

Abstract
Umbilical cord blood (CB) has emerged as an effective alternative donor source for hematopoietic stem cell transplantation. Despite this success, the prolonged duration of immune suppression following CB transplantation and the naiveté of CB T cells leave patients susceptible to viral infections. Adoptive transfer of ex vivo-expanded virus-specific T cells from CB is both feasible and safe. However, the manufacturing process of these cells is complicated, lengthy, and labor-intensive. We have now developed a simplified method to manufacture a single culture of polyclonal multivirus-specific cytotoxic T cells in less than 30 days. It eliminates the need for a live virus or transduction with a viral vector, thus making this approach widely available and GMP-applicable to target multiple viruses. The use of overlapping PepMixes as a source of antigen stimulation enable expansion of the repertoire of the T cell product to any virus of interest and make it available as a third party “off the shelf” treatment for viral infections following transplantation.
Keywords: cord blood, T cells, adoptive immunotherapy, cellular therapy, antiviral T cells, virus, cord blood transplantation
Graphical Abstract
Introduction
Umbilical cord blood (CB) transplantation (CBT) is emerging as an attractive alternative donor source for many hematologic malignancies, with outcomes comparable with matched related or unrelated bone marrow donors.1, 2, 3 CB stem cells are easily procured, require less stringent histocompatibility/human leukocyte antigen (HLA) matching criteria, possess a greater likelihood of matching for minorities,4 and cause fewer incidences of graft versus host disease (GvHD) compared with adult donor sources.1, 3, 5 These advantages of CBT, however, are offset by delayed immune reconstitution,6 making the recipient vulnerable to viral, bacterial, and fungal infections and consequent increased infectious disease morbidity and mortality.7, 8, 9 Several groups have shown that T cell immune reconstitution after double or single CBT (with or without serotherapy) is delayed,6, 10 and this, along with the naiveté of the infused CB T cells, correlates with an increased risk of viral reactivation or infection from latent and lytic viruses like cytomegalovirus (CMV), Epstein-Barr virus (EBV), and adenovirus (Adv) in the post-transplantation period.7, 11, 12
Like other latent viruses, BK virus (BKV) is present in most adults (up to 80%) and reactivates in the immune-compromised host, with rates as high as 60% in the allogeneic hematopoietic stem cell transplant (HSCT) setting,13 especially in recipients of CBT.14 Predisposing factors include myeloablative conditioning, positive pre-transplant serology, and the use of virus-naive donors such as CB as a stem cell source.14, 15, 16 Hemorrhagic cystitis (HC), a consequence of BKV infection, increases the median duration of hospitalization, the need for larger numbers of blood products, and costly pharmacologic treatments that are not always effective and can have unacceptable renal toxicities.13, 17 Although guidelines for surveillance and treatment of latent viruses like CMV with pharmacologic drugs have been well established, improvements in BKV therapy are still needed. The viremic load of BKV has been shown to affect overall survival. Patients with a high viral load of ≥10,000 copies/mL have an overall survival 1 year after HSCT of 48% compared with 89% in patients with a low virus burden.18 With the increasing use of CB as an acceptable source of stem cells even for adult patients,19 improvement of BKV therapies is warranted.
Adoptive T cell therapy using donor-derived ex vivo-expanded T cells has emerged as an effective strategy in preventing and treating viral infections.20, 21, 22, 23 Simplified methods for rapid production of multivirus-specific T cells from seropositive individuals have been validated and used for prophylaxis and treatment;24, 25, 26 however, this approach has not yet been successfully applied in the CBT setting because the only CB-derived multivirus-specific T cell approach currently in the clinic requires manufacturing times of 10+ weeks.27
We and others have shown that it is possible to expand virus-specific T cells (VSTs) even from seronegative23, 28, 29, 30 or naive donors such as CB.27, 31 Our previous methodology for the manufacture of trivirus-specific T cells from CB showed excellent in vitro and in vivo responses to CMV, EBV, and Adv;23, 27, 32 however, the process was complex, using viral vectors and live virus as the source of viral antigens, and because of the challenges associated with manufacturing these cells, it has not been widely adopted.
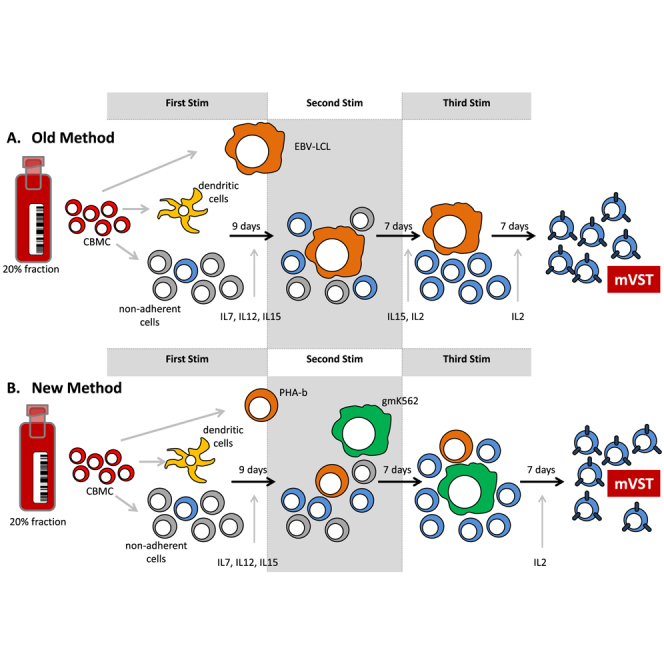
Here we developed a good manufacturing practices (GMP)-applicable methodology for the rapid manufacture of CB-derived multivirus-specific T cells that simplifies the manufacturing process, eliminates the need for live virus or viral vectors, and reduces the time required to produce multivirus-specific T cells (Figure 1). The use of commercially synthesized viral PepMixes instead of viral vectors not only reduced the manufacturing time but also simplified the process and enabled the introduction of other viral targets (such as BKV) to ensure that this approach will be more readily adaptable to include any virus of interest and broadly applicable for clinical use.
Figure 1.

Manufacturing Strategy
(A and B) The old methodology using EBV LCLs as antigen-presenting cells (A) and the new rapid production method using autologous PHA blasts and genetically modified K562s feeder cells (B).
Results
VSTs Specific for BKV Can Be Expanded from CB and Recognize Multiple Viral Antigens
Given the increasing clinical demand for BKV-specific T cells for CBT recipients and to establish that BKV-specific T cells can be generated from CB, we first tested the ability to generate BKV-specific T cells using our old methodology (Figure 1). The differences between the old and new method are outlined in Figure 1.
CB BKV VSTs were expanded from five different CB units. T cells were stimulated once with PepMix-pulsed dendritic cells (DCs), followed by two stimulations with PepMix-pulsed lymphoblastoid cell lines (LCLs). After three stimulations in the presence of interleukin-7 (IL-7), IL-15, or IL-2, T cells expanded a median of 15-fold (range, 6–24) by day 26 of culture (Figure 2A).
Figure 2.

Expansion and Specificity of BKV-VSTs
Dendritic cells were generated from CBMCs isolated from three cord bloods, and mature DCs were stimulated with BKV PepMixes, Large T, and VP-1 and then stimulated with irradiated EBV LCLs in the presence of IL-7 and IL-15. (A) Fold expansion of cytotoxic T lymphocytes (CTLs) after weekly stimulations and hypothetical cell counts after each stimulation. (B) ELISPOT assay demonstrating the antigen specificity of expanded CTLs to large T and VP1 after the third stimulation. (C) The surface marker phenotype of BKV-VSTs at week 3 (n = 3, mean) showed both CD4+ T cells and CD8+ T cells and absence of B and natural killer (NK) cells. (D) Intracellular cytokine staining of BKV-VSTs following restimulation with BKV PepMix, Large T, and VP1 showing polyfunctionality, producing IFN-γ.
Expanded CB-derived T cells showed specificity to BK antigens by interferon γ (IFN-γ) enzyme-linked immunospot (ELISPOT), recognizing both Large T (median, 369.5 spot-forming cells [SFCs]; range, 210–478.7; *p = 0.0015) and VP1 (median, 58 SFCs; range, 10–212; **p = 0.0915) compared with the negative control, Actin (Figure 2B). Final VST products were largely comprised of CD3+ T cells (mean, 89.5% ± 8.9%) (Figure 2C), with both CD4+ (mean, 42.4% ± 8.18%) and CD8+ (mean, 45.8% ± 2.5%) populations. DCs, B cells, and LCLs were not present in the final culture. Natural killer cells (CD3−CD56+) were detectable at low levels (mean, 5.17% of lymphocytes ± 2.96%). Intracellular cytokine staining of final VST products following stimulation with both BKV PepMixes showed consistent IFN-γ cytokine production (Figure 2D).
Rapid Generation of Multivirus-Specific T Cells in a Single Culture
Our original manufacturing strategy to generate tri-VSTs utilized EBV-LCLs as antigen-presenting cells (APCs). However, the approach took upward of 2–3 months because of the manufacture of EBV LCLs followed by three weekly stimulations with APC either transduced with an Ad5f35pp65 vector27 or viral PepMixes. To reduce the manufacturing time, we tested a more rapid approach to manufacture VSTs by pulsing mature DCs with six viral PepMixes from three viruses, CMV, adenovirus, and EBV, for the first stimulation and using autologous irradiated phytohemagglutinin (PHA) blasts as stimulators and modified K562 as feeder cells to provide costimulation for subsequent stimulations (Figure 1). After three stimulations, CB units (n = 4) had a median 14-fold expansion after the third stimulation (Figure 3A). Given that only a fraction of cells were used for each stimulation, when extrapolated, a mean of 1.47 × 108 (range, 0.67–3.17 × 108) VSTs could be generated from the 20% fraction of a CB unit (Figure 3B), with a manufacturing time of only 28 days (7 days for generation and mature DCs and 3 weeks for T cell expansion). This reduced the manufacturing time by almost 50%, with the old method requiring a minimum of 71 days.27 There was no difference in specificity for each of the three viruses by IFN-γ ELISPOT assay when comparing autologous LCLs or PHA-b with gmK562 cells as APCs (Figure 3C). Expansion in G-rex culture devices maintained or even improved the antigen recognition of all three viruses (Figure 3D), ensuring large-scale production of the multi-VSTs for clinical use. When tested for cytolytic potential in chromium release cytotoxicity assays, expanded T cells demonstrated specific killing of at least one virus when target cells (PHA blasts) were pulsed with CMV, EBV, and Adv (Figures 3E and 3F).
Figure 3.

Comparison of Virus-Specific T Cells that Were Expanded Using Irradiated Autologous LCLs at a Ratio of 4:1 for the Old Method or PHA Blasts with gmK562 at a Ratio of 1:1:4 for the New Method
(A) Fold expansion of CTLs after weekly stimulations and hypothetical cell counts after each stimulation. (B) Shown is the multivirus specificity as determined by IFN-γ ELISPOT assay. Each bar represents the mean of SFCs for four cell lines in response to stimulation with the respective viral PepMix or irrelevant PepMix (Actin) or positive control (SEB, data not shown). (C) IFN-γ ELISPOT responses of VST products produced by both methods showing no statistical difference in the recognition of the three viruses by the VSTs. (D) IFN-γ ELISPOT responses of VST products showing improved specificity when expanded in gas-permeable chambers compared with expansion in 24-well plates. (E and F) 1Cr release 4 hr after co-incubation of representative trivirus VSTs with PHA blasts pulsed with CMVpp65 and CMVie1 PepMix (CMV target), Adv-hexon and Adv-Penton (Adv target), PepMix (Adv-hexon target), and EBVbzlf1 and EBVlpm2 (EBV target). The data are the percentage of lysis of targets by representative VST products at E/T of 40:1, 20:1, 10:1, and 5:1.
VSTs Specific for BKV, CMV, Adv, and EBV Can Be Rapidly Produced in a Single Culture
After successfully generating trivirus-specific VSTs in a single culture using the rapid production method, we next sought to evaluate whether BKV antigens could be added to generate a four-virus product in a single culture. Using the rapid approach, we pulsed mature DCs with eight viral PepMixes derived from four viruses, used autologous irradiated PHA blasts as stimulators, and modified gmK562 cells as feeder cells. After three stimulations, nine CB units had a median 14-fold expansion after the third stimulation (Figure 4A). Extrapolating to include the entire T cell population at each expansion, a median 1.47 × 108 (67.5–317) VSTs could be expanded from the 20% CB fraction (Figure 4B) while retaining the same manufacturing time of 28 days as described above. The rapidly manufactured tetra-VST products specifically recognized BKV Large T and VP1 proteins (median, 145 (0–498) SFCs/1 × 105 cells) by IFN-γ ELISPOT and showed specificity for the three other viruses as well with median SFCs/1 × 105 cells as follows: CMVpp65 and IE1 (157 [6.5–363], Adv-hexon and Penton (331 [14–508], and EBVLMP2 and EBNA1 (79 [2–370]); p = 0.014 (Figure 4C). Moreover, there was no statistical difference in the specificity (p = 0.2) of the VSTs for each of the four viruses generated using the old method versus the rapid manufacture approach. The VSTs recognized each of the four viruses similarly by both methods (correlation coefficient r = 0.763, p = 0.0032) (Figure 4D). Of the nine multi-VSTs produced, 11% recognized one virus, 22% recognized two viruses, 33% were specific to three viruses, and 33% were specific to all four viruses (Figure 5).
Figure 4.

Expansion and Specificity of Multivirus VSTs by the New Method
(A) Fold expansion of four-virus VSTs after weekly stimulations. (B) Hypothetical cell counts after each stimulation extrapolated from cells plated and harvested and fold expansion. (C) Shown is the multivirus specificity of VSTs manufactured by the rapid method as determined by IFN-γ ELISPOT assay. Each bar represents the mean SFCs for nine cell lines in response to stimulation with respective viral PepMix or irrelevant PepMix (Actin) or positive control (SEB, data not shown). (D) IFN-γ ELISPOT responses of VST products produced by both methods showing no statistical difference in the recognition of the VSTs by the four viruses. (E and F) Surface phenotyping of rapidly produced multivirus VSTs at week 3 (n = 9, error bars show SD) showed CD4+ T cells and CD8+ cells and memory responses as shown in (F).
Figure 5.

Simultaneous Recognition of Multiple Viruses by VSTs by the Rapid Method as Evaluated by IFN-γ ELISPOT
Each slice represents the number of viruses recognized by the multi-VSTs.
All of the final rapid VST products were largely comprised of T cells (Figure 4E; Figure S1), with both CD4+ and CD8+ populations. DCs, B cells, and gmK562 cells were not present in the final culture. Natural killer cells were detectable at low levels (median, 0.2% [0%–6.23%]) in the lymphocyte gate. Intracellular cytokine staining of final VST products for BKV, Adv, CMV, and EBV in response to viral PepMixes showed consistent polyfunctionality, with both INF-γ and tumor necrosis factor alpha (TNF-α) cytokine production compared with Actin, with both CD4+ and CD8+ responses (Figure 4E; Figure S2).
Discussion
Here we describe a robust and simplified GMP-applicable manufacturing strategy for the generation of VSTs derived from CB that eliminates the need for live virus and viral vectors as the source of viral antigens.27 We have shown previously that VSTs can be primed in vitro from CB27 and that CB-derived T cells targeting CMV, EBV, and Adv can be manufactured using a GMP-applicable approach from the 20% fraction of CB units and administered to high-risk patients after CB transplantation28 (NCT00880789 and NCT01923766). This process, however, was relatively complex, which limited its widespread clinical use. To overcome these limitations, overlapping peptide pools (PepMixes) recognizing the full-length proteins of each of the target viruses were used to replace the viral vector and antigens expressed by the EBV LCL. As APCs, DCs, PHA blasts, and HLA-negative gmK562 cells modified to co-express CD80, CD83, CD86, and 4-1BBL were used.33 This dramatically reduced the manufacturing time in the laboratory from more than 70 days to approximately 30 days while still effectively stimulating and expanding T cells specific for multiple viral antigens in numbers sufficient for clinical use. Further, because this new strategy does not employ a viral vector, it allows the easy and rapid introduction of additional viral antigens; in this case, BKV antigens. Moreover, because the time-to-freeze was reduced by 50%, T cells are now available for infusion early after CBT (around day 30), when CBT recipients are most vulnerable to viral infections.8 Despite the shortened time to manufacture, the rapid multivirus VSTs maintained their antigen specificity, polyfunctionality, and cytolytic activity in vitro.
Since the initial report by Sun et al.29 showing the feasibility of expanding CB-derived VSTs ex vivo, many groups have used various methods for priming CB-derived DCs.34, 35 The report by Sun et al.29 using EBV lysate as a stimulator to pulse CB-derived DCs was a proof of principle for expanding virus-specific polyfunctional CD45RO+ T cells.29 The use of G-rex culture devices has overcome the limitation of expanding sufficient cells for clinical application; we have shown here that multivirus-specific T cells can be generated in numbers sufficient for multiple cell doses that appear more potent than those grown in flasks.36 Using our original manufacturing strategy, CB-derived T cells were infused to nine patients after CBT in doses of 5 × 10e6/m2 to 2.5 × 10e7/m2 with no infusion-related toxicities or GvHD within the first 45 days after VST infusion.23, 26, 28 Within 2 weeks of VST infusion, patients had detectable VSTs in their peripheral blood that persisted up to 1 year, suggesting that tri-VSTs from CB were safe and effective.26, 28
Our new manufacturing strategy further addressed the limitations of some of this prior work. The use of genetically modified K562s as artificial APCs provides the added co-stimulation with CD80, CD83, and CD86 that is usually reduced in CB DCs. The ability to use cytokines like IL-12 and TNF-α provided a more natural priming environment, and the ability to generate polyclonal cells with an ability to recognize multiple antigens minimizes viral escape to enhance the ability of CB-derived VSTs to produce anti-viral effects in vivo.
Although the strategy to develop a single culture of multi-VSTs is not novel per se, the clinical need for rapid manufacture of CB-derived multi-VSTs is highly significant and well established.25 The ability to rapidly expand polyclonal CD4+ and CD8+ T cells from the 20% fraction in numbers feasible for multiple doses makes it an attractive option for both adult and larger pediatric recipients of CBT, where the number of nucleated cells is a limiting factor.
Despite the faster approach, our method does have some limitations. The time to freeze for the rapidly produced VSTs is still three times longer than that required for multi-VSTs derived from seropositive donors. The cost of manufacturing also needs to be evaluated when considering scaling up GMP strategies to make them widely available. Although the cost of GMP-grade overlapping peptides is high, it is comparable with, if not less, than the cost of GMP-grade adenoviral vectors, which must undergo significant testing before release.
The possibility of antigen competition cannot be completely excluded but appears to be less than what is observed in similar multi-VSTs products derived from CMV-positive donors where there is skewing to a CMV-specific T cell response. Further, studies evaluating the persistence of rapid manufacture CB-derived multi-VSTs and their ability to mediate virus-specific killing in vivo are warranted.
Finally, given the large number of global inventories of frozen CB units, the development of CB-derived VST products also provides a novel platform for third-party use when adult donors are not readily available because we can select the HLA type to minimize GvHD. Adoptive immunotherapy using banked VSTs from partially HLA- matched donors has shown considerable success in treating EBV-related lymphoma and EBV, Adv, and CMV infections.26, 37, 38, 39, 40 Although transportation of cryopreserved cells has allowed this technology to be available to centers beyond the ones manufacturing the VST products, both nationally and internationally, the process of screening healthy donors and HLA selection can be time-consuming. The observed persistence of peripheral blood-derived third-party VSTs is 14–90 days, and multiple infusions are often required.41 CB-derived T cells exhibit less alloreactivity and therefore a decreased risk of GvHD.35 CB T cells are also known to have longer telomeres than adult blood, with a greater proliferative potential and a tendency for longer in vivo persistence; thus, multiple infusions may not be required. The ready availability of clinical-grade frozen CB units that are already screened and HLA-typed makes CB-derived VSTs an attractive and cost-saving option.
In summary, we have established a simple and rapid GMP-applicable methodology to generate multi-VSTs targeting CMV, EBV, Ad, and BKV from a small fraction of CB with the potential to affect the transplant-related morbidity and mortality in CB recipients.
Materials and Methods
Ethics Statement
All research involving human materials was approved by the respective institutional review boards at the Children’s National Medical Center and MD Anderson Cancer Center (MDACC). Fresh or frozen CB units were obtained from healthy donors who gave written informed consent.
Generation of DCs
CB mononuclear cells (CBMCs) were isolated by Ficoll/Hypaque centrifugation. CBMCs were washed twice, resuspended in CellGenix medium, and plated at approximately 1 × 107 cells/well in DC medium (CellGenix medium plus 2 mM L-glutamine; GlutaMax, Invitrogen) in a six-well plate for 1–2 hr at 37°C in a humidified CO incubator. Non-adherent T cells were removed by rinsing with PBS (Sigma) and cryopreserved. Cells adherent after 1 hr were cultured in DC medium with granulocyte-monocyte colony stimulating factor (GM-CSF; 800 U/mL) (sargramostim, Leukine; Immunex) and IL-4 (500 U/mL, R&D Systems) for 5 days.
Maturation of DCs and Pulsing of DCs with Viral PepMixes
On days 5–6, CB-derived DCs were matured using a cytokine cocktail consisting of LPS, GM-CSF, IL-4, IL-6, IL-1b, and prostaglandin E2 (Sigma) (“maturation cocktail”) for 1–2 days.
Ex Vivo Expansion of Multivirus-Specific VSTs Using PHA Blasts and gmK562 Cells
To generate rapidly manufactured CB-derived VSTs targeting EBV, CMV, Ad, and BKV, matured DCs were harvested and pulsed with overlapping 15-mer PepMixes spanning the entire length of the protein for each of the four viruses (CMV-IE1, CMV-pp65, Adv-hexon, Adv-penton, EBV-LMP2, EBV-EBNA1, BKV-VP1, and BKV-Large T; PepMixes, JPT Peptide Technologies) and used as APCs at a stimulator-to-responder ratio of 1:10–1:20 with non-adherent CBMCs. T cells were cultured in T cell medium containing RPMI 1640 supplemented with 40% Clicks medium, 10% human antibody (AB) serum (Valley), and 2 mM GlutaMax. For the first stimulation, a cytokine mix containing IL-7 (10 ng/mL), IL-12 (10 ng/mL), and IL-15 (5 ng/mL) (all from R&D Systems) was added.
To generate activated T cells, PHA blasts were used as APCs, and CBMCs or non-adherent T cells were stimulated with the mitogen PHA-L (5 μg/mL, Sigma-Aldrich). Cells were fed every 2–3 days with 100–200 U/mL of IL-2.
After 8–10 days, peptide-pulsed autologous irradiated (30 Gy) CB-derived PHA blasts were co-cultured with T cells at a T cell:PHA blast ratio of 1:1 and maintained in medium supplemented with IL-15 (5 ng/mL) on the second stimulation or medium supplemented with IL-2 (50 U/mL) for the third stimulation cycle. On the third stimulation, irradiated (200 Gy) K562 cells that were genetically modified to express the co-stimulatory molecules CD80, CD83, CD86, and 4-1BBL (gmK562) were added to the T cell:PHA-blast co-culture to provide further co-stimulation. GMP-grade gmK562 cells were a gift from Dr. Cliona Rooney at Baylor College of Medicine).
Generation of Multi-VSTs Using PepMix-Pulsed EBV-Transformed B Cell Lines from CBMCs
As the source of APCs for the second and third stimulations, 5 × 106 CBMCs were infected with concentrated supernatants from a B95-8 working cell bank as described previously.20 When the LCLs were in T-75 flasks and ready for use as stimulators, the cells were irradiated at 50 Gy, washed, resuspended at 5 × 105cells/mL of complete medium (RPMI medium [HyClone} plus human serum and GlutaMax) and then used as stimulators at a ratio of one T cell to four LCLs in a 24-well plate or five LCLs to one T cell in a G-Rex10 gas-permeable culture device as published.42
IFN-γ ELISPOT
Ninety-six-well filtration plates (MultiScreen, MSIPS4W10, Millipore) were coated overnight with 10 μg/mL anti-human IFN monoclonal antibody (mAb) (capture mAb, 1-DIK-purified, Mabtech). VSTs were plated at 1 × 105 cells/well and stimulated with overlapping 15-mer PepMixes encompassing proteins for each of the four viruses (for CMV-pp65, CMV-IE1, EBV-LMP2, EBV-EBNA1, Adv-hexon, Adv-Penton, BKV-Large T, and BKV-VP1, 0.5 nmol/peptide/well). Each condition was run in duplicate or triplicate. Staphylococcal enterotoxin-B (SEB) was used as positive control (1 μg/mL). A 15-mer PepMix encompassing the actin protein was used as a negative control. After 18–24 hr, the plates were washed and incubated with the secondary biotin-conjugated anti-human IFN-γ mAb (detector mAb, 7-B6-1 biotin, Mabtech) at 1 μg/mL. After incubation with avidin:biotinylated horseradish peroxidase (HRP) complex (Vectastain Elite ABC kit [standard], PK6100, Vector Laboratories), plates were developed with AEC substrate (A6926, Sigma-Aldrich), dried overnight, and quantified (Zellnet Consulting). The frequency of T cells specific for each peptide was expressed as SFCs per 1 × 105 cells. A correction for confluence was applied as follows: well count + 2 × (well count × (percent confluence / [1 − % confluence])).
Flow Cytometry
Staining of cell surface markers on CBMCs and T cells was performed with CD3-APC/Vio770, CD4-Vioblue, CD8-Viogreen, CD19-FITC, CD56-PE/Vio770, CD16-PE, CD62L-Vioblue, CD45RA-PE, CD45RO-APC, and CCR7-FITC (Miltenyi Biotec and BD Biosciences). 50,000 events per sample were acquired on a MACSQuant cytometer (Miltenyi), and the data were analyzed with Flow Jo (Tree Star).
Intracellular Cytokine Staining
For intracellular cytokine staining, T cells were rested with IL-2 (100 U/mL) overnight and then stimulated with PepMixes encompassing viral or control proteins (JPT) at 0.5 nmol/μL, along with anti-CD49d/CD28 antibodies (BD Biosciences) at 1:1,000 for co-stimulation and Golgi stop at 1 μL/mL. SEB and Actin were utilized as controls. T cells were stained with cell surface markers (CD3, CD4, and CD8), followed by permeabilization with Cytofix/Cytoperm (BD Biosciences), washing, and staining with IFN-γ-APC (BioLegend), IL-2-fluorescein isothiocyanate (FITC), and TNF-α-PE (BD Biosciences).
Cytotoxicity Assay
Cytotoxicity was measured by standard 51Cr release assay. Briefly, autologous or allogeneic PHA-stimulated lymphoblasts were produced and incubated with viral PepMixes (0.5 nmol/peptide/mL of each protein) for 1–2 hr and then pulsed with 51Cr (PerkinElmer) at 10 μCi per 1 × 105 cells for 1 hr and washed. Effector T cells rested overnight with IL-2 (100 U/mL) and were plated with radiolabeled target cells at multiple effector-to-target ratios. Maximum release was determined by lysis of radiolabeled targets with 1% Triton X-100 detergent. Targets and effectors were incubated at 37°C for 4–6 hr and centrifuged, and then supernatants were transferred to a 96-well scintillation plate and allowed to dry. Radioactivity was measured on a gamma counter. Specific lysis was determined as follows: (experimental CPM – background CPM) / (maximum CPM – background CPM). All experiments were performed in triplicate.
Data Analysis/Statistics
Results were evaluated using descriptive statistics (means, SDs, and ranges). Comparative analysis between ELISPOT and flow cytometry results for different epitopes was performed via comparison of row means and t tests. Comparisons between responses to viral peptides and irrelevant peptides were performed using a two-sided unpaired t test. Analysis was performed in GraphPad Prism (GraphPad).
Author Contributions
H.D. contributed to the conceptualization, methodology, investigation, formal analysis of data, writing of the original draft, and revision of the manuscript. M.L. contributed to the investigation. J.W.B. contributed to the formal analysis and investigation. S.P. contributed to the investigation and editing of the manuscript. C.B. contributed to the formal analyses of flow data. C.R.C. contributed to the methodology. E.J.S. contributed funding and resources, and contributed to editing of the manuscript. C.M.B. provided conceptualization, supervision, resources, formal analyses, and review and editing of the manuscript. P.J.H. contributed to the conceptualization, methodology, formal analysis, supervision, and review and editing of the manuscript.
Conflicts of Interest
The authors have no relevant conflicts of interest.
Acknowledgments
This research was funded by grants from the National Cancer Institute (PO1 CA148600e02 to C.M.B. and E.J.S.), a postdoctoral training grant from the American Cancer Society (PF-13-046-01-LIB to P.J.H.), a grant from the Children’s Cancer Foundation (to P.J.H.), and a New Investigator Award from The American Society for Blood and Marrow Transplantation (to H.D.). The authors would like to thank Dario Campana and St. Jude Children's Research Hospital for graciously providing the K562 cells.
Footnotes
Supplemental Information includes two figures and can be found with this article online at http://dx.doi.org/10.1016/j.omtm.2017.02.001.
Supplemental Information
References
- 1.Eapen M., Rubinstein P., Zhang M.J., Stevens C., Kurtzberg J., Scaradavou A., Loberiza F.R., Champlin R.E., Klein J.P., Horowitz M.M., Wagner J.E. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet. 2007;369:1947–1954. doi: 10.1016/S0140-6736(07)60915-5. [DOI] [PubMed] [Google Scholar]
- 2.Kurtzberg J., Laughlin M., Graham M.L., Smith C., Olson J.F., Halperin E.C., Ciocci G., Carrier C., Stevens C.E., Rubinstein P. Placental blood as a source of hematopoietic stem cells for transplantation into unrelated recipients. N. Engl. J. Med. 1996;335:157–166. doi: 10.1056/NEJM199607183350303. [DOI] [PubMed] [Google Scholar]
- 3.Rocha V., Cornish J., Sievers E.L., Filipovich A., Locatelli F., Peters C., Remberger M., Michel G., Arcese W., Dallorso S. Comparison of outcomes of unrelated bone marrow and umbilical cord blood transplants in children with acute leukemia. Blood. 2001;97:2962–2971. doi: 10.1182/blood.v97.10.2962. [DOI] [PubMed] [Google Scholar]
- 4.Hanley P.J., Cruz C.R., Shpall E.J., Bollard C.M. Improving clinical outcomes using adoptively transferred immune cells from umbilical cord blood. Cytotherapy. 2010;12:713–720. doi: 10.3109/14653249.2010.517518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gluckman E., Rocha V., Boyer-Chammard A., Locatelli F., Arcese W., Pasquini R., Ortega J., Souillet G., Ferreira E., Laporte J.P. Outcome of cord-blood transplantation from related and unrelated donors. Eurocord Transplant Group and the European Blood and Marrow Transplantation Group. N. Engl. J. Med. 1997;337:373–381. doi: 10.1056/NEJM199708073370602. [DOI] [PubMed] [Google Scholar]
- 6.Komanduri K.V., St John L.S., de Lima M., McMannis J., Rosinski S., McNiece I., Bryan S.G., Kaur I., Martin S., Wieder E.D. Delayed immune reconstitution after cord blood transplantation is characterized by impaired thymopoiesis and late memory T-cell skewing. Blood. 2007;110:4543–4551. doi: 10.1182/blood-2007-05-092130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballen K., Woo Ahn K., Chen M., Abdel-Azim H., Ahmed I., Aljurf M., Antin J., Bhatt A.S., Boeckh M., Chen G. Infection rates among acute leukemia patients receiving alternative donor hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2016;22:1636–1645. doi: 10.1016/j.bbmt.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lucchini G., Perales M.A., Veys P. Immune reconstitution after cord blood transplantation: peculiarities, clinical implications and management strategies. Cytotherapy. 2015;17:711–722. doi: 10.1016/j.jcyt.2015.03.614. [DOI] [PubMed] [Google Scholar]
- 9.Ruggeri A., Peffault de Latour R., Carmagnat M., Clave E., Douay C., Larghero J., Cayuela J.M., Traineau R., Robin M., Madureira A. Outcomes, infections, and immune reconstitution after double cord blood transplantation in patients with high-risk hematological diseases. Transpl. Infect. Dis. 2011;13:456–465. doi: 10.1111/j.1399-3062.2011.00632.x. [DOI] [PubMed] [Google Scholar]
- 10.Saliba R.M., Rezvani K., Leen A., Jorgensen J., Shah N., Hosing C., Parmar S., Oran B., Olson A., Rondon G. General and virus-specific immune cell reconstitution after double cord blood transplantation. Biol. Blood Marrow Transplant. 2015;21:1284–1290. doi: 10.1016/j.bbmt.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Escalón M.P., Komanduri K.V. Cord blood transplantation: evolving strategies to improve engraftment and immune reconstitution. Curr. Opin. Oncol. 2010;22:122–129. doi: 10.1097/cco.0b013e328335a56e. [DOI] [PubMed] [Google Scholar]
- 12.Geyer M.B., Jacobson J.S., Freedman J., George D., Moore V., van de Ven C., Satwani P., Bhatia M., Garvin J.H., Bradley M.B. A comparison of immune reconstitution and graft-versus-host disease following myeloablative conditioning versus reduced toxicity conditioning and umbilical cord blood transplantation in paediatric recipients. Br. J. Haematol. 2011;155:218–234. doi: 10.1111/j.1365-2141.2011.08822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giraud G., Priftakis P., Bogdanovic G., Remberger M., Dubrulle M., Hau A., Gutmark R., Mattson J., Svahn B.M., Ringden O. BK-viruria and haemorrhagic cystitis are more frequent in allogeneic haematopoietic stem cell transplant patients receiving full conditioning and unrelated-HLA-mismatched grafts. Bone Marrow Transplant. 2008;41:737–742. doi: 10.1038/sj.bmt.1705962. [DOI] [PubMed] [Google Scholar]
- 14.El-Zimaity M., Saliba R., Chan K., Shahjahan M., Carrasco A., Khorshid O., Caldera H., Couriel D., Giralt S., Khouri I. Hemorrhagic cystitis after allogeneic hematopoietic stem cell transplantation: donor type matters. Blood. 2004;103:4674–4680. doi: 10.1182/blood-2003-08-2815. [DOI] [PubMed] [Google Scholar]
- 15.Silva Lde.P., Patah P.A., Saliba R.M., Szewczyk N.A., Gilman L., Neumann J., Han X.Y., Tarrand J., Ribeiro R., Gulbis A. Hemorrhagic cystitis after allogeneic hematopoietic stem cell transplants is the complex result of BK virus infection, preparative regimen intensity and donor type. Haematologica. 2010;95:1183–1190. doi: 10.3324/haematol.2009.016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Megged O., Stein J., Ben-Meir D., Shulman L.M., Yaniv I., Shalit I., Levy I. BK-virus-associated hemorrhagic cystitis in children after hematopoietic stem cell transplantation. J. Pediatr. Hematol. Oncol. 2011;33:190–193. doi: 10.1097/MPH.0b013e3181fce388. [DOI] [PubMed] [Google Scholar]
- 17.Gilis L., Morisset S., Billaud G., Ducastelle-Leprêtre S., Labussière-Wallet H., Nicolini F.E., Barraco F., Detrait M., Thomas X., Tedone N. High burden of BK virus-associated hemorrhagic cystitis in patients undergoing allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2014;49:664–670. doi: 10.1038/bmt.2013.235. [DOI] [PubMed] [Google Scholar]
- 18.Haines H.L., Laskin B.L., Goebel J., Davies S.M., Yin H.J., Lawrence J., Mehta P.A., Bleesing J.J., Filipovich A.H., Marsh R.A., Jodele S. Blood, and not urine, BK viral load predicts renal outcome in children with hemorrhagic cystitis following hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2011;17:1512–1519. doi: 10.1016/j.bbmt.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 19.Milano F., Gooley T., Wood B., Woolfrey A., Flowers M.E., Doney K., Witherspoon R., Mielcarek M., Deeg J.H., Sorror M. Cord-Blood Transplantation in Patients with Minimal Residual Disease. N. Engl. J. Med. 2016;375:944–953. doi: 10.1056/NEJMoa1602074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rooney C.M., Smith C.A., Ng C.Y., Loftin S., Li C., Krance R.A., Brenner M.K., Heslop H.E. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. 1995;345:9–13. doi: 10.1016/s0140-6736(95)91150-2. [DOI] [PubMed] [Google Scholar]
- 21.Doubrovina E., Oflaz-Sozmen B., Prockop S.E., Kernan N.A., Abramson S., Teruya-Feldstein J., Hedvat C., Chou J.F., Heller G., Barker J.N. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119:2644–2656. doi: 10.1182/blood-2011-08-371971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peggs K.S., Verfuerth S., Pizzey A., Khan N., Guiver M., Moss P.A., Mackinnon S. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. 2003;362:1375–1377. doi: 10.1016/S0140-6736(03)14634-X. [DOI] [PubMed] [Google Scholar]
- 23.Hanley P.J., Bollard C.M., Brunstein C.G. Adoptive immunotherapy with the use of regulatory T cells and virus-specific T cells derived from cord blood. Cytotherapy. 2015;17:749–755. doi: 10.1016/j.jcyt.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerdemann U., Keirnan J.M., Katari U.L., Yanagisawa R., Christin A.S., Huye L.E., Perna S.K., Ennamuri S., Gottschalk S., Brenner M.K. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol. Ther. 2012;20:1622–1632. doi: 10.1038/mt.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papadopoulou A., Gerdemann U., Katari U.L., Tzannou I., Liu H., Martinez C., Leung K., Carrum G., Gee A.P., Vera J.F. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci. Transl. Med. 2014;6:242ra83. doi: 10.1126/scitranslmed.3008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naik S., Nicholas S.K., Martinez C.A., Leen A.M., Hanley P.J., Gottschalk S.M., Rooney C.M., Hanson I.C., Krance R.A., Shpall E.J. Adoptive immunotherapy for primary immunodeficiency disorders with virus-specific T lymphocytes. J. Allergy Clin. Immunol. 2016;137:1498–1505.e1. doi: 10.1016/j.jaci.2015.12.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanley P.J., Cruz C.R., Savoldo B., Leen A.M., Stanojevic M., Khalil M., Decker W., Molldrem J.J., Liu H., Gee A.P. Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood. 2009;114:1958–1967. doi: 10.1182/blood-2009-03-213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanley P.J., Melenhorst J.J., Nikiforow S., Scheinberg P., Blaney J.W., Demmler-Harrison G., Cruz C.R., Lam S., Krance R.A., Leung K.S. CMV-specific T cells generated from naïve T cells recognize atypical epitopes and may be protective in vivo. Sci. Transl. Med. 2015;7:285ra63. doi: 10.1126/scitranslmed.aaa2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Q., Burton R.L., Pollok K.E., Emanuel D.J., Lucas K.G. CD4(+) Epstein-Barr virus-specific cytotoxic T-lymphocytes from human umbilical cord blood. Cell. Immunol. 1999;195:81–88. doi: 10.1006/cimm.1999.1514. [DOI] [PubMed] [Google Scholar]
- 30.Szabolcs P., Niedzwiecki D. Immune reconstitution after unrelated cord blood transplantation. Cytotherapy. 2007;9:111–122. doi: 10.1016/j.bbmt.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savoldo B., Cubbage M.L., Durett A.G., Goss J., Huls M.H., Liu Z., Teresita L., Gee A.P., Ling P.D., Brenner M.K. Generation of EBV-specific CD4+ cytotoxic T cells from virus naive individuals. J. Immunol. 2002;168:909–918. doi: 10.4049/jimmunol.168.2.909. [DOI] [PubMed] [Google Scholar]
- 32.Hanley P.J., Shaffer D.R., Cruz C.R., Ku S., Tzou B., Liu H., Demmler-Harrison G., Heslop H.E., Rooney C.M., Gottschalk S., Bollard C.M. Expansion of T cells targeting multiple antigens of cytomegalovirus, Epstein-Barr virus and adenovirus to provide broad antiviral specificity after stem cell transplantation. Cytotherapy. 2011;13:976–986. doi: 10.3109/14653249.2011.575356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ngo M.C., Ando J., Leen A.M., Ennamuri S., Lapteva N., Vera J.F., Min-Venditti A., Mims M.P., Heslop H.E., Bollard C.M. Complementation of antigen-presenting cells to generate T lymphocytes with broad target specificity. J. Immunother. 2014;37:193–203. doi: 10.1097/CJI.0000000000000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serrano L.M., Pfeiffer T., Olivares S., Numbenjapon T., Bennitt J., Kim D., Smith D., McNamara G., Al-Kadhimi Z., Rosenthal J. Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy. Blood. 2006;107:2643–2652. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merindol N., Grenier A.J., Caty M., Charrier E., Duval A., Duval M., Champagne M.A., Soudeyns H. Umbilical cord blood T cells respond against the Melan-A/MART-1 tumor antigen and exhibit reduced alloreactivity as compared with adult blood-derived T cells. J. Immunol. 2010;185:856–866. doi: 10.4049/jimmunol.0902613. [DOI] [PubMed] [Google Scholar]
- 36.Vera J.F., Brenner L.J., Gerdemann U., Ngo M.C., Sili U., Liu H., Wilson J., Dotti G., Heslop H.E., Leen A.M., Rooney C.M. Accelerated production of antigen-specific T cells for preclinical and clinical applications using gas-permeable rapid expansion cultureware (G-Rex) J. Immunother. 2010;33:305–315. doi: 10.1097/CJI.0b013e3181c0c3cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leen A.M., Bollard C.M., Mendizabal A.M., Shpall E.J., Szabolcs P., Antin J.H., Kapoor N., Pai S.Y., Rowley S.D., Kebriaei P. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121:5113–5123. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haque T., McAulay K.A., Kelly D., Crawford D.H. Allogeneic T-cell therapy for Epstein-Barr virus-positive posttransplant lymphoproliferative disease: long-term follow-up. Transplantation. 2010;90:93–94. doi: 10.1097/TP.0b013e3181d7c424. [DOI] [PubMed] [Google Scholar]
- 39.Barker J.N., Doubrovina E., Sauter C., Jaroscak J.J., Perales M.A., Doubrovin M., Prockop S.E., Koehne G., O’Reilly R.J. Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood. 2010;116:5045–5049. doi: 10.1182/blood-2010-04-281873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uhlin M., Gertow J., Uzunel M., Okas M., Berglund S., Watz E., Brune M., Ljungman P., Maeurer M., Mattsson J. Rapid salvage treatment with virus-specific T cells for therapy-resistant disease. Clin. Infect. Dis. 2012;55:1064–1073. doi: 10.1093/cid/cis625. [DOI] [PubMed] [Google Scholar]
- 41.O’Reilly R.J., Prockop S., Hasan A.N., Koehne G., Doubrovina E. Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplant. 2016;51:1163–1172. doi: 10.1038/bmt.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanley P.J., Lam S., Shpall E.J., Bollard C.M. Expanding cytotoxic T lymphocytes from umbilical cord blood that target cytomegalovirus, Epstein-Barr virus, and adenovirus. J. Vis. Exp. 2012;3627:e3627. doi: 10.3791/3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.