

Abstract
Human pluripotent stem cells (hPSCs) are a promising source of regenerative material for clinical applications. However, hPSC transplant therapies pose the risk of teratoma formation and malignant transformation of undifferentiated remnants. These problems underscore the importance of developing technologies that completely prevent tumorigenesis to ensure safe clinical application. Research to date has contributed to establishing safe hPSC lines, improving the efficiency of differentiation induction, and indirectly ensuring the safety of products. Despite such efforts, guaranteeing the clinical safety of regenerative medicine products remains a key challenge. Given the intrinsic genome instability of hPSCs, selective growth advantage of cancer cells, and lessons learned through failures in previous attempts at hematopoietic stem cell gene therapy, conventional strategies are unlikely to completely overcome issues related to hPSC tumorigenesis. Researchers have recently embarked on studies aimed at locating and directly treating hPSC-derived tumorigenic cells. In particular, novel approaches to directly killing tumorigenic cells by transduction of suicide genes and oncolytic viruses are expected to improve the safety of hPSC-based therapy. This article discusses the current status and future perspectives of methods aimed at directly eradicating undifferentiated tumorigenic hPSCs, with a focus on viral vector transduction.
Keywords: adenoviral conditional targeting, conditionally replicating adenovirus, oncolytic virus, pluripotent stem cells, regenerative medicine, suicide gene, survivin, teratoma, tumorigenicity, viral vector
Graphical Abstract
Main Text
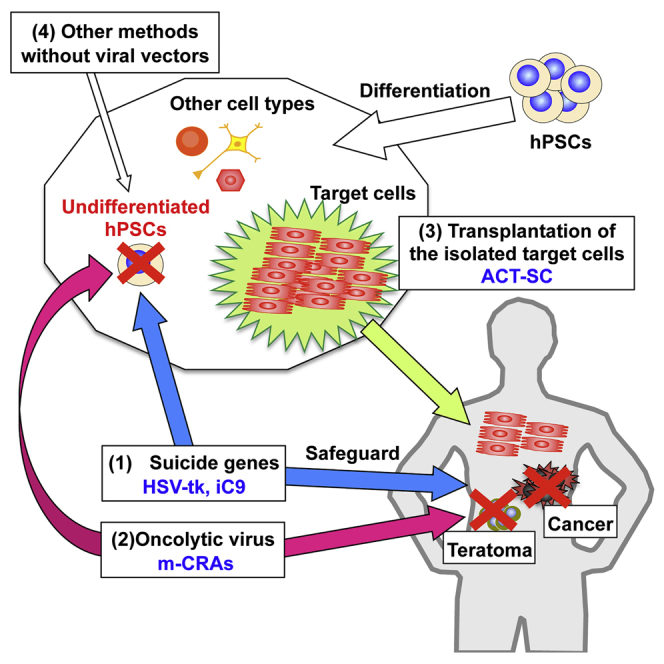
Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), are indefinitely self-renewing and pluripotent.1 These properties make hPSCs a promising platform for cell therapy, as well as a possible alternative to organ transplantation. Consequently, hPSCs have attracted increasing attention from scientists in the arenas of basic and applied biomedical research. hESCs have been employed in clinical trials involving human subjects in North America, Europe, and other regions of the world, and a clinical study using hiPSCs has been initiated in Japan.2, 3 However, the utility of hPSCs in regenerative medicine cannot be generalized on the basis of this small number of early clinical studies; instead, their applicability must be evaluated on an organ-specific basis. Concerns regarding the safety of hPSCs, in particular their tumorigenic potential, must be fully addressed before the hPSC technologies can be widely applied in clinical regenerative medicine. Transplanted cells containing undifferentiated hPSCs may give rise to benign teratomas or malignant tumors (Figure 1).4 Here we review the tumorigenic potential of hPSCs, its impact on the safety of regenerative medicine, and current technological limitations on the prevention of tumor formation. We also discuss new approaches for directly eliminating the tumorigenic potential of hPSCs, with a focus on the current status and future prospects of viral vector-based techniques (Figure 2).
Figure 1.

Conventional Strategy for Suppressing Tumorigenesis
Transplanted cells containing undifferentiated hPSCs may give rise to teratomas or cancers. Conventional methods, of which representatives are shown in the upper right box, aim to suppress tumorigenesis indirectly. Due to the biological properties of hPSCs, the issues shown in the lower right box remain to be solved.
Figure 2.

Novel Strategies for Directly and Specifically Eliminating Tumorigenic Cells
Two strategies that use virally transduced suicide genes (Figure 3) and oncolytic virus (Figure 4) specifically kill undifferentiated tumorigenic hPSCs. These systems not only prevent teratoma and cancer formation but also provide a safeguard against aberrant cells after transplantation. The latter represents a clinical advantage over other methods that do not involve viral vector manipulation. High-purity isolation of target cells improves their effectiveness and safety of the hPSC therapy, in part by decreasing the number of potentially tumorigenic cells. The adenoviral conditional targeting in stem cell (ACT-SC) method dramatically increases the homogeneity of transplanted cells (Figure 5).
Tumorigenic Potential of hPSCs
Historical Lessons from Clinical Applications
Safety should be the primary consideration in clinical and commercial application of innovative pharmaceutical and medical products. Agents with a high carcinogenic potential, no matter how beneficial they might be, must be eliminated from the development pipeline at an early stage. In the arena of regenerative medicine, stem cells are a double-edged sword. While stem cells are expected to provide a powerful means for innovative therapy that outdates conventional medicine, they pose a significant risk of tumorigenesis.
In practical terms, the unintended negative consequences of genetically manipulating stem cells were demonstrated in a clinical trial of ex vivo hematopoietic stem cell gene therapy in patients with X-linked severe combined immunodeficiency; this study provided biologists and clinicians with several important lessons about stem cell tumor formation.5 A high incidence of leukemia was documented in treated patients, although nonclinical studies had concluded otherwise. Nonclinical studies involving experimental animals may be useful for predicting the levels of tumor suppression in cell transplant therapy, but they can never ensure that the therapy is clinically safe and free from tumor risk in humans. In that study, the therapeutic gene (ϒ subunit of the interleukin-2 receptor gene) was introduced into hematopoietic stem cells via a retroviral vector. The vector also activated the locus associated with a known T cell oncogene, LIM domain only2 (LMO2) gene, which clinically manifested a few years after treatment. Due to its selective growth advantage, a single neoplastic stem cell can lead to the formation of a clinically significant cancer. Therefore, to ensure clinical safety, such tumorigenic cells must be completely eradicated from the transplanted cell population.
Biological Properties of hPSCs
Due to the fundamental biological properties of hPSCs, regenerative medicine technologies using hPSCs are associated with a higher risk of tumor generation than those using tissue stem cells (Figure 1).4 First, long-term culture in vitro makes undifferentiated hPSCs susceptible to mutations due to their genetic instability.4 Consequently, even if a master cell bank (MCB) is established from a clone whose genome has been completely sequenced, its cells may undergo mutations during the course of large-scale culture for clinical application. It is not practical to sequence the genome of individual cells in the final hPSC-derived transplant cell products. In addition, researchers have recently reported that, due to premature termination of reprogramming, hiPSCs undergo transformation via altered epigenetic regulation in the absence of genetic changes.6
To overcome the risk of tumorigenesis, researchers have tried to reduce the presence of undifferentiated hPSCs and tumorigenic cells in the cell culture. Those methods include improvements in the cell culture and reprogramming methods, establishment of safe hPSC MCBs through gene expression analysis and genome-wide sequencing, and development of efficient methods for inducing differentiation (Figure 1).4, 7 Notably, these conventional methods aim to suppress tumorigenesis indirectly. Given the possible presence of remnants with tumorigenic potential and the selective growth advantage of cancer cells, however, these techniques cannot fully suppress tumor development, although they can effectively decrease the chance of, or extend the time to, tumorigenesis. Therefore, in order to achieve a reliable hPSC-based regenerative medicine, a completely new and innovative strategy for directly and specifically killing or eliminating tumorigenic cells is urgently required.
hPSC Transduction Using Viral Vectors
Because this review discusses novel targeted techniques for destroying or removing tumorigenic cells, with a focus on viral vector-based approaches, we summarize the limitations of non-viral vectors and the characteristic features of representative viral vectors for hPSC transduction.
Non-viral Vectors
Non-viral gene transfection and expression techniques applied to hPSCs may be broadly categorized as physical (e.g., electroporation) or chemical (e.g., lipofection). Such methods are associated with considerable technical limitations. For example, depending on the size of genes to be transferred and the type of hPSC line, non-viral techniques often cannot achieve satisfactory transfection efficiencies. In addition, these techniques achieve short-lived gene expression that lasts for a few days, and they often lead to the death of hPSCs due to their susceptibility to transfection-induced cellular stress. By contrast, viral vectors can achieve high transduction efficiency and long-term gene expression, and they are, therefore, quite useful for genetic hPSC manipulation, although their production is technically demanding.
Retroviral and Lentiviral Vectors
The common types of vectors used for hPSC engineering are single-stranded RNA retroviruses and lentiviruses, which can be generated relatively easily.8, 9 Such vectors insert viral genes into the host DNA, theoretically enabling long-term expression of the inserted genes. However, these viral vectors have the noteworthy disadvantage that exogenous genes are silenced at a high frequency in undifferentiated pluripotent stem cells, including hPSCs, when viral long terminal repeats (LTRs) are used as their promoters.10, 11 This problem can be circumvented by using self-inactivating vectors that lack viral enhancers/promoters in the 3′ LTR; such vectors allow for exogenous promoters to control transgenic expression.12 In addition, pseudotyping with vesicular stomatitis virus G glycoprotein enables vector concentration, which, in combination with the broad tropism of the glycoprotein, increases transduction efficiency. Viral vectors that integrate exogenous genes into the host chromosome may possibly pose significant safety risks. The retrovirus-mediated stem cell gene therapy mentioned earlier caused a high incidence of leukemia in treated patients.5 Therefore, an MCB should probably be established from hPSC clones that have been shown to carry transgenes at safe genomic sites.
Adenoviral Vectors
The application of double-stranded DNA adenoviral vectors to pluripotent stem cells, including hPSCs, has been studied by a very small number of researchers, including the authors of this review.10, 13, 14 Because adenoviral vectors can be produced at much higher titers (1010–12 infectious units/mL) than retroviral and lentiviral vectors (106–7 infectious units/mL), the in vitro transduction efficiency of adenoviral vectors can be increased to nearly 100% by elevating the MOI.10 In addition, adenoviral DNA (including any exogenous genes) does not integrate into the host chromosome but persists as an episome in the nucleus, allowing stable long-term transgene expression under the control of exogenous promoters.10, 15 Because the host chromosome remains intact, the episomal nature of the adenoviral vector confers a significant advantage in clinical safety. It should be noted that the percentage of host hPSCs transduced with the episomal vector decreases rapidly with active cell division. Accurate knowledge and precise command of the properties of adenoviral vectors should facilitate discovery of new modalities for eradicating tumorigenic cells present in hPSC cultures.
Selective Killing of Tumorigenic hPSCs by Virally Transduced Suicide Genes
Suicide genes, a group of genes whose products induce cell death, have been extensively investigated in cancer gene therapy, and they have been used to kill or remove undifferentiated tumorigenic cells. There are two major approaches that utilize suicide genes.
Herpes Simplex Virus Thymidine Kinase
One approach is based on a well-studied gene therapy involving the herpes simplex virus thymidine kinase (HSV-tk) and ganciclovir (GCV).16, 17, 18 GCV, a nontoxic prodrug, is phosphorylated by intracellular HSV-tk and ultimately converted to GCV-triphosphate, which potently induces cell death by inhibiting DNA replication (Figure 3). This therapy can selectively kill actively proliferating cancer cells, including undifferentiated tumorigenic cells. Lentivirus-transduced and undifferentiated hPSCs expressing the HSV-tk gene are effectively eliminated in vitro in the presence of GCV.19, 20 In addition, in vivo teratomas derived from HSV-tk-transduced hPSCs shrink following the administration of GCV. HSV-tk + GCV therapy has the additional advantage of a bystander effect, in which the therapeutic effect of the drug is manifested in genetically unmodified cancer cells adjacent to the modified ones.16, 17, 18 However, it remains unclear to what extent the bystander effect occurs in hPSCs. The potential of the HSV-tk + GCV method to kill non-HSV-tk-transduced cells or HSV-tk-transduced cells whose HSV-tk expression has been silenced represents an additional advantage of this approach.
Figure 3.

Selective Killing of Tumorigenic hPSCs by Two Types of Suicide Genes
Herpes simplex virus thymidine kinase (HSV-tk) or inducible caspase 9 (iC9) gene is virally transduced into hPSCs. In the former, a nontoxic prodrug ganciclovir (GCV) is phosphorylated by HSV-tk expression, and ultimately GCV-triphosphate induces death in dividing tumorigenic hPSCs, but not in differentiated cells. In the latter, dimerizer treatment activates iC9, followed by caspase-3 activation. This ultimately results in apoptosis of the transduced cells.
Inducible Caspase 9
Another suicide gene system involves activation of a caspase, an endogenous apoptotic effector.21 Previous research using adoptive T cells and mesenchymal stromal cells showed that the inducible caspase 9 (iC9) protein, mediated by a small molecule dimerizer, activates caspase 3, which in turn eliminates damaged or unwanted cells (Figure 3). Recent work showed that the iC9 suicide system has the potential to kill tumorigenic cells and prevent teratoma formation in hPSC populations. Lentivirus-transduced hiPSCs expressing the iC9 gene mostly died by apoptosis within 24 hr of dimerizer treatment, and teratomas derived from iC9-expressing cells shrank in vivo following the administration of the dimerizer. These studies demonstrated that the iC9 suicide system effectively cleared tumorigenic stem cells. In addition, hiPSCs established from antigen-specific T cells and transduced by a lentivirus vector expressing the iC9 gene died by apoptosis after dimerizer treatment. Their findings suggested that the iC9 gene acts as a safety switch that eliminates tumor cells that develop over the course of clinical application, thereby improving the outcome of hiPSC-derived T cell therapy.22
Perspective
It should be noted, however, that these observations only show that this system can kill tumorigenic cells, prevent teratoma formation, and provide a safeguard against aberrant cells. More evidence is needed to validate the tumor-killing performance of this approach. No study to date has determined the level of suicide gene expression necessary to completely destroy all tumorigenic cells contained in a given hPSC population. In addition, it remains unknown whether the expression of suicide genes damages the differentiated cells of interest. Constitutively active promoters and the promoter of the pluripotency marker Nanog have been used to regulate suicide gene expression.19, 21, 22 However, little is known about the degrees of effectiveness of other promoters, as well as their specificity for the undifferentiated state, using this system.
In our own research on cancer gene therapy, we have identified an optimal therapeutic expression level for suicide gene, above which inducer (GCV)-independent nonspecific cell death occurs. When suicide genes are expressed above the optimal expression level in animal models, they tend to experience more severe adverse reactions, and the therapeutic effect of the suicide genes does not increase.18 Our previous work suggested the need to identify promoters that activate only undifferentiated cells (not the differentiated target cells) in a highly specific manner and induce optimal levels of suicide genes for maximum tumoricidal effects. To achieve this goal, we developed a tumorigenic cell-targeting lentiviral vector (TC-LV) platform that can incorporate promoter and fluorescent protein genes in the recombination cassette. We have used this platform to efficiently and simultaneously generate a variety of TC-LVs that encode different types of promoters, and these vectors are currently being used to identify the promoters that most effectively kill tumorigenic cells (K.I., unpublished data).
Selective Killing of Tumorigenic hPSCs by a Novel Oncolytic Virus Strategy
Oncolytic Virus and Conditionally Replicating Adenovirus Regulated with Multiple Tumor-Specific Factors for Cancer Therapy
Oncolytic virotherapy, using viruses engineered to selectively replicate in and kill tumor cells, has emerged as an innovative and promising strategy for cancer treatment.23, 24 A wide variety of oncolytic viruses have been developed that take advantage of the properties of the original viral species. An oncolytic virus expressing a cytokine was approved by the U.S. Food & Drug Administration and the European Medicines Agency in 2015.
We developed our unique vector by further modification of a conditionally replicating adenovirus (CRA), which was derived from a human adenovirus. This CRA, designated a conditionally replicating adenovirus regulated with multiple tumor-specific factors (m-CRA), is regulated by multiple tumor-specific factors (Figure 4).23 We devised a set of procedures to incorporate into m-CRA three independently constructed regulatory clusters that encode viral replication, therapeutic, and adenoviral backbone genes. This method enabled us to deliberately modify the properties of therapeutic adenoviral vectors.
Figure 4.

Selective Killing of Tumorigenic hPSCs by a Novel Oncolytic Virus Strategy
Survivin-responsive m-CRAs (Surv.m-CRAs), whose replication is controlled by the survivin promoter upstream of the adenoviral early region 1A (E1A), are promising anticancer agents that effectively and specifically treat a variety of cancers. The m-CRA platform technology is also applicable to selective eradication of tumorigenic cells in hPSC populations. Pre-infection of hPSCs with Surv.m-CRA results in efficient viral replication and cytocidal potential in undifferentiated cells, but not in differentiated cells.
We constructed a variety of m-CRAs expressing different properties, and we analyzed their therapeutic effect and cancer specificity (i.e., safety). The most favorable results were obtained with Surv.m-CRAs, whose replication is controlled by the survivin promoter (Figure 4).25, 26 Survivin (BIRC5), an inhibitor of apoptotic proteins, is overexpressed in most cancers. Its expression levels correlate with cancer prognosis, and survivin promoter activity is upregulated in cancer cells due to aberrant transcriptional activation.27 Comparison of the viral properties and therapeutic potentials of Surv.m-CRA and telomerase reverse transcriptase (Tert)-responsive m-CRA (Tert.m-CRA) revealed that Surv.m-CRA was safer and more effective.25 Another group employed a similar Tert-responsive CRA in a human cancer trial and reported a positive local effect and safety.28 Surv.m-CRA demonstrated elevated effectiveness against cancer stem cells, which are often resistant to conventional anticancer medications and radiotherapy.29 We are currently implementing a first-in-human investigator-initiated trial of Surv.m-CRA for the treatment of refractory malignant bone and soft tissue tumors.
A Novel Oncolytic Virus Strategy that Specifically Eliminates Tumorigenic hPSCs
We recently developed a method for employing oncolytic viruses to selectively and efficiently kill undifferentiated tumorigenic cells in hPSC culture using the Surv.m-CRA and Tert.m-CRA models.14 The results yielded multiple important findings (Figure 4). First, survivin, long considered a cancer-specific promoter, was unexpectedly more active in undifferentiated hPSCs than the Tert promoter, but it was minimally active in differentiated hPSCs. Second, both m-CRAs exhibited efficient viral replication and cytocidal potential in undifferentiated hPSCs (with Surv.m-CRA more potent than Tert.m-CRA), but not in co-cultured differentiated normal cells. Third, pre-infection of hPSCs with either m-CRA prevented teratoma formation in vivo. Thus, our m-CRA platform technology and Surv.m-CRA product, originally developed for cancer treatment, provide an effective means for abolishing undifferentiated tumorigenic hPSCs and producing tumor-free hPSC populations.
On the other hand, because transcriptional regulation of a viral gene by a cancer-specific promoter failed to achieve cancer-specific viral replication in certain types of viruses, not all oncolytic viruses are applicable to, or effective for, selective eradication of tumorigenic hPSCs. For instance, representative cancer-specific promoters, including the survivin and the Tert promoters that functioned well in Surv.m-CRAs and Tert.m-CRAs as well as in plasmids, drastically lost their cancer-specific potential in herpes simplex viruses.30, 31
Isolation of Target Cells from hPSCs
Limitations of Conventional Methodologies
Although many methods for inducing targeted differentiation have been reported in the literature, it is not currently feasible to obtain the target cells with 100% efficiency. Consequently, the transplant cell population contains a certain amount of unwanted types of differentiated and undifferentiated cells. To overcome this technical limitation, it is necessary to devise a purification method that isolates target cells from a heterogeneous culture. Currently, however, no surface antigen markers are available that could assist in identifying cardiomyocytes or many other types of cells. For many years, the only solution to this problem was to establish hPSCs that stably expressed reporter genes under the control of a tissue-specific gene promoter. This approach poses multiple challenges; it is time- and resource consuming to establish such stable cell lines, and the dependence of fluorescence intensity on target gene promoter activity means that fluorescence visualization cannot be achieved when activity is low.
Adenoviral Conditional Targeting in Stem Cell Method
We developed a technique for ensuring robust fluorescence protein expression, namely, the adenoviral conditional targeting in stem cell (ACT-SC) method (Figure 5).10 This method employs adenoviral vectors carrying two DNA constructs encoding the Cre protein and an expression-switching unit. The use of the adenoviral vector enables straightforward transduction of genes of interest into hPSCs by simply mixing the vector cocktail with the hPSC culture, even during the process of induction. Adenoviral vectors also facilitate stable expression of the virally transduced genes due to their episomal nature, as explained earlier. Even if the Cre expression level is extremely low, it excises the loxP-flanked stuffer gene only in the target cell, leading to transcription of the downstream fluorescent gene under the strong constitutively active promoter. Using the Nkx2.5 promoter, we successfully identified and purified primitive cells of the cardiac lineage for the first time. We also succeeded in separating mature cardiomyocytes from mouse embryonic stem cells using the sarcomeric α-myosin heavy chain promoter. In addition, we showed that ACT-SC is quite useful for isolating hPSC-derived target cells, and we have identified the optimal experimental conditions for doing so (K.M., unpublished data).
Figure 5.

Isolation of Target Cells by the ACT-SC Method
Infections of hPSCs with two adenoviral vectors carrying switching-expression DNA and regulatory DNA facilitate efficient transduction and stable expression of the transgenes. Following Cre-lox recombination, target cells are specifically visualized by robust fluorescence protein (e.g., EGFP), and they can be subsequently purified by cell sorting. UA-Pr, strong constitutively active promoter; CS-Pr, cell-specific promoter.
High-purity isolation of target cells is critical for improving the effectiveness and safety of the hPSC therapy (Figure 2). In addition to the aforementioned methods for abolishing tumorigenic cells, ACT-SC will play an important role in future clinical applications of hPSCs as a means to increase the homogeneity of transplanted cells. On the other hand, one potentially unfavorable feature of ACT-SC for clinical application is the persistence of remnant adenoviral genomes in the transplanted hPSC-derived target cells, which may induce some degree of cellular immunity against the transplanted cells. Nevertheless, this issue could be solved by optimization of the clinical conditions, e.g., the use of adenoviral vectors at an adequate MOI and subsequent culture to eliminate episomal adenoviral genomes.
Tumorigenic Cell Removal Not Involving Viral Vector Manipulation
Here we briefly describe several methods for eradicating tumorigenic cells that take advantage of the biological properties of undifferentiated cells (Figure 2). One group of researchers showed that prolonged methionine deprivation induces cell death more readily in undifferentiated than differentiated cells, because undifferentiated cells demand more methionine than differentiated cells.32 Another group of researchers developed a recombinant lectin-toxin fusion protein that combines an hPSC-specific lectin probe, rBC2LCN, with the catalytic domain of Pseudomonas aeruginosa exotoxin A.33 This fusion protein has the potential to completely eliminate hPSCs. Yet another group synthesized a molecular switch for microRNA-302a-5p, a molecule that is highly and specifically expressed in undifferentiated hPSCs but gradually decreases to basal levels during differentiation.34 This switch sensitively detected undifferentiated and partially differentiated hiPSCs. The early success of these techniques warrants further detailed investigation of their tumor selectivities and impacts on differentiated target cells.
It should be noted that these techniques are generally applicable to undifferentiated pre-transplant cells, but they cannot address post-transplant cell tumorigenesis or cancer development (Figure 2). In this regard, we believe that viral transduction of suicide genes has a clinical advantage over other methods, because such genes serve as a safeguard against possible tumorigenesis.
Conclusions and Future Prospects
Safe and robust techniques to prevent hPSC tumorigenesis are indispensable for the advancement of regenerative medicine. In addition to existing indirect methods for reducing tumorigenic cell contamination, new techniques should be developed that can directly, securely, and specifically eradicate tumorigenic cells. The innovative viral vector-based technologies reviewed in this article promise to have far-reaching impacts that supersede conventional approaches. Future development and optimization of these and other new technologies will help to overcome the problems of hPSC tumorigenesis in the context of regenerative medicine.
References
- 1.Wu J., Izpisua Belmonte J.C. Stem Cells: A Renaissance in Human Biology Research. Cell. 2016;165:1572–1585. doi: 10.1016/j.cell.2016.05.043. [DOI] [PubMed] [Google Scholar]
- 2.Trounson A., DeWitt N.D. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016;17:194–200. doi: 10.1038/nrm.2016.10. [DOI] [PubMed] [Google Scholar]
- 3.Shi Y., Inoue H., Wu J.C., Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 2017;16:115–130. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee A.S., Tang C., Rao M.S., Weissman I.L., Wu J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013;19:998–1004. doi: 10.1038/nm.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCormack M.P., Rabbitts T.H. Activation of the T-cell oncogene LMO2 after gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2004;350:913–922. doi: 10.1056/NEJMra032207. [DOI] [PubMed] [Google Scholar]
- 6.Ohnishi K., Semi K., Yamamoto T., Shimizu M., Tanaka A., Mitsunaga K., Okita K., Osafune K., Arioka Y., Maeda T. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014;156:663–677. doi: 10.1016/j.cell.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Chen K.G., Mallon B.S., McKay R.D., Robey P.G. Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell. 2014;14:13–26. doi: 10.1016/j.stem.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kosai K.I., Finegold M.J., Thi-Huynh B.T., Tewson M., Ou C.N., Bowles N., Woo S.L., Schwall R.H., Darlington G.J. Retrovirus-mediated in vivo gene transfer in the replicating liver using recombinant hepatocyte growth factor without liver injury or partial hepatectomy. Hum. Gene Ther. 1998;9:1293–1301. doi: 10.1089/hum.1998.9.9-1293. [DOI] [PubMed] [Google Scholar]
- 9.Ikoma T., Takahashi T., Nagano S., Li Y.M., Ohno Y., Ando K., Fujiwara T., Fujiwara H., Kosai K. A definitive role of RhoC in metastasis of orthotopic lung cancer in mice. Clin. Cancer Res. 2004;10:1192–1200. doi: 10.1158/1078-0432.ccr-03-0275. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi T., Kawai T., Ushikoshi H., Nagano S., Oshika H., Inoue M., Kunisada T., Takemura G., Fujiwara H., Kosai K. Identification and isolation of embryonic stem cell-derived target cells by adenoviral conditional targeting. Mol. Ther. 2006;14:673–683. doi: 10.1016/j.ymthe.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Schlesinger S., Goff S.P. Retroviral transcriptional regulation and embryonic stem cells: war and peace. Mol. Cell. Biol. 2015;35:770–777. doi: 10.1128/MCB.01293-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooray S., Howe S.J., Thrasher A.J. Retrovirus and lentivirus vector design and methods of cell conditioning. Methods Enzymol. 2012;507:29–57. doi: 10.1016/B978-0-12-386509-0.00003-X. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki K., Mitsui K., Aizawa E., Hasegawa K., Kawase E., Yamagishi T., Shimizu Y., Suemori H., Nakatsuji N., Mitani K. Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proc. Natl. Acad. Sci. USA. 2008;105:13781–13786. doi: 10.1073/pnas.0806976105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitsui K., Ide K., Takayama A., Wada T., Irie R., Kosai K. Conditionally replicating adenovirus prevents pluripotent stem cell-derived teratoma by specifically eliminating undifferentiated cells. Mol. Ther. Methods Clin. Dev. 2015;2:15026. doi: 10.1038/mtm.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khai N.C., Takahashi T., Ushikoshi H., Nagano S., Yuge K., Esaki M., Kawai T., Goto K., Murofushi Y., Fujiwara T. In vivo hepatic HB-EGF gene transduction inhibits Fas-induced liver injury and induces liver regeneration in mice: a comparative study to HGF. J. Hepatol. 2006;44:1046–1054. doi: 10.1016/j.jhep.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 16.Chen S.H., Chen X.H., Wang Y., Kosai K., Finegold M.J., Rich S.S., Woo S.L. Combination gene therapy for liver metastasis of colon carcinoma in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:2577–2581. doi: 10.1073/pnas.92.7.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen S.H., Kosai K., Xu B., Pham-Nguyen K., Contant C., Finegold M.J., Woo S.L. Combination suicide and cytokine gene therapy for hepatic metastases of colon carcinoma: sustained antitumor immunity prolongs animal survival. Cancer Res. 1996;56:3758–3762. [PubMed] [Google Scholar]
- 18.Terazaki Y., Yano S., Yuge K., Nagano S., Fukunaga M., Guo Z.S., Komiya S., Shirouzu K., Kosai K. An optimal therapeutic expression level is crucial for suicide gene therapy for hepatic metastatic cancer in mice. Hepatology. 2003;37:155–163. doi: 10.1053/jhep.2003.50018. [DOI] [PubMed] [Google Scholar]
- 19.Cheng F., Ke Q., Chen F., Cai B., Gao Y., Ye C., Wang D., Zhang L., Lahn B.T., Li W., Xiang A.P. Protecting against wayward human induced pluripotent stem cells with a suicide gene. Biomaterials. 2012;33:3195–3204. doi: 10.1016/j.biomaterials.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 20.Tieng V., Cherpin O., Gutzwiller E., Zambon A.C., Delgado C., Salmon P., Dubois-Dauphin M., Krause K.H. Elimination of proliferating cells from CNS grafts using a Ki67 promoter-driven thymidine kinase. Mol. Ther. Methods Clin. Dev. 2016;6:16069. doi: 10.1038/mtm.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yagyu S., Hoyos V., Del Bufalo F., Brenner M.K. An Inducible Caspase-9 Suicide Gene to Improve the Safety of Therapy Using Human Induced Pluripotent Stem Cells. Mol. Ther. 2015;23:1475–1485. doi: 10.1038/mt.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ando M., Nishimura T., Yamazaki S., Yamaguchi T., Kawana-Tachikawa A., Hayama T., Nakauchi Y., Ando J., Ota Y., Takahashi S. A Safeguard System for Induced Pluripotent Stem Cell-Derived Rejuvenated T Cell Therapy. Stem Cell Reports. 2015;5:597–608. doi: 10.1016/j.stemcr.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagano S., Oshika H., Fujiwara H., Komiya S., Kosai K. An efficient construction of conditionally replicating adenoviruses that target tumor cells with multiple factors. Gene Ther. 2005;12:1385–1393. doi: 10.1038/sj.gt.3302540. [DOI] [PubMed] [Google Scholar]
- 24.Ungerechts G., Bossow S., Leuchs B., Holm P.S., Rommelaere J., Coffey M., Coffin R., Bell J., Nettelbeck D.M. Moving oncolytic viruses into the clinic: clinical-grade production, purification, and characterization of diverse oncolytic viruses. Mol. Ther. Methods Clin. Dev. 2016;3:16018. doi: 10.1038/mtm.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamizono J., Nagano S., Murofushi Y., Komiya S., Fujiwara H., Matsuishi T., Kosai K. Survivin-responsive conditionally replicating adenovirus exhibits cancer-specific and efficient viral replication. Cancer Res. 2005;65:5284–5291. doi: 10.1158/0008-5472.CAN-04-2657. [DOI] [PubMed] [Google Scholar]
- 26.Horikawa Y., Wang Y., Nagano S., Kamizono J., Ikeda M., Komiya S., Kosai K.I. Assessment of an altered E1B promoter on the specificity and potency of triple-regulated conditionally replicating adenoviruses: implications for the generation of ideal m-CRAs. Cancer Gene Ther. 2011;18:724–733. doi: 10.1038/cgt.2011.44. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda S., Pelus L.M. Survivin, a cancer target with an emerging role in normal adult tissues. Mol. Cancer Ther. 2006;5:1087–1098. doi: 10.1158/1535-7163.MCT-05-0375. [DOI] [PubMed] [Google Scholar]
- 28.Nemunaitis J., Tong A.W., Nemunaitis M., Senzer N., Phadke A.P., Bedell C., Adams N., Zhang Y.A., Maples P.B., Chen S. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol. Ther. 2010;18:429–434. doi: 10.1038/mt.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanoue K., Wang Y., Ikeda M., Mitsui K., Irie R., Setoguchi T., Komiya S., Natsugoe S., Kosai K. Survivin-responsive conditionally replicating adenovirus kills rhabdomyosarcoma stem cells more efficiently than their progeny. J. Transl. Med. 2014;12:27. doi: 10.1186/1479-5876-12-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang C.T., Song J., Bu X., Cong Y.S., Bacchetti S., Rennie P., Jia W.W. Herpes simplex virus type-1 infection upregulates cellular promoters and telomerase activity in both tumor and nontumor human cells. Gene Ther. 2003;10:1494–1502. doi: 10.1038/sj.gt.3302005. [DOI] [PubMed] [Google Scholar]
- 31.Glass M., Söling A., Messerle M. Tumor-specific activity of cellular regulatory elements is down-regulated upon insertion into the herpes simplex virus genome. J. Neurovirol. 2008;14:522–535. doi: 10.1080/13550280802348214. [DOI] [PubMed] [Google Scholar]
- 32.Shiraki N., Shiraki Y., Tsuyama T., Obata F., Miura M., Nagae G., Aburatani H., Kume K., Endo F., Kume S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014;19:780–794. doi: 10.1016/j.cmet.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 33.Tateno H., Onuma Y., Ito Y., Minoshima F., Saito S., Shimizu M., Aiki Y., Asashima M., Hirabayashi J. Elimination of tumorigenic human pluripotent stem cells by a recombinant lectin-toxin fusion protein. Stem Cell Reports. 2015;4:811–820. doi: 10.1016/j.stemcr.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parr C.J., Katayama S., Miki K., Kuang Y., Yoshida Y., Morizane A., Takahashi J., Yamanaka S., Saito H. MicroRNA-302 switch to identify and eliminate undifferentiated human pluripotent stem cells. Sci. Rep. 2016;6:32532. doi: 10.1038/srep32532. [DOI] [PMC free article] [PubMed] [Google Scholar]