

Abstract
Atomic structures of adeno-associated virus (AAV)-DJ, alone and in complex with fondaparinux, have been determined by cryoelectron microscopy at 3 Å resolution. The gene therapy vector, AAV-DJ, is a hybrid of natural serotypes that was previously derived by directed evolution, selecting for hepatocyte entry and resistance to neutralization by human serum. The structure of AAV-DJ differs from that of parental serotypes in two regions where neutralizing antibodies bind, so immune escape appears to have been the primary driver of AAV-DJ’s directed evolution. Fondaparinux is an analog of cell surface heparan sulfate to which several AAVs bind during entry. Fondaparinux interacts with viral arginines at a known heparin binding site, without the large conformational changes whose presence was controversial in low-resolution imaging of AAV2-heparin complexes. The glycan density suggests multi-modal binding that could accommodate sequence variation and multivalent binding along a glycan polymer, consistent with a role in attachment, prior to more specific interactions with a receptor protein mediating entry.
Keywords: AAV, adeno-associated virus, glycan, heparan sulfate, attachment, receptor, structure, electron microscopy, gene therapy, vector
Graphical Abstract
Introduction
Interest in adeno-associated virus (AAV) stems mostly from its experimental and clinical use as a vector in gene therapy of (for example) hemophilia B and lipoprotein lipase deficiency.1, 2, 3 The virus’s interactions with host cells are therefore of fundamental interest and of interest as a foundation for development of more specific means of gene delivery.
AAVs have single-stranded DNA (ssDNA) genomes that are mostly replaced in gene therapy vectors. The DNA is surrounded by a protein shell composed of 60 copies of a capsid protein (in three variant forms), whose structure was first solved by X-ray crystallography for the type species, AAV-2.4 Functions such as cell attachment, entry, and immune evasion are associated with this capsid shell.5 Several serotypes, including AAV-2, bind to cell surface heparan sulfate proteoglycan (HSPG) that was considered, until recently, to serve as the “primary” entry receptor.6 Structural studies of complexes with the analog heparin were limited to electron microscopy at 2 and 1 nm, respectively,7, 8 which were insufficient to arbiter between different characterizations of the binding site and the magnitude of glycan-induced conformational changes. Higher resolution required a shift from natural heparins of mixed sequence to synthetic analogs. AAV-DJ complexed with sucrose octasulfate (SOS) at 5 Å resolution showed arginines interacting with sulfates,9 consistent with the earlier 1 nm AAV-2/heparin study.8 Both showed the glycan bound to heparan-binding domains (HBDs)10 on the sides of prominent surface spikes, the polymer winding loosely between adjacent HBDs related by a 3-fold axis of symmetry.8 However, arguably, the AAV-2 studies7, 8 have not been at a resolution sufficient for robust characterization of induced conformational changes, as proposed, motivating the current AAV-DJ studies at crystallographic-like resolution.
AAV-DJ is a recombinant variant selected for resistance to human neutralizing sera and for improved liver tropism, which is a chimeric mix of serotypes 2, 8, and 9.11 A low-resolution (5 Å) structure by cryoelectron microscopy (cryo-EM), as well as functional properties, indicated gross similarity to AAV-2.11, 12 AAV-DJ provided somewhat higher-resolution cryo-EM data than other AAV2-like natural serotypes (for which it serves as a model), likely because it was prepared as a virus-like particle (VLP), devoid of the ssDNA in natural virions, thereby offering greater EM contrast and superior alignment.
Other serotypes have analogous but different interactions with glycans. AAV-3B was co-crystallized with SOS. Diffraction to 6.5 Å resolution provided no detail, but interacting arginines could be identified by overlaying the difference map13 on the 2.6 Å resolution uncomplexed structure.14 Lying in a depression surrounding the 3-fold axis, they were different from those interacting in AAV-2.13 Both gain- and loss-of-function mutants could be created, with stronger or weaker heparin binding and cell transduction, by mixing and matching glycan-binding residues from AAV-2 and AAV-3B in chimeric mutant viral vectors.13 This established that the interactions were additive in glycan binding and less specific than anticipated. Structure-inspired mutation of amino acids in AAV-6 has implicated yet another cast of positively charged amino acids in heparin binding, only partly overlapping with the AAV-2 site,15 whereas analogous studies implicated a distinct pocket of mostly polar amino acids in the galactose binding of AAV-9.16 Although AAV-5, the most divergent in sequence, binds to a different type of glycan, mutation implicates a site similar to that of AAV-3B.17 The emerging picture is of surprising diversity in orthologous mechanisms for cell surface interactions.
Structural studies have been complemented by biophysical chemistry, measuring the specificity of binding by surface plasmon resonance (SPR) with heparinoid libraries18 or using glycan array technology.19 These studies showed modest specificity for different types of heparinoid, their sequences, and sites of sulfation. They revealed a stepwise dependence of binding on length of oligosaccharide, rationalized in terms of the length needed to fill one HBD site, and the longer length that enables bridging to a symmetry-related site with dramatic increasing in avidity.18 Thus, studies were needed of a more realistic heparinoid analog than the highly sulfonated SOS disaccharide.
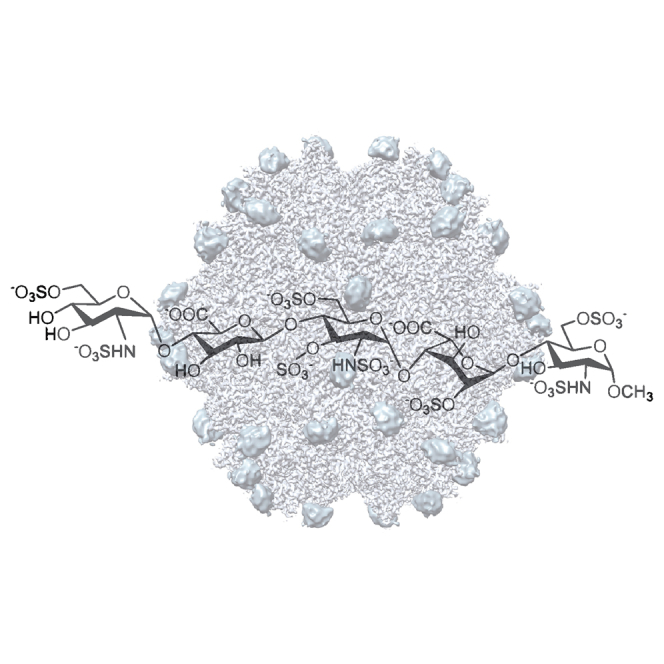
Fondaparinux is a synthetic five-ring heparinoid, terminated with an O-methyl group, but otherwise identical to sequences prevalent in heparin and heparan sulfate (Figure 1). It is an anticoagulant marketed by GlaxoSmithKline under the trade name Arixtra, indirectly inhibiting factor Xa by binding antithrombin III.20 Fondaparinux offered a close analog with similar sulfation to the natural HSPG receptor, but without its sequence heterogeneity. It would be long enough to fill a single HBD site, but not to bridge between symmetric sites, avoiding heterogeneity in the ways that sites might be bridged.
Figure 1.

Glycan Analog Structure
The chemical structure of fondaparinux (Arixtra) is compared with the most common disaccharide units within the heparan sulfate and heparin polysaccharides. Heparan sulfate also contains regions of higher sulfonation, like heparin,43 and the stronger binding to AAV-2 by heparin over desulfated polysaccharides indicates that it is sulfonated regions that interact with AAV.18, 19
Here, the binding of fondaparinux is analyzed and structure is characterized through a cryo-EM reconstruction of the AAV-DJ complex at 2.8 Å resolution and the unbound virus at 3.5 Å resolution.
Results
Fondaparinux Binding by SPR
The competition assay measured inhibition by fondaparinux of the binding of AAV-2 to a chip coated with heparin (Figure 2). Sensorgrams are similar to those of short heparin fragments (degree of polymerization [dp] ≤ 12) that cannot bridge between symmetry-related sites on the virus.18 Short oligosaccharides compete poorly with the high-avidity multi-site binding of heparin, precluding quantitative KD determination by curve fitting. Nevertheless, fondaparinux yields a similar reduction in RU as short heparin oligosaccharides (2 ≤ dp ≤ 12). The effect is dose dependent, with a 5% to 30% reduction in RU for fondaparinux as the concentration is increased from 1 to 20 μM. For commensurate inhibition of AAV binding to heparin, ∼10-fold more fondaparinux is needed than HSPG, the native receptor,6 or the more highly charged SOS (Figure 2).18
Figure 2.

Binding of Fondaparinux, Arixtra, to AAV-2
(A–C) Assays were by inhibition of binding to heparin, measured by SPR (A and B), or (C) inhibition of HeLa cell transduction. (A) SPR sensorgrams of AAV-2 binding to surface heparin in competition with solution of heparin/fondaparinux. The AAV-2 concentration was 0.5 nM, and solution concentrations of heparin or fondaparinux were 1,000 nM; (B) SPR sensorgrams of AAV-2 binding to the heparin surface in different solution concentrations of fondaparinux are shown. (C) Transduction by AAV-2 vector is inhibited in a dose-dependent manner by fondaparinux. Cells were transduced with an AAV-2 mCherry-encoding vector in the presence of increasing concentrations of fondaparinux, with transduction assayed by fluorescence, measured by flow cytometry. Error bars show SD of triplicate measurements.
Fondaparinux inhibition of AAV-2 HeLa cell transduction is also dose dependent (Figure 2). In the absence of fondaparinux, scAAV2-CMV-mCherry at 1.2 × 104 viral genomes per cell (vg/cell) transduces 90% of cells. Transduction decreases to 20% at 2 mM fondaparinux. Concentrations exceeding 10 mM result in loss of cell viability (data not shown) presumably because of increased osmolarity. (Transduction and viability were not diminished in sucrose controls at 10–30 μM [Figure S1].)
High-Resolution EM Structures
The cryo-EM reconstructions of the bound and unbound forms represent the best of several attempts. For the complex, 3.5 Å (Fourier shell correclation [FSC]0.143)21, 22 was achieved with conventional processing and 3.3 Å with frame-dependent motion correction. The 2.8 Å reconstruction was achieved with empirically optimized dose compensation, attenuating high-frequency contributions in later frames, and using 107,454 particles. The native dataset yielded 38,922 particles and a resolution of 3.5 Å. Detail in the reconstruction allows interpretation at a level hitherto only possible with X-ray crystallography (Figure 3).23
Figure 3.

Quality of the Structure
(A) Representative fit of the AAV-DJ structure to the reconstruction of the fondaparinux complex. The density is contoured at 9 σ. (B) The occupancy-weighted difference map was calculated to best show the ligand-bound form of the viral protein (colored by atom type). The map at 5 Å resolution is shown at the 5 σ level. Density for the fondaparinux is noisier than in the conventional difference map (Figure 4A), as expected for non-unit difference coefficients. The bound protein structure is shown in green (backbone plus two arginines), whereas the unbound backbone is shown in red. Symmetry-equivalent subunits are colored purple.
Fondaparinux Binding by Difference Map Analysis
The binding site was apparent from a standard (unweighted) Δρ difference map, calculated by subtracting the reconstruction of the unbound form from that of the complex (see Materials and Methods). Difference maps were improved by first filtering the two datasets to the same resolution. A 5 Å limit gave the best difference map, with density enveloping the fondaparinux at 13 σ (Figure 4). It is not clear whether the additional features at 4 Å are real or noise (Figure 4). Thus, refinement of the fondaparinux structure used a conservative density-gradient optimization (not molecular dynamics) into the 5 Å map, using strong restraints to enforce excellent stereochemistry. This resolution is not sufficient to visualize functional groups that would resolve modeling ambiguities. Thus, the model is representative of a family of possible configurations, whose average corresponds to the observed density. The maximum difference density at the fondaparinux is at 33 σ. The next highest peak is 11 σ (on a 3-fold axis, where noise is higher). Resolving the controversy between two lower-resolution AAV-2/heparin complexes,7, 8 the fondaparinux density superimposes on the heparin of the 8.3 Å complex8 (Figure 4) and on the SOS in the AAV-DJ complex9 (Figure 4).
Figure 4.

Density for Fondaparinux in Its Complex with AAV-DJ Compared with Related Structures
(A) As in other panels, a standard difference map for fondaparinux at 5 Å resolution is shown at the 13 σ level in orange, the ligand is colored by atom type; the trace of the viral protein backbone (and arginines of the HBD) is colored green or purple for symmetry-equivalent subunits. (B) The higher-resolution (4 Å) difference map, shown at the 14 σ level, was not as readily interpretable. (C) Difference density at 7 σ from the 8.3 Å AAV-2/heparin complex is superimposed in pink.8 (D) The atomic model and map of the SOS complex are overlaid on the AAV-DJ structure as found in the fondaparinux complex (4.8 Å resolution; 7 σ; pink).9
The site may not be occupied simultaneously in all 60 symmetry-equivalent locations on the virus capsid. The reconstruction is averaged between particles and over each particle’s icosahedral symmetry. Average ligand occupancy is estimated, two ways, to be equal to or greater than ∼0.3. An estimate of 0.3–0.4 comes by subtracting progressively more unbound map from complex until the density of ligand and virus are equally strong. This underestimates occupancy, because the glycan is less constrained than the protein and higher atomic displacements (B factors) weaken the density. A second estimate comes from dual-conformer atomic refinement of the viral protein structure in bound and unbound states (see later), yielding a bound-state occupancy of 0.33. This is also an approximation, because it is difficult to distinguish similar bound and unbound protein conformations in the summed density of the experimental reconstruction.
Atomic Refinement versus EM Data and Comparison with Homology Modeling
The atomic structure of the viral protein was optimized in several steps. First, it was refined against the highest resolution reconstruction, that of the fondaparinux complex, after seeing that ligand affected only local regions. Starting from the 5 Å native structure,12 the atomic model was fit into the 2.8 Å map, refining by stereochemically restrained real-space torsion angle dynamics.24 The 1.0 Å root-mean-square (RMS) change in backbone reflected mostly the improved resolution, rather than effects of the ligand (see later). The correlation coefficient improved from 0.61 to 0.84, when calculated using all grid points within 3.4 Å of any atom.
AAV-DJ is a chimeric variant with components of sequence from AAV-2, -8, and -9. At one point, we had hoped to bypass experimental structure through homology modeling using Modeler25 and the then-known structures of AAV-2 and -84, 26 as templates. The predicted structure was optimized by gas-phase annealing, surrounding a subunit with its nine symmetry-equivalent neighbors to ensure loop conformations consistent with the viral quaternary structure. The subsequent 5 Å EM structure12 differed from the prediction by 1.7 Å (RMS, all atom) and 1.2 Å (Cα). Even though a glycan complex, the 2.8 Å structure is closer to the prediction, with RMS deviations (RMSDs) of 1.3 Å (all atom) and 1.0 Å (Cα), indicating that, overall, a careful homology model can match in quality a 5 Å experimental structure. Now, with the 2.8 Å experimental AAV-DJ structure, we see that variable region 1 (VR-I) differs from AAV-2 and AAV-8 by up to 8 and 11 Å, and is locally closer (1 Å maximum) to AAV-9 (Table 1). A homology model with an AAV9-like VR-I, built using only AAV-8 and AAV-9 as templates, is in closer agreement with the experimental structure (1.1 Å, RMS all atom, and 0.4 Å, Cα), but it was only in hindsight that we knew that these and only these templates should be used.
Table 1.
Differences between AAV-DJ and Parental Strains
| AAV-2 | AAV-8 | AAV-9 | |
|---|---|---|---|
| Complete Structure | |||
| Residues | 217–735 | 220–738 | 219–736 |
| Sequence identity (%) | 93 | 89 | 85 |
| RMSDall (Å) | 1.1 | 1.2 | 1.0 |
| RMSDCα (Å) | 0.8 | 1.0 | 0.8 |
| Maximal Cα difference (Å) | 7.7 | 11.0 | 8.2 |
| VR-Ia | |||
| Residues | 262–268 | 263–271 | 262–270b |
| Sequence identity (%) | 33 | 67 | 100b |
| RMSDall (Å) | 4.0 | 6.7 | 0.8b |
| RMSDCα (Å) | 3.6 | 6.3 | 0.8b |
| Maximal Cα difference (Å) | 7.7 | 11.0 | 1.1b |
| VR-IVa | |||
| Residues | 449–468 | 452–471b | 451–469 |
| Sequence identity (%) | 45 | 95b,c | 45 |
| RMSDall (Å) | 1.9 | 1.2b | 2.9 |
| RMSDCα (Å) | 2.0 | 1.0b | 3.3 |
| Maximal Cα difference (Å) | 6.4 | 2.8b | 8.2 |
| All Regions Except VR-I and VR-IVa | |||
| Sequence identity (%) | 95 | 89 | 86 |
| RMSDall (Å) | 1.0 | 0.9 | 0.9 |
| RMSDCα (Å) | 0.6 | 0.5 | 0.4 |
| Maximal Cα difference (Å) | 1.7 | 1.5 | 1.4 |
Statistics were calculated with Superpose.71
Variable regions (VRs) are defined according to structural differences between AAV serotypes.72
The natural serotype, locally closest to AAV-DJ.
VR-IV shares greatest sequence identity with AAV-8, but at least one residue (457) and perhaps as many as three residues (455–457) have been derived from the sequence of AAV-2.
The 2.8 Å experimental structure provides detail beyond the homology model. Relative to the original AAV-2/8-based homology model, side-chain rotamers were changed at 121 (23%) amino acids. Fifty-four solvent waters were modeled peaks into three σ peaks with good hydrogen-bonding interactions (0.1 waters/amino acid, as expected of an 2.8 Å X-ray structure27). Backbone differences from the AAV-8/-9 homology model, exceeding the RMSD, are restricted to several loops: near Thr456, with a maximal Cα shift of 1.8 Å, Gly267 (1.5 Å), Asn705 (1.5 Å), Gly548 (1.3 Å), Gly522 (1.3 Å), Thr493 (1.1 Å), and Asn717 (1.1 Å); these regions are key to AAV-DJ’s properties, as discussed later.
Comparison of AAV-DJ with Parental Structures of AAV-2, -8, and -9
Differences between AAV-DJ and parental serotypes are summarized in Table 1 and Figures 5 and S2. Core regions are highly conserved, with differences limited to surface loops encoding the characteristic functional properties of AAV-DJ. Conservation of the core is to be expected with overall sequence identities for AAV-DJ VP1 of 93% for AAV-2, 89% for AAV-8, and 85% for AAV-9. Structural differences are in a subset of the VR loops, previously noted as sites of structural difference and sequence diversity between serotypes.5 Cα differences between AAV-DJ and any of the parental serotypes exceed 1 Å at VR-I, -II, -III, -IV, -V, and -IX. In VR-I (residues 262–270), AAV-DJ approximates most closely AAV-9 (maximal Cα deviation = 1.1 Å), with which it shares the same local sequence, and is quite different from AAV-2 and -8 (maximal Cα deviations of 8 and 11 Å) that have 33% and 67% sequence identity, respectively (Table 1). In VR-IV (residues 451–470), the structure most closely follows AAV-8 (maximal Cα deviation = 2.8 Å; 95% local sequence identity), deviating by 6 and 8 Å from AAV-2 and AAV-9 (∼50% local sequence identity). All of the VRs are surface exposed, and some have among the weakest density in the AAV-DJ reconstruction: VR-IV, VR-IX (near residue Tyr707), VR-I, and VR-II (Glu329-Gly330). As in the crystal structures of the parental serotypes, B factors in these regions refine to high values. Even though some of these regions are somewhat disordered, their structures are distinct. For example, at Gly454-Gly455 in VR-IV, AAV-DJ and AAV-8 differ by up to 3 Å, and both are distinct from AAV-2 or AAV-9.
Figure 5.

Comparison of Natural Serotypes to AAV-DJ
Roadmap73 surface projections show 1 of the 60 equivalent icosahedrally symmetric triangular regions (bounded by a 5-fold axis and two 3-fold axes) of each structure. They are colored by Cα difference with AAV-DJ from blue (≤0.5 Å) to red (≥2.5 Å). Outlined are binding footprints for mostly neutralizing monoclonal antibodies, determined by cryo-EM and other methods (listed in Table S1). (AAV-6, not shown, is very similar to AAV-1/5H7.) (A–D) AAV-2 (B) and AAV-8 (D) are serotypes whose sequences are represented in the AAV-DJ hybrid; AAV-5 (C) was selected out during directed evolution by IVIg escape; and AAV-1 (A) was not part of the pool from which AAV-DJ was evolved, but it shows additional regions where neutralizing antibodies can bind.
Dual-Conformer Refinement Shows Only Small and Local Differences on Glycan Binding
Conformational changes potentially driven by the glycan binding were characterized in the following way. The structure, at this point in the refinement, represented an “average” of bound and unbound conformations weighted by glycan occupancy. An unbound structure was now obtained by density gradient refinement of this structure into the native 3.5 Å reconstruction. Where the “average” and unbound models differed, the standard difference map was examined. Where evidence of conformational change was lacking in the map, model differences were assumed to have resulted from lower precision in the 3.5 Å native reconstruction, and the model was reset to the 2.8 Å structure. Where supported by the difference density, dual conformers were used. The fully bound conformation was modeled and refined into an occupancy-weighted difference map (see Materials and Methods) designed to subtract contributions to the map from un-liganded protein. Regions far from conformational changes were fixed in their structures refined at 2.8 Å resolution. Finally, dual-conformer refinement allowed the sum of bound and unbound structures to be refined against the full 2.8 Å data, avoiding limitations that come with the lower-resolution native data. The increase in degrees of freedom was mitigated by using dual conformers only locally for 10 of 517 residues (2%), where indicated by the difference map. The structure was refined with torsion angle-simulated annealing, ignoring close approaches of atoms between bound and unbound states. Local dual-conformer refinement improved the density correlation from 0.84 to 0.86.
Discussion
Structural Rationalization for Changed Phenotype on Directed Evolution of AAV-DJ
Because the effects of the glycan upon the virus structure are local and subtle, the 2.8 Å structure can provide detailed insights into the makings of AAV-DJ from the parental serotypes under the selection of directed evolution.11, 28, 29 In so doing, we compare AAV-DJ with the X-ray structures of AAV-2,4 AAV-8,26 and AAV-9,30 all at circa 3 Å resolution. Predominantly, we see that the tertiary loop structure comes from the underlying sequence and is little affected by contacts from a neighboring background of different parental strain. Indeed, most changed regions are surface exposed and have fewer contacts anyway. We also see that the effects on structure of a sequence substitution are local, with no evidence that changes are propagated away from the site. Perhaps directed evolution11 selects for conservative changes without long-range disruptions.
AAV-DJ was selected both for transduction of human liver cells and then resistance to neutralization by pooled human antisera (intravenous immunoglobulin [IVIg]).11 Liver transduction had selected hybrids from five of eight starting serotypes (2, 4, 5, 8, and 9); then IVIg-escape selected a single hybrid of serotypes 2, 8, and 9. The most prominent difference between AAV-DJ and AAV-2 is in VR-I at the epitope of neutralizing mouse monoclonal antibody (mAb) A20.8, 12, 31, 32 This explains the lack of recognition of AAV-DJ by mAb A20, in spite of 93% capsid sequence identity with AAV-211 (Table 1; Figure S2). Now, with a 2.8 Å AAV-DJ EM structure and crystal structures for all parental strains, AAV-2,4 AAV-8,26 and AAV-9,30 detailed comparisons can be made. Germane to the IVIg selection, antibody binding sites have been mapped for two parental serotypes (AAV-2 and AAV-8) and one (AAV-5) that was selected out. Mappings have used cryo-EM at resolutions of 8–19 Å, scanning mutagenesis, and/or peptide scanning.32, 33, 34, 35, 36, 37, 38 Structural differences (>1.5 Å) between AAV-DJ and natural serotypes represented in the hepatocyte-selected hybrids are predominantly within footprints of neutralizing mAb (Figure 5). Conversely, there are differences >1.5 Å in all neutralizing mAb footprints mapped at nanometer or better resolution. Thus, it is reasonable to propose that predominant structural features selected during the directed evolution of AAV-DJ have allowed it to escape the binding of neutralizing antibodies. (It is quite possible that changes selected under IVIg pressure have also contributed pleiotropically to altered cell tropism.)
A number of important caveats should be listed. First, the logic implicitly assumes that mouse monoclonals (like A20) are representative of dominant neutralizing antibodies in human serum, which seems plausible, but there is no evidence. Second, the antibody footprints on AAV have significant uncertainties, because of sometimes limited EM resolution, and known challenges in homology modeling of VRs in antibody loops. Third, the mAb footprints cover, in aggregate, a significant fraction of the outer surface. Finally, Cα differences are just one (easy) metric to survey, but sequence differences and side-chain structure will also mediate antibody escape.
Glycan Binding
There are technical reasons (see Results) why the fondaparinux occupancy might be underestimated, but weak affinity is consistent with the SPR and inhibition assays. Although both fondaparinux and SOS are bound by the arginines of the HBD9 that are implicated in heparin binding,4, 8, 10, 39 binding is weaker than that of heparin.18 The lower-bound fondaparinux occupancy ≈0.3 estimate corresponds to a KD ≈10 mM upper bound,40 although binding might actually be stronger. Thus, the IC50 ≈0.7 mM from transduction inhibition (Figure 2) is not inconsistent. However, how can approximate millimolar fondaparinux binding be reconciled with a stronger sub-nanomolar KD for heparin?18 The likely explanation is that glycan polymers achieve strong effective binding (avidity) by simultaneously binding to several symmetry-related sites, as no short oligomer can.18 The shortest distance between sites on the virus corresponds to ∼13 saccharide units,18 so an ∼17 kDa heparin polymer could, in principle, bind at up to six sites. Three or more would be required to achieve nanomolar avidity with millimolar affinity at each site.
The ligand was visualized at lower resolution than the viral protein, including the HBD loop and side chains of the binding site. There are both chemical and technological (EM) factors that contribute to blurriness, and indeed, ligands are generally less well resolved than protein in EM structures.41 However, it is noteworthy that fondaparinux is not as well resolved as SOS. SOS has a higher sulfate content, perhaps leading to tighter electrostatic interactions with binding site arginines. SOS is less flexible42 and more pseudo-symmetrical, perhaps yielding fewer distinct binding configurations and less disorder. Fondaparinux has sulfation that is more typical of heparin or the high-sulfation regions of heparan sulfate,43 so its apparently varied configurations may be biologically relevant. Higher order in the surrounding protein suggests a site that is able to bind to the various glycan configurations and sequence elements that AAV would encounter within a heparinoid polymer. It would also explain the observed low specificity, with in vitro binding to a variety of heparinoids, including some chondroitin sulfates.18, 19 Biological implications are that there might be a variety of cell surface glycans to which each AAV serotype might bind, and that there might also be connective tissue or extracellular matrix with glycans that decoy AAV away from productive infections.
Individual sulfates are not well resolved in the density for fondaparinux, nor were sulfate interactions used as a constraint in model-building. However, the 2-N-sulfo group of the middle pyranose ring and the 6-O-sulfo group of the fifth ring are both in proximity to the arginines previously implicated in binding, Arg587 and Arg590, respectively.4, 8, 10, 39 SPR using heparinoid libraries and glycan array binding studies have both indicated that sulfate groups at 2-N and 6-O are more critical than at 2-O.19, 44 The four modeled fondaparinux sulfates, close to these arginines, superimpose within 2–4 Å of sulfates that were resolved in the SOS complex.9 This modest correspondence is another indication that the viral binding site, particularly the arginine side chains, is adaptable to different glycan structures.
The Question of Glycan-Induced Conformational Change in AAV
The prior AAV2/heparin structures differed in the exact location of perceived heparin density and the need7 (or not8) for conformational changes that would allow interactions with genetically implicated arginines.10, 39 In the study at 18 Å resolution,7 1 σ difference peaks, interpreted as heparin, required a hypothesized glycan-induced protein conformational change for interaction with the arginines. Difference density at ∼0.5 σ near the tips of the 3-fold spikes and near the HI loop was cited in support of a postulated heparin-triggered allosteric change that would widen a channel along the 5-fold axis for subsequent DNA release.7 Even though the fondaparinux might have partial occupancy in the current study, its density is 10-fold stronger and more robust statistically. Differences with the 18 Å heparin study7 reflect not only improved resolution, but, we believe, the on-grid preparation of the complex used here and in the 8 Å heparin study.8 This minimizes aggregation and increases the fraction of EM-observable particles that are in the bound state. The fondaparinux location is consistent with the 8 Å heparin and 5 Å SOS studies,8, 9 contacting directly the genetically implicated arginines without the conformational changes proposed in the 18 Å study.7 The current 2.8 Å resolution should be sufficient for such conformational changes to be clear. However, no evidence is seen of a substantial shift of the refined model, or of paired positive and negative difference density peaks that would be expected.
Subtle, rather than large, conformational change is reminiscent of the crystal structures of foot-and-mouth disease viruses (FMDVs) complexed with heparinoid oligosaccharides.45, 46, 47 So too are pre-formed sulfate-binding sites containing arginines, and natural or in vitro modulation of binding affinity and infectivity upon mutation of these residues.13, 46 The findings with AAV and FMDV are all in contrast with HIV with its CD4-induced changes to gp120 that modulate binding to heparan sulfate, and the structural changes in heparan sulfate binding domains that are key to HIV co-receptor interactions.48 Thus, among viral proteins, a wide spectrum of glycan interactions is emerging, from rigid to flexible. AAV joins FMDV, in a class shown to have more rigid interactions. It remains to be seen whether this proves to be more common among non-enveloped viruses with no need for attachment to trigger membrane fusion.
The absence of widespread conformational changes in AAV is consistent with recently changed perception of the role of glycan-binding during infection. HSPG was originally thought to be a primary cell receptor6 with various membrane proteins suggested to have secondary roles during endocytic entry. Recently, the gene product KIAA0319L (also known as AAVR) has been identified as a protein essential for AAV entry, with many of the properties classically associated with viral receptors.49 Furthermore, at high multiplicity of infection, overexpression of KIAA0319L can overcome the inhibitory effect of a heparan synthesis cell knockout.49 The results of this research add to the emerging picture that the glycan binding site can tolerate lower specificity interactions than once anticipated. Glycan binding might serve a more accessory role in adherence, so that the virus is anchored in close proximity to the cell surface, thereby maximizing the chance of binding to a receptor that is transiently exposed on the surface.
Materials and Methods
Expression and Purification of AAV-DJ
Empty capsids of AAV-DJ were expressed in SF9 insect cells using the Bac-to-Bac Baculovirus Expression Vector System (Invitrogen) as previously described, except that AAV-DJ substituted for AAV-2.12, 50 The VLPs were purified, as before, using three rounds of CsCl density gradient ultracentrifugation, followed by heparin affinity chromatography, eluting with a NaCl gradient. Capsids were then diluted in 50 mM HEPES, 25 mM MgCl2, 25 mM NaCl (pH 7.4), or, for SPR, into 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 (pH 7.4).
Other Materials
Fondaparinux (Arixtra; molecular weight [MW] = 1,728) was from GlaxoSmithKline and Mylan. Heparin was a porcine intestinal 16 kDa fraction from Celsus Laboratories. SOS was from Toronto Biochemicals. Heparin oligosaccharides, classified by dp, included tetrasaccharide (dp4) and hexasaccharide (dp6), prepared by partial hydrolysis using heparin lyase 1 of bovine lung heparin (Sigma) followed by size fractionation. SPR Sensor SA chips were from GE Healthcare. EM copper grids were R2/2 200 mesh from Quantifoil.
SPR
SPR measurements were performed on a BIAcore 3000 and analyzed with the BIAevaluation software (version 4.0.1).
Preparation of the Heparin Biochip
Biotinylated heparin was prepared by reaction of sulfo-N-hydroxysuccinimide long-chain biotin (Thermo Scientific) with unsubstituted glucosamine amino groups in the polysaccharide chain51 and was immobilized to a streptavidin (SA) chip based on the manufacturer’s protocol. In brief, 20 μL of the heparin-biotin conjugate (0.1 mg/mL) in HBS-EP running buffer (GE Healthcare) was injected over flow cell 2 (FC2) of the SA chip, at 10 μL/min. Immobilization was confirmed by an ∼50 RU increase in the sensor chip. The control flow cell (FC1) was prepared by 1 min injection with saturated biotin.
Heparin-Binding Inhibition Assay
AAV at 8.3 pM (equivalent to capsid protein at 0.5 nM) was diluted in HBS-EP buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, 0.005% surfactant P20 [pH 7.4]) and injected at 30 μL/min. The same buffer was flowed through, during 3 min of dissociation. The sensor surface was regenerated with 30 μL of 0.5% SDS, then with 2 M NaCl. SPR measurements were at 25°C. Inhibition was measured by pre-mixing the AAV sample with fondaparinux at 1–20 μM, or, for comparison, tetrasaccharide (dp4), hexasaccharide (dp6) at 1 μM, or SOS or heparin at 0.1 μM, and the binding to heparin was re-measured as above. After each competition assay, AAV binding was re-measured to ensure that the surface had been completely regenerated.
Transduction Inhibition Assays
HeLa S3 cells (ATCC CCL-2.2) were plated at 1.1 × 103 cells/well in 96-well plates (Falcon, Becton Dickinson) and cultured overnight in F-12K medium (HyClone, GE Life Sciences), supplemented with 10% fetal bovine serum. Fondaparinux (Arixtra) was purchased from Mylan Institutional. The specified concentrations of fondaparinux were introduced to the medium 10–15 min before cells were transduced with the self-complementary vector scAAV2-CMV-mCherry (University of North Carolina Vector Core) at an MOI of 1.2 × 104 viral genomes per cell (vg/cell). Cells were incubated for 25 hr at 37°C, at which time they were trypsinized and assayed for transgene expression by flow cytometry tuned to PE/Cy-5 on a MACSQuant Analyzer 10 (Miltenyi Biotec). Results were analyzed using FlowJo v10 software (FlowJo).
Cryo-EM Sample Preparation
AAV-DJ was complexed with ligand on the EM grid to avoid aggregation. Aggregation was observable when mixed in bulk solution, presumably because of oligosaccharide cross-linking polyvalent AAV particles. AAV-DJ (3 μL) was applied to copper grids that had been glow discharged in 75%/25% Ar/O at a concentration of 0.6 mg/mL. For complex, the grid was hand blotted before adding 3 μL 5.7 mM fondaparinux in ultrapure water for 15 s, giving a >600-fold excess of ligand over virus subunits. All grids were vitrified in liquid nitrogen-cooled ethane using an FEI Vitrobot (FEI) with a single 3 s blot of force 1 at 100% humidity and 4°C. Grids were stored in liquid nitrogen until used.
Image Acquisition
For uncomplexed AAV-DJ, a dataset was collected using a magnification of 29,000. Images were acquired using an FEI Titan Krios (FEI) using Leginon52 with a total dose of 52.63 e−/Å2. The images were recorded on a DE-20 direct electron detector (Direct Electron), dose fractionated across 18 frames with a dose rate of 2.9 e−/Å2 per frame. Defocus was randomly set between 2.0 and 3.5 μm, with defocus estimates made continually throughout image acquisition using the automated contrast transfer function (CTF) estimator ACE53 and CTFFIND3.54
For the AAV-DJ/fondaparinux complex, two datasets were obtained from the same grid at a magnification of 29,000×, but with differing defocus ranges. Images were acquired using an FEI Titan Krios (FEI) using Leginon52 with a total dose of ∼66 e−/Å2, fractionated across 45 frames at 1.5 e−/Å2 per frame. The first dataset was collected with defocus between 1.5 and 3.0 μm, whereas the second was between 0.75 and 1.75 μm (using defocus estimates as above).
Dose Compensation and Frame Alignment
Frame alignment and dose compensation55 were performed with version 2.5.1 of software provided by Direct Electron. In brief, rolling frame averages are iteratively aligned to the current sum of frames. Dose compensation is through Gaussian low-pass filtering to a frame resolution that is dependent on the cumulative dose, using resolutions calibrated from attenuation of the electron diffraction of catalase crystals (Direct Electron).55 Following alignment and dose compensation for entire frames, individual particles were picked and the alignment and dose compensation repeated.
Image Processing
The AAV-DJ native dataset yielded 522 images and 38,922 particles. The first datasets for the AAV/fondaparinux complex yielded 503 images and 65,978 particles, and the second yielded 548 images and 54,188 particles. Particle picking was semi-automatic using FindEM56 within Appion57 and using a template that was a rotational average of manually picked particles. Particles over carbon were manually deselected. Initial orientations were generated using EMAN158 by refining against the structure of AAV, low-pass-filtered to 20 Å resolution, with two iterations and an angular increment of 1°. The Euler angles were then further refined with nine iterations of FREALIGN,59 constraining 60-fold icosahedral symmetry throughout image processing. Image classification was attempted to differentiate particles with low and high ligand occupancy. It was expected to be challenging, because the ligand constitutes only 2.5% of the mass. Indeed, the reconstruction for the class with higher occupancy was only marginally different, so the full dataset was used thereafter. Reconstructions were sharpened to correct for high-frequency attenuation using EM-B factor60 with default parameters.
Atomic Modeling and Fitting
Envelope corrections, together with the effective EM magnifications and resolutions, were least-squares refined, using RSRef,24 to maximize agreement between the prior 4.5 Å AAV-DJ atomic structure12 and the new 2.8 and 3.5 Å maps. (Resolution is refined by adjusting the limit on a Butterworth low-pass filter applied to the atomic model, until its density optimally agrees with the experimental reconstruction, thereby cross-checking the FSC-derived estimates.) Through the calibration to the atomic model, reconstructions of the fondaparinux complex and uncomplexed AAV-DJ were put on uniform scales in map values and in magnification. Structure factors for the 3.5 Å native were scaled in reciprocal space to the 2.8 Å reconstruction of the complex by resolution shell using EMAN,58 after low-pass filtering both reconstructions to 6, 5, or 4 Å resolution. The 5 Å difference map was used to model the fondaparinux, starting with the structure from PDB: 4X7R.61 Because the occupancy of the glycan site was less than unity, density for the viral protein in the complex represented a mixture of bound and unbound conformations. The bound state was revealed through a difference map weighted by the ligand occupancy, O62:
An initial estimate for the occupancy came by adjusting O until density for the glycan and viral protein were commensurate. After modeling the glycan and the glycan-bound configuration of the HBD loop, the estimate of the occupancy was refined using RSRef24 such that the weighted sum of bound and unbound atomic models gave best agreement with the observed reconstruction of the complex. The process was repeated through iterations of atomic refinement, because an improved model allowed an improved estimate of the occupancy and hence improvement in the weighted difference map.
Models were manually adjusted to fit reconstructions using Coot.63 Structures for the viral protein were refined by simulated annealing torsion angle optimization into the native and weighted difference reconstructions. Refinement used a real-space objective function that calculates the density of each map grid point from the contributions of neighboring atoms, accounting for the effects of resolution and the EM envelope.24 This objective function was embedded in the crystallographic refinement program CNS v1.364 to take advantage of its stereochemical restraints and a low-parameter torsion angle optimizer65 that mitigates over-fitting. An additional restraint was added through a flat-bottom potential to ensure that φ and ψ backbone dihedrals did not deviate from the allowed areas of a Ramachandran plot.
Stereochemical restraints for the fondaparinux were based on the NTO entry in the library of the CCP4 suite,66 with the addition of restraints toward preferred glycosidic torsion angles.67 Pucker of the pyranose rings was checked post facto through calculation of Cremer-Pople parameters, but not enforced as an explicit restraint68, 69 (http://www.ric.hi-ho.ne.jp/asfushi/). The figures were generated using PyMol.70
Author Contributions
Conceptualization, M.S.C.; Methodology, S.M.S. and R.J.L.; Software, S.M.S. and M.S.C.; Formal Analysis, Q.X., S.M.S., and F.Z.; Investigation, Q.X., N.L.M., O.D., J.M.S., A.J.N., and D.R.S.; Data Curation, Q.X. and S.M.S.; Writing – Original Draft, Q.X., R.J.L., S.M.S., and M.S.C.; Writing – Review & Editing, M.S.C.; Visualization, Q.X.; Supervision, S.M.S., R.J.L., and M.S.C.; Project Administration, M.S.C.; Funding Acquisition, S.M.S., R.J.L., and M.S.C.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
Data processing and reconstruction was performed with help from the Florida State University (FSU) shared High-Performance Computing facility. This work was supported by National Institutes of Health grants R01-GM66875 (to M.S.C.), R01-GM86892 (to S.M.S.), S10-RR025080 (to Kenneth A. Taylor, FSU), and R01-GM38060 (R.J.L.).
Footnotes
Supplemental Information includes two figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.omtm.2017.02.004.
Accession Numbers
An atomic model of both unbound and fondaparinux complex conformations is available from the Protein Data Bank (PDB): 5UF6 (http://www.rcsb.org). EM reconstructions are available from the Electron Microscopy Data Bank (http://www.ebi.ac.uk/pdbe/emdb/) under accession number EMD: 8574 for the native, complex, and difference maps. Structure refinement software is available from Oregon Health & Science University (http://xtal.ohsu.edu).
Supplemental Information
References
- 1.Carter B.J., Burstein H., Peluso R.W. Adeno-Associated Virus and AAV Vectors for Gene Delivery. In: Templeton N.S., editor. Gene and cell therapy: Therapeutic mechanisms and strategies. CRC Press; 2008. pp. 115–156. [Google Scholar]
- 2.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaudet D., Méthot J., Déry S., Brisson D., Essiembre C., Tremblay G., Tremblay K., de Wal J., Twisk J., van den Bulk N. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20:361–369. doi: 10.1038/gt.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie Q., Bu W., Bhatia S., Hare J., Somasundaram T., Azzi A., Chapman M.S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA. 2002;99:10405–10410. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agbandje-McKenna M., Chapman M.S. Correlating Structure with Function in the Viral Capsid. In: Kerr J.R., Cotmore S.F., Bloom M.E., Linden R.M., Parrish C.R., editors. Parvoviruses. Hodder Arnold, Ltd.; 2006. pp. 124–139. [Google Scholar]
- 6.Summerford C., Samulski R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy H.C., Bowman V.D., Govindasamy L., McKenna R., Nash K., Warrington K., Chen W., Muzyczka N., Yan X., Baker T.S., Agbandje-McKenna M. Heparin binding induces conformational changes in Adeno-associated virus serotype 2. J. Struct. Biol. 2009;165:146–156. doi: 10.1016/j.jsb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Donnell J., Taylor K.A., Chapman M.S. Adeno-associated virus-2 and its primary cellular receptor--Cryo-EM structure of a heparin complex. Virology. 2009;385:434–443. doi: 10.1016/j.virol.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Q., Spilman M., Meyer N.L., Lerch T.F., Stagg S.M., Chapman M.S. Electron microscopy analysis of a disaccharide analog complex reveals receptor interactions of adeno-associated virus. J. Struct. Biol. 2013;184:129–135. doi: 10.1016/j.jsb.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kern A., Schmidt K., Leder C., Müller O.J., Wobus C.E., Bettinger K., Von der Lieth C.W., King J.A., Kleinschmidt J.A. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grimm D., Lee J.S., Wang L., Desai T., Akache B., Storm T.A., Kay M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lerch T.F., O’Donnell J.K., Meyer N.L., Xie Q., Taylor K.A., Stagg S.M., Chapman M.S. Structure of AAV-DJ, a retargeted gene therapy vector: cryo-electron microscopy at 4.5 Å resolution. Structure. 2012;20:1310–1320. doi: 10.1016/j.str.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lerch T.F., Chapman M.S. Identification of the heparin binding site on adeno-associated virus serotype 3B (AAV-3B) Virology. 2012;423:6–13. doi: 10.1016/j.virol.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lerch T.F., Xie Q., Chapman M.S. The structure of adeno-associated virus serotype 3B (AAV-3B): insights into receptor binding and immune evasion. Virology. 2010;403:26–36. doi: 10.1016/j.virol.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Q., Lerch T.F., Meyer N.L., Chapman M.S. Structure-function analysis of receptor-binding in adeno-associated virus serotype 6 (AAV-6) Virology. 2011;420:10–19. doi: 10.1016/j.virol.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bell C.L., Gurda B.L., Van Vliet K., Agbandje-McKenna M., Wilson J.M. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J. Virol. 2012;86:7326–7333. doi: 10.1128/JVI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Afione S., DiMattia M.A., Halder S., Di Pasquale G., Agbandje-McKenna M., Chiorini J.A. Identification and mutagenesis of the adeno-associated virus 5 sialic acid binding region. J. Virol. 2015;89:1660–1672. doi: 10.1128/JVI.02503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F., Aguilera J., Beaudet J.M., Xie Q., Lerch T.F., Davulcu O., Colón W., Chapman M.S., Linhardt R.J. Characterization of interactions between heparin/glycosaminoglycan and adeno-associated virus. Biochemistry. 2013;52:6275–6285. doi: 10.1021/bi4008676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mietzsch M., Broecker F., Reinhardt A., Seeberger P.H., Heilbronn R. Differential adeno-associated virus serotype-specific interaction patterns with synthetic heparins and other glycans. J. Virol. 2014;88:2991–3003. doi: 10.1128/JVI.03371-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters R.J.G., Joyner C., Bassand J.-P., Afzal R., Chrolavicius S., Mehta S.R., Oldgren J., Wallentin L., Budaj A., Fox K.A., Yusuf S., OASIS-6 investigators The role of fondaparinux as an adjunct to thrombolytic therapy in acute myocardial infarction: a subgroup analysis of the OASIS-6 trial. Eur. Heart J. 2008;29:324–331. doi: 10.1093/eurheartj/ehm616. [DOI] [PubMed] [Google Scholar]
- 21.Böttcher B., Wynne S.A., Crowther R.A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;386:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- 22.Rosenthal P.B., Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Bartesaghi A., Merk A., Banerjee S., Matthies D., Wu X., Milne J.L., Subramaniam S. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science. 2015;348:1147–1151. doi: 10.1126/science.aab1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chapman M.S., Trzynka A., Chapman B.K. Atomic modeling of cryo-electron microscopy reconstructions--joint refinement of model and imaging parameters. J. Struct. Biol. 2013;182:10–21. doi: 10.1016/j.jsb.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eswar N., Eramian D., Webb B., Shen M., Sali A. Protein structure modeling with MODELLER. In: Baxevanis A.D., Stein L.D., Stormo G.D., Yates J.R. III, editors. Current Protocols in Bioinformatics. John Wiley & Sons; 2006. pp. 5.6.1–5.6.30. [Google Scholar]
- 26.Nam H.J., Lane M.D., Padron E., Gurda B., McKenna R., Kohlbrenner E., Aslanidi G., Byrne B., Muzyczka N., Zolotukhin S., Agbandje-McKenna M. Structure of adeno-associated virus serotype 8, a gene therapy vector. J. Virol. 2007;81:12260–12271. doi: 10.1128/JVI.01304-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weichenberger C.X., Afonine P.V., Kantardjieff K., Rupp B. The solvent component of macromolecular crystals. Acta Crystallogr. D Biol. Crystallogr. 2015;71:1023–1038. doi: 10.1107/S1399004715006045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koerber J.T., Jang J.H., Schaffer D.V. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol. Ther. 2008;16:1703–1709. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W., Asokan A., Wu Z., Van Dyke T., DiPrimio N., Johnson J.S. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol. Ther. 2008;16:1252–1260. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 30.DiMattia M.A., Nam H.J., Van Vliet K., Mitchell M., Bennett A., Gurda B.L., McKenna R., Olson N.H., Sinkovits R.S., Potter M. Structural insight into the unique properties of adeno-associated virus serotype 9. J. Virol. 2012;86:6947–6958. doi: 10.1128/JVI.07232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wistuba A., Kern A., Weger S., Grimm D., Kleinschmidt J.A. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J. Virol. 1997;71:1341–1352. doi: 10.1128/jvi.71.2.1341-1352.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wobus C.E., Hügle-Dörr B., Girod A., Petersen G., Hallek M., Kleinschmidt J.A. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 2000;74:9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huttner N.A., Girod A., Perabo L., Edbauer D., Kleinschmidt J.A., Büning H., Hallek M. Genetic modifications of the adeno-associated virus type 2 capsid reduce the affinity and the neutralizing effects of human serum antibodies. Gene Ther. 2003;10:2139–2147. doi: 10.1038/sj.gt.3302123. [DOI] [PubMed] [Google Scholar]
- 34.Lochrie M.A., Tatsuno G.P., Christie B., McDonnell J.W., Zhou S., Surosky R., Pierce G.F., Colosi P. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J. Virol. 2006;80:821–834. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCraw D.M., O’Donnell J.K., Taylor K.A., Stagg S.M., Chapman M.S. Structure of adeno-associated virus-2 in complex with neutralizing monoclonal antibody A20. Virology. 2012;431:40–49. doi: 10.1016/j.virol.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurda B.L., Raupp C., Popa-Wagner R., Naumer M., Olson N.H., Ng R., McKenna R., Baker T.S., Kleinschmidt J.A., Agbandje-McKenna M. Mapping a neutralizing epitope onto the capsid of adeno-associated virus serotype 8. J. Virol. 2012;86:7739–7751. doi: 10.1128/JVI.00218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gurda B.L., DiMattia M.A., Miller E.B., Bennett A., McKenna R., Weichert W.S., Nelson C.D., Chen W.J., Muzyczka N., Olson N.H. Capsid antibodies to different adeno-associated virus serotypes bind common regions. J. Virol. 2013;87:9111–9124. doi: 10.1128/JVI.00622-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tseng Y.S., Gurda B.L., Chipman P., McKenna R., Afione S., Chiorini J.A., Muzyczka N., Olson N.H., Baker T.S., Kleinschmidt J., Agbandje-McKenna M. Adeno-associated virus serotype 1 (AAV1)- and AAV5-antibody complex structures reveal evolutionary commonalities in parvovirus antigenic reactivity. J. Virol. 2015;89:1794–1808. doi: 10.1128/JVI.02710-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu P., Xiao W., Conlon T., Hughes J., Agbandje-McKenna M., Ferkol T., Flotte T., Muzyczka N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 2000;74:8635–8647. doi: 10.1128/jvi.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danley D.E. Crystallization to obtain protein-ligand complexes for structure-aided drug design. Acta Crystallogr. D Biol. Crystallogr. 2006;62:569–575. doi: 10.1107/S0907444906012601. [DOI] [PubMed] [Google Scholar]
- 41.Borgnia M.J., Banerjee S., Merk A., Matthies D., Bartesaghi A., Rao P., Pierson J., Earl L.A., Falconieri V., Subramaniam S., Milne J.L. Using cryo-EM to map small ligands on dynamic metabolic enzymes: studies with glutamate dehydrogenase. Mol. Pharmacol. 2016;89:645–651. doi: 10.1124/mol.116.103382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desai U.R., Vlahov I.R., Pervin A., Linhardt R.J. Conformational analysis of sucrose octasulfate by high resolution nuclear magnetic resonance spectroscopy. Carbohydr. Res. 1995;275:391–401. doi: 10.1016/0008-6215(95)00159-q. [DOI] [PubMed] [Google Scholar]
- 43.Conrad H.E. Academic Press; 1998. Heparin-Binding Proteins. [Google Scholar]
- 44.Zhang F., Walcott B., Zhou D., Gustchina A., Lasanajak Y., Smith D.F., Ferreira R.S., Correia M.T., Paiva P.M., Bovin N.V. Structural studies of the interaction of Crataeva tapia bark protein with heparin and other glycosaminoglycans. Biochemistry. 2013;52:2148–2156. doi: 10.1021/bi400077b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fry E.E., Lea S.M., Jackson T., Newman J.W., Ellard F.M., Blakemore W.E., Abu-Ghazaleh R., Samuel A., King A.M., Stuart D.I. The structure and function of a foot-and-mouth disease virus-oligosaccharide receptor complex. EMBO J. 1999;18:543–554. doi: 10.1093/emboj/18.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fry E.E., Newman J.W., Curry S., Najjam S., Jackson T., Blakemore W., Lea S.M., Miller L., Burman A., King A.M., Stuart D.I. Structure of Foot-and-mouth disease virus serotype A10 61 alone and complexed with oligosaccharide receptor: receptor conservation in the face of antigenic variation. J. Gen. Virol. 2005;86:1909–1920. doi: 10.1099/vir.0.80730-0. [DOI] [PubMed] [Google Scholar]
- 47.Han S.-C., Guo H.-C., Sun S.-Q. Three-dimensional structure of foot-and-mouth disease virus and its biological functions. Arch. Virol. 2015;160:1–16. doi: 10.1007/s00705-014-2278-x. [DOI] [PubMed] [Google Scholar]
- 48.Connell B.J., Lortat-Jacob H. Human immunodeficiency virus and heparan sulfate: from attachment to entry inhibition. Front. Immunol. 2013;4:385. doi: 10.3389/fimmu.2013.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pillay S., Meyer N.L., Puschnik A.S., Davulcu O., Diep J., Ishikawa Y., Jae L.T., Wosen J.E., Nagamine C.M., Chapman M.S., Carette J.E. An essential receptor for adeno-associated virus infection. Nature. 2016;530:108–112. doi: 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urabe M., Ding C., Kotin R.M. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- 51.Hernaiz M., Liu J., Rosenberg R.D., Linhardt R.J. Enzymatic modification of heparan sulfate on a biochip promotes its interaction with antithrombin III. Biochem. Biophys. Res. Commun. 2000;276:292–297. doi: 10.1006/bbrc.2000.3453. [DOI] [PubMed] [Google Scholar]
- 52.Suloway C., Pulokas J., Fellmann D., Cheng A., Guerra F., Quispe J., Stagg S., Potter C.S., Carragher B. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 53.Mallick S.P., Carragher B., Potter C.S., Kriegman D.J. ACE: automated CTF estimation. Ultramicroscopy. 2005;104:8–29. doi: 10.1016/j.ultramic.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 54.Mindell J.A., Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 55.Spear J.M., Noble A.J., Xie Q., Sousa D.R., Chapman M.S., Stagg S.M. The influence of frame alignment with dose compensation on the quality of single particle reconstructions. J. Struct. Biol. 2015;192:196–203. doi: 10.1016/j.jsb.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roseman A.M. Particle finding in electron micrographs using a fast local correlation algorithm. Ultramicroscopy. 2003;94:225–236. doi: 10.1016/s0304-3991(02)00333-9. [DOI] [PubMed] [Google Scholar]
- 57.Lander G.C., Stagg S.M., Voss N.R., Cheng A., Fellmann D., Pulokas J., Yoshioka C., Irving C., Mulder A., Lau P.W. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ludtke S.J., Baldwin P.R., Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 59.Grigorieff N. FREALIGN: high-resolution refinement of single particle structures. J. Struct. Biol. 2007;157:117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 60.Fernández J.J., Luque D., Castón J.R., Carrascosa J.L. Sharpening high resolution information in single particle electron cryomicroscopy. J. Struct. Biol. 2008;164:170–175. doi: 10.1016/j.jsb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 61.Sobhanifar S., Worrall L.J., Gruninger R.J., Wasney G.A., Blaukopf M., Baumann L., Lameignere E., Solomonson M., Brown E.D., Withers S.G., Strynadka N.C. Structure and mechanism of Staphylococcus aureus TarM, the wall teichoic acid α-glycosyltransferase. Proc. Natl. Acad. Sci. USA. 2015;112:E576–E585. doi: 10.1073/pnas.1418084112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith T.J., Kremer M.J., Luo M., Vriend G., Arnold E., Kamer G., Rossmann M.G., McKinlay M.A., Diana G.D., Otto M.J. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science. 1986;233:1286–1293. doi: 10.1126/science.3018924. [DOI] [PubMed] [Google Scholar]
- 63.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 65.Rice L.M., Brünger A.T. Torsion angle dynamics: reduced variable conformational sampling enhances crystallographic structure refinement. Proteins. 1994;19:277–290. doi: 10.1002/prot.340190403. [DOI] [PubMed] [Google Scholar]
- 66.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mulloy B., Forster M.J., Jones C., Davies D.B. N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 1993;293:849–858. doi: 10.1042/bj2930849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cremer D., Pople J.A. A general definition of ring puckering coordinates. J. Am. Chem. Soc. 1975;97:1354–1358. [Google Scholar]
- 69.Jeffrey G.A., Yates J.H. Stereographic representation of the Cremer-Pople right-puckering parameters for pyranoid rings. Carbohydr. Res. 1979;74:319–322. [Google Scholar]
- 70.DeLano W.L. DeLano Scientific; 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- 71.Chapman B.K., Davulcu O., Skalicky J.J., Brüschweiler R.P., Chapman M.S. Parsimony in protein conformational change. Structure. 2015;23:1190–1198. doi: 10.1016/j.str.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Padron E., Bowman V., Kaludov N., Govindasamy L., Levy H., Nick P., McKenna R., Muzyczka N., Chiorini J.A., Baker T.S., Agbandje-McKenna M. Structure of adeno-associated virus type 4. J. Virol. 2005;79:5047–5058. doi: 10.1128/JVI.79.8.5047-5058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chapman M.S. Mapping the surface properties of macromolecules. Protein Sci. 1993;2:459–469. doi: 10.1002/pro.5560020318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.