

Abstract
Central resistance to the actions of insulin and leptin is associated with the onset of obesity and type 2 diabetes mellitus, whereas leptin and insulin signaling is essential for both glucose and energy homeostasis. Although it is known that leptin resistance can lead to attenuated insulin signaling, whether insulin resistance can lead to or exacerbate leptin resistance is unknown. To investigate the molecular events underlying crosstalk between these signaling pathways, immortalized hypothalamic neuronal models, rHypoE-19 and mHypoA-2/10, were used. Prolonged insulin exposure was used to induce cellular insulin resistance, and thereafter leptin-mediated regulation of signal transduction and gene expression was assessed. Leptin directly repressed agouti-related peptide mRNA levels but induced urocortin-2, insulin receptor substrate (IRS)-1, IRS2, and IR transcription, through leptin-mediated phosphatidylinositol 3-kinase/Akt activation. Neuronal insulin resistance, as assessed by attenuated Akt phosphorylation, blocked leptin-mediated signal transduction and agouti-related peptide, urocortin-2, IRS1, IRS2, and insulin receptor synthesis. Insulin resistance caused a substantial decrease in insulin receptor protein levels, forkhead box protein 1 phosphorylation, and an increase in suppressor of cytokine signaling 3 protein levels. Cellular insulin resistance may cause or exacerbate neuronal leptin resistance and, by extension, obesity. It is essential to unravel the effects of neuronal insulin resistance given that both peripheral, as well as the less widely studied central insulin resistance, may contribute to the development of metabolic, reproductive, and cardiovascular disorders. This study provides improved understanding of the complex cellular crosstalk between insulin-leptin signal transduction that is disrupted during neuronal insulin resistance.
Resistance to the central actions of leptin and insulin, key signals involved in regulating whole-body energy homeostasis, often underlie metabolic disorders through mechanisms that remain unclear. Within the central nervous system, the hypothalamus is a crucial site for the metabolic actions of insulin and leptin (1, 2). Unlike peripheral organs composed of comparatively homogeneous cell types, within the hypothalamus, diverse neuronal subpopulations contribute to the maintenance of energy homeostasis. The central melanocortin system represents a crucial point of action for the regulatory roles of insulin and leptin in body weight maintenance. Anorexigenic pro-opiomelanocortin-expressing neurons and orexigenic neuropeptide Y (NPY)/agouti-related peptide (AgRP)-expressing neurons form the basis of the melanocortin system in regulating energy homeostasis. Acting in opposition to the pro-opiomelanocortin/cocaine- and amphetamine-regulated transcript neurons, activation of the NPY/AgRP neurons by leptin or insulin results in a reduction of orexigenic neuropeptide expression, which leads to a decrease in food intake and an increase in energy expenditure (3).
At the cellular level, insulin and leptin signal transduction are essential in mediating peripheral processes, such as feeding suppression and glucose homeostasis (4). Insulin-mediated hypothalamic regulation of feeding and glucose homeostasis is mediated largely through the insulin receptor substrate (IRS)–phosphatidylinositol-3-kinase (PI3K) pathway (5, 6). Under normal conditions, insulin action results in the activation of Akt (protein kinase B), which phosphorylates the transcription factor forkhead box protein 1 (FoxO1) to decrease the transcription of orexigenic neuropeptides (7). Thus, when insulin action is impaired, the inhibitory pressure on FoxO1 action is relieved, and orexigenic neuropeptides are up-regulated. The regulation of neuropeptide gene expression in hypothalamic neurons essentially underlies hypothalamic maintenance of energy homeostasis. Impairment of central insulin action has been shown to be a primary factor involved in the development of obesity (8–11). Similarly, leptin administered centrally or peripherally decreases food intake and increases energy expenditure (12, 13). Leptin signaling occurs primarily through the Janus tyrosine kinase (JAK)–signal transducer and activator of transcription (STAT) pathway, although it is also known to activate the PI3K and the mitogen-activated protein kinase (MAPK)–extracellular signal–regulated kinases (ERK) pathway in the hypothalamus (14–18). Mice lacking functional leptin receptors or with attenuated leptin signaling exhibit severe obesity (19, 20). Central administration of leptin in obese, leptin-deficient mice can ameliorate the obese phenotype (21, 22). Although both hormones function through distinct signal transduction pathways, recent evidence has proven substantial crosstalk between insulin and leptin signaling at various levels of the IRS-PI3K pathway, perhaps as an adaptive mechanism to ensure tight regulation of nutrient homeostatic processes (23–26). Although the role of the PI3K pathway in leptin action is not as well understood, it has been shown that intracerebroventricular administration of PI3K inhibitors hinders the ability of leptin to reduce food intake (27). Given that insulin and leptin signaling in the hypothalamus is strongly linked and integral to the maintenance of whole-body energy homeostasis, understanding the molecular mediators linking these pathways is critical (23).
Comprehensive details on the cellular mechanisms underlying central insulin and leptin signaling are limited, particularly in the hypothalamus (28). Given the evidence supporting the overlap of insulin and leptin signal transduction, it is postulated that inhibitory crosstalk between these pathways is likely to significantly contribute to the progression of obesity and related morbidities such as type 2 diabetes mellitus (23–26, 29). Based on this notion, numerous studies have focused on potential mechanisms that underlie leptin and insulin resistance, and progress in this field has identified suppressor of cytokine signaling 3 (SOCS3) as a key cytoplasmic negative regulator of leptin and insulin signaling (30–33). SOCS3 may underlie putative negative crosstalk between the insulin and leptin signaling pathways (34, 35). In accordance with this, recent in vivo studies provide credence to the idea that insulin and leptin modulate each another's actions at the neuronal level via the PI3K pathway (36, 37). Through the use of leptin-deficient and leptin receptor-deficient mice, it was shown that compromised leptin signaling results in decreased hypothalamic insulin sensitivity (37). Similarly, a recent study determined that chronic leptin infusion resulted in attenuated leptin signaling, which in turn attenuated hypothalamic insulin sensitivity (36). These studies clearly demonstrate that exposure to chronically high leptin levels modulates intracellular signaling components of the insulin-signaling pathway in the hypothalamus. Thus, chronic exposure to elevated leptin levels in the hypothalamus can negatively impact insulin signaling. However, the mechanisms involved in the role of insulin in sensitizing hypothalamic neurons to leptin action remain to be elucidated (37). Hyperinsulinemia is often the driver of insulin resistance, and it has been suggested, although not yet confirmed, that central insulin resistance may contribute to or exacerbate central leptin resistance (11). Recent evidence suggests that hyperinsulinemia, resulting from environmental factors as well as genetic susceptibility, is the primary cause of insulin resistance, which leads to the development of diabetes and obesity in mammals (8, 38). Thus, whether insulin resistance can hinder leptin modulation of hypothalamic neurons is a pressing question that remains to be determined.
Insulin resistance (or the attenuated cellular response to insulin) is a hallmark feature in altered energy homeostasis leading to obesity. The mechanisms involved in the onset of insulin resistance have been well established in the periphery (28, 39). However, much less is known about the mechanisms involved in the onset of central insulin resistance, resulting in perturbed glucose and energy homeostasis (40). In addition, the molecular consequences of insulin resistance, including the central effects of resistance on transcriptional regulation of key components involved in signal transduction or hypothalamic feeding-related neuropeptides remain unknown. In the present study, we aimed to determine whether cellular insulin resistance alters leptin signal transduction or leptin-mediated control of hypothalamic neuropeptide expression to determine the effects of chronic exposure to increased insulin levels on cellular leptin sensitivity. We used a novel, immortalized neuronal cell model, rHypoE-19, which was derived from embryonic rat hypothalamus (41) as well as the mHypoA-2/10 cell, derived from adult mouse hypothalamus (42) to corroborate key findings. These neuronal models display functional characteristics similar to those of in vivo models and express the appropriate cellular machinery, enabling detailed investigations of the molecular mechanisms underlying normal and perturbed hormonal modulation of a hypothalamic feeding-related neuronal subtype.
Materials and Methods
Cell culture and reagents
rHypoE-19 and mHypoA-2/10 neurons, generated as described previously (41–43), were cultured in monolayer in Dulbecco's modified Eagle's medium (Sigma-Aldrich, Oakville, Ontario, Canada) supplemented with 5% fetal bovine serum and 1% penicillin-streptomycin (GIBCO, Burlington, Ontario, Canada). Cells were grown in 5% CO2 at 37°C, as described previously. We used human biosynthetic insulin (a gift from Novo-Nordisk Canada Inc., Mississauga, Ontario, Canada), which was diluted in phosphate-buffered saline (PBS). Lyophilized mouse recombinant leptin was obtained from the National Hormone and Peptide Program (A. F. Parlow, Torrance, California) and reconstituted in PBS. The G protein β antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, California). The anti-Akt, phospho-specific Akt (Ser473), ERK1/2 (Thr202/Tyr204), phospho-specific ERK1/2 (p44/p42), anti-STAT3, phospho-specific STAT3 (Tyr705), anti-JAK2, phospho-specific JAK2 (Tyr1007/1008), phospho-specific IRβ, anti-FoxO1, phospho-specific FoxO1 (Ser256), and SOCS3 antibodies were obtained from Cell Signaling Technology Inc (Danvers, Massachusetts). The PI3K inhibitors wortmannin and LY294002 were purchased from Tocris Bioscience (Ellisville, Missouri) and diluted in dimethyl sulfoxide (DMSO).
Semiquantitative RT-PCR
rHypoE-19 neurons were grown in 60-mm plates to approximately 80% to 85% confluence. The guanidinium thiocyanate-phenol-chloroform extraction protocol was used to extract total RNA (44). The purity ratio measurements and concentration of RNA samples were quantified with the NanoDrop 2000c spectrophotometer (Thermo Scientific, Nepean, Ontario, Canada). RNA was treated with Turbo DNase (Ambion, Streetsville, Ontario, Canada) to remove genomic DNA in the samples. A OneStep RT-PCR was then performed (QIAGEN, Mississauga, Ontario, Canada) by combining 200 to 400 ng of DNase-treated RNA template with OneStep RT-PCR buffer, dNTPs, enzyme mix, and primers according to the manufacturer's instructions. Next, PCR products were separated on an agarose gel and visualized under UV light. PCR products were sequenced to confirm fragment identity.
Quantitative RT-PCR (qRT-PCR)
RNA (DNase-treated, 2–4 mg) was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Streetsville, Ontario, Canada). Then 50 ng of cDNA was amplified by real-time PCR using an SYBR Green Master Mix and 0.2 U of Platinum Taq DNA polymerase (Invitrogen, Burlington, Ontario, Canada), with specific primers to each gene (see Supplemental Figure 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org.doc). Each sample was run in triplicate on the Applied Biosystems Prism 7900 Sequence Detection System and amplified according to the following cycle sequences: 2 minutes at 50°C, 10 minutes at 95°C; 40 cycles of 15 seconds at 95°C, 1 minute at 60°C; and 15 seconds at 95°C, 15 seconds at 60°C, and finally 15 seconds at 95°C. Real-time, quantitative PCR data was quantified using the standard curve method and normalized to the housekeeping gene, 18S rRNA.
Western blot analysis
rHypoE-19 and mHypoA-2/10 cells were grown to 90% to 95% confluence, serum-starved for 4 to 16 hours, and then treated with 10 nM insulin, leptin, or PBS vehicle. The cells were harvested at 0, 5, 15, 30, and/or 60 minutes after treatment using a 1× lysis buffer (Cell Signaling Technology Inc.) supplemented with 1 mM phenylmethylsulfonyl fluoride. Total protein (30 μg) was run on an 8% sodium dodecyl sulfate-polyacrylamide gel and transferred onto an Immobilon-P polyvinylidene difluoride membrane (Bio-Rad, Mississauga, Ontario, Canada). The membranes were incubated with 5% bovine serum albumin in Tris-buffered saline with 0.1% Tween for 1 hour to prevent the nonspecific binding of antibodies. Next, the blots were incubated overnight at 4°C with primary antibody after which they were complexed with secondary horseradish peroxidase antibody and visualized using the enhanced chemiluminescence method captured using a KODAK Image Station. Western blot experiments were normalized using the relative value of the phosphorylated samples divided by the total protein control values for the specific protein analyzed.
Statistics
Data are presented as the mean ± SEM and were analyzed by GraphPad Prism (GraphPad Software Inc., San Diego, California) software. A t test, 2-way ANOVA, or 1-way ANOVA was used, as indicated, followed by a Bonferroni post hoc test to ascertain statistical significance.
Results
Characterization of the neuronal models
The rHypoE-19 neurons were immortalized and generated from primary cultures of hypothalamic cells derived from embryonic day 18 rats, as reported previously (41, 43). This cell line exhibits neuronal morphology and expresses neuronal markers with a unique array of receptors and neuropeptides (41). To evaluate whether this cell line was appropriate for use in the present study, a detailed gene expression profile was conducted using RT-PCR (Supplemental Figure 1). Robust expression of insulin receptor (IR), the long form of the leptin receptor (ObRb), and essential components of the insulin and leptin signaling cascades, including IRS1, IRS2, and SOCS3 (35), were expressed in the rHypoE-19 neurons. The expression of feeding-related neuropeptides, such as AgRP, NPY, and UCN2, was also detected, along with prohormone convertases PC1/3 and PC2, previously found to be coexpressed in AgRP neurons (45). To corroborate key results, the mHypoA-2/10 neurons, an adult cell line derived from mouse hypothalamus, was used (42). The mHypoA-2/10 neuronal cell line has been characterized previously as a leptin- and insulin-responsive cell line with an expression profile similar to that of the rHypoE-19 neurons (Supplemental Figure 1) (46).
Leptin-mediated regulation of gene expression in rHypoE-19 neuronal model
After treatment with 10 nM leptin or PBS vehicle, total mRNA was isolated at 1, 2, 4, 8, and 24 hours and leptin-dependent modifications in IR, IRS1, IRS2, AgRP, and UCN2 mRNA levels were determined using qRT-PCR. Given the well-established repression of hypothalamic AgRP gene expression by leptin in vivo (26, 47, 48), we assessed changes in AgRP on leptin treatment. Indeed, leptin rapidly and significantly repressed AgRP mRNA levels, with changes in expression occurring 1 hour after treatment (Figure 1a). In accordance with the central anorexigenic actions of leptin, we found that treatment with 10 nM leptin significantly up-regulated transcript levels of the anorexigenic neuropeptide UCN2 (Figure 1b) at 8 and 24 hours. Similarly, significant up-regulation of transcription levels for insulin-sensitizing genes, the IR (Figure 1e), and other key signaling molecules of the PI3K pathway, such as IRS1 (Figure 1c) and IRS2 (Figure 1d) also occurred at 8 and 24 hours after treatment with 10 nM leptin. Overall, our results indicate that leptin exhibits a significant role in the regulation of neuronal gene expression linked to insulin signaling in the rHypoE-19 hypothalamic model.
Figure 1.

Leptin modulates mRNA expression. rHypoE-19 neurons were treated with 10 nM leptin or PBS vehicle over a 24-h time course. RNA was isolated at 1, 2, 4, 8, and 24 hours after treatment, and mRNA expression was determined by qRT-PCR primers for AgRP (a), UCN2 (b), IRS1 (c), IRS2 (d), and IR (e). mRNA levels were normalized to 18S rRNA. Data are shown with SEMs (n = 3–6 independent experiments). *, P < .05; **, P < .01; ***, P < .001, vs vehicle control, as per 2-way ANOVA with a Bonferroni post hoc test.
Leptin induces phosphorylation of JAK2, STAT3, and Akt in rHypoE-19 neuronal cells
It is well known that leptin action in the hypothalamus occurs predominantly via the JAK-STAT pathway (16–18). However, whether leptin activates Akt in the hypothalamus remains to be confirmed. Some studies have shown leptin-induced phosphorylation of Akt (26, 36), whereas others indicate that leptin does not significantly modulate the phospho status of this protein phosphorylation of Akt, suggesting that crosstalk may occur upstream or outside of the classic PI3K pathway (37, 49). To determine whether the classic leptin-signaling pathway, in addition to the PI3K-Akt pathway, is activated by leptin in the rHypoE-19 neuronal cell line, neurons were treated with 10 nM leptin over a 60-minute time course. The phosphorylation status of Akt, JAK2, and STAT3 were then analyzed. Indeed, leptin-induced phosphorylation of JAK2 (Figure 2a) and STAT3 (Figure 2b) 15 minutes after treatment. We found that leptin also induced phosphorylation of Akt (Figure 2c) after 15 minutes of 10 nM leptin treatment. Our findings demonstrate that leptin activates the classic JAK-STAT, as well as the PI3K-Akt pathway in the rHypoE-19 model.
Figure 2.
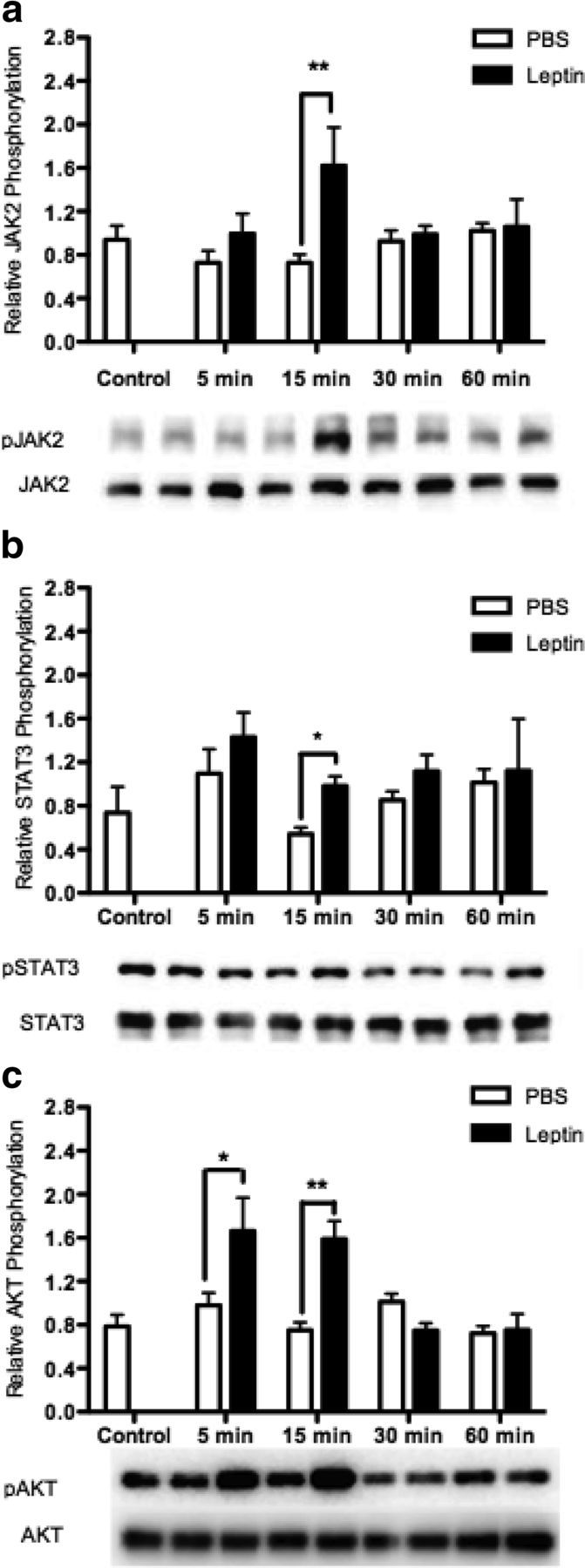
Leptin induces phosphorylation of JAK3, STAT3, and AKT. rHypoE-19 neurons were treated with 10 nM leptin or PBS vehicle, and protein was isolated at 0 (nontreatment control), 5, 15, 30, and 60 minutes after treatment, and analyzed using Western blot analysis with phospho-specific antibodies against STAT3 (a), JAK2 (b), and AKT (c). All blots were probed with total STAT, total JAK, or total AKT antibody as a loading control. Data are shown with SEMs (n = 4–7 independent experiments). *, P < .05; **, P < .01, vs vehicle control, as per 2-way ANOVA with a Bonferroni post hoc test.
Insulin pretreatment attenuates insulin-mediated phosphorylation of Akt and ERK1/2 in the rHypoE-19 neuronal model
As expected in the rHypoE-19 neurons, we found that insulin activates PI3K, indicated by continuous induction of Akt phosphorylation at 5, 15, 30, and 60 minutes after insulin treatment (Figure 3a). Furthermore, it was shown that insulin phosphorylates and activates ERK1/2, 15 minutes after treatment (Figure 3b). Similarly, this short-term induction of phospho-ERK1/2 has been detected previously at 5 minutes after insulin treatment in a hepatic and hypothalamic cell line (50, 51). This temporal activation of ERK1/2, in comparison with Akt, may be due to the formation of a negative feedback loop by insulin-induced activation of ERK phosphatases (51).
Figure 3.

Insulin induces phosphorylation of AKT and ERK1/2. rHypoE-19 neurons were treated with 10 nM insulin or PBS vehicle, and protein was isolated at 0 (control), 5, 15, 30, and 60 minutes after treatment and analyzed using Western blot analysis with phospho-specific antibodies against AKT (a) and ERK1/2 (b). All blots were probed with either total AKT or total ERK antibody as a loading control. Data are shown with SEMs (n = 3–4 independent experiments). ***, P < .001; **, P < .01 as per 2-way ANOVA with a Bonferroni post hoc test.
To determine the minimum dose of insulin pretreatment required to induce a state of insulin resistance in the rHypoE-19 neurons, cells were pretreated with PBS vehicle or 10 nM, 100 nM, and 1000 nM insulin for 24 hours. Subsequent to pretreatment, the cells were washed with PBS, placed in fresh medium containing no insulin for 1 hour, and then rechallenged with 10 nM insulin to determine whether or not insulin signaling was attenuated, as assessed by Akt and/or ERK1/2 phosphorylation. Protein was then isolated 15 minutes after insulin rechallenge. With the use of Western blot analysis, it was determined that a minimum pretreatment of 100 nM insulin for 24 hours was required to significantly attenuate insulin signaling, as demonstrated by the loss of insulin-induced phosphorylation of Akt (Figure 4a) as well as ERK1/2 (Figure 4b) in the rHypoE-19 neurons, which had not been documented previously.
Figure 4.

Chronic insulin exposure attenuates insulin-induced activation of PI3K and MAPK pathways. rHypoE-19 neurons were pretreated with PBS or 10 nM, 100 nM, or 1000 nM insulin for 24 hours. The cells were then washed with PBS and allowed to recover in fresh medium for 1 hour before being rechallenged with 10 nM insulin or PBS vehicle. Relative phosphorylated AKT (a) or ERK1/2 (b) levels were normalized to total AKT or total ERK levels, respectively. Data are shown with SEMs (n = 3 independent experiments). ***, P < .001, as per 2-way ANOVA with a Bonferroni post hoc test.
Pretreatment with insulin inhibits leptin-induced phosphorylation of key signaling molecules in rHypoE-19 and mHypoA-2/10 neurons
To determine whether the induction of insulin resistance had any ramifications on the signaling cascade of leptin, cells were once again pretreated with 100 nM insulin or vehicle control for 24 hours. After insulin pretreatment, cells were washed with PBS, incubated in new medium for 1 hour, and then treated with leptin to determine leptin receptor responsiveness. With use of Western blot analysis, it was determined that insulin resistance prevented leptin-induced induction of phospho-STAT3 in both cell lines (Figure 5, c and d) and blocked the induction of phospho-Akt (Figure 5a) and the induction of phospho-ERK (Figure 5b) in the rHypoE-19 and mHypoA-19 neurons, respectively. This result indicates that endogenous leptin signaling in the hypothalamic neuronal models may be impaired after the induction of insulin resistance. Our findings also demonstrate that leptin activation of signaling pathways is cell type specific. In addition to the activation of the classic JAK-STAT pathway, leptin activates the PI3K-Akt pathway (Figure 5a) although not the ERK pathway in the rHypoE-19 model, whereas the ERK pathway (Figure 5b) and not the PI3K pathway was activated in the mHypoA-2/10 cell model. In addition, it should be noted that for leptin-induced Akt activation in the rHypoE-19 cell line, the possibility that the changes seen at the protein level can at least be partially attributable to chronic insulin exposure itself cannot be ruled out, given the increase in phospho-Akt levels (Figure 5a) after the induction of insulin resistance.
Figure 5.

Chronic insulin exposure attenuates leptin-induced phosphorylation of key signaling molecules. rHypoE-19 and mHypoA-2/10 neurons were pretreated with PBS vehicle or 100 nM insulin for 24 hours. The cells were then washed and allowed to recover in fresh medium for 1 hour before being rechallenged with 10 nM leptin or PBS vehicle. Protein was isolated at 15 minutes after treatment and analyzed using Western blot analysis with phospho-specific antibodies against AKT (a), ERK1/2 (b), or STAT3 (c and d), and protein levels were normalized to total AKT, total ERK1/2, or total STAT3 levels, respectively. Data are shown with SEMs (n = 3–5 independent experiments). *, P < .05; **, P < .001; as per t tests.
Pretreatment with insulin inhibits leptin-mediated regulation of gene expression in rHypoE-19 neuronal model
To determine the effects of prolonged insulin exposure on leptin modulation of the rHypoE-19 neurons, the transcriptional regulation of leptin was analyzed before and after the induction of insulin resistance. As described previously, rHypoE-19 neurons were pretreated with 100 nM insulin for 24 hours after treatment with 10 nM leptin or PBS vehicle. RNA was isolated at 1 and 8 hours after leptin treatment, and transcript levels were analyzed using qRT-PCR. It was determined that pretreatment with insulin for 24 hours prevented the leptin-induced reduction of AgRP transcript levels (Figure 6a). In addition, 24-hour insulin pretreatment prevented leptin-induced up-regulation of IRS1 (Figure 6c), IRS2 (Figure 6d), and IR (Figure 6e). Similarly, insulin resistance hindered leptin-mediated effects on UCN2 transcript levels (Figure 6b). Therefore, these data indicate that chronic insulin exposure impairs the transcriptional regulation of genes by leptin in the rHypoE-19 neurons. However, the possibility that insulin resistance may at least partially contribute to the changes in transcript levels independently of leptin effects could not be ruled out, because up-regulation of certain transcript levels was observed with insulin pretreatment alone (UCN2 [Figure 6b], IRS1 [Figure 6c], and IR [Figure 6e]).
Figure 6.
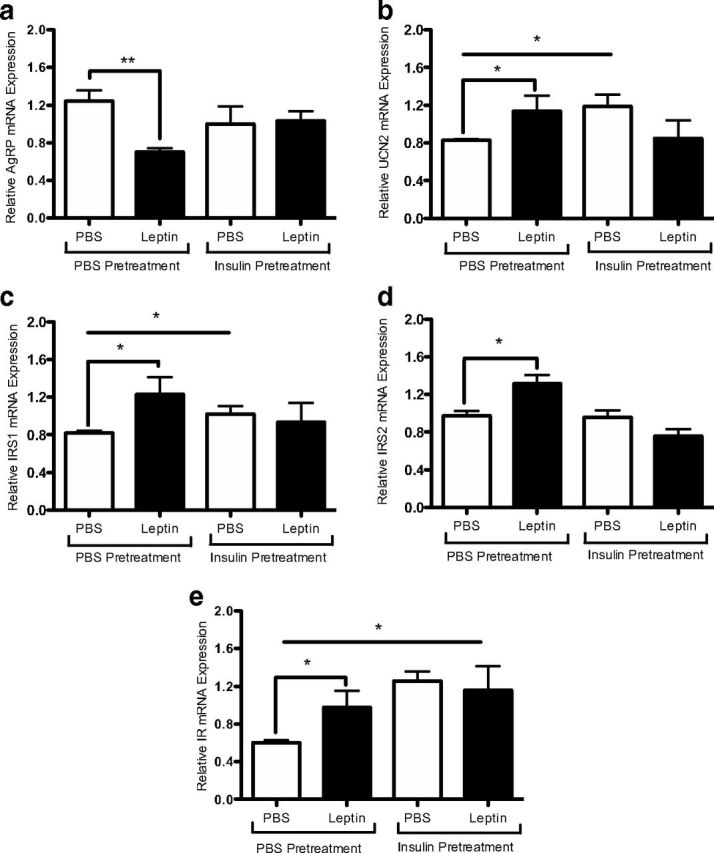
Leptin modulation of neuronal transcript levels was abolished after induction of insulin resistance. rHypoE-19 neurons were pretreated with either PBS or 100 nM insulin for 24 hours. The cells were then washed and allowed to recover in fresh medium for 1 hour before 10 nM leptin treatment for 1 hour (AgRP only) or 8 hours. Insulin resistance attenuates a leptin-induced decrease of AgRP (a) and increases in UCN2 (b), IRS1 (c), IRS2 (d), and IR (e) transcript levels. mRNA levels were determined by qRT-PCR and normalized to 18S rRNA and shown with SEMs (n = 3–4 independent experiments). *, P < .05; **, P < .01, vs vehicle control, as per t tests.
LY294002 and wortmannin inhibit leptin-mediated regulation of gene expression in rHypoE-19 neuronal model
To confirm that the PI3K-Akt pathway is involved with the attenuation of leptin-mediated regulatory control over hypothalamic neurons, the PI3K-specific inhibitors, LY294002 and wortmannin, were used. The appropriate inhibitor concentrations were determined previously using comparable hypothalamic cell lines (51). To ensure the efficacy and specificity of each inhibitor, rHypoE-19 cells were pretreated with either PBS vehicle, DMSO vehicle, 25 μM LY294002, or 1 μM wortmannin for 1 hour, followed by insulin treatment. Protein was isolated and analyzed using Western blot analysis with phospho-specific antibodies for Akt. Both LY294002 and wortmannin inhibited the phosphorylation of Akt by insulin, whereas pretreatment with PBS and DMSO vehicle maintained the insulin-induced phosphorylation of Akt (Figure 7a). Next, to determine whether attenuation of the PI3K pathway accounts for the loss of regulatory control by leptin in neurons, rHypoE-19 cells were pretreated with PBS vehicle, DMSO vehicle, 25 μM LY294002, or 10 μM wortmannin for 1 hour, followed by 10 nM leptin treatment. Total mRNA was isolated 1 hour or 8 hours after treatment as indicated and analyzed using qRT-PCR. Both LY294002 and wortmannin abolished the decrease of AgRP mRNA expression by leptin, whereas pretreatment with PBS or DMSO vehicle retained the leptin-induced decrease of AgRP transcript levels (Figure 7b). Furthermore, the increases in UCN2 (Figure 7c), IRS1 (Figure 7d), IRS2 (Figure 7e), and IR (Figure 7f) transcript levels were diminished after inhibitor treatment, whereas PBS and DMSO pretreatment did not hinder the previously determined effects of leptin to increase these transcript levels at the time points examined (refer to Figure 2, a–d). Taken together, these results suggest that the transcriptional regulation mediated by leptin in the rHypoE-19 neurons requires intact PI3K signal transduction.
Figure 7.

Leptin modulation of neuronal transcript levels is abolished after treatment with LY294002 or wortmannin. rHypoE-19 neurons were pretreated with PBS vehicle, DMSO vehicle, 25 μM LY294002 (LY), or 1 μM wortmannin (WORT) for 1 hour before treatment with 10 nM leptin or PBS vehicle. First, it was ensured that induction of phospho-AKT after 15 minutes of insulin treatment is blocked by LY294002 and wortmannin (a). mRNA was isolated 1 hour (for AgRP only) or 8 hours after treatment as indicated and analyzed using qRT-PCR. Pretreatment with inhibitors attenuates leptin-induced increase of UCN2 (b), IR (c), IRS1 (d), IRS2 (e), and decrease in AgRP (f) transcript levels. mRNA levels were normalized to 18S rRNA and shown with SEMs (n = 4–9 independent experiments). *, P < .05; **, P < .01; **, P < .001, as per 2-way ANOVA with a Bonferroni post hoc test.
Chronic insulin exposure modulates intracellular signaling components of the insulin and leptin signaling pathway in rHypoE-19 and mHypoA-2/10 neurons
Cells were pretreated with PBS vehicle or 100 nM insulin for 24 hours to induce insulin resistance (Figure 8, a and b). The neurons were then washed and allowed to recover in fresh medium for 1 hour before a rechallenge with 10 nM insulin or PBS vehicle. Protein was isolated at 15 minutes after treatment and analyzed using Western blot analysis. It was found that chronic insulin exposure decreased protein levels of the β subunit of the IR (Figure 8, c and d) and prevented the increase in the phosphorylation of FoxO1, seen with 10 nM insulin treatment (Figure 8, e and f). In addition, chronic exposure to insulin significantly up-regulated the protein levels of SOCS3 (Figure 8, g and h), a known inhibitor of both insulin and leptin signal transduction. Taken together, these results indicate that the induction of cellular insulin resistance can alter overall cellular function and normal leptin signaling through specific alterations in components common to both pathways as observed in 2 distinct hypothalamic cell lines.
Figure 8.

Effects of chronic insulin treatment on intracellular signaling components of the leptin- and insulin-signaling pathways. rHypoE-19 and mHypoA-2/10 neurons were pretreated with PBS vehicle (−) or 100 nM insulin (+) for 24 hours. The cells were then washed and allowed to recover in fresh medium for 1 hour before a rechallenge with 10 nM insulin or PBS control. Protein was isolated at 15 minutes after treatment and analyzed using Western blot analysis with phospho-specific antibodies against AKT (a and b), IRβ (c and d), FoxO1 (e and f), and SOCS3 (g and h). Protein levels were normalized to total AKT and FoxO1 and G protein (Gβ). Data are shown with SEMs (n = 3–9 independent experiments). **, P < .01; ***, P < .001, as per 1-way ANOVA with a Bonferroni post hoc test.
Discussion
Central leptin and insulin signaling is critical in maintaining nutrient homeostasis through feeding suppression. Resistance to the biological effects of both hormones is strongly linked to the development of obesity and type 2 diabetes mellitus. Within the hypothalamus, there is substantial evidence that leptin and insulin levels are reflective of energy homeostasis and adiposity and that they both exert their central regulatory effects, at least in part, through the PI3K pathway (23, 52–55). Although it is known that leptin resistance can lead to decreased hypothalamic insulin sensitivity (36, 37), whether central insulin resistance can contribute to leptin resistance is not known. Our data have implicated hyperinsulinemia as a proximate cause or contributing factor in the development or progression of both cellular insulin and leptin resistance, suggesting that insulin can modulate leptin signaling (8–10). In the present study, we determined that induction of cellular insulin resistance does indeed attenuate leptin modulation of hypothalamic neurons at the protein signaling and transcriptional levels as seen in the rHypoE-19 and in the mHypoA-2/10 hypothalamic cell model.
Consistent with the literature, we demonstrated that leptin significantly repressed AgRP mRNA levels rapidly, with changes in expression occurring 1 hour after treatment in the rHypoE-19 model. This rapid effect is probably attributed to changes in AgRP mRNA stability rather than an inhibition of transcription. Furthermore, we analyzed transcript levels of UCN2, a member of the corticotropin-releasing hormone family of peptides, which are novel regulators of energy homeostasis and metabolism (56). Within the hypothalamus, urocortins have an important role in decreasing food intake (56–58), but there is limited information about the mediators of the anorexigenic actions of urocortins. In line with its central anorexigenic actions, we found that leptin could directly up-regulate the transcript levels of the potent satiety hormone UCN2 in the rHypoE-19 hypothalamic neuronal line. This finding directly implicates UCN2 in leptin-mediated control of food intake for the first time.
Leptin is known to be a potent insulin sensitizer in the central nervous system, although the mechanisms through which it might accomplish this effect are unclear (26, 36, 37, 59). The increase in IR transcript levels, in addition to crucial insulin signaling molecules, such as IRS1 and IRS2, may serve as a mechanism through which leptin sensitizes neurons to insulin by mediating more efficient insulin signaling and thus heightening insulin sensitivity. It was shown previously that in mature adipocytes, increased expression of IR and IRS proteins leads to improved cellular sensitivity to insulin (60). We also showed that ERK activation is attenuated in response to chronic insulin exposure, which has not been known to occur previously, as it is thought that insulin resistance affects the PI3K and MAPK signaling pathways in the periphery differentially (61, 62). Given the heterogeneity of hypothalamic neuronal phenotypes, the molecular effects of insulin resistance may be cell type specific as has been suggested recently (62).
Efforts thus far have not addressed the changes in gene expression that may accompany defects in the insulin and leptin signaling cascades and have failed to investigate a potentially fundamental aspect of central resistance to leptin action (63). It was found that leptin-dependent regulation of neuropeptide expression, specifically of AgRP and UCN2, was abolished upon the induction of insulin resistance. In addition, the loss of transcriptional regulation mediated by leptin over IRS and IR mRNA and protein expression may contribute to the reduction in PI3K activity, ultimately leading to a decrease in insulin sensitivity. Chronic insulin treatment decreases IRS2 mRNA and IRS1 protein levels by proteosomal degradation in cultured hepatocytes, which may contribute to the onset of insulin resistance (64–66). However, not much is known about the actions of leptin in insulin resistance, specifically in the regulation of IRS1, IRS2, and IR expression. Although leptin signals mainly through the JAK-STAT3 pathway, it also utilizes the PI3K pathway to modulate metabolism (23, 26, 27, 36, 37, 67). If the ability of leptin to decrease feeding is dependent on central PI3K signaling, then defective activation of this pathway due to insulin resistance may contribute to the attenuation of leptin-dependent modulation of neuropeptide transcription, key signaling molecules, and receptors within hypothalamic neurons. Accordingly, leptin-induced up-regulation of these important mediators of insulin action may underlie its established role in increasing neuronal sensitivity to insulin (36, 37). Taken together, our data suggest that chronic insulin exposure impairs the ability of leptin to appropriately regulate transcription. This probably has important physiological ramifications, and these results should be subsequently validated in an animal model.
PI3K-specific inhibitors abolished transcriptional regulation by leptin, mimicking chronic insulin exposure and thereby implicating PI3K activity in downstream neuropeptide transcription. Leptin requires activation of mammalian target of rapamycin signaling, which is in turn triggered by the activation of the PI3K/Akt cascade for its inhibition of food intake (4, 68, 69). Indeed, it was shown previously that intracerebroventricular injection of PI3K inhibitors abolishes the effect of leptin in the reduction of food intake (27, 70), inhibits the central actions of leptin in the increase of peripheral insulin sensitivity (26), and inhibits the central action of leptin in the enhancement of hypothalamic insulin sensitivity (37). On the other hand, it was shown that pretreatment of human embryonic kidney 293 cells with insulin induces a state of leptin resistance by abolishing the effects of leptin on JAK2 phosphorylation and reducing JAK2-associated PI3K activity (54). Although the mechanisms of negative crosstalk remain unknown, it is suggested that leptin signaling may be attenuated as a result of defective insulin signaling in peripheral cells (54). Akt phosphorylation can be abolished as a result of numerous postreceptor defects, which include serine phosphorylation or degradation of IRS proteins, leading to decreased PI3K activity and as a result of decreased Akt phosphorylation (51). These results provide evidence that an attenuation of PI3K pathway activity can abolish the transcriptional regulation of neuropeptides by leptin.
We have shown that insulin resistance interferes with leptin-induced activation of key signaling molecules. Interestingly, leptin induced activation of the PI3K-Akt pathway in the rHypoE-19 neurons but failed to do so in the mHypoA-2/10 neurons. In contrast, the MAPK-ERK pathway was activated in the mHypoA-2/10 neurons but not in the rHypoE-19 neurons. When these results are evaluated, it is important to remember that the hypothalamus is composed of numerous subpopulations of neurons, and, as a result, different cell types may respond differentially to specific stimuli. In vivo studies assess the overall integrated response of the hypothalamus in response to stimuli; the cell models used in this article are able to address the questions in the emerging area of neuron-specific and pathway-specific changes that accompany insulin resistance (62). It is also important to note that the induction of insulin resistance alone may, at least partially, account for altered gene and protein regulation in addition to or independent of leptin effects. These data provide credence to the notion of cell type–specific responses to hormonal stimulation and thus resistance (62, 71). Given the heterogeneity of the hypothalamic architecture, in contrast with the largely uniform cell types of peripheral organs, it is reasonable that distinct leptin-responsive neuronal subtypes may respond differentially to hormonal stimulation. However, leptin activation of the JAK-STAT pathway occurred in both hypothalamic cell lines and was attenuated after the induction of insulin resistance.
As a possible mechanism for the reduction of both STAT3 and Akt phosphorylation, we hypothesized that chronic stimulation of the IR increases expression of SOCS3, a cytoplasmic key negative regulator of leptin and insulin signaling (31). We found that there was a significant up-regulation of SOCS3 protein after pretreatment with insulin. SOCS3 may underlie substantial crosstalk between the insulin and leptin signaling pathways (34–36). Like leptin, insulin induces expression of SOCS3, and it has been shown that decreased neuronal SOCS3 expression enhances sensitivity to both hormones (33, 72). Accordingly, it was recently suggested that increased ObRb stimulation increases SOCS3 gene expression causing SOCS3 protein levels to reach a threshold concentration that causes reduced STAT3 activation, contributing to the development of diet-induced obesity (73). Based on these findings, it is possible that overactivation of the IR, which is also known to increase expression of SOCS3 (72), may result in decreased activation of STAT3 and Akt, leading to hindered insulin and leptin signaling. Although leptin controls feeding by its transcriptional regulation of hypothalamic neuropeptides, the mechanism through which it modulates gene expression remains unclear (7). It has been shown that impaired FoxO1 nuclear exclusion in hypothalamic neurons renders leptin unable to decrease food intake and suppress AgRP transcription, which in turn contributes to the development of metabolic diseases (7, 40, 74). We provide evidence that insulin resistance abolishes insulin-induced phosphorylation and thus cytoplasmic sequestration of FoxO1, which may also contribute to the decreased transcriptional regulation of hypothalamic neurons by leptin.
Ultimately, it is essential to characterize the cellular and molecular ramifications of insulin resistance on hypothalamic neurons to develop effective therapeutic agents for hormone resistance. In particular, to determine how insulin resistance interferes with leptin action in neurons, given the crosstalk between these hormone pathways, is requisite. Whether these signaling changes induced by insulin resistance translate to gene expression changes in these neurons and therefore interfere with this principal aspect of central leptin and insulin action has not been addressed before this. Investigation of the rHypoE-19 and mHypoA-19 hypothalamic cell models suggests that chronic exposure to insulin causes a cross-desensitization of the leptin signaling cascade and modifies the neuronal responses to leptin at the protein and transcriptional levels (Figure 9). It is particularly noteworthy that SOCS3 protein levels were significantly up-regulated with prolonged insulin exposure. The rHypoeE-19 and the mHypoA-2/10 neurons present as a useful tool to investigate the molecular basis of normal and perturbed hormone action such as identifying dysregulated microRNAs that have been recently defined as a cause of cell type–specific and pathway-specific insulin resistance (62). Perturbation of the regulatory control by leptin of key signaling molecules, receptors, and neuropeptides that mediate its anorexigenic effects through disruption of the PI3K pathway may contribute to or perpetuate the unfavorable metabolic profile associated with obesity and its comorbidities.
Figure 9.

Model of the putative role of central insulin resistance in attenuating leptin modulation of rHypoE-19 hypothalamic neurons. Chronic insulin stimulation attenuates activity of the PI3K pathway by hindering phosphorylation of Akt that may be required leptin-mediated transcriptional regulation of neurons. In addition, chronic stimulation of the insulin receptor may lead to an increase in SOCS3 protein levels, which are known to hinder both leptin and insulin signal transduction by dephosphorylation of the insulin receptor, JAK, STAT, and IRS proteins. This may, in part, account for the observed decrease in leptin-induced phosphorylation of Akt and STAT3 after induction of insulin resistance.
Acknowledgments
We thank to Dr. Leigh Wellhauser for critical reading of the manuscript.
This work was supported by the Canadian Institutes for Health Research (CIHR), Canadian Diabetes Association, Canada Foundation for Innovation, and Canada Research Chairs Program. A.N.-A. was supported by a CIHR MSc Studentship, and J.A.M. was supported by a Banting and Best Diabetes Centre Studentship.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AgRP
- agouti-related peptide
- DMSO
- dimethyl sulfoxide
- ERK
- extracellular signal-regulated kinase
- FoxO1
- forkhead box protein 1
- IR
- insulin receptor
- IRS
- insulin receptor substrate
- JAK
- Janus tyrosine kinase
- MAPK
- mitogen-activated protein kinase
- NPY
- neuropeptide Y
- PI3K
- phosphatidylinositol 3-kinase
- PBS
- phosphate-buffered saline
- qRT-PCR
- quantitative RT-PCR
- SOCS3
- suppressor of cytokine signaling 3
- STAT
- signal transducer and activator of transcription.
References
- 1. Morton GJ , Cummings DE , Baskin DG , Barsh GS , Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. [DOI] [PubMed] [Google Scholar]
- 2. Schwartz MW , Woods SC , Porte D , Seeley RJ , Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. [DOI] [PubMed] [Google Scholar]
- 3. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. [DOI] [PubMed] [Google Scholar]
- 4. Woods SC , D'Alessio DA. Central control of body weight and appetite. J Clin Endocrinol Metab. 2008;93:S37–S50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gelling RW , Morton GJ , Morrison CD , et al. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 2006;3:67–73. [DOI] [PubMed] [Google Scholar]
- 6. Niswender KD , Morrison CD , Clegg DJ , et al. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–231. [DOI] [PubMed] [Google Scholar]
- 7. Kitamura T , Feng Y , Kitamura YI , et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. [DOI] [PubMed] [Google Scholar]
- 8. Corkey BE. Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes. 2012;61:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lustig RH. Hypothalamic obesity: causes, consequences, treatment. Pediatr Endocrinol Rev. 2008;6:220–227. [PubMed] [Google Scholar]
- 10. Weyer C , Hanson RL , Tataranni PA , Bogardus C , Pratley RE. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: evidence for a pathogenic role of relative hyperinsulinemia. Diabetes. 2000;49:2094–2101. [DOI] [PubMed] [Google Scholar]
- 11. Lustig RH. Childhood obesity: behavioral aberration or biochemical drive? Reinterpreting the First Law of Thermodynamics. Nat Clin Pract Endocrinol Metab. 2006;2:447–458. [DOI] [PubMed] [Google Scholar]
- 12. Ahima RS , Prabakaran D , Mantzoros C , et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. [DOI] [PubMed] [Google Scholar]
- 13. Halaas JL , Gajiwala KS , Maffei M , et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. [DOI] [PubMed] [Google Scholar]
- 14. Cui H , Cai F , Belsham DD. Leptin signaling in neurotensin neurons involves STAT, MAP kinases ERK1/2, and p38 through c-Fos and ATF1. FASEB J. 2006;20:2654–2656. [DOI] [PubMed] [Google Scholar]
- 15. Bjørbaek C , Buchholz RM , Davis SM , et al. Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem. 2001;276:4747–4755. [DOI] [PubMed] [Google Scholar]
- 16. Banks AS , Davis SM , Bates SH , Myers MG. Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–14572. [DOI] [PubMed] [Google Scholar]
- 17. Bjørbaek C , Uotani S , da Silva B , Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. [DOI] [PubMed] [Google Scholar]
- 18. Gautron L , Elmquist JK. Sixteen years and counting: an update on leptin in energy balance. J Clin Invest. 2011;121:2087–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen H , Charlat O , Tartaglia LA , et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. [DOI] [PubMed] [Google Scholar]
- 20. Margetic S , Gazzola C , Pegg GG , Hill RA. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. 2002;26:1407–1433. [DOI] [PubMed] [Google Scholar]
- 21. Campfield LA , Smith FJ , Guisez Y , Devos R , Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. [DOI] [PubMed] [Google Scholar]
- 22. Thornton JE , Cheung CC , Clifton DK , Steiner RA. Regulation of hypothalamic proopiomelanocortin mRNA by leptin in ob/ob mice. Endocrinology. 1997;138:5063–5066. [DOI] [PubMed] [Google Scholar]
- 23. Carvalheira JB , Torsoni MA , Ueno M , et al. Cross-talk between the insulin and leptin signaling systems in rat hypothalamus. Obes Res. 2005;13:48–57. [DOI] [PubMed] [Google Scholar]
- 24. Duan C , Li M , Rui L. SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J Biol Chem. 2004;279:43684–43691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hill JW , Williams KW , Ye C , et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118:1796–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morton GJ , Gelling RW , Niswender KD , Morrison CD , Rhodes CJ , Schwartz MW. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–420. [DOI] [PubMed] [Google Scholar]
- 27. Niswender KD , Morton GJ , Stearns WH , Rhodes CJ , Myers MG , Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. [DOI] [PubMed] [Google Scholar]
- 28. Ceddia RB , Koistinen HA , Zierath JR , Sweeney G. Analysis of paradoxical observations on the association between leptin and insulin resistance. FASEB J. 2002;16:1163–1176. [DOI] [PubMed] [Google Scholar]
- 29. Berthou F , Rouch C , Gertler A , Gerozissis K , Taouis M. Chronic central leptin infusion differently modulates brain and liver insulin signaling. Mol Cell Endocrinol. 2011;337:89–95. [DOI] [PubMed] [Google Scholar]
- 30. Björnholm M , Münzberg H , Leshan RL , et al. Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest. 2007;117:1354–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Howard JK , Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. 2006;17:365–371. [DOI] [PubMed] [Google Scholar]
- 32. Howard JK , Cave BJ , Oksanen LJ , Tzameli I , Bjørbaek C , Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. [DOI] [PubMed] [Google Scholar]
- 33. Mori H , Hanada R , Hanada T , et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. [DOI] [PubMed] [Google Scholar]
- 34. Niswender KD , Schwartz MW. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocrinol. 2003;24:1–10. [DOI] [PubMed] [Google Scholar]
- 35. Morrison CD , Huypens P , Stewart LK , Gettys TW. Implications of crosstalk between leptin and insulin signaling during the development of diet-induced obesity. Biochim Biophys Acta. 2009;1792:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burgos-Ramos E , Chowen JA , Arilla-Ferreiro E , Canelles S , Argente J , Barrios V. Chronic central leptin infusion modifies the response to acute central insulin injection by reducing the interaction of the insulin receptor with IRS2 and increasing its association with SOCS3. J Neurochem. 2011;117:175–185. [DOI] [PubMed] [Google Scholar]
- 37. Koch C , Augustine RA , Steger J , et al. Leptin rapidly improves glucose homeostasis in obese mice by increasing hypothalamic insulin sensitivity. J Neurosci. 2010;30:16180–16187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mehran AE , Templeman NM , Brigidi GS , et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16:723–737. [DOI] [PubMed] [Google Scholar]
- 39. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Nutrition 1997;13:65; discussion 64, 66. [DOI] [PubMed] [Google Scholar]
- 40. Plum L , Belgardt BF , Brüning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest. 2006;116:1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gingerich S , Wang X , Lee PK , et al. The generation of an array of clonal, immortalized cell models from the rat hypothalamus: analysis of melatonin effects on kisspeptin and gonadotropin-inhibitory hormone neurons. Neuroscience. 2009;162:1134–1140. [DOI] [PubMed] [Google Scholar]
- 42. Belsham DD , Fick LJ , Dalvi PS , et al. Ciliary neurotrophic factor recruitment of glucagon-like peptide-1 mediates neurogenesis, allowing immortalization of adult murine hypothalamic neurons. FASEB J. 2009;23:4256–4265. [DOI] [PubMed] [Google Scholar]
- 43. Belsham DD , Cai F , Cui H , Smukler SR , Salapatek AM , Shkreta L. Generation of a phenotypic array of hypothalamic neuronal cell models to study complex neuroendocrine disorders. Endocrinology. 2004;145:393–400. [DOI] [PubMed] [Google Scholar]
- 44. Chomczynski P , Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. [DOI] [PubMed] [Google Scholar]
- 45. Creemers JW , Pritchard LE , Gyte A , et al. Agouti-related protein is posttranslationally cleaved by proprotein convertase 1 to generate agouti-related protein (AGRP)83–132: interaction between AGRP83–132 and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology. 2006;147:1621–1631. [DOI] [PubMed] [Google Scholar]
- 46. Dalvi PS , Erbiceanu FD , Irwin DM , Belsham DD. Direct regulation of the proglucagon gene by insulin, leptin, and cAMP in embryonic versus adult hypothalamic neurons. Mol Endocrinol. 2012;26:1339–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Korner J , Savontaus E , Chua SC , Leibel RL , Wardlaw SL. Leptin regulation of Agrp and Npy mRNA in the rat hypothalamus. J Neuroendocrinol. 2001;13:959–966. [DOI] [PubMed] [Google Scholar]
- 48. Morrison CD , Morton GJ , Niswender KD , Gelling RW , Schwartz MW. Leptin inhibits hypothalamic Npy and Agrp gene expression via a mechanism that requires phosphatidylinositol 3-OH-kinase signaling. Am J Physiol Endocrinol Metab. 2005;289:E1051–E1057. [DOI] [PubMed] [Google Scholar]
- 49. Tups A , Anderson GM , Rizwan M , et al. Both p110α and p110β isoforms of phosphatidylinositol 3-OH-kinase are required for insulin signalling in the hypothalamus. J Neuroendocrinol. 2010;22:534–542. [DOI] [PubMed] [Google Scholar]
- 50. Keeton AB , Bortoff KD , Franklin JL , Messina JL. Blockade of rapid versus prolonged extracellularly regulated kinase 1/2 activation has differential effects on insulin-induced gene expression. Endocrinology. 2005;146:2716–2725. [DOI] [PubMed] [Google Scholar]
- 51. Mayer CM , Belsham DD. Central insulin signaling is attenuated by long-term insulin exposure via insulin receptor substrate-1 serine phosphorylation, proteasomal degradation, and lysosomal insulin receptor degradation. Endocrinology. 2010;151:75–84. [DOI] [PubMed] [Google Scholar]
- 52. Carvalheira JB , Ribeiro EB , Folli F , Velloso LA , Saad MJ. Interaction between leptin and insulin signaling pathways differentially affects JAK-STAT and PI 3-kinase-mediated signaling in rat liver. Biol Chem. 2003;384:151–159. [DOI] [PubMed] [Google Scholar]
- 53. Carvalheira JB , Siloto RM , Ignacchitti I , et al. Insulin modulates leptin-induced STAT3 activation in rat hypothalamus. FEBS Lett. 2001;500:119–124. [DOI] [PubMed] [Google Scholar]
- 54. Kellerer M , Lammers R , Fritsche A , et al. Insulin inhibits leptin receptor signalling in HEK293 cells at the level of Janus kinase-2: a potential mechanism for hyperinsulinaemia-associated leptin resistance. Diabetologia. 2001;44:1125–1132. [DOI] [PubMed] [Google Scholar]
- 55. Szanto I , Kahn CR. Selective interaction between leptin and insulin signaling pathways in a hepatic cell line. Proc Natl Acad Sci USA. 2000;97:2355–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuperman Y , Chen A. Urocortins: emerging metabolic and energy homeostasis perspectives. Trends Endocrinol Metab. 2008;19:122–129. [DOI] [PubMed] [Google Scholar]
- 57. Pan W , Kastin AJ. Urocortin and the brain. Prog Neurobiol. 2008;84:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Skelton KH , Owens MJ , Nemeroff CB. The neurobiology of urocortin. Regul Pept. 2000;93:85–92. [DOI] [PubMed] [Google Scholar]
- 59. Fujikawa T , Chuang JC , Sakata I , Ramadori G , Coppari R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci USA. 2010;107:17391–17396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pederson T , Rondinone CM. Regulation of proteins involved in insulin signaling pathways in differentiating human adipocytes. Biochem Biophys Res Commun. 2000;276:162–168. [DOI] [PubMed] [Google Scholar]
- 61. Cusi K , Maezono K , Osman A , et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Könner AC , Brüning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012;16:144–152. [DOI] [PubMed] [Google Scholar]
- 63. Marino JS , Xu Y , Hill JW. Central insulin and leptin-mediated autonomic control of glucose homeostasis. Trends Endocrinol Metab. 2011;22:275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hirashima Y , Tsuruzoe K , Kodama S , et al. Insulin down-regulates insulin receptor substrate-2 expression through the phosphatidylinositol 3-kinase/Akt pathway. J Endocrinol. 2003;179:253–266. [DOI] [PubMed] [Google Scholar]
- 65. Shimomura I , Matsuda M , Hammer RE , Bashmakov Y , Brown MS , Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- 66. Ueki K , Kondo T , Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol. 2004;24:5434–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Roman EA , Reis D , Romanatto T , et al. Central leptin action improves skeletal muscle AKT, AMPK, and PGC1α activation by hypothalamic PI3K-dependent mechanism. Mol Cell Endocrinol. 2010;314:62–69. [DOI] [PubMed] [Google Scholar]
- 68. Cota D , Proulx K , Smith KA , et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. [DOI] [PubMed] [Google Scholar]
- 69. Woods SC , Seeley RJ , Cota D. Regulation of food intake through hypothalamic signaling networks involving mTOR. Annu Rev Nutr. 2008;28:295–311. [DOI] [PubMed] [Google Scholar]
- 70. Rahmouni K , Haynes WG , Morgan DA , Mark AL. Intracellular mechanisms involved in leptin regulation of sympathetic outflow. Hypertension. 2003;41:763–767. [DOI] [PubMed] [Google Scholar]
- 71. Vogt MC , Brüning JC. CNS insulin signaling in the control of energy homeostasis and glucose metabolism—from embryo to old age. Trends Endocrinol Metab. 2013;24:76–84. [DOI] [PubMed] [Google Scholar]
- 72. Emanuelli B , Peraldi P , Filloux C , Sawka-Verhelle D , Hilton D , Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. 2000;275:15985–15991. [DOI] [PubMed] [Google Scholar]
- 73. Gamber KM , Huo L , Ha S , Hairston JE , Greeley S , Bjorbaek C. Over-expression of leptin receptors in hypothalamic POMC neurons increases susceptibility to diet-induced obesity. PLoS One. 2012;7:e30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fukuda M , Jones JE , Olson D , et al. Monitoring FoxO1 localization in chemically identified neurons. J Neurosci. 2008;28:13640-13648. [DOI] [PMC free article] [PubMed] [Google Scholar]