

Characterization of a cutis laxa mouse model reveals distinct requirements for elastic fiber formation along the arterial tree.
Keywords: Fibluin-4, extracellular matrix, cutis laxa, ascending aortic aneurysm, arterial stiffness, elastic fiber
Abstract
Homozygous or compound heterozygous mutations in fibulin-4 (FBLN4) lead to autosomal recessive cutis laxa type 1B (ARCL1B), a multisystem disorder characterized by significant cardiovascular abnormalities, including abnormal elastin assembly, arterial tortuosity, and aortic aneurysms. We sought to determine the consequences of a human disease–causing mutation in FBLN4 (E57K) on the cardiovascular system and vascular elastic fibers in a mouse model of ARCL1B. Fbln4E57K/E57K mice were hypertensive and developed arterial elongation, tortuosity, and ascending aortic aneurysms. Smooth muscle cell organization within the arterial wall of large conducting vessels was abnormal, and elastic fibers were fragmented and had a moth-eaten appearance. In contrast, vessel wall structure and elastic fiber integrity were normal in resistance/muscular arteries (renal, mesenteric, and saphenous). Elastin cross-linking and total elastin content were unchanged in large or small arteries, whereas elastic fiber architecture was abnormal in large vessels. While the E57K mutation did not affect Fbln4 mRNA levels, FBLN4 protein was lower in the ascending aorta of mutant animals compared to wild-type arteries but equivalent in mesenteric arteries. We found a differential role of FBLN4 in elastic fiber assembly, where it functions mainly in large conduit arteries. These results suggest that elastin assembly has different requirements depending on vessel type. Normal levels of elastin cross-links in mutant tissue call into question FBLN4’s suggested role in mediating lysyl oxidase–elastin interactions. Future studies investigating tissue-specific elastic fiber assembly may lead to novel therapeutic interventions for ARCL1B and other disorders of elastic fiber assembly.
INTRODUCTION
Extracellular matrix (ECM) proteins play an integral role in maintaining arterial wall integrity. Perturbations or deficiencies of several ECM proteins have been shown to alter the structure, biomechanical properties, and/or cell signaling within the arterial wall, leading to disease. For instance, mutations in fibrillin 1, transforming growth factor–β (TGFβ) receptor type 2, collagen 3A1, and lysyl oxidase (LOX) cause Marfan syndrome, Loeys-Dietz syndrome, vascular Ehlers-Danlos syndrome, and familial thoracic aortic aneurysm, respectively, all of which have weakening of large arterial wall and development of ascending aortic aneurysm as features. Recently, mutations in fibulin-4 (FBLN4), also known as epidermal growth factor (EGF)–containing fibulin-like extracellular matrix protein 2 (EFEMP2), were described in humans with autosomal recessive cutis laxa type 1B (ARCL1B) (1–9). In addition to inelastic or redundant skin, some of the hallmark features of this disease include arterial tortuosity, aortic aneurysm, and pulmonary emphysema. Other more variable presentations include abnormalities of the skeletal system (10, 11).
FBLN4 belongs to a seven-member family of ECM proteins that share significant sequence and structural homology characterized by a C-terminal fibulin domain preceded by repeated Ca2+-binding EGF (cbEGF)–like motifs (12–14). Originally discovered through sequence homology to fibulin-1, fibulin-2, and fibulin-3 (15), FBLN4 is known to play a critical role in elastic fiber formation. Fbln4 knockout (KO) mice die perinatally from severe lung and vascular abnormalities, including failure of alveolar septation, arterial tortuosity, aneurysms, and rupture due to the absence of intact elastic fibers (16). In contrast, smooth muscle–specific KO and hypomorphic mouse models (17–19) of Fbln4 show elastic fiber fragmentation, arterial tortuosity, and aneurysmal dilation of the ascending aorta (20, 21), suggesting that elastic fiber assembly is negatively affected when FBLN4 levels fall below a critical level.
How FBLN4 functions in elastic fiber assembly is not known. In vitro data show that the N terminus of FBLN4 can bind the propeptide of LOX and promote the association of LOX with tropoelastin (18, 22). This, in addition to the fact that the Lox null mouse model has features in common with the Fbln4 KO mouse model (23, 24), has led to the currently accepted hypothesis that FBLN4 functions to deliver LOX to tropoelastin to facilitate cross-linking.
To study the effects of FBLN4 missense mutations in vivo, Igoucheva et al. (25) recently generated a knock-in mouse carrying the E57K mutation identified in humans. This was the first mutation identified in a human with ARCL1B (4). Skin biopsy from this patient showed fewer elastic fibers, and studies using dermal fibroblasts suggested that the mutant protein is poorly secreted, is unstable, or is unable to bind matrix, leading to aberrant elastic fiber formation and the resultant clinical phenotype (4). Initial characterization of Fbln4E57K/E57K mice showed that the mouse model recapitulates the clinical manifestations seen in humans, including loose skin, bent forelimbs, pulmonary emphysema, arterial tortuosity, and ascending aortic aneurysms (25). In addition to elastic fiber abnormalities in the skin and large arteries, collagen fibrils were irregularly shaped, with many large fibrils noted in the dermis of Fbln4E57K/E57K mice (25).
Here, we examined the functional consequences of the E57K mutation in Fbln4 on the cardiovascular system and noted significant hypertension and large artery stiffness in mutant mice. Because blood pressure is primarily regulated by resistance/muscular arteries, we investigated the effect of the E57K mutation in Fbln4 on the structure of muscular arteries and noted surprising differences between conduit and resistance arteries. Whereas elastic lamellae within the wall of large vessels of the mutant mouse were severely fragmented and vascular smooth muscle cells (SMCs) were disorganized, resistance arteries were seemingly unaffected by the mutation. Furthermore, unlike the skin (25), where elastin levels were decreased, we observed no change in elastin content, as measured by desmosine levels, in large conduit or small resistance arteries. Our data suggest tissue-specific roles for FBLN4 in maintaining arterial wall integrity and raise the interesting possibility that the requirements for elastic fiber assembly differ depending on vessel and tissue type.
RESULTS
Adult Fbln4E57K/E57K mice have ascending aortic aneurysms, arterial tortuosity, and elastic fiber fragmentation
Because arterial aneurysms and tortuosity are hallmark features of humans carrying recessive mutations in FBLN4 (4, 5, 10, 11), we used a mouse model carrying a known human FBLN4 mutation (Fbln4E57K/E57K) to evaluate how the mutant protein influences cardiovascular development and integrity. Mice recessive for the mutation were born at the expected Mendelian ratio (25), were similar in size to their wild-type (WT) littermates (fig. S1), and had no increased mortality by 1 year of age. As shown in Fig. 1A, arterial elongation and tortuosity, as well as variable branching angles of arteries from the aortic arch, were noted in all Fbln4E57K/E57K mice, but not in WT or heterozygous littermates. Ascending aortic aneurysms and/or aortic root dilations were noted in approximately half of the Fbln4E57K/E57K animals. No aneurysms or dilations were seen in any other arterial segment or in WT or heterozygous littermates.
Fig. 1. Fbln4E57K/E57K mice develop arterial elongation, vascular tortuosity, ascending aortic aneurysms, elastic fiber fragmentation, and SMC disarray.
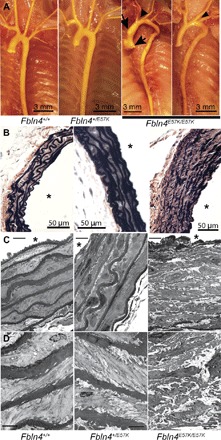
(A) Gross morphology of the thoracic aorta and its branches in adult Fbln4E57K/E57K mice and control (Fbln4+/+ and Fbln4+/E57K) littermates injected with yellow latex to visualize the vasculature. Arrows indicate aortic root dilatation and ascending aortic aneurysm seen in approximately half of Fbln4E57K/E57K mice; arrowheads indicate the different angles at which arterial branches come off the aortic arch. Arterial tortuosity and elongation are noted in all Fbln4E57K/E57K mice. (B to D) VVG (Verhoeff–van Gieson) stain (B) and transmission electron micrographs of the ascending aorta of 3-month-old Fbln4E57K/E57K mice and littermate controls at two different magnifications (C versus D). In addition to severe elastic fiber fragmentation, there is increased medial wall thickness and SMC disarray in the aorta of Fbln4E57K/E57K mice. At higher magnification (D), the moth-eaten or spongy appearance of the fragmented elastic fibers is appreciated in the aorta of Fbln4E57K/E57K mice. In addition, there is loss of smooth muscle contact with elastic fibers in Fbln4E57K/E57K vessels. Scale bars, 8 μm (C) and 4 μm (D). Asterisks indicate vessel lumen.
VVG staining of the ascending aorta of Fbln4E57K/E57K mice showed extensive elastic fiber fragmentation and disarray (Fig. 1B) as well as increased wall thickness (fig. S2) due mainly to thickening of the medial layer. Ultrastructural examination of the large vessels by electron microscopy showed moth-eaten or spongy appearance of elastin and round-shaped SMCs reminiscent of dedifferentiated cells that are detached from the elastic lamellae (Fig. 1C). Notably, the severity of elastic fiber fragmentation and disarray was variable in different regions of the same vessel cross section. The arterial wall abnormalities were seen in both aneurysmal and nonaneurysmal areas of the ascending aorta, and there was no evidence of arterial calcification, as shown by negative von Kossa staining (fig. S3), up to 1 year of age. Similar ultrastructural changes were evident in the wall of the descending thoracic aorta (Fig. 2, A and B), although this vascular segment did not develop aneurysms. In addition to the elastic fiber and SMC abnormalities, collagen fibril size was slightly variable, with significantly larger fibrils in the adventitia of Fbln4E57K/E57K aorta (Fig. 2C).
Fig. 2. Elastic fiber fragmentation and SMC disarray in descending aorta of Fbln4E57K/E57K mice.
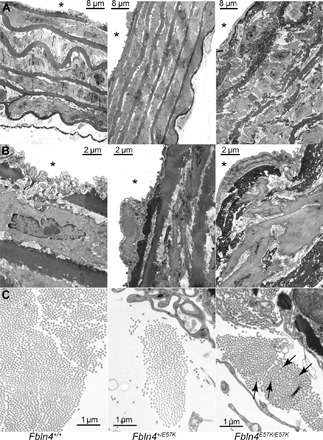
Transmission electron micrographs of descending aorta of 3-month-old Fbln4E57K/E57K and littermate mice (Fbln4+/+ and Fbln4+/E57K). (A and B) In addition to severe elastic fiber fragmentation, there is increased medial wall thickness and SMC disarray in the aorta of Fbln4E57K/E57K mice. At higher magnification, the moth-eaten or spongy appearance of the fragmented elastic fibers (B) and variable (arrows = larger) collagen fibril size (C) are appreciated in the aorta of Fbln4E57K/E57K mice. Asterisks indicate vessel lumen.
Fbln4E57K/E57K mice have large artery stiffness and systolic hypertension
Perturbations in elastin/collagen content or elastic fiber integrity often lead to changes in arterial stiffness. When the mechanical properties of large arteries in adult Fbln4E57K/E57K mice were characterized, we found that the ascending aortae and carotid arteries reached maximal dilation at lower pressures than those of heterozygous or WT mice, indicating that they have stiffer large arteries (Fig. 3, A and B). Because of significant differences in the outer diameter of aneurysmal versus nonaneurysmal Fbln4E57K/E57K ascending aortae, the data were separated into two groups. As shown in Fig. 3A, although the aneurysmal Fbln4E57K/E57K ascending aortae have a larger diameter at 0 mmHg, their pressure-diameter curve has the same compliance as that of nonaneurysmal Fbln4E57K/E57K aortae (that is, parallel curves), suggesting that the material properties or mechanics of Fbln4E57K/E57K ascending aortae are not influenced by the presence of an aneurysm.
Fig. 3. Fbln4E57K/E57K mice develop large artery stiffness and systolic hypertension.
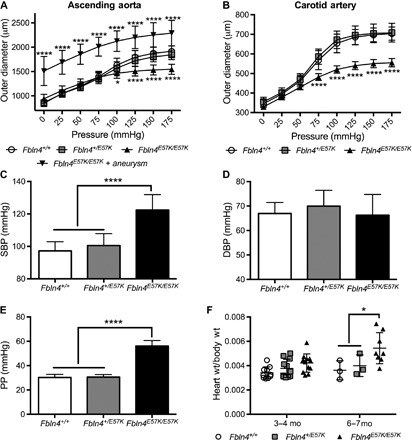
Pressure-diameter relationships of ascending aorta (A) and carotid arteries (B) from 3- to 6-month-old Fbln4E57K/E57K male mice and littermates (Fbln4+/+ and Fbln4+/E57K). At each pressure, the vessel outer diameter was compared to that of WT vessels using two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test. n = 7 to 10 for ascending aorta except for aneurysmal Fbln4E57K/E57K, where n = 3 (A) and 8 to 9 (B) for carotid arteries. Arterial systolic blood pressure (SBP) (C), diastolic blood pressure (DBP) (D), and pulse pressure (PP) (E) of 3- to 4-month-old Fbln4E57K/E57K male and female mice and littermates. PP was calculated as the difference between measured SBP and DBP. Pressure was compared among all genotypes using one-way ANOVA with Tukey’s multiple comparisons test. n = 10 to 12 per genotype. (F) Heart weight–to–body weight ratios between the three genotypes presented in two distinct age groups. At 6 to 7 months of age, mice with aortic root dilatation developed cardiac hypertrophy. Data are means ± SD. *P < 0.05, ****P < 0.0001.
To assess the physiologic consequences of elastic fiber fragmentation and large artery stiffness, we measured arterial blood pressure and found that Fbln4E57K/E57K mice have significant systolic hypertension with widened PP, but no elevation in DBP compared to WT or heterozygous littermates (Fig. 3, C to E). The systolic hypertension and widened PP were completely penetrant and independent of aneurysm formation. There were no differences in circumferential arterial stiffness or blood pressure between WT and heterozygous (Fbln4+/E57K) mice (Fig. 3, A to E). Heart rate was not different among all three genotypes (fig. S4). As shown in Fig. 3F, we noted differences in Fbln4E57K/E57K heart size at 6 months of age. Echocardiographic evaluation of Fbln4E57K/E57K mice with aortic root dilation at 6 weeks of age showed normal shortening fraction. However, by 6 months of age, the mice with the larger heart sizes had aortic root dilation and aortic root insufficiency, leading to worse shortening fraction, suggesting that cardiomyopathy develops over time (table S1).
Small resistance arteries have normal elastic fibers and normal wall structure in Fbln4E57K/E57K mice
Blood pressure is largely regulated by small resistance vessels. To determine whether these vessels were also affected by the E57K mutation, we examined the ultrastructure of saphenous arteries and second-order mesenteric arteries of 3-month-old Fbln4E57K/E57K mice and littermate controls. Surprisingly, these vessels showed none of the structural abnormalities evident in the large vessels and were similar to vessels from heterozygous mutant or WT mice (Fig. 4C, a and b, and fig. S5). The internal elastic lamina of Fbln4E57K/E57K mesenteric and saphenous arteries showed the normal amorphous appearance of elastin on electron microscopy, as opposed to the moth-eaten and fragmented appearance noted in large vessels. In addition, the medial layer of Fbln4E57K/E57K mesenteric and saphenous arteries consisted of the typical two to three SMC layers and was not thickened compared to WT arteries.
Fig. 4. The E57K mutation in Fbln4 has differential effects on elastic versus resistance artery integrity.
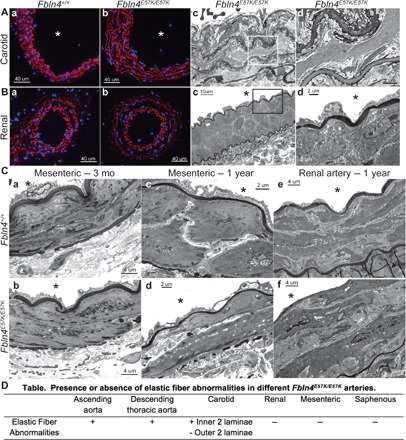
Alexa Fluor 633 hydrazide staining of carotid (A, a and b) and renal arteries (B, a and b) from 3-month-old Fbln4E57K/E57K (b) and WT littermate mice (a). Red indicates elastic fibers, and blue depicts nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI). Mutant vessels are indistinguishable from WT vessels using Alexa Fluor 633 hydrazide staining. Transmission electron micrographs of carotid (A, c and d) and renal arteries (B, c and d) from 3-month-old Fbln4E57K/E57K mice are shown. Micrographs in (d) are higher magnification of the inset demarcated in (c). Note the moth-eaten appearance of the inner two elastic fibers compared to the normal appearance of the outer two elastic fibers in the carotid artery (A, d). (C) Transmission electron micrographs of second-order mesenteric arteries from 3-month-old (a and b) and 1-year-old (c and d) Fbln4E57K/E57K and WT littermate mice in addition to 1-year-old renal arteries. Asterisks indicate vessel lumen. (D) Table summarizing the presence or absence of elastic fiber abnormalities in the different Fbln4E57K/E57K arteries examined.
The elastic laminae of Fbln4E57K/E57Karteries are adversely affected in conducting but not muscular arteries
The normal elastic fibers present in mesenteric and saphenous arteries suggested that the E57K mutation has different effects on elastin assembly in muscular arteries as compared to elastic arteries. Visualization of elastic fibers with Alexa Fluor 633 hydrazide in the carotid (Fig. 4A, a and b) and renal (Fig. 4B, a and b) arteries did not indicate any significant structural differences between WT and Fbln4E57K/E57K vessels. However, ultrastructural examination of Fbln4E57K/E57K common carotid artery, which has four elastic lamellae, by transmission electron microscopy showed moth-eaten appearance of the two inner elastic lamellae, whereas the two outer laminae closest to the adventitia were unaffected (Fig. 4A, c and d). In contrast, ultrastructural examination of Fbln4E57K/E57K main renal artery, which only has an internal and an external elastic lamina (muscular artery), showed intact elastic laminae and normal arterial wall structure (Fig. 4B, c and d). To assess whether elastic fiber abnormalities developed over time with aging in Fbln4E57K/E57K muscular arteries, we examined the ultrastructure of mesenteric and renal arteries at 1 year of age. As shown in Fig. 4C, the elastic laminae of Fbln4E57K/E57K muscular arteries remained intact at 1 year of age. A table summarizing elastic fiber abnormalities noted by electron microscopy in the different Fbln4E57K/E57K arteries examined is available in Fig. 4D.
Elastic fiber fragmentation and medial wall thickness of Fbln4E57K/E57K ascending aortae are present at birth
To determine whether elastic fiber fragmentation in the Fbln4E57K/E57K ascending aorta is a problem with fiber assembly in early development or is a postnatal degenerative process that occurs with aging, we examined the ultrastructure of ascending aorta right after birth [postnatal day 1 (P1)]. As shown in Fig. 5, there is significant elastic fiber fragmentation and medial wall thickening in Fbln4E57K/E57K ascending aorta compared to WT vessels, suggesting that mutant FBLN4 leads to abnormal elastic fiber formation during fetal development. In addition, SMCs in the mutant aorta were less organized and did not show the tightly layered structure seen with WT animals.
Fig. 5. Large artery elastic fiber fragmentation and medial wall thickening occur developmentally in Fbln4E57K/E57K mice.
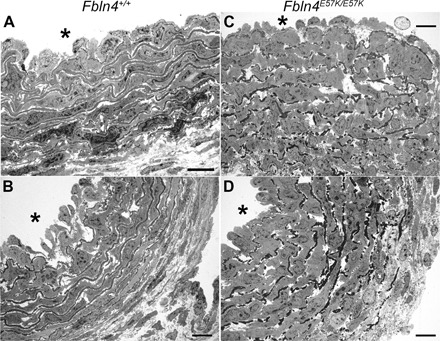
Transmission electron micrographs of P1 ascending aorta from Fbln4+/+ (A and B) and Fbln4E57K/E57K (C and D) mice. Asterisks indicate vessel lumen. Scale bars, 10 μm.
The E57K mutation in Fbln4 does not affect arterial collagen or elastin content or elastic fiber gene expression
To determine the effect of E57K mutation in Fbln4 on elastin cross-linking and collagen concentration in arteries, we measured desmosine and hydroxyproline content in ascending aorta (Fig. 6, A and B) and carotid (fig. S6) and mesenteric arteries (Fig. 6, C and D) of 3- to 4-month-old Fbln4E57K/E57K and littermate mice. Desmosine and hydroxyproline content was equivalent in vessels from all three genotypes, indicating normal elastin and collagen cross-linking and protein levels. There were also no significant differences in the expression of the elastic fiber genes Fbln4, Fbln5, elastin (Eln), Lox, or fibrillin 1 (Fbn1) in the ascending aorta or mesenteric arteries of all three genotypes (Fig. 7, A and B). Similar results were seen in the lung (fig. S7). Because elastic fiber formation starts in late gestation and continues until maturation at ~P21, we sought to determine the effect of the E57K mutation in Fbln4 on elastic fiber gene expression while elastin assembly is ongoing. As shown in Fig. 7 (C and D) and similar to what was seen in adult vessels, the E57K mutation in Fbln4 did not affect elastic fiber gene expression in P7 aorta or mesenteric arteries.
Fig. 6. E57K mutation in Fbln4 does not alter arterial elastin or collagen content.
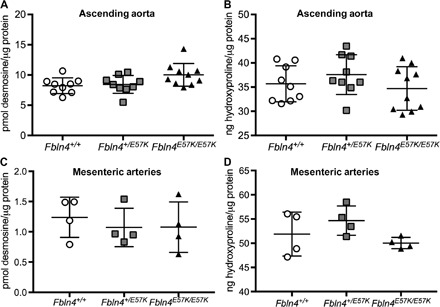
Desmosine (A and C) and hydroxyproline (B and D) content were determined in ascending aortae (A and B) and mesenteric arteries (C and D) of 3- to 4-month-old Fbln4E57K/E57K (closed triangles) male and female mice and littermates (open circles, Fbln4+/+; gray squares, Fbln4+/E57K).
Fig. 7. E57K mutation in Fbln4 does not affect arterial elastic fiber gene expression.
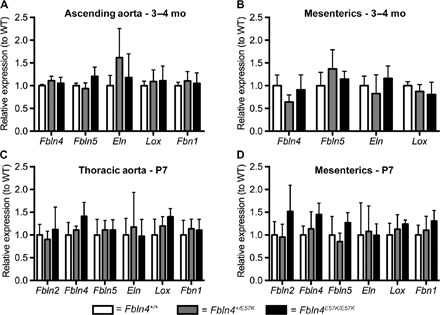
Expression of indicated elastic fiber–associated genes (Fbln2, fibulin-2; Fbln4, fibulin-4; Fbln5, fibulin-5; Eln, elastin; Lox, lysyl oxidase; Fbn1, fibrillin 1) in ascending aortae (A and C) and mesenteric arteries (B and D) of 3- to 4-month-old (A and B) and P7 (C and D) Fbln4E57K/E57K (closed bars) and littermate mice (open bars, Fbln4+/+; gray bars, Fbln4+/E57K). Gene expression was normalized to that of Gapdh. Data are means ± SD and were compared using one-way ANOVA with Tukey’s multiple comparisons test. n = 3 to 8 per genotype.
FBLN4(E57K) protein level is reduced in extracts of Fbln4E57K/E57K ascending aorta, but not mesenteric arteries
Western blot analysis of urea extracts from the adult ascending aorta of Fbln4E57K/E57K and littermate control mice showed significantly reduced protein in the aorta of mutant mice, an observation confirmed by immunofluorescence staining of FBLN4 protein in ascending aorta and carotid artery (Fig. 8, A and C). However, in mesenteric arteries, the amount of FBLN4(E57K) protein was equivalent to WT (Fig. 8B). Similar trends of FBLN4 protein levels were observed in aorta and mesenteric arteries of P7 Fbln4E57K/E57K and littermate mice (fig. S8). The effect of the E57K mutation on FBLN4 secretion was also tested in dermal fibroblasts. As shown in fig. S9 and consistent with previous results (25), there was significantly more protein in the cell lysates of Fbln4E57K/E57K cells and less in the culture medium compared to cells from WT or heterozygous mice, suggesting reduced mutant protein secretion.
Fig. 8. FBLN4(E57K) protein level is reduced in ascending aortae, but not in mesenteric arteries of adult mice.
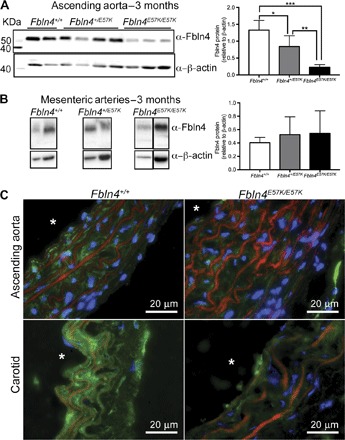
Western blot analysis of urea extracts from ascending aortae (A) and mesenteric arteries (B) of Fbln4E57K/E57K mice and littermate controls using a polyclonal rabbit anti-FBLN4 antibody. FBLN4 level was normalized to that of β-actin. The normalized data are represented in the bar graphs on the right. n = 4 to 7 per genotype. Data are means ± SD and were compared using one-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Immunofluorescence of ascending aorta and carotid artery sections of 3-month-old Fbln4E57K/E57K and WT control mice. Green, FBLN4; red, autofluorescence of elastic fibers; blue, DAPI. Asterisks indicate vessel lumen.
DISCUSSION
Homozygous or compound heterozygous mutations in FBLN4 (EFEMP2) lead to ARCL1B, a devastating multisystem disorder characterized by loose inelastic skin, arterial tortuosity and aneurysms, pulmonary emphysema, and skeletal abnormalities (1–11). Recently, a mouse model carrying a missense mutation in Fbln4 (E57K) found in two independent infants with ARCL1B was generated (25). Focusing primarily on the cutaneous and skeletal features, initial characterization of Fbln4E57K/E57K mice identified abnormalities in elastic fibers and collagen fibrils as the basis for the observed phenotypes (25). By characterizing the cardiovascular phenotype of this mouse, we identified several novel findings that extend our knowledge and raise further questions on the role of FBLN4 in elastic fiber formation and in maintaining arterial wall integrity, not only between different tissues, such as the skin and the vasculature, but also along the arterial tree.
All Fbln4E57K/E57K mice had systolic hypertension with widened PP and large artery stiffness. Higher systolic pressure contrasts with the Fbln4SMKO and the hypomorphic Fbln4 mouse models that exhibited normal SBP but lower DBP (20, 21). This difference, coupled with the fact that Fbln4E57K/E57K mice live to over 1 year of age, suggests that the E57K mutation is not a loss-of-function mutation but may alter or interfere with normal FBLN4 function or availability. Cardiac dysfunction and hypertrophy were only seen in Fbln4E57K/E57K mice with aortic insufficiency secondary to aortic root dilation at 6 months of age, but not at 6 weeks of age, indicating that cardiac dysfunction is exacerbated by the aortic insufficiency, arterial stiffness, and systolic hypertension seen with mutant FBLN4.
Fbln4E57K/E57K mice developed arterial tortuosity and elongation, as well as ascending aortic aneurysms that were confined to the ascending aorta. No dilation or narrowing was noted anywhere else along the arterial tree. The ascending aorta is the initial vessel segment that senses the volume/pressure ejected with each ventricular contraction; therefore, with an abnormal wall, it may be at greatest risk for dilation. Alternatively, the restriction of aneurysm formation to the ascending aorta may be a consequence of the differing developmental origins of SMCs in the ascending aorta (neural crest), as opposed to SMCs derived from the somatic or paraxial mesoderm that make up the rest of the arterial tree (26). Although the variable severity of elastic fiber fragmentation may explain why only 50% of the homozygous mutant mice developed ascending aortic aneurysms (25), one cannot rule out the presence of potential modifiers. It is important to note that the presence of fragmented elastic fibers and elevated blood pressure alone do not explain aneurysm formation. Humans or mice with elastin insufficiency (ELN+/−), or mice completely lacking elastin (Eln−/−), are hypertensive and develop vascular stenosis but their ascending aortae do not dilate (27–29). Similarly, mice lacking fibulin-5 or humans with FBLN5 mutations (autosomal recessive cutis laxa type 1A) have fragmented elastic fibers, arterial tortuosity and elongation, and elevated SBP but do not develop aneurysms (30, 31). Therefore, changes in cell-cell or cell-ECM signaling may be responsible for aneurysm development. Increased TGFβ signaling has been implicated in aneurysm formation in the hypomorphic and SMKO Fbln4 mouse models, as well as mouse models of Marfan and Loeys Dietz syndromes (17, 20, 32–34). Future studies will determine whether altered TGFβ signaling plays a role in aneurysm formation in Fbln4E57K/E57K mice.
An interesting finding in this study was the unchanged elastin content, as assessed by desmosine levels, in both the ascending aorta and carotid arteries of Fbln4E57K/E57K mice despite extensive elastic fiber fragmentation. Even with normal elastin levels, the mutant vessels are significantly stiffer than WT, which indicates that the integrity and organization of elastin, and not quantity alone, are important for normal vessel function. A significant increase in desmosine content was recently reported in the ascending aorta of SMC-specific Fbln4 KO (SMKO) mice (35). Normal or increased elastin cross-linking in the mutant aortae is in sharp contrast to the Fbln4E57K/E57K skin, where desmosine was decreased (25), and to the global Fbln4 KO mouse model, where elastin cross-links were decreased by 94% in the aorta (16). These tissue-specific differences are intriguing. Although not well elucidated, evidence for tissue-specific differences in elastic fiber assembly were found in a mouse model of autosomal dominant cutis laxa (ADCL). In this mouse model, a bacterial artificial chromosome carrying the human elastin gene with a single base pair deletion known to result in ADCL was expressed as a transgene in mice. In mice carrying the transgene, skin and lung incorporated mutant elastin, whereas the aorta incorporated very low levels of the mutant protein, suggesting that elastin assembly was different in the aorta than in the skin and lung (36).
Another striking example of tissue-specific differences in FBLN4 function is the normal appearance of elastin in the Fbln4E57K/E57K muscular arteries, specifically mesenteric, saphenous, and renal arteries. To our knowledge, all current data in the literature presume that the process of elastic fiber formation, particularly arterial elastic lamellae, is similar along the arterial tree. The data presented herein suggest that elastic lamellae may develop differently in different arterial beds. The finding that the muscular vessels are unaffected by the Fbln4 mutation suggests either that FBLN4 is not necessary for resistance or muscular artery elastic fiber formation or that there is functional compensation by another protein.
FBLN4’s presumed function in elastic fiber assembly is to initiate cross-linking by guiding LOX to tropoelastin (18). The fact that desmosine levels are essentially normal suggests that the cross-linking function of LOX is intact in Fbln4E57K/E57K mice. This suggests either that the E57K mutation affects secretion or amount of the protein rather than its function or that FBLN4 is not necessary for cross-linking but rather serves another function important for elastic fiber formation and integrity that is altered by the E57K mutation. This latter possibility is supported by the normal cross-linking seen when FBLN4 is deleted from arterial SMCs (35). Fibulins have been shown to play a role in maintaining cell morphology, growth, and adhesion (14, 37–41). To that end, SMC contractile genes were down-regulated in the ascending aorta of Fbln4SMKO mice, suggesting that FBLN4 may play a role in SMC differentiation and maturation (19). It is clear that FBLN4 is crucial developmentally because Fbln4E57K/E57K mice had elastic fiber fragmentation and increased large vessel medial wall thickness as early as P1, but its exact role in vessel wall development or homeostasis remains unclear.
In conclusion, we have characterized the structural and functional consequences of an FBLN4 mutation associated with ARCL1B on the cardiovascular system. In addition to ascending aortic aneurysms, arterial tortuosity, and elongation, homozygous mutant mice have stiff large arteries and systolic hypertension, a finding not described or reported in ARCL1B patients (1–11). Although previously known to be associated with elastic fiber formation in large arteries, data presented herein indicate that FBLN4 is not required for elastic fiber formation in small muscular arteries, thereby calling into question the assumed requirement of FBLN4 in elastic fiber assembly. This model will provide a useful tool to study tissue-specific differences in elastic fiber formation and to better understand the role of FBLN4 in elastic fiber assembly.
MATERIALS AND METHODS
Mice
Generation of mice carrying a knock-in mutation (G to A) in exon 4, leading to substitution of glutamate to lysine at amino acid 57, was previously described (25). Briefly, C57BL/6 embryonic stem cells were targeted with a vector carrying the point mutation in Fbln4 and loxP/FRT-flanked Neo cassette in intron 4. After identification of germline transmission of the targeted allele, the neomycin resistance gene was removed by crossing the resulting mice with mice ubiquitously expressing FLP1 recombinase on the C57BL/6 background. Mice heterozygous for knock-in allele were intercrossed to generate mice of all three genotypes (Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K) used in the experiments, as described in this article. Tail DNA was used to genotype the mice, as previously described (25). The mice were housed under standard conditions with free access to food and water. All protocols were approved by the Animal Studies Committee of Washington University School of Medicine.
Gross morphology
After euthanasia with CO2, the abdominal and chest walls of Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K adult mice were dissected away. To drain the blood from the vasculature, 5 ml of 1× phosphate-buffered saline (PBS) (pH 7.4) was flushed through the apex of left ventricle after transecting the left common iliac (32). Yellow latex (1 to 2 ml) (Ward’s Science) was then injected through the left ventricle. The yellow latex was allowed to set by keeping the mice moist for 3 to 4 hours at 4°C. Mice were then fixed in 10% neutral buffered formalin (Fisher Scientific) at 4°C overnight, after which they were stored in 70% ethanol at 4°C until dissection was performed.
Vessel dissection for histology or electron microscopy
After euthanasia with CO2, the abdominal and chest walls of Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice were dissected away, and an incision was made through the right atrium. Five milliliters of 1× PBS (pH 7.4) was flushed through the apex of the left ventricle. Maintaining the needle in the same position, we exchanged the PBS for 10% neutral buffered formalin (Fisher Scientific), which was allowed to flow through the left ventricle at 95-cm H2O, equivalent to the mean arterial pressure. After pressure fixation, vessels (ascending aorta, descending aorta, carotid arteries, and/or mesenteric arteries) were dissected and placed at 4°C either in 10% neutral buffered formalin for histology or in a solution containing 2.5% glutaraldehyde and 0.1 M sodium cacodylate for electron microscopy.
VVG and von Kossa staining
After fixation with neutral buffered formalin, vessels were washed with 1× PBS (twice for 15 min) and then dehydrated by running through an ethanol gradient (30, 50, and then 70% for 30 min each), after which they were taken to the Histology Core Facility, where dehydration was completed and the samples were paraffin-embedded and sectioned. Sections were deparaffinized with xylene and hydrated by running through an ethanol gradient (100, 90, 80, 70, 50, and then 30%), followed by distilled water. Von Kossa staining was performed by the Histology Core Facility, whereas VVG staining was performed following the IHC World protocol. Briefly, sections were stained in Verhoeff’s solution for 30 min, differentiated in 2% ferric chloride for 1 to 2 min, treated with 5% sodium thiosulfate, and then counterstained with van Gieson’s solution for 5 min. Solutions were purchased from American MasterTech. The sections were then dehydrated to 100% ethanol and treated with xylene before mounting with VectaMount (Vector Laboratories Inc.) and coverslip placement. Images were obtained using a Zeiss Axioskop 50 microscope and QCapture Pro software (Media Cybernetics Inc.).
Transmission electron microscopy
After isolation, vessels were fixed in 2.5% glutaraldehyde and 0.1 M sodium cacodylate at 4°C overnight. Vessels were then sent to Washington University’s Center for Cellular Imaging for processing and thin sectioning. Images were taken using a JEOL JEM-1400Plus transmission electron microscope that is equipped with an Advanced Microscopy Techniques XR111 high-speed, 4000 × 2000–pixel, phosphor-scintillated, 12-bit charge-coupled device (CCD) camera.
Alexa Fluor 633 hydrazide staining
Vessels (left common carotid artery and right renal artery) from Fbln4+/+, Fbln4+/E57K, and Fbln4E57K/E57K mice were dissected and frozen in optimal cutting temperature (OCT) compound (Sakura Finetek) at −80°C. With a cryostat, 3-μm sections were obtained and fixed in 4% paraformaldehyde for 10 min at 4°C. Sections were washed twice with 1× PBS for 5 min each and then incubated in 1:1000 of a 2 mM Alexa Fluor 633 hydrazide (Life Technologies) stock in 1% bovine serum albumin (BSA)/1% fish gelatin/0.05% Triton-X in 1× PBS for 5 min at room temperature. Sections were then washed twice with 1× PBS for 5 min each. Slides were mounted with DAPI Fluoromount-G (SouthernBiotech) and coverslipped. Images were obtained using a Zeiss Axioskop 50 microscope and QCapture Pro software (Media Cybernetics Inc.).
Blood pressure measurement
Mice were anesthetized with 1.5% isoflurane and maintained on a heating pad to keep their body temperature at 37°C, monitored via a rectal thermometer. A 2- to 3-mm midline incision was made in the neck; the lobes of the thymus were separated to expose the right common carotid artery. After clamping and making a small nick in the right common carotid artery, a Millar pressure transducer (model SPR-671) was introduced and advanced to the ascending aorta. SBP, DBP, and heart rate were recorded using the PowerLab data acquisition system (ADInstruments), and data were analyzed using LabChart 7 for Mac software (ADInstruments).
Compliance studies
Arterial pressure-diameter curve measurements were performed as previously described (42). Briefly, after blood pressure measurement, mice were euthanized under isoflurane anesthesia, and the ascending aorta and the left common carotid artery were excised and placed in physiology saline solution (PSS) composed of 130 mM NaCl, 4.7 mM KCl, 1.6 mM CaCl2, 1.18 mM MgSO4-7H2O, 1.17 mM KH2PO4, 14.8 mM NaHCO3, 5.5 mM dextrose, and 0.026 mM EDTA (pH 7.4). After the vessels were cleaned of surrounding fat, they were mounted on a pressure arteriograph (Danish Myo Technology) and maintained in PSS at 37°C. Vessels were visualized with an inverted microscope connected to a CCD camera and a computerized system, which allows continuous recording of vessel diameter. Because intravascular pressure was increased from 0 to 175 mmHg by 25-mmHg increments, the vessel outer diameter was recorded at each step (12 s per step). The average of three measurements at each pressure was reported.
Echocardiography
Noninvasive cardiac ultrasound examination of 6-week-old and 6-month-old Fbln4+/+ and Fbln4E57K/E57K was performed under light anesthesia by the Mouse Cardiovascular Phenotyping Core at Washington University School of Medicine in St. Louis. A VisualSonics echocardiography machine (FUJIFILM VisualSonics Inc.) with a 15-MHz linear transducer was used to characterize the structure and function of the heart and great vessels.
Desmosine and hydroxyproline assays
After completion of compliance studies, ascending aorta and carotid arteries were dried of excess physiologic saline solution and kept at −20°C until protein quantification, and desmosine/hydroxyproline measurement assays were performed as previously described (42). All mesenteric artery branches from each mouse were dissected and included as one sample. Briefly, each sample was hydrolyzed with 20μl of 6 N hydrochloric acid (Thermo Scientific) at 105°C for 48 hours. Samples were dried at 65°C for 90 min in SpeedVac, and each pellet was dissolved in 400 μl of water and filtered with a 0.45-μm filter.
A ninhydrin-based assay was used to determine the protein content of each sample (43). An aliquot of each hydrolyzed sample was reacted with 100 μl of 100 mM ninhydrin [dissolved in 75% ethylene glycol containing SnCl2 (2.5 mg/ml) and 1 M sodium acetate (pH 5.5)] at 85°C for 10 min. The protein content of each sample was determined by measuring the absorbance at 575 nm and comparing it to a calibration standard for protein hydrolysis (Pickering Laboratories).
A chloramine-T colorimetric assay was used to determine hydroxyproline content (44). An aliquot of each hydrolyzed sample was oxidized with 1.5 mg of chloramine-T for 20 min at room temperature and then reacted with 20 mg of p-dimethylaminobenzaldehyde dissolved in propanol containing 20% perchloric acid for 20 min at 65°C. Samples were done in duplicate. Absorbance at 550 nm was measured using a Synergy H4 Multi-Mode plate reader, and hydroxyproline content was determined by comparison to a standard curve. Reagents for this assay were purchased from Sigma-Aldrich.
To determine desmosine content in the hydrolyzed samples, a competitive enzyme-linked immunosorbent assay was used (45). Briefly, an aliquot of hydrolysate was added to a well of a desmosine-ovalbumin–coated 96-well plate (Elastin Products Company). Rabbit anti-desmosine antibody (1:4000) (Elastin Products Company) was added to each well and incubated at room temperature for 1 hour. After unbound antibodies were washed off, a goat anti-rabbit secondary antibody conjugated to horseradish peroxidase (HRP) (NA934V, GE Health Sciences) was added. After 1 hour of incubation, SureBlue TMB peroxidase substrate was added to determine peroxidase activity by measuring the absorbance at 650 nm. Desmosine content was determined by extrapolation from a desmosine standard curve (Elastin Products Company) run on the same plate.
RNA isolation and quantitative real-time reverse transcription polymerase chain reaction
After euthanasia with CO2, the chest cavity was exposed, and 5 ml of 1× PBS was flushed through the apex of the left ventricle after snipping the right atrium. Ascending aorta (from the root up to, but before, the first branch of the aortic arch) for adult mice and all thoracic aorta for P7 mice, left lung, and mesenteric arteries (all branches for each mouse) were isolated and stored in RNAlater at −80°C until mRNA was isolated using TRIzol following the manufacturer’s protocol (Life Technologies). Total RNA (1 μg) was reverse-transcribed using High-Capacity RNA-to-cDNA Kit per the manufacturer’s protocol (Life Technologies). Real-time polymerase chain reaction (PCR) was done using 1 μl of complementary DNA template, TaqMan Fast Universal PCR Master Mix, and TaqMan assays (primers/probes) obtained from Life Technologies. Reactions were run in duplicate on the ViiA real-time PCR system, and experimental gene expression was normalized to that of Gapdh. TaqMan assays used in this study are Mm00514670_m1 (Eln), Mm00445429_m1 (Efemp2), Mm00488601_m1 (Fbln5), Mm00495386_m1 (Lox), Mm00514908_m1 (Fbn1), Mm99999915_g1 (Gapdh), Mm02342430_g1 (Ppia or cyclophilin), and Mm00484266_m1 (Fbln2).
Mouse tissue protein extraction
Ascending aorta and most branches of mesenteric arteries were dissected after flushing the vasculature with 1× PBS through the apex of the left ventricle. Tissues were stored at −80°C until protein extraction was performed. Tissues were homogenized using TissueLyser II (Qiagen) at 30 Hz for 5 min in an 8 M urea solution in 16 mM Na2HPO4 (pH 7) containing protease inhibitors, and homogenates were incubated at 4°C overnight. After centrifugation, the supernatant was diluted to 2 M urea using 16 mM Na2HPO4, and bicinchoninic acid (BCA) assay (Bio-Rad) was performed to measure protein concentration. Protein was then precipitated using 10% trichloroacetic acid at 4°C for 1 hour. After centrifugation, the pellet was washed three times with ice-cold acetone and resuspended in Laemmli buffer containing dithiothreitol (DTT). The suspension was boiled for 5 min and used for SDS–polyacrylamide gel electrophoresis (SDS-PAGE).
Primary mouse dermal fibroblast preparation and protein isolation
A 1 cm × 1 cm piece of skin from the back of P1 mice was cut into 1 mm × 1 mm fragments and incubated in 1× Dulbecco’s modified Eagle’s medium containing 15% fetal bovine serum, 1% nonessential amino acids, 1% glutamate, and 1× penicillin-streptomycin solution in a 10-cm tissue culture dish.
Once confluent in a 10-cm dish (passage #2 or #3), cells were placed in serum-free culture media for 24 hours. Supernatant was removed and concentrated using Ultracel-30K centrifugal filters (EMD Millipore). Radioimmunoprecipitation assay buffer (1 ml) (Cell Signaling Technology) containing protease inhibitors was added to the cells and incubated at 4°C for 10 min. Cells were then scraped off and incubated at 4°C overnight. After centrifugation, the protein concentration of the supernatant was determined using the Bio-Rad BCA assay. DTT-containing Laemmli buffer was added to 11 μg of protein, and the sample was boiled for 5 min and used for SDS-PAGE.
FBLN4 antibody generation and Western blotting
Protein sequences for all mouse fibulin family members (fibulin-1 to fibulin-7) were aligned using the online software Clustal Omega. A unique sequence in the N terminus of FBLN4 was identified as potentially antigenic (TDGYEWDADSQHC). Peptide synthesis and keyhole limpet hemocyanin conjugation, rabbit immunization, bleeding, and antigen affinity purification of the antibody were performed by GenScript. The resulting polyclonal rabbit anti-mouse FBLN4 antibody was used as the primary antibody for Western blotting. Protein from tissues (ascending aorta or mesenteric arteries), cell lysates (primary mouse dermal fibroblasts), or supernatants was subjected to SDS-PAGE on a 10% gel. Protein was transferred to a polyvinylidene difluoride membrane, which was then blocked in 5% milk/0.1% Tween 20 in 1× PBS at room temperature (RT) for 1 hour. Rabbit anti-mouse FBLN4 antibody in blocking solution (1:800) was then added and incubated overnight at 4°C. The membrane was washed three times with 0.1% Tween 20/1× PBS and incubated with a secondary antibody [1:4000 donkey anti-rabbit immunoglobulin G (IgG)–HRP, GE Healthcare] in blocking solution for 1 hour at RT. Immobilon Western HRP Substrate (EMD Millipore) was used for protein detection via the ChemiDoc MP imaging system (Bio-Rad). After blocking for 1 hour at RT, the membrane was then incubated in 1:4000 mouse anti–β-actin antibody (Sigma-Aldrich), followed by 1:10,000 goat anti-mouse IgG-HRP (KPL) to assess protein loading.
Immunofluorescence
After euthanasia with CO2, the chest cavity was exposed, the right atrium was clipped, and 5 ml of 1× PBS was flushed through the apex of the left ventricle. Ascending aorta and carotid arteries were dissected and frozen in OCT medium (Tissue-Tek, Sakura Finetek) at −80°C. With a cryostat, 3-μm sections were placed on slides, fixed in ice-cold acetone for 10 min, and washed with 1× PBS. To minimize autofluorescence of elastic fibers in the green channel, sections were then treated with Chicago Sky Blue (Sigma, 0.5% solution in 1× PBS) for 5 min at RT. Sections were washed with 1× PBS, blocked in a solution containing 1% BSA + 1% fish gelatin + 0.05% Triton X-100 for 1 hour at RT, and incubated in 1:500 rabbit anti-mouse FBLN4 antibody in blocking solution overnight at 4°C. The sections were then washed with 1× PBS and incubated in 1:2000 goat anti-rabbit–Alexa Fluor 488 antibody (Life Technologies) and 1:10,000 DAPI in blocking solution at RT for 1 hour. Slides were washed with 1× PBS, mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific), and coverslipped. Images were obtained using a Zeiss Axioskop 50 microscope and QCapture Pro software (Media Cybernetics Inc.).
Statistical analysis
One-way or two-way ANOVA with Tukey’s multiple comparisons test was used to determine differences between genotypes, as indicated in each figure legend. Statistical analyses were run using Prism 6 for Mac OS X (GraphPad Software Inc.). Data are presented as means ± SD. Differences were considered statistically significant when P was equal to or less than 0.05.
Supplementary Material
Acknowledgments
We thank T. Hall for administrative assistance. We also thank B. Coleman and M. Scott from the Elvie L. Taylor Histology Core Facility for paraffin embedding, sectioning, and performing von Kossa staining of vessel samples; M. Levy and R. Roth from the Washington University Center for Cellular Imaging for processing and thin sectioning samples for transmission electron microscopy; and A. Kovacs from the Mouse Cardiovascular Phenotyping Core for performing and analyzing mouse echocardiograms. Funding: C.M.H. is a Scholar of the Child Health Research Center at Washington University School of Medicine (NIH K12-HD076224). C.M.H. also received support from the NIH (training grant T32-HD043010-11) and the Mallinckrodt Foundation Physician-Scientist Training Program Fellow support. V.S.L. received support from the NIH (training grants T32-EB18266 and T32-HL125241). This work was funded by NIH grants R01-HL53325 and R01-HL105314 to R.P.M. Funding for electron microscopy was provided by the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital. Author contributions: C.M.H. and R.P.M. conceptualized and designed the research; C.M.H., T.J.B., M.L., and V.S.L. performed experiments and analyzed data; C.M.H., T.J.B., M.L., V.S.L., and R.P.M. interpreted results of experiments; C.M.H. prepared figures and drafted the manuscript; C.M.H. and R.P.M. edited and revised the manuscript; and C.M.H., T.J.B., M.L., V.S.L., M.-L.C., and R.P.M. approved the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Fbln4E57K/E57K mice must be obtained through a material transfer agreement. They are also available for purchase from the Jackson Laboratory. Readers may purchase the Fbln4(E57K) mice from the Jackson Laboratory website at www.jax.org/strain/027945. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602532/DC1
table S1. Echocardiographic data of Fbln4E57K/E57K and littermate WT mice.
fig. S1. Fbln4E57K/E57K mice are similar in size to their littermates.
fig. S2. Fbln4E57K/E57K ascending aortae have increased medial wall thickness.
fig. S3. Fbln4E57K/E57K ascending aortae do not develop calcification.
fig. S4. Fbln4E57K/E57K mice have similar heart rates to their heterozygous and WT littermates.
fig. S5. Internal elastic lamina is unaffected in Fbln4E57K/E57K saphenous arteries.
fig. S6. Homozygous E57K mutation in Fbln4 does not alter arterial elastin or collagen content.
fig. S7. Homozygous E57K mutation in Fbln4 does not alter ECM gene expression in lungs.
fig. S8. FBLN4(E57K) protein level is reduced in P7 ascending aorta, but not in mesenteric arteries.
fig. S9. FBLN4(E57K) protein secretion is impaired compared to WT FBLN4.
REFERENCES AND NOTES
- 1.Dasouki M., Markova D., Garola R., Sasaki T., Charbonneau N. L., Sakai L. Y., Chu M.-L., Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am. J. Med. Genet. A 143A, 2635–2641 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Hebson C., Coleman K., Clabby M., Sallee D., Shankar S., Loeys B., Van Laer L., Kogon B., Severe aortopathy due to fibulin-4 deficiency: Molecular insights, surgical strategy, and a review of the literature. Eur. J. Pediatr. 173, 671–675 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Hoyer J., Kraus C., Hammersen G., Geppert J.-P., Rauch A., Lethal cutis laxa with contractural arachnodactyly, overgrowth and soft tissue bleeding due to a novel homozygous fibulin-4 gene mutation. Clin. Genet. 76, 276–281 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Hucthagowder V., Sausgruber N., Kim K. H., Angle B., Marmorstein L. Y., Urban Z., Fibulin-4: A novel gene for an autosomal recessive cutis laxa syndrome. Am. J. Hum. Genet. 78, 1075–1080 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iascone M., Sana M. E., Pezzoli L., Bianchi P., Marchetti D., Fasolini G., Sadou Y., Locatelli A., Fabiani F., Mangili G., Ferrazzi P., Extensive arterial tortuosity and severe aortic dilation in a newborn with an EFEMP2 mutation. Circulation 126, 2764–2768 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Kappanayil M., Nampoothiri S., Kannan R., Renard M., Coucke P., Malfait F., Menon S., Ravindran H. K., Kurup R., Faiyaz-Ul-Haque M., Kumar K., De Paepe A., Characterization of a distinct lethal arteriopathy syndrome in twenty-two infants associated with an identical, novel mutation in FBLN4 gene, confirms fibulin-4 as a critical determinant of human vascular elastogenesis. Orphanet J. Rare Dis. 7, 61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajeshkannan R., Kulkarni C., Kappanayil M., Nampoothiri S., Malfait F., De Paepe A., Moorthy S., Imaging findings in a distinct lethal inherited arteriopathy syndrome associated with a novel mutation in the FBLN4 gene. Eur. Radiol. 24, 1742–1748 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Renard M., Holm T., Veith R., Callewaert B. L., Adès L. C., Baspinar O., Pickart A., Dasouki M., Hoyer J., Rauch A., Trapane P., Earing M. G., Coucke P. J., Sakai L. Y., Dietz H. C., De Paepe A. M., Loeys B. L., Altered TGFβ signaling and cardiovascular manifestations in patients with autosomal recessive cutis laxa type I caused by fibulin-4 deficiency. Eur. J. Hum. Genet. 18, 895–901 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawyer S. L., Dicke F., Kirton A., Rajapkse T., Rebeyka I. M., McInnes B., Parboosingh J. S., Bernier F. P., Longer term survival of a child with autosomal recessive cutis laxa due to a mutation in FBLN4. Am. J. Med. Genet. A 161, 1148–1153 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Berk D. R., Bentley D. D., Bayliss S. J., Lind A., Urban Z., Cutis laxa: A review. J. Am. Acad. Dermatol. 66, 842.e1–842.e17 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Urban Z., Davis E. C., Cutis laxa: Intersection of elastic fiber biogenesis, TGFβ signaling, the secretory pathway and metabolism. Matrix Biol. 33, 16–22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi N., Kostka G., Garbe J. H. O., Keene D. R., Bächinger H. P., Hanisch F.-G., Markova D., Tsuda T., Timpl R., Chu M.-L., Sasaki T., A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J. Biol. Chem. 282, 11805–11816 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Papke C. L., Yanagisawa H., Fibulin-4 and fibulin-5 in elastogenesis and beyond: Insights from mouse and human studies. Matrix Biol. 37, 142–149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Timpl R., Sasaki T., Kostka G., Chu M.-L., Fibulins: A versatile family of extracellular matrix proteins. Nat. Rev. Mol. Cell Biol. 4, 479–489 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Giltay R., Timpl R., Kostka G., Sequence, recombinant expression and tissue localization of two novel extracellular matrix proteins, fibulin-3 and fibulin-4. Matrix Biol. 18, 469–480 (1999). [DOI] [PubMed] [Google Scholar]
- 16.McLaughlin P. J., Chen Q., Horiguchi M., Starcher B. C., Stanton J. B., Broekelmann T. J., Marmorstein A. D., McKay B., Mecham R., Nakamura T., Marmorstein L. Y., Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol. Cell. Biol. 26, 1700–1709 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanada K., Vermeij M., Garinis G. A., de Waard M. C., Kunen M. G. S., Myers L., Maas A., Duncker D. J., Meijers C., Dietz H. C., Kanaar R., Essers J., Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ. Res. 100, 738–746 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Horiguchi M., Inoue T., Ohbayashi T., Hirai M., Noda K., Marmorstein L. Y., Yabe D., Takagi K., Akama T. O., Kita T., Kimura T., Nakamura T., Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc. Natl. Acad. Sci. U.S.A. 106, 19029–19034 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang J., Davis E. C., Chapman S. L., Budatha M., Marmorstein L. Y., Word R. A., Yanagisawa H., Fibulin-4 deficiency results in ascending aortic aneurysms: A potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 106, 583–592 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang J., Yamashiro Y., Papke C. L., Ikeda Y., Lin Y., Patel M., Inagami T., Le V. P., Wagenseil J. E., Yanagisawa H., Angiotensin-converting enzyme–induced activation of local angiotensin signaling is required for ascending aortic aneurysms in fibulin-4–deficient mice. Sci. Transl. Med. 5, 183ra58 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moltzer E., te Riet L., Swagemakers S. M. A., van Heijningen P. M., Vermeij M., van Veghel R., Bouhuizen A. M., van Esch J. H. M., Lankhorst S., Ramnath N. W. M., de Waard M. C., Duncker D. J., van der Spek P. J., Rouwet E. V., Danser A. H. J., Essers J., Impaired vascular contractility and aortic wall degeneration in fibulin-4 deficient mice: Effect of angiotensin II type 1 (AT1) receptor blockade. PLOS ONE 6, e23411 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirai M., Ohbayashi T., Horiguchi M., Okawa K., Hagiwara A., Chien K. R., Kita T., Nakamura T., Fibulin-5/DANCE has an elastogenic organizer activity that is abrogated by proteolytic cleavage in vivo. J. Cell Biol. 176, 1061–1071 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hornstra I. K., Birge S., Starcher B., Bailey A. J., Mecham R. P., Shapiro S. D., Lysyl oxidase is required for vascular and diaphragmatic development in mice. J. Biol. Chem. 278, 14387–14393 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Maki J. M., Sormunen R., Lippo S., Kaarteenaho-Wiik R., Soininen R., Myllyharju J., Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am. J. Pathol. 167, 927–936 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Igoucheva O., Alexeev V., Halabi C. M., Adams S. M., Stoilov I., Sasaki T., Arita M., Donahue A., Mecham R. P., Birk D. E., Chu M.-L., Fibulin-4 E57K knock-in mice recapitulate cutaneous, vascular and skeletal defects of recessive cutis laxa 1B with both elastic fiber and collagen fibril abnormalities. J. Biol. Chem. 290, 21443–21459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Majesky M. W., Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 27, 1248–1258 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Faury G., Pezet M., Knutsen R. H., Boyle W. A., Heximer S. P., McLean S. E., Minkes R. K., Blumer K. J., Kovacs A., Kelly D. P., Li D. Y., Starcher B., Mecham R. P., Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J. Clin. Invest. 112, 1419–1428 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D. Y., Brooke B., Davis E. C., Mecham R. P., Sorensen L. K., Boak B. B., Eichwald E., Keating M. T., Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276–280 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Li D. Y., Faury G., Taylor D. G., Davis E. C., Boyle W. A., Mecham R. P., Stenzel P., Boak B., Keating M. T., Novel arterial pathology in mice and humans hemizygous for elastin. J. Clin. Invest. 102, 1783–1787 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura T., Lozano P. R., Ikeda Y., Iwanaga Y., Hinek A., Minamisawa S., Cheng C.-F., Kobuke K., Dalton N., Takada Y., Tashiro K., Ross J. Jr, Honjo T., Chien K. R., Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 415, 171–175 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Yanagisawa H., Davis E. C., Starcher B. C., Ouchi T., Yanagisawa M., Richardson J. A., Olson E. N., Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 415, 168–171 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Gallo E. M., Loch D. C., Habashi J. P., Calderon J. F., Chen Y., Bedja D., van Erp C., Gerber E. E., Parker S. J., Sauls K., Judge D. P., Cooke S. K., Lindsay M. E., Rouf R., Myers L., ap Rhys C. M., Kent K. C., Norris R. A., Huso D. L., Dietz H. C., Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J. Clin. Invest. 124, 448–460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holm T. M., Habashi J. P., Doyle J. J., Bedja D., Chen Y., van Erp C., Lindsay M. E., Kim D., Schoenhoff F., Cohn R. D., Loeys B. L., Thomas C. J., Patnaik S., Marugan J. J., Judge D. P., Dietz H. C., Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 332, 358–361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramnath N. W. M., Hawinkels L. J. A. C., van Heijningen P. M., te Riet L., Paauwe M., Vermeij M., Danser A. H. J., Kanaar R., ten Dijke P., Essers J., Fibulin-4 deficiency increases TGF-β signalling in aortic smooth muscle cells due to elevated TGF-β2 levels. Sci. Rep. 5, 16872 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papke C. L., Tsunezumi J., Ringuette L.-J., Nagaoka H., Terajima M., Yamashiro Y., Urquhart G., Yamauchi M., Davis E. C., Yanagisawa H., Loss of fibulin-4 disrupts collagen synthesis and maturation: Implications for pathology resulting from EFEMP2 mutations. Hum. Mol. Genet. 24, 5867–5879 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugitani H., Hirano E., Knutsen R. H., Shifren A., Wagenseil J. E., Ciliberto C., Kozel B. A., Urban Z., Davis E. C., Broekelmann T. J., Mecham R. P., Alternative splicing and tissue-specific elastin misassembly act as biological modifiers of human elastin gene frameshift mutations associated with dominant cutis laxa. J. Biol. Chem. 287, 22055–22067 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clinton G. M., Rougeot C., Derancourt J., Roger P., Defrenne A., Godyna S., Argraves W. S., Rochefort H., Estrogens increase the expression of fibulin-1, an extracellular matrix protein secreted by human ovarian cancer cells. Proc. Natl. Acad. Sci. U.S.A. 93, 316–320 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura T., Ruiz-Lozano P., Lindner V., Yabe D., Taniwaki M., Furukawa Y., Kobuke K., Tashiro K., Lu Z., Andon N. L., Schaub R., Matsumori A., Sasayama S., Chien K. R., Honjo T., DANCE, a novel secreted RGD protein expressed in developing, atherosclerotic, and balloon-injured arteries. J. Biol. Chem. 274, 22476–22483 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Pfaff M., Sasaki T., Tangemann K., Chu M.-L., Timpl R., Integrin-binding and cell-adhesion studies of fibulins reveal a particular affinity for αIIbβ3. Exp. Cell Res. 219, 87–92 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Qing J., Maher V. M., Tran H., Argraves W. S., Dunstan R. W., McCormick J. J., Suppression of anchorage-independent growth and matrigel invasion and delayed tumor formation by elevated expression of fibulin-1D in human fibrosarcoma-derived cell lines. Oncogene 15, 2159–2168 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Schiemann W. P., Blobe G. C., Kalume D. E., Pandey A., Lodish H. F., Context-specific effects of fibulin-5 (DANCE/EVEC) on cell proliferation, motility, and invasion. Fibulin-5 is induced by transforming growth factor-β and affects protein kinase cascades. J. Biol. Chem. 277, 27367–27377 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Halabi C. M., Broekelmann T. J., Knutsen R. H., Ye L., Mecham R. P., Kozel B. A., Chronic antihypertensive treatment improves pulse pressure but not large artery mechanics in a mouse model of congenital vascular stiffness. Am. J. Physiol. Heart Circ. Physiol. 309, H1008–H1016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Starcher B., A ninhydrin-based assay to quantitate the total protein content of tissue samples. Anal. Biochem. 292, 125–129 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Reddy G. K., Enwemeka C. S., A simplified method for the analysis of hydroxyproline in biological tissues. Clin. Biochem. 29, 225–229 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Gunja-Smith Z., An enzyme-linked immunosorbent assay to quantitate the elastin crosslink desmosine in tissue and urine samples. Anal. Biochem. 147, 258–264 (1985). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602532/DC1
table S1. Echocardiographic data of Fbln4E57K/E57K and littermate WT mice.
fig. S1. Fbln4E57K/E57K mice are similar in size to their littermates.
fig. S2. Fbln4E57K/E57K ascending aortae have increased medial wall thickness.
fig. S3. Fbln4E57K/E57K ascending aortae do not develop calcification.
fig. S4. Fbln4E57K/E57K mice have similar heart rates to their heterozygous and WT littermates.
fig. S5. Internal elastic lamina is unaffected in Fbln4E57K/E57K saphenous arteries.
fig. S6. Homozygous E57K mutation in Fbln4 does not alter arterial elastin or collagen content.
fig. S7. Homozygous E57K mutation in Fbln4 does not alter ECM gene expression in lungs.
fig. S8. FBLN4(E57K) protein level is reduced in P7 ascending aorta, but not in mesenteric arteries.
fig. S9. FBLN4(E57K) protein secretion is impaired compared to WT FBLN4.