister.
Cell culture
HEK-293, Cos-7, and HeLa cells were cultured in high glucose (4500 mg/L) DMEM with GlutaMAX I (Invitrogen, Cergy Pontoise, France), supplemented with 10% fetal calf serum, 100-U/mL penicillin and 100-μg/mL streptomycin (Invitrogen), and 25mM HEPES. Cells were incubated in a humidified 95% air, 5% CO2-controlled atmosphere at 37°C. Medium was changed every 3 or 4 days. Passages were performed once per week, using 1× trypsin/EDTA (Invitrogen).
Construction of the expression vectors
The PRR insertion was introduced by PCR into the WT KISS1R sequence cloned in a pcDNA3.1 expression vector (WT-KISS1R) (Table 1) as described elsewhere (14). Briefly, the mutation was reproduced by PCR, then the PCR fragment was cut by the restriction enzymes SbfI and PpumI (New England Biolabs, Evry, France) and subcloned into a KISS1R-expressing vector in place of the WT sequence. The resulting vector was called PRR-KISS1R. To study cell surface expression of KISS1R, a human influenza hemagglutinin (HA) epitope (YPYDVPDYA) was added at the N-terminal end of KISS1R after the initial methionine (11). The resulting plasmid was called WT-HA-KISS1R. The vector expressing a HA-tagged PRR-mutated KISS1R (PRR-HA-KISS1R) was constructed by exchanging a HindIII-BsrGI fragment from PRR-KISS1R into WT-HA-KISS1R.
Table 1.
Name and Sequence of the Proline-Arginine-Rich Region of the Different KISS1R Construction
| KISS1R plasmid | Amino acid sequence of interest |
|---|---|
| WT | PRR PRR PRR |
| (PRR)4 | PRR PRR PRR PRR |
| (AAA)4 | AAA AAA AAA AAA |
| AAA(PRR)3 | AAA PRR PRR PRR |
| (PRR)3AAA | PRR PRR PRR AAA |
| ΔPRR |
For the construction of (AAA)4-, AAA(PRR)3-, and (PRR)3AAA-KISS1R-expressing vectors, in which the 4 PRR repeats were replaced, respectively, by AAAAAAAAAAAA, AAAPRRPRRPRR, and PRRPRRPRRAAA, a fragment with SbfI, BamHI, and KpnI, HindIII restriction sites were synthesized and inserted in pUC57 cloning vector between KpnI and HindIII restriction sites (Eurogentec, Angers, France). Next, fragments of interest were cut by the restriction enzymes SbfI and BamHI (New England Biolabs) and subcloned into KISS1R- or HA-KISS1R-expressing vectors in place of the WT sequence.
To construct the vector expressing a KISS1R without the PRR repeats, ΔPRR-HA-KISS1R or ΔPRR-KISS1R, a SbfI-BamHI fragment containing a SfaAI restriction site was amplified by PCR from the WT receptor and subcloned into HA-KISS1R- or KISS1R-expressing vector at SbfI and BamHI sites. This plasmid was cut by SfaAI, incubated with T4 DNA polymerase (New England Biolabs) to remove the 3′ cohesive extremity and generate blunted extremities and then incubated with T4 DNA ligase (New England Biolabs) to obtain ΔPRR-HA-KISS1R and ΔPRR-KISS1R plasmids.
Plasmid constructs were validated by direct sequencing of the inserted fragments and by restriction-enzyme mapping.
Transfection
HEK-293, Cos-7, and HeLa cells were seeded in a 35-mm dish. Twenty-four hours after plating, cells were transfected with expression vectors using FuGENE HD (Roche Diagnostics, Meylan, France) or using the calcium phosphate transfection kit (Invitrogen) according to the manufacturer's instructions.
Inositol phosphate (IP) measurements
Twenty-four hours after transfection, cells were seeded in 96-well plates at a density of 20,000 cells/well. After 24 hours (48 h after transfection), total IP was measured using the IP-One HTRF Assay kit (Cisbio Bioassays, Bagnols-sur-Cèze, France) according to the manufacturer's instructions. Briefly, cells were stimulated with Kp10 during 1 hour at 37°C. After stimulation, the fluorophore d2-labeled IP1 and anti-IP1 antibody conjugated to cryptate (IP-One Tb) were added for 1 hour at room temperature, and the plate was read at 620 nm and 665 nm using PARADIGM Detection Platform (Beckman Coulter, Brea, California). Each functional analysis was performed in triplicates.
MAPK activation
Twenty-four hours after transfection, HeLa cells were serum starved for 18 hours. Cells were treated with Kp10 at 10−5M in DMEM supplemented with 0.2% BSA during 10 minutes. Total proteins were extracted with Cell Lysis buffer (Cell Signaling, Danvers, Massachusetts) according to the manufacturer's instructions. Thirty micrograms of proteins were resolved in 10% SDS-PAGE and electroblotted for 1 hour at 100 V, onto Hybon-P membranes (GE Healthcare, Velizy, France). Membranes were blocked 3 hours at room temperature, by using 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) and incubated overnight at 4°C with a mouse monoclonal antiphospho-p44/42MAPK antibody (Cell Signaling) diluted in blocking solution. Membranes were washed 3 times 10 minutes at room temperature in TBST, incubated for 1 hour with a peroxidase-conjugated fragment antigen binding-specific goat antimouse IgG (Sigma-Aldrich, Saint-Quentin Fallavier, France) in blocking solution, and washed again with TBST. Bound antibodies were revealed by chemiluminescence with the Immun-Star WesternC Chemiluminescent kit (Bio-Rad, Marne-la-Coquette, France) using ChemiDoc XRS (Bio-Rad). After stripping of bound antibodies, membranes were probed again with a monoclonal mouse anti-p44/42MAPK antibody (Cell Signaling) according to the same procedure.
KISS1R quantification by ELISA
Twenty-four hours after transfection, cells were seeded in 96-well plates at a density of 30,000 cells/well. Twenty-four hours later, cells were washed with PBS, fixed with 2% paraformaldehyde for 10 minutes at room temperature. To quantify the total cellular expression of KISS1R, cells were permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) for 10 minutes, washed, and then blocked with PBS supplemented with 2% BSA (Sigma-Aldrich). Cells were incubated with the monoclonal rat anti-HA antibody 3F10 (Roche Diagnostics) at 0.5 μg/mL in PBS supplemented with 1% BSA for 1 hour at room temperature. After 3 washes with PBS, cells were incubated with a goat antirat antibody coupled to horseradish peroxidase (Jackson ImmunoResearch, Suffold, England) at 0.2 μg/mL in PBS-1% BSA. Signal was detected and quantified with BM Chemiluminescence ELISA Substrate POD (Roche Diagnostics) using Paradigm counter (Beckman Coulter). For cell surface quantification, the protocol was the same, with the exception that cells were not permeabilized. Each analysis was performed in triplicates.
Molecular dynamics of WT-KISS1R and PRR-KISS1R fragment
Protein structure fragments encompassing PRR repetitions of WT-KISS1R and PRR-KISS1R have been analyzed through molecular dynamics approach. A detailed description of the method may be found in the Supplemental Data, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org. Fragments were of 24-residue length for the WT sequence and 27-residue length for the PRR sequence and were, respectively, named (PRR)3 and (PRR)4. Five structural models for (PRR)3 and 5 for (PRR)4 were built using I-Tasser webserver (19). In addition, each model was constrained to add polyproline II (PPII) helix conformation (20), leading to 20 independent structural models (10 for (PRR)3 and 10 for (PRR)4). Molecular dynamics simulations were performed with GROMACS 4.0.5 software (21) for each protein fragment. The structural models were also analyzed using secondary structure assigned with DSSP software (22) and a refined approach named protein blocks (PBs) analysis (23). This latter approach allowed a precise comparative analysis of conformations of (PRR)3 and (PRR)4 (24). It was also used to compute the entropy termed Neq, which quantifies the stability of local protein conformations.
Statistical analysis
Data were analyzed using GraphPad Prism software (GraphPad, San Diego, California) by 1-way ANOVA with a Bonferroni test if number of groups was superior to 2 (see figures 2, 4, 6, and 7 below) or by a 2-tail Student's t test when 2 groups were compared (see figure 3 below). Differences were considered significant when P < .05. For IPs measurements and MAPK activation, results are expressed as percentage of maximal stimulation of WT-KISS1R, which was set at 100%. In all figures reporting KISS1R quantification, the expression of mutated receptors was expressed as percentage of WT-KISS1R expression.
Results
Sequencing analysis
Sequencing of the coding exons of KISS1R revealed a heterozygous in-frame insertion of 9 nucleotides at position 1023 (c.1023Ins9) (Figure 1A). The c.1023Ins9 allele encoded for a KISS1R with an additional PRR repeat in a proline-arginine-rich region of the intracellular domain at residue 342. It led to the synthesis of a receptor with 4 consecutive PRR repeats instead of 3 (Figure 1B). This mutant receptor was called PRR-KISS1R. The DNA of the parents and the sister of the proband were not available for analysis.
Figure 1.

KISS1R heterozygous mutation found in an IHH patient. DNA was extracted from blood lymphocytes. The KISS1R exons were sequenced as explained in Materials and Methods. A, Chromatograms showing double peaks after nucleotide 1023. Inserted nucleotides are indicated in bold and underlined. B, Protein sequence of KISS1R showing the 3 PRR repeats and the additional PRR in bold.
Functional analyses
To study the effect of the PRR insertion on KISS1R function, a PRR-KISS1R-encoding plasmid was transiently transfected in HeLa cells, and Kp10-induced generation of intracellular IP was quantified. Forty-eight hours after transfection, no Kp10-induced increase in IP was observed in control cells transfected with the empty vector. In cells transiently expressing WT-HA-KISS1R, Kp10 increased intracellular IPs (Figure 2A). In PRR-HA-KISS1R-expressing cells, Kp10 also induced an intracellular increase of IPs (Figure 2A). However, the maximal effect of Kp10 stimulation was lowered by 46 ± 1% in PRR-HA-KISS1R-expressing cells as compared with WT-HA-KISS1R-expressing cells. Moreover, the EC50 in PRR-HA-KISS1R-expressing cells was slightly increased by 1.8-fold (9.8 ± 1.0 × 10−7M for PRR-HA-KISS1R vs 5.3 ± 1.3 × 10−7M for WT-HA-KISS1R). To ensure that the HA-tag did not modify the activity of the receptors, Kp10-induced generations of IP in tagged and untagged WT-KISS1R and PRR-KISS1R transiently transfected cells were compared. Similar Kp10-induced IP increase was observed, indicating that the HA-tag did not alter KISS1R activation (data not shown).
Figure 2.

The additional PRR disturbs the maximal Kp10-induced activation of KISS1R in HeLa cells. A, IP production in WT-HA-KISS1R (black line), PRR-HA-KISS1R (gray line), and pcDNA3.1-transfected cells (dotted line) induced by 1-hour incubation with Kp10. Data are shown as the mean ± SEM of the 2 experiments, each performed in triplicates. B, MAPK pathway activation by a 10-minute treatment with Kp10 at 10−5M. The density of Phospho-MAPK (P-MAPK) bands was quantified and normalized by the density of MAPK bands. Data are the mean of triplicates (WT-HA-KISS1R in black and PRR-HA-KISS1R in gray); **P < .01 and ***P < .001.
Kp was known to activate the MAPK pathway (25). To study Kp10-induced activation of the MAPK pathway, WT-HA-KISS1R and PRR-HA-KISS1R transiently transfected HeLa cells were incubated with 10−5M of Kp10 for 10 minutes. Total proteins were extracted, immunoblotted with antiphospho-MAPK antibody (called P-MAPK in Figure 2) and then with anti-MAPK antibody. Kp-induced MAPK phosphorylation was significantly lower in PRR-HA-KISS1R-expressing cells than in WT-HA-KISS1R-expressing cells (Figure 2B). To verify that the difference observed between WT and PRR-KISS1R was not cell-type specific, we reproduced these functional analyses in HEK293 and Cos-7 cell lines (data not shown). Data confirmed that the maximal Kp10 stimulation of intracellular IP accumulation and of the MAPK pathway was significantly lower in PRR-HA-KISS1R-expressing cells than in WT-HA-KISS1R-expressing cells.
Cell surface quantification by ELISA
Functional analyses showed that the PRR insertion impaired the activation of the receptor and the maximal stimulation of KISS1R by Kp10. The difference between WT-KISS1R and PRR-KISS1R could thus be due to a lower amount of PRR-KISS1R expressed at the cell surface. To analyze cell surface expression of PRR-HA-KISS1R, the HA-tag was quantified by ELISA in nonpermeabilized (cell surface expression) and in permeabilized (total expression) transiently transfected HeLa cells. Cell surface expression of PRR-HA-KISS1R was significantly decreased by 2-fold compared with WT-HA-KISS1R (Figure 3A). Total expression did not significantly change between PRR-HA-KISS1R and WT-HA-KISS1R (Figure 3B). These results indicate that the additional PRR impaired the intracellular trafficking of KISS1R. We also tested the effect of the PRR insertion on the internalization of KISS1R. In HEK293 cells, the PRR insertion did not modify the constitutive internalization of KISS1R as well as Kp10-induced internalization (data not shown).
Figure 3.

PRR-KISS1R is poorly expressed at the cell surface. KISS1R expression was quantified by an ELISA against the HA-tag in HeLa cells transiently expressing WT-HA-KISS1R (black bars) and PRR-HA-KISS1R (gray bars). A, Cell surface expression in the absence of triton. B, Total expression in triton-permeabilized cells. Data are reported as the mean ± SEM of 3 independent experiments, each performed in triplicates; ***P < .001.
Effect of PRR-KISS1R on WT-HA-KISS1R expression
Because the PRR insertion was found heterozygous in the affected patient, and isolated gonadotropic deficiency due to KISS1R mutation is usually transmitted as a recessive trait, we suspected that the PRR-KISS1R could decrease the expression of the WT receptor at the cell surface and therefore act as a dominant negative receptor on WT-KISS1R. For that purpose, WT-HA-KISS1R was coexpressed in HeLa cells with untagged PRR-KISS1R by transient transfection. The expression of WT-HA-KISS1R was subsequently quantified by ELISA. The coexpression of WT-HA-KISS1R with PRR-KISS1R in HeLa cells resulted in a reduced amount of WT-HA-KISS1R at the cell surface (Figure 4A). This reduction was significantly different with 2-fold more of untagged PRR-KISS1R than WT-HA-KISS1R alone. Cell surface expression of PRR-HA-KISS1R coexpressed with increasing amounts of untagged PRR receptor was also quantified. Coexpression of PRR-HA-KISS1R with PRR-KISS1R only slightly reduced its cell surface expression (Figure 4A). The total expression of WT-HA-KISS1R cotransfected with 2-fold more PRR-KISS1R was significantly reduced by 2-fold (Figure 4B), whereas the total expression of PRR-HA-KISS1R was only slightly perturbed by the increasing amount of cotransfected PRR-KISS1R. The ratio of the cell surface expression to the total expression of the WT-KISS1R was therefore not modified by the cotransfection with PRR-KISS1R (Figure 4C). In addition to have an altered intracellular trafficking, PRR-KISS1R also disturbed the total expression of WT-KISS1R.
Figure 4.
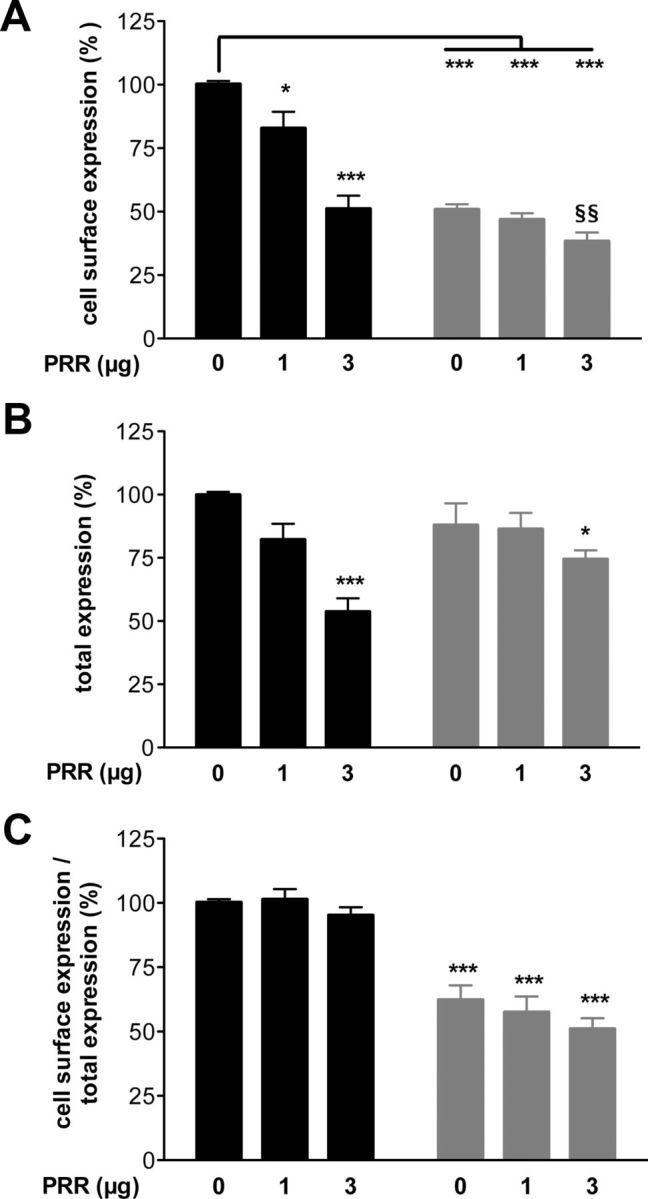
PRR-KISS1R exerts a dominant negative effect on the expression of WT-KISS1R. A, Cell surface expression in the absence of triton. B, Total expression in triton-permeabilized cells. C, Ratio of cell surface expression on total expression. HeLa cells were cotransfected with 1.5 μg of WT-HA-KISS1R alone or with 1 or 3 μg of nontagged PRR-KISS1R (black bars), or with 1.5 μg of PRR-HA-KISS1R alone or with 1 or 3 μg of nontagged PRR-KISS1R (gray bars). ELISA was performed as previously described (see Materials and Methods and Figure 3). Data are reported as the mean ± SEM of 3 independent experiments, each performed in triplicates (*, compared with WT-HA-KISS1R expression in the absence of PRR-KISS1R; §, compared with PRR-HA-KISS1R expression alone); *P < .05, ***P < .001, and §§P < .01.
Molecular dynamics
To understand the impact of the additional PRR in the conformation of the intracellular domain, we analyzed the protein dynamics using classical approaches (26). No structure was available in the Protein Data Bank (27), and no related proteins with available structure can be found. We have thus focused the analysis on protein fragments encompassing the PRR repeats, ie, a fragment of 24 residues for WT-KISS1R ((PRR)3) and 27 residues for PRR-KISS1R ((PRR)4) (Supplemental Figure 1A). No available structure or homologue was found for this sequence. Secondary structure prediction methods (eg, Position Specific Iterated-Prediction, PSI-PRED) (28) predicted coil with a poor accuracy but also a flexible region (data not shown) (29).
I-Tasser webserver (30) was used to propose structural models for both fragments. According to the I-Tasser's C-scores, the best 5 structural models for (PRR)3 and for (PRR)4 have been conserved. Because the proline-rich region was often seen as a PPII helix (31), we also tested models with PRR repeats constrained as PPII helices using the Modeler software (32). The structural models with the lowest Discrete Optimized Protein Energy (DOPE) score were selected. Hence, each protein fragment was represented by 5 free structural models, ie, without PPII helices, and 5 constrained structural models, ie, with PPII helices. A first analysis in terms of secondary content showed that 95% of the structure corresponded to a coil state according to DSSP assignment.
A molecular dynamic analysis was performed for the 20 different structural models for at least 50 nsec. As expected, the 2 protein fragments were highly dynamic. The first visual analysis showed a compaction of the WT fragment. To quantify this compaction, distances between the extremities of (PRR)3 or (PRR)4 were computed. At first, all structural models of (PRR)3 (free and PPII constrained) showed both a rapid and irreversible decrease in the distance between extremities (Figure 5A). On average, the distance was 5 Å. In contrast, (PRR)4 structural models (free and PPII constrained) showed a higher distance between extremities that remained around 20 Å.
Figure 5.
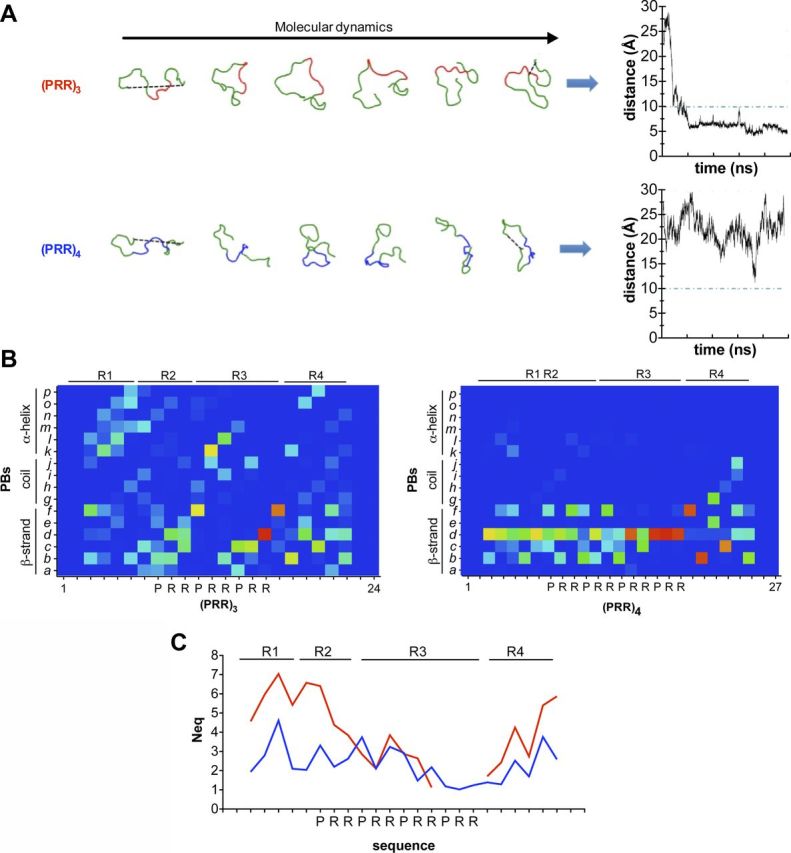
The additional PRR constrains the proline-arginine-rich region into a PPII helix. Two protein fragments of 24 or 27 residues were first modeled with I-Tasser. The best 5 structural models were selected and constrained to adopt a PPII helix conformation. Ten structural models were selected for each protein fragment. Molecular dynamic simulations were then performed with GROMACS software as described in Supplemental data. A, Distances between extremities of both protein fragments were measured (hatched line) during 50 nsec. B, PBs analysis of both fragments, (PRR)3 in the left panel and (PRR)4 in the right panel. a to p correspond to 16 local prototypes (see Supplemental data) with PB, d corresponds to a PPII helix. Color ranges indicated the probability to adopt a local conformation (dark blue, ie, 0%, to red, ie, 100%). (C) Neq for each residue in (PRR)3 (red line) and in (PRR)4 (blue line).
To analyze the conformations of the peptide more in depth, a structural alphabet was used (33), ie, a set of small local protein structures that can be used to approximate every part of a protein structure. The structural alphabet used for this analysis, namely PBs (23, 24), is composed of 16 distinct prototypes that are 5 residues in length. The analysis of (PRR)3 by the structural alphabet revealed 4 consecutive regions (R1 to R4) (Figure 5B, left panel). R1 was highly fuzzy, mainly associated to helical PBs (PBs m is the core of helix). R2 was associated to PBs related to β-strands (PBs b to d). R3 was less fuzzy, but not associated to β conformations, only 2 residues preferred PBs c, whereas most of the others were more associated to turns (succession of PBs fl and d). R4 was fuzzier than R3 with tendencies to β-strands PBs. The analysis of (PRR)4 by the structural alphabet showed a strong preference for the PBs d (Figure 5B, right panel). Both R1 and R2 were in β conformation, but not β-sheets. The adjunction of 1 PRR repeat showed a striking constraint of R3 with only PBs d for the last PRR repeat. It was the core of the PPII helix. R4 showed an accented transition (PBs fbe/gc).
To quantify constraints, the PB distribution was transformed in a quantitative value (Neq). If only 1 PB was observed for a residue, Neq equals to 1; if all PBs were predicted with the same probability, Neq equals to 16. At the N terminus of the analyzed sequences (R1 and R2), Neq were lower for (PRR)4 fragment than for (PRR)3 (Figure 5C). Then, Neq were roughly comparable for the 2 first PRR repeats. The addition of 1 repeat induced a high constraint with a Neq close to 1. The C terminus of both sequences had higher Neq, but again, Neq was lower for (PRR)4 than for (PRR)3.
From these results, it should be noted that (PRR)3 was highly flexible and did not possess a PPII helix, having only few positions that were stable, which were comparable with turns. The adjunction of an extra PRR repeat created a strong rigidity of the peptide, with conformation into a PPII helix. The content of PPII helix of structural models of (PRR)3 (free and PPII constrained) was very limited (<3%), whereas the PPII helix content of (PRR)4 structural models (free and PPII constrained) was always high and especially for the last PRR repetition (>85%).
Implication of the proline- and arginine-rich domain in KISS1R expression
To study how the PRR insertion interferes with the normal function of KISS1R, mutants of the PRR repeat were constructed (Table 1). We tested the nature of the inserted residues as well as the position of the insertion of the triplet in regards to the normal PRR repeat. Substitution of the PRR insertion by AAA at the N terminus of the PRR repeat (AAA(PRR)3) slightly increased the EC50 by 1.2-fold (1.5 ± 1.1 × 10−7M for WT-HA vs 1.9 ± 1.0 × 10−7M for AAA(PRR)3) and decreased maximal stimulation by 30% (Figure 6A). This maximal decrease of IP generation was associated with a decrease of the total and cell surface expression (Figure 6B). Interestingly, addition of an AAA at the C-terminal end of the PRR repeat ((PRR)3AAA) did not change the EC50 value nor the maximal stimulation of the mutant receptor (Figure 6A). A slight decrease of cell surface and total expression was observed for (PRR)3AAA-KISS1R when compared with WT-KISS1R (Figure 6B). To definitively affirm the functional importance of the PRR repeats, the 4 PRR repeats were substituted by alanines ((AAA)4) or deleted (ΔPRR; only the last arginine was conserved). The absence of PRR repeats by deletion or AAA substitution induced an increase of the EC50 (1.5 ± 1.1 × 10−7M for WT-HA vs 8.2 ± 0.8 × 10−7M for (AAA)4 and 1.1 ± 0.8 × 10−6M for ΔPRR) and a decrease of the maximal stimulation. Overall, ratios of cell surface expression/total expression of these mutated receptors were not changed (Figure 6B).
Figure 6.
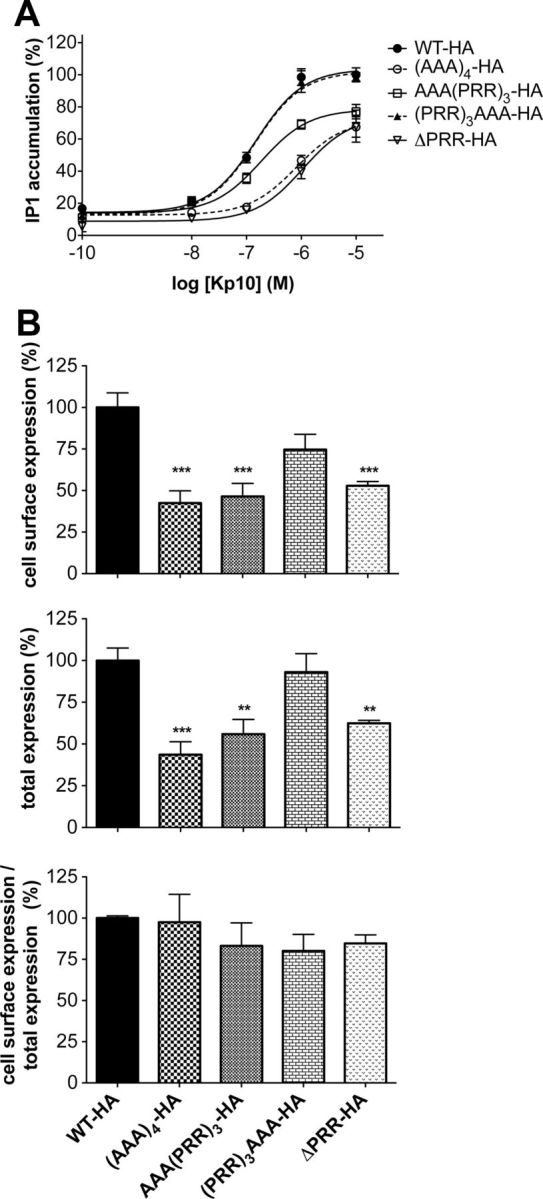
The dominant negative effect of the additional PRR is not due to the biochemical nature of inserted residues. A, Kp10-induced IP production in HeLa cells transiently expressing (AAA)4, AAA(PRR)3, (PRR)3AAA, and ΔPRR-mutated KISS1R. Data represent 2 experiments, performed in triplicates. B, Quantification of cell surface and total expression of mutated KISS1R and ratio of cell surface expression on total expression. Data are reported as the mean ± SEM of 2 experiments performed in triplicates; **P < .01 and ***P < .001.
The dominant negative effect of each mutant on WT-KISS1R expression was then tested by coexpression in HeLa cells. (AAA)4, AAA(PRR)3, and ΔPRR-KISS1R decreased the total expression of WT-HA-KISS1R (64 ± 16%, 72 ± 16%, and 69 ± 10% of the total expression in absence of the mutated receptors, respectively) (Figure 7). Cell surface expression of WT-HA-KISS1R also decreased in similar proportions (60 ± 2%, 64 ± 5%, and 48 ± 3% for WT-HA + (AAA)4, WT-HA + AAA(PRR)3, and WT-HA + ΔPRR, respectively). The relative expression at the cell surface to the total expression was therefore not changed by the coexpression of a mutated receptor (Figure 7). It is important to note that (PRR)3AAA-KISS1R had no significant effect on neither total nor cell surface expression of WT-HA-KISS1R.
Figure 7.
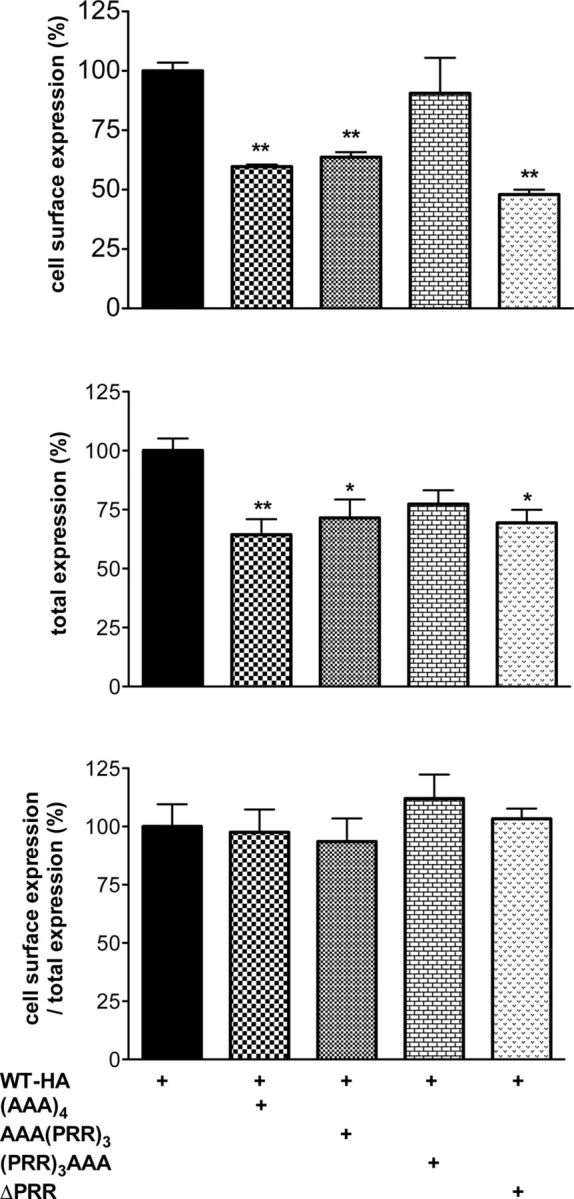
Ala-KISS1R-mutants exert a dominant negative effect on the WT-KISS1R cell surface expression except the (PRR)3AAA mutant. HeLa cells were transfected with 1.5 μg of HA-KISS1R and 1.5 μg of nontagged KISS1R mutants. ELISA was performed as previously described. Data are reported as the mean ± SEM of 2 experiments performed in triplicates; *P < .5 and **P < .01.
Discussion
Natural inactivating mutations of GPCR have long been considered as a cause of endocrine diseases (receptors of LH, TSH, FSH, GnRH, GHRH, melanocortin 2, melanocortin 3, melanocortin 4, PTH, calcium sensing receptor [CaSR], and KISS1R), retinis pigmentosa (Rhodopsin), nephrogenic diabetes insipidus (arginine vasopression receptor 2), and Hischsprung disease (endothelin receptor type B). They were also associated to susceptibility to HIV infection (chemokine receptor 5), determination of skin and hair colors (melanocortin 3 receptor), or increased susceptibility to melanoma (melanocortin 1 receptor) (34, 35). Most of these diseases or physiological traits are transmitted as a recessive trait. In few cases, a dominant transmission has been reported (34). Loss-of-function mutation of KISS1R leading to isolated gonadotropic deficiency has been recently described (4, 5). Few homozygous or compound heterozygous mutations have been described in patients with recessive transmission of IHH (4, 5, 10–15). In the present study, we showed that the insertion of a PRR sequence in the intracellular domain of KISS1R disturbed the normal expression of the receptor at cell surface. The KISS1R bearing the additional PRR repeat exerts a dominant negative effect on the WT receptor expression. This additional PRR within a repetition of 3 consecutive PRR sequence constrained this sequence into a PPII helix and increases its rigidity. Altogether, this study highlights the possible dominant transmission of IHH due to KISS1R mutations. It also underscores the importance of a proline-arginine-rich region within the intracellular domain potentially involved in the normal folding and intracellular signaling pathways of KISS1R.
IHH due to KISS1R mutations is transmitted as a recessive trait (4, 5, 10–15). Several KISS1R-mutated cases have now been described. However, the understanding of the genotype-phenotype correlation of KISS1R mutations is not obvious, which underscores the complexity of the regulation of the gonadotropic axis by Kp. For instance, the complete inactivation of KISS1R may lead to a severe gonadotropic deficiency with complete absence of pubertal onset (11). Similar dramatic inactivation of KISS1R may be associated to partial phenotype with incomplete pubertal progression (4, 5, 14). In one case, a pubertal delay was reported in one heterozygous parent (4). The neuroendocrine reactivation of the gonadotropic axis is progressive, starting before the appearance of the clinical signs of puberty (36, 37). This reactivation is related to the increase of Kp synthesis but also to the capacity of KISS1R to be activated by Kp (38). Because the PRR insertion reduced cell surface expression of the mutant KISS1R but also the expression of WT-KISS1R by a dominant negative effect, we propose that the IHH phenotype observed in this patient is attributable to the PRR insertion. Such dominant negative effect of natural GPCR mutants on the WT receptor has already been described in TSH receptor, melanocortin 4 receptor, chemokine receptor 5, and CaSR (34).
The repetition of 3 PRRs is unique to the human KISS1R. However, this part of the intracellular domain is arginine-rich in other species (Figure 8). The arginine-rich motif RXR is considered as the core motif of endoplasmic reticulum (ER)-exit or ER-retention signals (39). Natural mutations of the RXR motif favored cell surface localization of CaSR, which suggests that it may constitute an ER-retention signal for this receptor (40). Initially, we suspected that the additional PRR repeat might lead to the formation of an additional RXR motif leading to an ER retention of the mutant receptor. Our in vitro mutagenesis of the PRR region did not support this hypothesis. Deletion of the complete PRR repetition did not lead to a higher cell surface expression of KISS1R. This arginine-rich region of the intracellular domain thus does not function as a retention signal in KISS1R.
Figure 8.

The alignment of sequence of the PRR-repeat region indicates that Arg344 and Arg346 (in bold) are conserved among species and in mutant receptors.
It is now well known that during GPCR biosynthesis, dimer formation is required to pass quality control (QC) checkpoints in the ER (41). The role of GPCR dimerization in the ER QC is complex. In some instances, the heterodimerization is required for a normal expression, whereas in other situations, heterodimerization between GPCR is deleterious. The latter has been well described for several mutated GPCRs, which generate asymmetric structures with their WT receptor and are then recognized as misfolded proteins and degraded (42). The coexpression of WT-HA-KISS1R with mutant receptors highly supports that the dimer symmetry is important for KISS1R to pass the ER QC. Our results also show that the dominant negative effect of PRR-KISS1R on WT-KISS1R was observed for a mutant receptor bearing 4 alanine triplets in place of PRR repeats. The dominant negative effect of PRR-KISS1R was thus not due to the presence of a RXR motif in the heterodimer, which had not been masked by the WT-KISS1R as described for the glutamate metabotropic receptor (43).
The lower expression at the cell surface of PRR-KISS1R homodimers without the change of the total expression suggests that PRR-KISS1R homodimers disturbed the intracellular traffic of the receptor. In silico molecular dynamics indicates that the PRR insertion strongly alters the flexibility of the proline- and arginine-rich region. It underlines that WT-KISS1R proline- and arginine-rich region does not possess stable PPII conformation and is highly flexible, whereas the addition of 1 PRR repeat in this region forms a quite rigid sequence and a true PPII helix. We suspect that the PRR insertion could modify the interaction of the proline-arginine-rich region of KISS1R with an unknown partner protein. To date, the catalytic subunit of protein phosphatase 2A is the only protein known to physically interact with the intracellular domain of KISS1R. This interaction occurs between Arg335 and Ala358, including the PRR repetitions, and seems to be involved in the antimetastatic role of Kp (44). Additional experiments are necessary to test whether the additional PRR repeat disturbs the interaction of KISS1R with the catalytic subunit of protein phosphatase 2A.
Surprisingly, our results showed that the PRR region is also involved in the signaling pathway of KISS1R. Indeed, the EC50 values of (AAA)4-KISS1R or ΔPRR-KISS1R were both increased. The sequence alignment among species of the proline- and arginine-rich region indicates that prolines are poorly conserved (Figure 8). In contrast, the Arg344 is completely conserved but also in (PRR)3-AAA, which has the same EC50 as WT-KISS1R, suggesting that this arginine is particularly important for coupling KISS1R to Gq proteins. Additional functional analyses are needed to precisely delineate residues of this PRR region critical for the transduction pathway of KISS1R.
The present study provides the first description of a mutant KISS1R having a dominant negative effect on the WT receptor, ultimately causing IHH. It also highlights the functional importance of a proline- and arginine-rich region in the intracellular domain that appears to be involved in the folding of this domain. In respect with the abundance of arginine residues in the region of KISS1R, where the PRR insertion is located, with the important role of arginine-rich motifs in protein interactions, it would be particularly interesting to characterize proteins interacting with KISS1R in GnRH neurons. Identification of such KISS1R protein partners could yield a better understanding of the activation of the Kp-KISS1R system at the onset of puberty.
Acknowledgments
This work was supported by the Institut National de la Santé et de la Recherche Médicale and the French National Research Agency Grant FrenchKiss ANR-07-BLAN-0056-04.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CaSR
- calcium sensing receptor
- ER
- endoplasmic reticulum
- GPCR
- G protein-coupled receptor
- HA
- human influenza hemagglutinin
- IHH
- isolated hypogonadotropic hypogonadism
- IP
- inositol phosphate
- KISS1R
- KISS-1 receptor
- Kp
- kisspeptin
- PB
- protein block
- PPII
- polyproline II
- PRR
- proline-arginine-arginine
- QC
- quality control
- TBST
- Tris-buffered saline containing 0.1% Tween 20
- WT
- wild type.
References
- 1. de Roux N , Young J , Misrahi M , et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337:1597–1602. [DOI] [PubMed] [Google Scholar]
- 2. Bouligand J , Ghervan C , Tello JA , et al. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–2748. [DOI] [PubMed] [Google Scholar]
- 3. Topaloglu AK , Tello JA , Kotan LD , et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N Engl J Med. 2012;366:629–635. [DOI] [PubMed] [Google Scholar]
- 4. de Roux N , Genin E , Carel JC , Matsuda F , Chaussain JL , Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seminara SB , Messager S , Chatzidaki EE , et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. [DOI] [PubMed] [Google Scholar]
- 6. Topaloglu AK , Reimann F , Guclu M , et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. García-Galiano D , van Ingen Schenau D , Leon S , et al. Kisspeptin signaling is indispensable for neurokinin B, but not glutamate, stimulation of gonadotropin secretion in mice. Endocrinology. 2012;153:316–328. [DOI] [PubMed] [Google Scholar]
- 8. Dungan HM , Gottsch ML , Zeng H , et al. The role of kisspeptin-GPR54 signaling in the tonic regulation and surge release of gonadotropin-releasing hormone/luteinizing hormone. J Neurosci. 2007;27:12088–12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oakley AE , Clifton DK , Steiner RA. Kisspeptin signaling in the brain. Endocr Rev. 2009;30:713–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lanfranco F , Gromoll J , von Eckardstein S , Herding EM , Nieschlag E , Simoni M. Role of sequence variations of the GnRH receptor and G protein-coupled receptor 54 gene in male idiopathic hypogonadotropic hypogonadism. Eur J Endocrinol. 2005;153:845–852. [DOI] [PubMed] [Google Scholar]
- 11. Nimri R , Lebenthal Y , Lazar L , et al. A novel loss-of-function mutation in GPR54/KISS1R leads to hypogonadotropic hypogonadism in a highly consanguineous family. J Clin Endocrinol Metab. 2011;96:E536–E545. [DOI] [PubMed] [Google Scholar]
- 12. Semple RK , Achermann JC , Ellery J , et al. Two novel missense mutations in g protein-coupled receptor 54 in a patient with hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2005;90:1849–1855. [DOI] [PubMed] [Google Scholar]
- 13. Teles MG , Trarbach EB , Noel SD , et al. A novel homozygous splice acceptor site mutation of KISS1R in two siblings with normosmic isolated hypogonadotropic hypogonadism. Eur J Endocrinol. 2010;163:29–34. [DOI] [PubMed] [Google Scholar]
- 14. Tenenbaum-Rakover Y , Commenges-Ducos M , Iovane A , Aumas C , Admoni O , de Roux N. Neuroendocrine phenotype analysis in five patients with isolated hypogonadotropic hypogonadism due to a L102P inactivating mutation of GPR54. J Clin Endocrinol Metab. 2007;92:1137–1144. [DOI] [PubMed] [Google Scholar]
- 15. Breuer O , Abdulhadi-Atwan M , Zeligson S , et al. A novel severe N-terminal splice site KISS1R gene mutation causes hypogonadotropic hypogonadism but enables a normal development of neonatal external genitalia. Eur J Endocrinol. 2012;167:209–216. [DOI] [PubMed] [Google Scholar]
- 16. d'Anglemont de Tassigny X , Fagg LA , et al. Hypogonadotropic hypogonadism in mice lacking a functional Kiss1 gene. Proc Natl Acad Sci USA. 2007;104:10714–10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lapatto R , Pallais JC , Zhang D , et al. Kiss1−/− mice exhibit more variable hypogonadism than Gpr54−/− mice. Endocrinology. 2007;148:4927–4936. [DOI] [PubMed] [Google Scholar]
- 18. Sykiotis GP , Plummer L , Hughes VA , et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA. 2010;107:15140–15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fitzkee NC , Fleming PJ , Gong H , Panasik N , Street TO , Rose GD. Are proteins made from a limited parts list? Trends Biochem Sci. 2005;30:73–80. [DOI] [PubMed] [Google Scholar]
- 21. Hess B , Kutzner C , van der Spoel D , Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theor Comp. 2008;4:435–447. [DOI] [PubMed] [Google Scholar]
- 22. Kabsch W , Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 23. de Brevern AG , Etchebest C , Hazout S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins. 2000;41:271–287. [DOI] [PubMed] [Google Scholar]
- 24. Joseph AP , Agarwal G , Mahajan S , et al. A short survey on protein blocks. Biophys Rev. 2010;2:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Castellano JM , Navarro VM , Fernandez-Fernandez R , et al. Ontogeny and mechanisms of action for the stimulatory effect of kisspeptin on gonadotropin-releasing hormone system of the rat. Mol Cell Endocrinol. 2006;257–258:75–83. [DOI] [PubMed] [Google Scholar]
- 26. Altschul SF , Madden TL , Schäffer AA , et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berman HM , Westbrook J , Feng Z , et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. [DOI] [PubMed] [Google Scholar]
- 29. Bornot A , Etchebest C , de Brevern AG. Predicting protein flexibility through the prediction of local structures. Proteins. 2011;79:839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roy A , Kucukural A , Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williamson MP. The structure and function of proline-rich regions in proteins. Biochem J. 1994;297:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sali A , Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. [DOI] [PubMed] [Google Scholar]
- 33. Offmann B , Tyagi M , de Brevern AG. Local protein structures. Curr Bioinform. 2007;3:165–202. [Google Scholar]
- 34. Tao YX. Inactivating mutations of G protein-coupled receptors and diseases: structure-function insights and therapeutic implications. Pharmacol Ther. 2006;111:949–973. [DOI] [PubMed] [Google Scholar]
- 35. Vassart G , Costagliola S. G protein-coupled receptors: mutations and endocrine diseases. Nat Rev Endocrinol. 2011;7:362–372. [DOI] [PubMed] [Google Scholar]
- 36. Manasco PK , Umbach DM , Muly SM , et al. Ontogeny of gonadotropin, testosterone, and inhibin secretion in normal boys through puberty based on overnight serial sampling. J Clin Endocrinol Metab. 1995;80:2046–2052. [DOI] [PubMed] [Google Scholar]
- 37. Manasco PK , Umbach DM , Muly SM , et al. Ontogeny of gonadotrophin and inhibin secretion in normal girls through puberty based on overnight serial sampling and a comparison with normal boys. Hum Reprod. 1997;12:2108–2114. [DOI] [PubMed] [Google Scholar]
- 38. Clarkson J , Han SK , Liu X , Lee K , Herbison AE. Neurobiological mechanisms underlying kisspeptin activation of gonadotropin-releasing hormone (GnRH) neurons at puberty. Mol Cell Endocrinol. 2010;324:45–50. [DOI] [PubMed] [Google Scholar]
- 39. Michelsen K , Yuan H , Schwappach B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 2005;6:717–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stepanchick A , McKenna J , McGovern O , Huang Y , Breitwieser GE. Calcium sensing receptor mutations implicated in pancreatitis and idiopathic epilepsy syndrome disrupt an arginine-rich retention motif. Cell Physiol Biochem. 2010;26:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bulenger S , Marullo S , Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26:131–137. [DOI] [PubMed] [Google Scholar]
- 42. Williams D , Devi LA. Escorts take the lead molecular chaperones as therapeutic targets. Prog Mol Biol Transl Sci. 2010;91:121–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Margeta-Mitrovic M , Jan YN , Jan LY. A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron. 2000;27:97–106. [DOI] [PubMed] [Google Scholar]
- 44. Evans BJ , Wang Z , Mobley L , et al. Physical association of GPR54 C-terminal with protein phosphatase 2A. Biochem Biophys Res Commun. 2008;377:1067–1071. [DOI] [PubMed] [Google Scholar]