

Abstract
Despite rapid progress, many problems and limitations persist and limit the applicability of gene-editing techniques. Making use of meganucleases, TALENs, or CRISPR/Cas9-based tools requires an initial step of pre-screening to determine the efficiency and specificity of the designed tools. This step remains time consuming and material consuming. Here we propose a simple, cheap, reliable, time-saving, and highly sensitive method to evaluate a given gene-editing tool based on its capacity to induce chromosomal translocations when combined with a reference engineered nuclease. In the proposed technique, designated engineered nuclease-induced translocations (ENIT), a plasmid coding for the DNA-editing tool to be tested is co-transfected into carefully chosen target cells along with that for an engineered nuclease of known specificity and efficiency. If the new enzyme efficiently cuts within the desired region, then specific chromosomal translocations will be generated between the two targeted genomic regions and be readily detectable by a one-step PCR or qPCR assay. The PCR product thus obtained can be directly sequenced, thereby determining the exact position of the double-strand breaks induced by the gene-editing tools. As a proof of concept, ENIT was successfully tested in different cell types and with different meganucleases, TALENs, and CRISPR/Cas9-based editing tools.
Keywords: CRISPR/Cas9, TALEN, translocations, PCR, assay
Graphical Abstract
Introduction
The field of in vivo gene editing using engineered nucleases is in strong development with the constantly increasing panel of available tools and techniques for testing their efficiency. Those gene-editing tools include meganucleases, transcription activator-like (TAL) effector nucleases (TALENs),1 and the clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system2 (reviewed by Saada et al.3).
Whatever the tools used, editing the genome is based on the generation of double-strand breaks (DSBs), which will trigger DNA repair, resulting in genome modifications either through homologous recombination (HR) or error-prone non-homologous end joining (NHEJ) processes. The modifications thus obtained include gene disruptions, insertions, substitutions, and chromosomal rearrangements.4
The first and most important step in gene editing requires testing the efficiency and precision of the engineered nuclease. This is mostly based on detection of mismatches introduced in the target region by NHEJ. Various methods are employed to this aim, but they are all time-consuming and technically challenging.
The method of choice for the identification of precise mutations remains direct detection via DNA sequencing. This requires multiple steps prior to sequencing: establishment of a clonal cell population, DNA extraction, amplification of the target sequence by PCR, its purification, and cloning. Aligning the DNA sequence eventually produced with the wild-type homologous sequence allows for mutations to be screened at the cutting site. Easier and faster alternatives have been proposed, such as screening for DNA mismatches by single-stranded conformational polymorphism (SSCP) analysis5, 6 or denaturing high-performance liquid chromatography (DHPLC).7, 8 Those techniques are based on the property of single-stranded DNAs to adopt specific conformations that can be altered by a single base change, resulting in modified migration in gel electrophoretic assays (SSCP) or through a chromatographic column (DHPLC). Even though more rapid, those methods still require multiple steps (a PCR amplification, the amplicon denaturation, and annealing and detection of differences in the mobility of the single-stranded DNAs). Moreover, it is currently impossible to use this technique to analyze fragments longer than 1,000 bp and also to locate the mutation within the DNA sequence.9 Other techniques, like restriction enzyme digestion-suppressed PCR (RE-PCR), T7 endonuclease I (T7EI) endonuclease assay, and the Surveyor assay, that are based on detection of mismatches have been developed to circumvent the abovementioned limitations. In RE-PCR, the genomic DNA is submitted to digestion with a restriction enzyme recognizing a sequence that contains the cleavage site target of the gene-editing tool. If the restriction site is mutated via NHEJ, then the enzyme will not cut it, and amplicons can be detected by PCR. In the absence of introduced mutations, the restriction enzyme will recognize and cleave the targeted DNA, and amplification will be suppressed.10 The problems with RE-PCR are that it is a two-step technique that requires the use of a specific restriction enzyme, and it is only qualitative and therefore not suitable for assessing the cutting efficiency of engineered nucleases to be tested. The T7EI assay and the Surveyor assay are both based on recognition and cleavage of heteroduplex DNA at mismatches by mismatch-specific nucleases: the T7 endonuclease or the Surveyor enzyme.11 Both techniques are widely used because they are less time- and money-consuming compared with sequencing, but they are not very reliable in terms of reproducibility. Indeed, the presence of random mutations or polymorphisms in cell lines can yield false positives. Moreover, there is an extreme variability in existing protocols that induces the problem of reproducibility (e.g., compare Fu et al.,12 Hou et al.,13 and Sedlak et al.14).
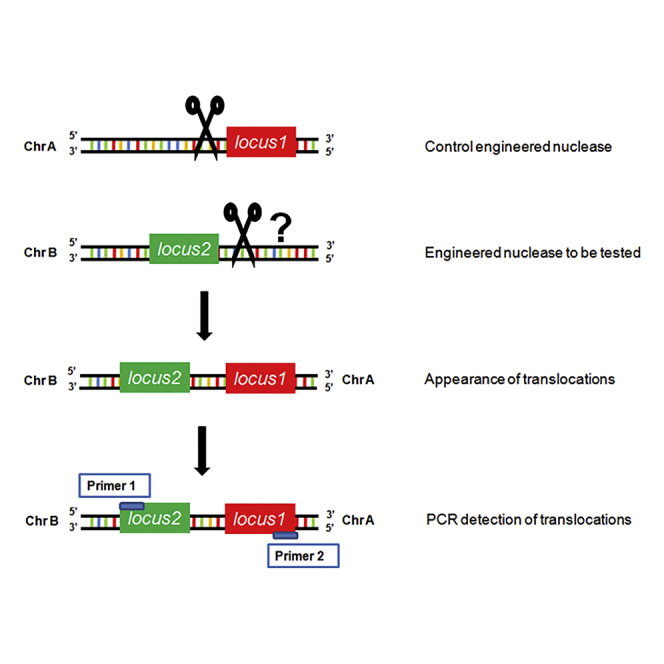
DNA DSBs produced at two separate sites can induce chromosomal translocations via NHEJ that can erroneously join the ends of two broken chromosomes15, 16 (reviewed by Iarovaia et al.17). Recently, simultaneous expression of engineered nucleases targeting different loci has been reported to induce specific chromosomal translocations in human mesenchymal precursor and epithelial cells.18, 19 Here we propose to take advantage of this capacity to induce chromosomal translocations to evaluate the efficiency and precision of engineered nucleases. A one-step method referred to as engineered nuclease-induced translocations (ENIT) is described. Based on PCR, this technique is rapid, sensitive, reproducible, and cheap. Using engineered nucleases targeting two different genomic loci and specific primers, the described technique allows the visualization and reliable quantification of genomic cleavage in fewer than 1,000 transfected cells.
Results
PCR Detection of a Specific Chromosomal Translocation Induced by Two Engineered Nucleases
ENIT is based on cellular co-expression of two engineered nucleases, one of known specificity and proven efficiency and the other to be tested. The expected result is a specific chromosomal translocation that should be detectable by a simple PCR assay. To specifically induce DSBs in two target loci of choice, we used two pairs of TALEN subunits, one recognizing the MYC gene locus at 8q24 (TAL8F + TAL8R) and the other recognizing the IGH gene locus at 14q32 (TAL14F + TAL14R), cloned into GFP-expressing plasmids. To this aim, HeLa cells were simultaneously transfected with the four TALEN subunits and kept for 72 hr in culture. The obtained transfection efficiency (ranging from 4% to 20% in different experiments) was checked via GFP expression prior to extraction of genomic DNA. PCR was then performed with primers surrounding the DNA sequences targeted by the TALENs. The two pairs of primers corresponding to the MYC (MYC F + MYC R) and IGH (IGH F + IGH R) gene loci produced the expected PCR bands in both non-transfected (Figure 1A, lanes 2 and 3, respectively) and transfected HeLa cells (Figure 1B, lanes 2 and 3, respectively). When the MYC forward primer (MYC F) was used together with the IGH reverse primer (IGH R), a single, ∼500-bp-long specific amplicon was observed specifically in HeLa cells transfected with the TALEN plasmids (Figure 1B, lane 4, versus Figure 1A, lane 4), suggesting that a t(8;14) chromosomal rearrangement had occurred in transfected cells, producing a recombined MYC-IGH locus. For further characterization, the amplicon was purified from the agarose gel and sequenced. Aligning the obtained sequence with the NCBI reference sequences of IGH and MYC (NG_001019.5 and NG_007161.1, respectively) revealed a perfect match up to the beginning of the TALEN cutting site where several mismatches were detected (Figures 1C and 1D, highlighted in bold; Figures S1A and S1B), as expected.
Figure 1.

PCR Detection of a Rearranged DNA Sequence Resulting from t(8;14) Chromosomal Translocation
(A) PCR on DNA extracted from untransfected HeLa cells. The represented amplicons were obtained using the following primer pairs: lane 1, GAPDH F + R; lane 2, MYC F + R; lane 3, IGH F + R; lane 4, MYC F + IGH R. M is the molecular weight marker. (B) PCR on DNA extracted from transfected HeLa cells with the four TALENS recognizing the MYC and IGH genes. The represented amplicons were obtained using the following primer pairs: lane 1, GAPDH F + R; lane 2, MYC F + R; lane 3, IGH F + R; lane 4, MYC F + IGH R. The experiments were repeated three times and representative gels are displayed. (C and D) Alignment of IGH (C) or MYC (D) wild-type NCBI reference sequences (NG_001019.5 and NG_007161.1, respectively) with the sequence of the amplicon obtained from DNA extracted from transfected HeLa cells and amplified with MYC F + IGH R primers. The part of the alignment where the sequences are perfectly aligned is highlighted in light gray, whereas the parts of the sequences containing mismatches (in bold) next to the TALENs cutting site and next to the place where the translocation occurred are highlighted in dark gray.
The t(8;14) translocation thus generated was further ascertained in one experiment of fluorescence in situ hybridization (FISH) performed on chromosome spreads from the TALEN-transfected HeLa cells using probes for the MYC and IGH loci. We observed one chromosome with colocalized MYC and IGH signals in one spread of 100. A spread with metaphase chromosomes from one cell containing the translocation (indicated by an arrow) is represented in Figure 2. No IGH-MYC translocation was ever observed in untransfected HeLa cells used as controls. From these results, ENIT thus appears to be a suitable approach for evaluating the capacity of engineered nucleases to specifically target DNA sequences and trigger testable chromosomal translocations.
Figure 2.

Chromosome Spread of HeLa Cells
Representative FISH image of a chromosome spread of one HeLa cell transfected with the four TALENS targeting the MYC and IGH genes. The chromosome indicated by the red arrow exhibits the t(8:14) translocation, as evidenced by colocalized MYC (red) and IGH (green) signals. Left: enlarged image of the metaphase chromosome containing the t(8:14) translocation. At least 100 metaphases were analyzed to detect one translocation.
ENIT Sensitivity
The cutting efficiency of TALENs is known to be relatively low.20 Further experiments were thus carried out to determine the minimum number of transfected cells needed for the expected chromosomal translocation to be detectable by the PCR assay. DNA samples prepared from non-transfected and transfected HeLa cells were serially diluted from 1,000 to 0.8 ng, followed by PCR amplification using the MYC F and IGH R primers as described above. A minimum of 2,000 transfected cells (corresponding to ∼13.4 ng of DNA at 6.7 pg/cell21) is thus required for detection of the translocation using ENIT (Figure 3A).The accuracy of dilution was checked by PCR on the same samples using MYC F + MYC R primers (Figure 3B). The primers are listed in Table 1.
Figure 3.

Sensitivity of the PCR-Based Detection of Translocations
(A) Intensity of bands obtained by the PCR amplification of serial dilutions of the DNA obtained from HeLa cells transfected with the four TALENS recognizing the MYC and IGH regions. The amplicons were obtained with MYC F and IGH R primers. (B) The intensity of each band was related to the number of transfected cells from which we obtained the DNA used for PCR. The reliability of dilutions was calculated by amplifying the same samples with MYC F + R and applying the same method of quantification. Error bars represent the variation of band intensities obtained in one transfection experiment. The experiment was repeated twice, and representative graphs from one experiment are displayed.
Table 1.
PCR Primers Used in the Current Study
| Forward Primer | Reverse Primer | |
|---|---|---|
| MYC (TAL-8) | GGCCGTTTTAGGGTTTGTTGG | CTTTTCTCTCCCTCCACCACC |
| MYC_in (TAL-8) | AGGAGGTGGCTGGAAACTTGT | CGCTATGCTGGATTTTGCTGCA |
| IGH (TAL-14) | CCCAGCCCTTGTTAATGGACT | AGGTCCCCTTGCTCTAGAAGT |
| IGH_in(TAL-14) | CTGAATGGGGATGGCATGGGT | TCCCCTCCCTTCTGAGTCTGC |
| 4q35 (TAL-4) | AGTTTGCACTGGAGCAGAGATG | GGGATACCGACAGCAATAGTCC |
| EZH2 | CCATGCACAATATTTAGTTGGCTCT | AAAGATTTCAGAGCAATCCTCAAGC |
We confirmed this result with a real-time PCR approach. Because of the sensitivity of the technique and to avoid the possible detection of false positives, we made an adjustment to the ENIT and carried out a nested PCR. The first PCR was carried out with the same primer pairs that were used in the other experiments, MYC F + IGH R and MYC F + MYC R. The second PCR step was performed with MYC_in (F and R) or MYC_in F + IGH_in R primer sets (Table 1). Amplification curves of the corresponding products were assessed after the second step PCR (Figures S2A and S2B).
In this case, we were able to detect an amplicon using 50 ng DNA at a transfection efficiency of 10%, which corresponds to ∼700 transfected cells. The specificity of the detected amplicon was confirmed by the absence of any amplification in the untransfected sample amplified with MYC F + IGH R primers (Figure S2C) while the MYC amplicon was present (Figure S2D). As expected, a nested real-time PCR is more sensitive than an endpoint PCR and may be used for detection of translocations in a quantitative way.
Applying the same method of calculation, we found that approximately at least ∼7,000 cells were needed for the Surveyor assay and twice as many for the T7 assay (Table S1). From these data, we can conclude that the ENIT approach is at least as sensitive as the currently commercially available techniques.
ENIT Efficiency in Other Experimental Systems
We then wished to determine whether the described method could be employed with other TALEN combinations. Plasmids encoding the TAL8F and TAL8R TALEN subunits were transfected into HeLa cells together with another couple of subunits targeting the q35 locus on chromosome 4 (TAL4F + TAL4R). Using the ENIT technique as described above, an amplicon was obtained by PCR on transfected but not untransfected cells (Figures 4A and 4B). We then tested ENIT on MRC5-SV40 immortalized fibroblasts that were transfected with TAL8 and TAL14 expression vectors. The expected t(8;14) PCR amplification product was detected specifically in transfected but not in non-transfected cells (Figure S3, lane 4).
Figure 4.

ENIT Can Be Used to Detect the Efficiency of Various Gene-Editing Tools and Targets
(A) PCR on DNA extracted from HeLa cells transfected with the four TALENS recognizing the MYC and 4q (FSHD) regions. The represented amplicons were obtained using the following primer pairs: lane 1, 4q F+R; lane 2, MYC F + R; lane 3, MYC F + 4q R. (B) PCR on DNA extracted from untransfected HeLa cells. The represented amplicons were obtained using the following primer pairs: lane 1, 4q F + R; lane 2, MYC F + R; lane 3, MYC F + 4q R. (C) PCR on DNA extracted from HeLa cells transfected with the two TALENS recognizing the MYC region and the CRISPR/Cas9 for the EZH2 gene region. The represented amplicons were obtained using the following primer pairs: lane 1, MYC F + R; lane 2, EZH2 F + R; lane 3, EZH2 F + MYC R. (D) PCR on DNA extracted from untransfected HeLa cells. The represented amplicons were obtained using the following primer pairs: lane 1, MYC F + R; lane 2, EZH2 F + R; lane 3, EZH2 F + MYC R.
As a further proof of concept, HeLa cells were co-transfected with the TAL8F and TAL8R vectors and with CRISPR/Cas9 constructs designed to target the EZH2 gene locus on chromosome 7. Again, a translocation-specific amplicon was found only in cells transfected with the TAL8 plasmids and the CRISPR/Cas9 vector but not in the controls (Figures 4C and 4D, lane 3). Similar results were obtained with a combination of TAL8F + TAL8R and a plasmid vector expressing a meganuclease targeting the 4q35 chromosomal region (data not shown).
Following the same experimental protocol, we finally tested a CRISPR/Cas9 system targeted to the 4q35 locus but with a guide already tested by our group and known to be non-functional. In this case, no specific band was observed in transfected HeLa cells regardless of the DNA concentration and primer combinations used (data not shown).
Together, these results demonstrate that the ENIT technique can be used with engineered nucleases of various types and in different cellular contexts. Compared with the conventional techniques, the method reported here saves time and is cheaper to perform.
Discussion
ENIT Inherent Advantages
Engineered nucleases such as TALENs and CRISPR/Cas9 are currently employed in a variety of applications, including cancer research and gene therapy (reviewed by Barrangou and Doudna22). They can be used to create targeted DSBs, point mutations, deletions, or insertions in a given DNA sequence. One crucial step is to evaluate their potential in terms of cutting efficiency and specificity. The methods currently used are either not highly reproducible or time-consuming and expensive. A fast, reliable, and time- and money-saving method remained to be developed. Here we propose to exploit the fact that induction of two DSBs in different chromosomes should result in inter-chromosomal translocations.18, 19 When combining one nuclease with a known target site and efficacy and one to be tested, specific translocations should occur only if both nucleases cleaved specifically in the given cell. Translocations can then be readily detected by PCR amplification, yielding products that can be directly sequenced without prior cloning. In addition, the ENIT method described here is both much cheaper and less time-consuming than existing multi-step approaches.
ENIT Compared with Other Methods
DNA sequencing is probably the most precise and reliable approach, but the time required to obtain the expected result (clonal cell populations must be established before sequencing, which is not feasible for all cell types) is often suboptimal. Other techniques, like SSCP and DHPLC, are faster, but they can neither evaluate fragments longer than 1,000 bp nor determine the location of the mutation produced within the DNA sequence. The RE-PCR approach does not allow determination of the efficiency of engineered nucleases. The T7 assay and the Surveyor assay, based on the recognition and cleavage of mismatches induced by the introduction of mutation because of the error-prone NHEJ DNA repair pathway, are the current standards. Compared with our technique, they are more expensive, time-consuming, and less or as sensitive. ENIT therefore is an attractive and powerful tool to evaluate newly engineered nucleases because it is simple, reproducible, and money-saving while providing reliable results in just a few hours.
ENIT Limitations and Guidelines for Efficiency Optimization
Which cells are chosen as recipients may affect the sensitivity of the method proposed here. Because of their higher genome instability, tumor cell lines are more susceptible to chromosomal translocations than normal cells. Indeed, it has been demonstrated that tumor cells spontaneously display chromosome instability (deletions, translocations, and duplications); they also proliferate faster than normal cells.23, 24 For these reasons, translocations induced by engineered nucleases are more likely to occur in tumor cell lines. We thus recommend using cell lines such as HeLa or MRC5-SV40, as in the present study (Figures 1 and S3, respectively). However, in principle, ENIT should also be applicable to non-tumoral transformed cell lines.
For reasons that require more investigation, we have been unable so far to detect translocations when PCR primers were located in close proximity (around 200 bp) to each other (data not shown) unless a nested PCR is performed. We therefore suggest to design primers on sequences located more than 200 bp away from the cleavage site targeted by TALENs or the guide RNA in the CRISPR/Cas9 system or to use a nested qPCR approach.
In ENIT, one TALEN or one CRISPR/Cas9 must be used with a known cleavage site and efficiency to test the efficiency of the untested engineered nuclease. In the present work, we have made use of a control TALEN targeting the promoter region of the MYC gene at 8q24. Designed in our laboratory, this TALEN has induced chromosomal translocations with high efficiency, probably because its target is localized within a transcribed region with an open chromatin structure.25 These specifics should be taken into account when designing other control nucleases.
In summary, ENIT is a very powerful method to test the cutting efficiency of a newly designed gene editing tool, simplifying and accelerating the very first steps in exponentially growing numbers of biological applications of genomic editing.
Materials and Methods
TALENs and Guide RNA-CRISPR-Cas9 Design and Assembly
TALENS targeting genomic locus regions 8q24 (MYC), 14q32 (IGH), and 4q35-facioscapulohumeral muscular dystrophy (FSHD) region were designed in our lab and produced by Life Technologies using Gateway cloning for genome editing.26
The sequences of interest were cloned into pcDNA6.2_N-EmGFP-DEST_A341 vectors. The CRISPR guide RNA (gRNA) targeting the locus 7q36.1 (EZH2) was designed in our laboratory using the online CRISPR design tool (http://crispr.mit.edu/). The targeted sequences are listed in Table 2.
Table 2.
Genomic Sequences Targeted by TALENs
| Targeted Sequence 5′–3′ | |
|---|---|
| MYC (TAL-8) | tcccccgctggaaaccttgcacctcggacgctcctgctcctgcccccacctga |
| IGH (TAL-14) | tggcatcgcccttgtctaa |
| 4q35 (TAL4) | tgctacagcactagtgaaactgcaataccacagacagccaactggggaagaaa |
Briefly, DNA sequences of gRNAs with the highest scores were selected for further cloning in a plasmid kindly provided by Dr. Khaled Mehdi (Institut Gustave Roussy). DNA oligonucleotides were then ligated into the BsmBI-digested plasmid containing the U6 promoter and a Cas9 expression cassette (kindly provided by Dr. Khaled Mehdi) using T4 DNA ligase (New England Biolabs) following the manufacturer’s instructions. The following gRNA was used for EZH2: 5′-TTCATACGCTTTTCTGTAGG-3′.
Cell Lines and Transfection
HeLa cells (ATCC #CCL-2) were seeded at 2 × 106cells/25-cm2 flask 24 hr before transfection and cultured in 5 mL of DMEM (Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin (Gibco) (complete medium) at 37°C with 5% CO2. Transfections were done with GFP-labeled TALENs designed for the MYC gene (TAL-8F-GFP and TAL-8R-GFP), the IGH gene (TAL-14F-GFP and TAL-14R-GFP), 4q35-specific TALENs (TAL-4F-GFP and TAL-4R-GFP), and EZH2-specific CRISPR-Cas9 plasmids. Cells were transfected with two couples of TALENs or a couple of TALENs and a CRISPR/Cas9 simultaneously.
For each co-transfection, 3 μg of each plasmid were diluted in one tube with sterile 150 mM NaCl to a final volume of 500 μL. In another tube, 3 μL of JetPEI (Polyplus-transfection) reagent per each microgram of DNA were diluted with sterile 150 mM NaCl to a final volume of 500 μL. Both mixes were combined and incubated for 30 min at room temperature. The mixture was added to the cell culture with 5 mL complete medium, and cells were cultured for 72 hr at 37°C with 5% CO2.
MRC5 SV40 fibroblasts were kindly provided by Patricia Kannouche (Institut Gustave Roussy). They were seeded at 0.7 × 106 cells/25-cm2 flask 24 hr before transfection and cultured in RPMI medium supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO2. For TAL8 and TAL14 co-transfection, 3 μg of each plasmid were mixed in one tube with Opti-MEM (Thermo Fisher Scientific) to a final volume of 390 μL. In another tube, 2.6 μL of Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) per each microgram of DNA was mixed with Opti-MEM to a final volume of 390 μL. Both mixes were combined and incubated for 20 min at room temperature. The mixture was added to the cell culture with 4.4 mL of complete medium, and cells were cultured for 72 hr at 37°C with 5% CO2.
To assess the rate of transfection, after 72 hr, the cells were trypsinized and resuspended in 5 mL complete medium, and the percentage of GFP-positive cells was analyzed by flow cytometry (BD Accuri C6 cytometer, BD Biosciences).
PCR Amplification and DNA Electrophoresis
DNA was extracted from transfected and untransfected cells with the NucleoSpin tissue kit (Macherey-Nagel) according to the manufacturer’s instructions. The extracted DNA was used for PCR, which was performed in a 20-μL reaction mixture containing 10 μL of 2XFastStart SYBR Green Master mix Rox (Roche), 1 μL of 10 μM forward primer, 1 μL of 10 μM reverse primer, and, if not stated otherwise, 250 ng of DNA. Primers were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/) from the following NCBI reference sequences: NG_007161.1 (MYC), NG_001019.5 (IGH), AY028079.1 (4q35), and NG_032043.1 (EZH2); their sequences are listed in Table 1. All possible primer combinations (gene1 forward + gene2 reverse, gene1 reverse + gene2 forward, gene1 forward + gene2 forward, and gene1 reverse + gene2 reverse) were tested to detect translocation.
PCRs were performed in a Geneamp PCR System 9700 (Applied Biosystems) with the following cycling conditions: one cycle of 95°C for 10 min, 40 cycles of 95°C for 30 s, 57°C for 1 min, 72°C for 1 min, and one cycle of 72°C for 10 min. Samples were loaded on 1% agarose gel, and the size of amplicons was determined by comparison with a 1Kb Plus DNA ladder (Thermo Fisher Scientific).
Sequencing of PCR Products
PCR products were run and isolated from 1% agarose gel using the NucleoSpin gel and PCR clean-up kit (Macherey-Nagel), according to the manufacturer’s instructions, and analyzed by sequencing (Eurofins Genomics, Cochin Sequencing Platform). Sequence data were aligned using Seqman, version II (DNASTAR).
Preparation of Chromosome Spreads
Adherent HeLa cells (approximately 5 × 106) were synchronized in G2/M phase by overnight treatment with the CDK1 inhibitor RO-3306 (Sigma-Aldrich) at a final concentration of 5 μM. After rinsing in 1× PBS and mild trypsinization, cells were blocked in metaphase through a 45-min treatment with colcemid (final concentration, 0.1 μg/mL) at 37°C. After one wash in 1× PBS, cells were gently resuspended in 18 mL of a preheated 75 mM KCl hypotonic solution and incubated for 13 min at 37°C. Swollen cells were then pre-fixed by addition of 1 mL of freshly prepared fixation solution (3:1 methanol:acetic acid) and incubated for 5 min at room temperature (RT). After centrifugation (5 min, 150 relative centrifugal force), cells were well resuspended in 6.5 mL of the fixing solution and incubated for 20 min at RT. Cells were then pelleted by centrifugation for 10 min at 400 × g, fixed again in 6 mL of the ice-cold fixing solution for 15 min at 4°C, centrifuged for 10 min at 400 × g (4°C), and resuspended in 0.5 mL of the ice-cold fixing solution.
Mitotic spreads were prepared by dropping the fixed swollen cells onto clean and dry microscopic slides from a height of approximately 10 cm. The resulting chromosome slides were air-dried and processed immediately for FISH or stored at −20°C until use.
FISH of Chromosome Spreads
“RainbowFISH” probes used for hybridization and staining of human chromosome loci 8q24 (MYC gene locus) and 14q32 (IGH gene locus) were provided by Empire Genomics. The MYC probe was labeled with SpectrumOrange and the IGH probe with SpectrumGreen. Slides with chromosome spreads prepared as described previously were incubated in 20% glycerol (Euromedex) in 1× PBS for at least 1 hr at RT. Slides were then submitted three times to freezing/thawing in liquid nitrogen and further incubated in 0.1 M HCl (20 min, RT). Possible traces of RNA were digested by incubation (1 hr, 37°C) in 200 μg/mL RNase A (Sigma-Aldrich) diluted in 2× saline sodium citrate (SSC) (Euromedex). After three washes in the 2× SSC buffer, the slides were stored at 4°C in 50% deionized formamide (Bio Basic) in 2× SSC. For hybridization, probes were denatured (5 min, 75°C) in a Thermomixer (Eppendorf) in the hybridization buffer provided with the probes. Simultaneously, the slides with the chromosomes spreads were denatured by incubation in 70% formamide for 5 min at 75°C in a dry block heater (Bio-Techne). Hybridization was carried out by incubating the slides with the probes for 5 days at 37°C in a humid chamber. The slides were then rinsed in 50% formamide in 2× SSC (5 min, 37°C), followed by washing twice in 2× SSC (5 min each, RT). Slides were then washed three times in 1× PBS (5 min each, RT), mounted using DAPI-containing Vectashield (Vector Laboratories), covered with a coverslip, and analyzed immediately or after storage at 4°C.
DNA Dilutions and Gel Band Intensity Quantification
DNA from transfected cells was serially diluted starting from 1,000 ng DNA to 500, 100, 50, 20, and 0.8 ng DNA. Genomic PCR was carried out on each DNA dilution in duplicate as described above. PCR products were then collected and loaded in 1% agarose gel for electrophoresis. The size of amplicons was determined by comparison with the 1Kb Plus DNA ladder (Thermo Fisher Scientific).
Bands were quantified using ImageJ software (NIH); the integrated density parameter of each band was measured and adjusted by eliminating the integrated density value of the background. The minimum number of cells for detecting a translocation (n) was calculated considering the amount of DNA used for PCR, the average amount of DNA present in one HeLa cell21 (6.7 pg), and the percentage of transfected cells as follows:
Those parameters were plotted together with the intensity of each visible band. Graphs were obtained using GraphPad prism version 5.00 (GraphPad).
The accuracy of DNA dilutions was controlled by amplifying the same DNA using MYC F and MYC R primers. The PCR products were then collected and loaded on a 1% agarose gel. The size of amplicons was determined by comparison with the 1Kb Plus DNA ladder (Thermo Fisher Scientific).
Nested Real-Time PCR
DNA was extracted from transfected and untransfected HeLa cells with the NuceloSpin tissue kit (Macherey-Nagel) according to the manufacturer’s instructions. The extracted DNA was used for the first PCR (PCR1) step that was performed with the MYC F + IGH R or MYC F + MYC R primer pairs as in previous experiments. The PCR1 product at a final dilution of 1/100 was used for the second amplification step (PCR2). PCR2 was carried out using inner primer sets. MYC_in F + IGH_in R or MYC_in F + MYC_in R was used to amplify the amplicons derived from the MYC F + IGH R or MYC F + MYC R amplifications, respectively. PCR1 and PCR2 reaction mixtures were prepared as described previously. Real-time PCR was performed in a StepOnePlus real-time PCR system (Applied Biosystems) with the following cycling conditions: PCR1: 95°C, 10 min followed by 25 cycles (95°C, 15 s; 60°C; 30 s; 72°C, 1 min); PCR2: 95°C, 3 min followed by 25 cycles (95°C, 15 s; 60°C; 30 s; 72°C, 30 s). Melting curves were also assessed to determine the absence of false amplicons.
Author Contributions
M.L. and Y.V. conceived and designed the study. D.G. and Y.B.S. designed and performed the experiments and interpreted the data. K.O. performed the PCR experiments. T.T. and N.L. performed the nested PCR experiments. C.R. performed the FISH experiments. N.S. and M.R. critically interpreted the data. M.L., D.G., Y.B.S., and Y.V. wrote the manuscript.
Conflicts of Interest
The authors declare that they have no competing interests.
Acknowledgments
This work was supported by the AFM (MEGAFSHD), INSERM (ENVIBURKITT), and ANRS (to Y.V.). T.T. acknowledges a fellowship from the ANRS. Funding for the open access charge was provided by INSERM.
Footnotes
Supplemental Information includes three figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.omtm.2017.03.001.
Supplemental Information
References
- 1.Christian M., Cermak T., Doyle E.L., Schmidt C., Zhang F., Hummel A., Bogdanove A.J., Voytas D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 3.Saada Y.B., Dib C., Lipinski M., Vassetzky Y.S. Genome- and cell-based strategies in therapy of muscular dystrophies. Biochemistry (Mosc.) 2016;81:678–690. doi: 10.1134/S000629791607004X. [DOI] [PubMed] [Google Scholar]
- 4.Gaj T., Gersbach C.A., Barbas C.F., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayashi K., Yandell D.W. How sensitive is PCR-SSCP? Hum. Mutat. 1993;2:338–346. doi: 10.1002/humu.1380020503. [DOI] [PubMed] [Google Scholar]
- 6.Inazuka M., Wenz H.M., Sakabe M., Tahira T., Hayashi K. A streamlined mutation detection system: multicolor post-PCR fluorescence labeling and single-strand conformational polymorphism analysis by capillary electrophoresis. Genome Res. 1997;7:1094–1103. doi: 10.1101/gr.7.11.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Underhill P.A., Jin L., Lin A.A., Mehdi S.Q., Jenkins T., Vollrath D., Davis R.W., Cavalli-Sforza L.L., Oefner P.J. Detection of numerous Y chromosome biallelic polymorphisms by denaturing high-performance liquid chromatography. Genome Res. 1997;7:996–1005. doi: 10.1101/gr.7.10.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu B., Sawyer N.A., Chiu C., Oefner P.J., Underhill P.A. DNA mutation detection using denaturing high-performance liquid chromatography (DHPLC) Curr. Protoc. Hum. Genet. 2006;Chapter 7 doi: 10.1002/0471142905.hg0710s48. Unit 7.10. [DOI] [PubMed] [Google Scholar]
- 9.Qiu P., Shandilya H., D’Alessio J.M., O’Connor K., Durocher J., Gerard G.F. Mutation detection using Surveyor nuclease. Biotechniques. 2004;36:702–707. doi: 10.2144/04364PF01. [DOI] [PubMed] [Google Scholar]
- 10.Xie K., Yang Y. RNA-guided genome editing in plants using a CRISPR-Cas system. Mol. Plant. 2013;6:1975–1983. doi: 10.1093/mp/sst119. [DOI] [PubMed] [Google Scholar]
- 11.Vouillot L., Thelie A., Pollet N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 2015;5:407–415. doi: 10.1534/g3.114.015834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou P., Chen S., Wang S., Yu X., Chen Y., Jiang M., Zhuang K., Ho W., Hou W., Huang J., Guo D. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015;5:15577. doi: 10.1038/srep15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sedlak R.H., Liang S., Niyonzima N., De Silva Feelixge H.S., Roychoudhury P., Greninger A.L., Weber N.D., Boissel S., Scharenberg A.M., Cheng A. Digital detection of endonuclease mediated gene disruption in the HIV provirus. Sci. Rep. 2016;6:20064. doi: 10.1038/srep20064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mani R.-S., Chinnaiyan A.M. Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat. Rev. Genet. 2010;11:819–829. doi: 10.1038/nrg2883. [DOI] [PubMed] [Google Scholar]
- 16.Richardson C., Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405:697–700. doi: 10.1038/35015097. [DOI] [PubMed] [Google Scholar]
- 17.Iarovaia O.V., Rubtsov M., Ioudinkova E., Tsfasman T., Razin S.V., Vassetzky Y.S. Dynamics of double strand breaks and chromosomal translocations. Mol. Cancer. 2014;13:249–258. doi: 10.1186/1476-4598-13-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brunet E., Simsek D., Tomishima M., DeKelver R., Choi V.M., Gregory P., Urnov F., Weinstock D.M., Jasin M. Chromosomal translocations induced at specified loci in human stem cells. Proc. Natl. Acad. Sci. USA. 2009;106:10620–10625. doi: 10.1073/pnas.0902076106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piganeau M., Ghezraoui H., De Cian A., Guittat L., Tomishima M., Perrouault L., René O., Katibah G.E., Zhang L., Holmes M.C. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. 2013;23:1182–1193. doi: 10.1101/gr.147314.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison M.M., Jenkins B.V., O’Connor-Giles K.M., Wildonger J. A CRISPR view of development. Genes Dev. 2014;28:1859–1872. doi: 10.1101/gad.248252.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serth J., Kuczyk M.A., Paeslack U., Lichtinghagen R., Jonas U. Quantitation of DNA extracted after micropreparation of cells from frozen and formalin-fixed tissue sections. Am. J. Pathol. 2000;156:1189–1196. doi: 10.1016/S0002-9440(10)64989-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrangou R., Doudna J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016;34:933–941. doi: 10.1038/nbt.3659. [DOI] [PubMed] [Google Scholar]
- 23.Landry J.J.M., Pyl P.T., Rausch T., Zichner T., Tekkedil M.M., Stütz A.M., Jauch A., Aiyar R.S., Pau G., Delhomme N. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda) 2013;3:1213–1224. doi: 10.1534/g3.113.005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roschke A.V., Rozenblum E. Multi-layered cancer chromosomal instability phenotype. Front. Oncol. 2013;3:302. doi: 10.3389/fonc.2013.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gombert W.M., Farris S.D., Rubio E.D., Morey-Rosler K.M., Schubach W.H., Krumm A. The c-myc insulator element and matrix attachment regions define the c-myc chromosomal domain. Mol. Cell. Biol. 2003;23:9338–9348. doi: 10.1128/MCB.23.24.9338-9348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katzen F. Gateway(®) recombinational cloning: a biological operating system. Expert Opin. Drug Discov. 2007;2:571–589. doi: 10.1517/17460441.2.4.571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.