

SUMMARY
Charcot-Marie-Tooth (CMT) disease is a clinically and genetically heterogeneous group of inherited polyneuropathies. Mutations in 80 genetic loci can cause forms of CMT, resulting in demyelination and axonal dysfunction. The clinical presentation, including sensory deficits, distal muscle weakness, and atrophy, can vary greatly in severity and progression. Here, we used mouse models of CMT to demonstrate genetic interactions that result in a more severe neuropathy phenotype. The cell adhesion molecule Nrcam and the Na+ channel Scn8a (NaV1.6) are important components of nodes. Homozygous Nrcam and heterozygous Scn8a mutations synergized with both an Sh3tc2 mutation, modeling recessive demyelinating Charcot-Marie-Tooth type 4C, and mutations in Gars, modeling dominant axonal Charcot-Marie-Tooth type 2D. We conclude that genetic variants perturbing the structure and function of nodes interact with mutations affecting the cable properties of axons by thinning myelin or reducing axon diameter. Therefore, genes integral to peripheral nodes are candidate modifiers of peripheral neuropathy.
Graphical abstract
INTRODUCTION
Charcot-Marie-Tooth (CMT) disease comprises a heterogeneous collection of heritable polyneuropathies. Its hallmarks include muscle weakness, atrophy, fatigue, and loss of sensory function in the distal extremities, resulting from demyelination and/or axon degeneration in the peripheral nervous system (Saporta and Shy, 2013; Dyck and Thomas, 2005).
CMT is considered a Mendelian disorder, with over 80 associated loci in humans (Timmerman et al., 2014). However, the severity of neuropathy can vary even within alleles (Pareyson and Marchesi, 2009). Therefore, it is likely that many pathways can influence the severity of peripheral neuropathy, and one presumed basis for the variability is the presence of genetic modifiers (Gonzaga-Jauregui et al., 2015).
Here, we have used mouse models of two forms of CMT to investigate genetic modifiers of neuropathy. First, we identified a spontaneous mutation in Sh3tc2 that creates a model of recessive CMT-4C, a demyelinating neuropathy (Arnaud et al., 2009; Senderek et al., 2003). We also use two different point mutations in glycyl tRNA synthetase (Gars) (Seburn et al., 2006; Achilli et al., 2009) that are models of CMT2D, a dominant axonal neuropathy (Antonellis et al., 2003). In the present study, we demonstrate that the phenotype of these CMT mouse models becomes more severe in the presence of either of two other mutations that cause “subclinical” changes at peripheral nodes.
The Nav1.6 (Scn8a) sodium channel is the primary voltage-gated sodium channel at peripheral nodes of Ranvier and axon initial segments (Caldwell et al., 2000). The immunoglobulin (Ig) superfamily-member cell adhesion molecule Nrcam (Neuroglial-related cell adhesion molecule) is part of a transmembrane complex at nodes. During development, NRCAM and gliomedin in the Schwann cell establish a heminode in the axon at the boundary of the maturing Schwann cell, which then fuses into a mature node (Eshed-Eisenbach and Peles, 2013; Rasband and Peles, 2015; Feinberg et al., 2010). The loss of Nrcam results in delayed formation of nodes, and occasional “split nodes” in adult peripheral nerves, but no obvious signs of neuromuscular dysfunction (Amor et al., 2014; Custer et al., 2003). NRCAM’s role is somewhat redundant, as Nrcam and Gliomedin double-mutant mice fail to maintain nodes (Amor et al., 2014). Similarly, mutations in the paranodal Contactin-associated protein-like1 (CASPR) are more severe in combination with Nrcam mutations (Feinberg et al., 2010). In the axon, NRCAM binds to ankyrins intracellularly, and ankyrins bind sodium channels (Davis and Bennett, 1994; Jenkins and Bennett, 2001). Similarly, Scn8a homozygous mice have extensive neurological problems, but heterozygotes are behaviorally and histologically normal (Burgess et al., 1995). However, we found that introducing either Nrcam homozygous or Scn8a heterozygous mutations into our CMT models results in a more severe neuropathy.
Our results demonstrate that mutations that reduce myelin thickness or cause axon atrophy act synergistically with mutations that alter nodes and/or reduce sodium currents in peripheral axons. These results have implications for CMT genetic diagnosis and prognosis and suggest possible treatments.
RESULTS
The Identification of Spontaneous Nrcam and Sh3tc2 Mutations
The synergy of demyelinating peripheral neuropathy and impairments in peripheral node organization was made evident in a spontaneous mouse mutation identified and characterized at The Jackson Laboratory. This strain—nm4302—arose in the background of a knockout of the cannabinoid receptor 2. Some mice displayed impaired hindlimb function soon after weaning, which progressed to near complete hindlimb paralysis and death by 5 months of age. This was inconsistent with the Cnr2 knockout phenotype (Tschöp et al., 2009) and was not linked to the knockout allele, indicating a spontaneous mutation had arisen. This mutation was recessive, since unaffected parents produced litters with a subset of affected offspring. Hindlimb muscles in affected mice showed severe atrophy. Angular fibers and foci of atrophy without signs of degeneration/regeneration, fibrosis, or fatty infiltration were consistent with neurogenic etiology (Figures 1A and 1B).
Figure 1. Nm4302 Mutations in Sh3tc2 and Nrcam.
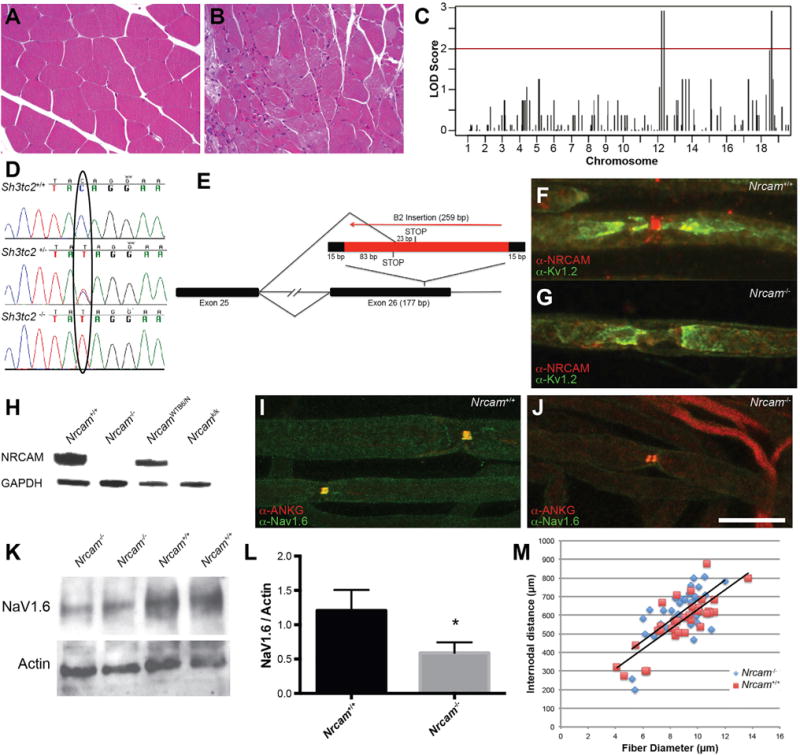
(A and B) Muscle histology from the gastrocnemius of control (A) and an affected Nm4302 mouse (B).
(C) Genetic mapping associations on both Chr12 and Chr18. The logarithm of the odds score (LOD, y axis) of SNPs is plotted versus their chromosomal location (x axis).
(D) Chromatograms of sequencing genomic DNA of a wild-type (top), Sh3tc2 heterozygote (middle), and Sh3tc2 homozygote (bottom) show the C to T conversion.
(E) The B2 element insertion in exon 26 of Nrcam results in a 259-bp insertion with a duplication of the flanking 15 bp of the exon. Splicing from exon 25 to 26 results in a premature stop codon in the B2 element sequence, and splicing into the B2 element itself results in a premature stop codon 23 bp downstream of the new splice junction.
(F and G) Immunolabeling of NRCAM (red) and KV1.2 (green) at nodes of Ranvier in teased sciatic nerve axons from wild-type mice (F) or Nrcam mutant mice (G) reveals an absence of signal in the mutant, confirmed in teased nerves from three mice per genotype.
(H) Western blotting protein from brain extracts revealed an absence of signal in Nrcam mutant mice (Nrcam−/−). Mice from the KOMP2 program carrying an Nrcam mutation (Nrcamk/k) and a strain-matched littermate control (NrcamWTB6/N) were used as controls. Wild-type littermates from each Nrcam mutant strain contain protein at the predicted size, and anti-GAPDH was used as a loading control (below). Three independent blots were probed, and three mice per genotype were examined.
(I) Wild-type nodes of Ranvier have a single, colocalized site of NaV1.6 and ankyrinG immunoreactivity.
(J) In Nrcam mutant mice, nodes “split” with two sites of NaV1.6 and ankyrinG labeling. Four mice of each genotype were examined.
(K) Western blot of sciatic nerve lysates probed with antibodies against NaV1.6 revealed reduced signal relative to beta-actin loading control. Proteins migrated at their anticipated molecular weights of ~225 and ~42 kDa for NaV1.6 and beta-actin, respectively.
(L) Quantification of (K) revealed a 51% decrease in NaV1.6 intensity (three Nrcam−/− and three C57BL/6J control analyzed, p = 0.03), mean ± SD.
(M) The relationship between internodal distance and fiber diameter was not changed in Nrcam−/− mice compared to C57BL/6 controls (n = 3 mice per genotype, 37 Nrcam−/− and 34 Nrcam+/+ axons measured).
Scale bar in (J), 7 μm for (F) and (G) and 14 μm for (I) and (J).
Using genetic mapping and exome sequencing, we identified two mutations in these mice. Genetic mapping revealed regions on chromosomes 12 (Chr12) and Chr18 as associated with the paralysis phenotype (Figure 1C and Experimental Procedures). Affected mice were recovered not at 1:4, but at closer to 1:16 in the mapping cross, consistent to two recessive loci, and mildly affected mice were also observed, presumably carrying only one of the mutations. We also performed exome sequencing on an affected mouse, which revealed a single base pair C to T conversion in exon 3 of Sh3tc2 on Chr18, creating a premature stop codon at amino acid 71 (G71X, Figure 1D). Mutations in Sh3tc2 cause a recessive demyelinating neuropathy (CMT 4C), and knockout mice have reduced myelin thickness and progressive retraction of Schwann cells from nodes of Ranvier (Arnaud et al., 2009; Senderek et al., 2003). The Sh3tc2 gene is within the mapping interval on Chr18, making Sh3tc2 a strong candidate. On Chr12, we identified a B2 element insertion in exon 26 of Nrcam (see Experimental Procedures). The Nrcam gene is within our mapping interval and is a candidate gene for its role in Na+ channel localization at nodes of Ranvier and axon initial segments, and for its previously reported genetic interactions with Lpin1, causing transient hindlimb paralysis (Douglas et al., 2009). The B2 element disrupts coding sequence and splicing of Nrcam, resulting in premature stop codons (Figure 1E).
NRCAM is 1,256 amino acids, with a single transmembrane domain at amino acids 1120–1142. Both of the B2-element-associated stop codons terminate translation before the transmembrane domain, resembling a previously reported allele, Q1033X, which is a protein null (Douglas et al., 2009). Consistent with this, anti-NRCAM immunolabeling with an antibody raised against the entire extracellular domain was eliminated at nodes of Ranvier in the sciatic nerve in Nrcam mutant samples (Figures 1F and 1G), and no NRCAM protein was detected by western blotting from brain homogenates (Figure 1H).
Demyelinating Sh3tc2 Mutations Strongly Synergize with Nrcam Mutations
The phenotype of the Sh3tc2 mutation on its own is consistent with previous reports (Arnaud et al., 2009), and the mice develop a tremor and demyelinating neuropathy. The Nrcam mutation on its own is also consistent with previous reports, with occasional split nodes, having two distinct bands of NaV1.6 and AnkyrinG (Figures 1I, 1J, and S1) (Amor et al., 2014). This occurred at 6.6% ± 2.8% of the Nrcam−/− nodes, versus zero at control nodes (n = 4 mice of each genotype, average of 99 nodes per nerve scored). Levels of NaV1.6 in the sciatic nerve were reduced on western blots (Figures 1K and 1L). Normalizing to beta-actin as a loading control, NaV1.6 levels in Nrcam−/− mice were reduced to 49% of wild-type levels (n = 3 mutant and three C57BL/6J control mice, 6 weeks of age). Since Na+ channels may localize to internodal axon membranes (Shrager, 1989), we performed semiquantitative immunofluorescence with NaV1.6 and neurofilament light chain as an internal control but did not detect a decrease in NaV1.6 labeling at nodes of Nrcam−/− nerves (Figure S1). Localization of paranodal proteins such as CASPR were unaffected (Feinberg et al., 2010; Amor et al., 2014; data not shown).
Despite the changes in NaV1.6 localization and abundance, the Nrcam−/− mice do not have an overt neuromuscular phenotype or obvious deficits in motor behavior. Other axonal parameters, such as the internodal distance were unchanged (Figure 1M). The average internodal distance divided by fiber diameter (axon plus myelin) was 70.3 ± 12.4 for Nrcam−/− axons and 64.9 ± 10.0 for C57BL/6J control mice (n = 3 mice per genotype, a total of 37 Nrcam−/− and 34 Nrcam+/+ internodal distances measured).
The mild neuromuscular dysfunction of the Nrcam or Sh3tc2 mutant mice is further substantiated by phenotyping performed by the knockout mouse program, with Nrcam−/− mice having poor performance on a rotarod test and Sh3tc2−/− mice having reduced grip strength, but otherwise no significant (p < 0.05) neuromuscular phenotypes (KOMP2; https://www.mousephenotype.org/data/search). However, in combination, these two mutations cause paralysis in the spontaneous double-mutant strain nm4302.
The basis for this progressive paralysis was explored by examining peripheral nerve anatomy and function in control, Nrcam−/−, Sh3tc2−/−, and double-mutant mice. Cross-sections of the motor branch of the femoral nerve were examined (Figures 2A–2D). No changes in axon diameter, myelin thickness, or axon number were observed for Nrcam mutant samples (Figures 2B and 2E–2G). As expected, Sh3tc2−/− samples had reduced myelin thickness (p < 0.0001) and supernumerary Schwann cell processes, but no decrease in myelinated axon number, and no change in axon diameter (Figures 2C and 2E–2G). Axons in double-mutant mice essentially mirrored the Sh3tc2−/− reduction myelin thickness (p < 0.0001 versus control and p = 0.27 versus Sh3tc2−/−), but no change in axon number or size (Figures 2D and 2E–2G). Thus, the Nrcam mutation does not exacerbate the demyelinating effects of Sh3tc3−/−. In functional assays, nerve conduction velocity (NCV) was slightly decreased in Nrcam−/− mice (p = 0.04). A previous study of Nrcam mutant mice reported normal NCV, but at P2, an early stage of myelination, whereas we studied adult mice (Custer et al., 2003). NCV was further decreased in Sh3tc2−/− mice (p < 0.001) and even more severely reduced in double-mutant animals (p < 0.001) (Figure 2H). Myelin packing was examined by transmission electron microscopy (Figure S2). Myelin wraps on Nrcam−/− axons were slightly closer (12.9 ± 1.4 nm between wraps) than control and Sh3tc2−/− axons (14.7 ± 1.6 and 14.1 ± 0.7, respectively, p < 0.05), but this was not additionally reduced in the double-mutant mice (12.3 ± 2.0). The reduced overall myelin thickness in Sh3tc2−/− and double-mutant mice was therefore due primarily to a reduced number of myelin wraps, not changes in myelin packing. The relationship between axon diameter and myelin thickness (the “g ratio”) is comparably reduced compared to control in both Sh3tc2−/− and double-mutant mice, but unchanged in Nrcam mutant nerves (Figure S2).
Figure 2. Peripheral Nerve Anatomy and Function in Sh3tc2 and Nrcam Mutations.
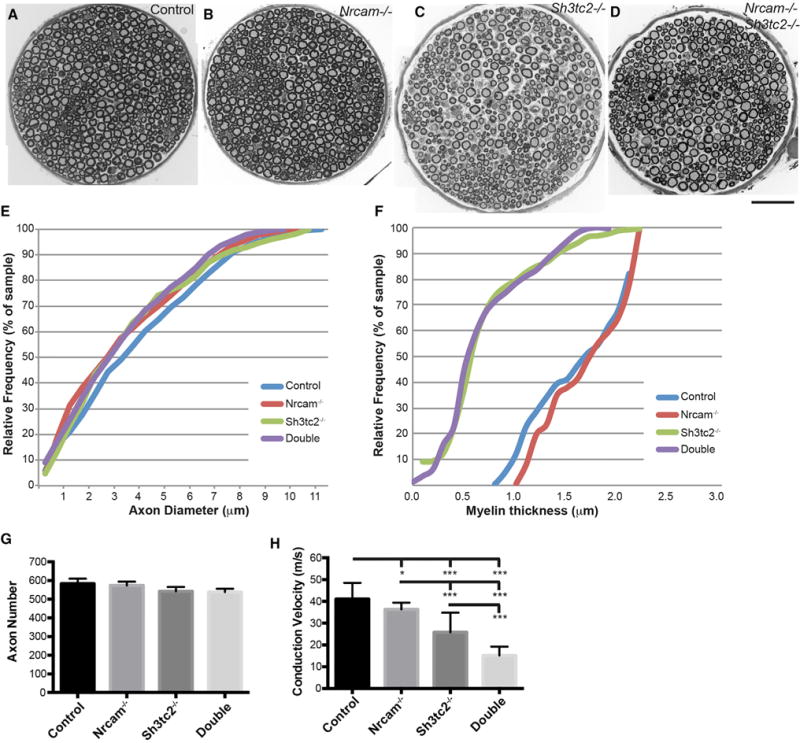
(A–D) Montages of cross-sections of the motor branch of the femoral nerve from wild-type (A), Nrcam−/− (B), Sh3tc2−/− (C), and double-mutant (D) mice are shown.
(E) A cumulative histogram of axon diameters indicates no change in the single or double mutants.
(F) Cumulative histograms of myelin thickness reveal no change in Nrcam−/− nerves, a significant decrease in Sh3tc2−/− nerves, but no additional decrease in double-mutant nerves.
(G) Axon number was not changed in any genotype.
(H) NCV was reduced in Nrcam−/− mice, was reduced to a greater degree in Sh3tc2−/− mice, and was further reduced in double-mutant mice.
Data from eight mice per genotype are presented in (E)–(G); (H) includes ten control, 14 Nrcam−/−, ten Sh3tc2−/−, and 14 double-mutant mice. Values in (G) and (H) are mean ± SD. Mice were from 73 to 145 days old (average 104 days) in (E)–(G) and 57–125 days old (average 103 days) in (H). *p < 0.05, **p < 0.01. Scale bar in (D), 50 μm for (A)–(D).
Nrcam mutant nerves have no changes in myelination, axon diameter, or internodal distance, so their reduced NCV may result from Na+ channel dysfunction at nodes (following the equation: ∂V/∂T = i/C). However, NRCAM also interacts with paranodal proteins such as CASPR, and Sh3tc2 mutations also cause the Schwann cells to retract from the node, so changes in other nodal properties and channel types could also contribute to the phenotype of both mutants.
Changes in peripheral nerves typically caused by CMT such as axon loss do not explain the muscle atrophy and paralysis in double-mutant mice. Examining neuromuscular junctions (NMJs) did not show evidence of denervation or die-back neuropathy (Seburn et al., 2006; Spaulding et al., 2016), but NMJs did show a phenotype. The NMJ morphology of the plantaris muscle in Nrcam−/− and Sh3tc2−/− single-mutant animals was normal (Figures 3A–3C). However, in double-mutant mice by 3.5 months of age (~100 days), NMJs showed extensive fragmentation, sprouting of nerve terminals, and extrasynaptic acetylcholine receptor expression (Figures 3D and 3E). These changes were not evident at 2.5 months of age (Figure S3).
Figure 3. Neuromuscular Junction Sprouting and Fragmentation in Nrcam;Sh3tc2 Double-Mutant Mice.
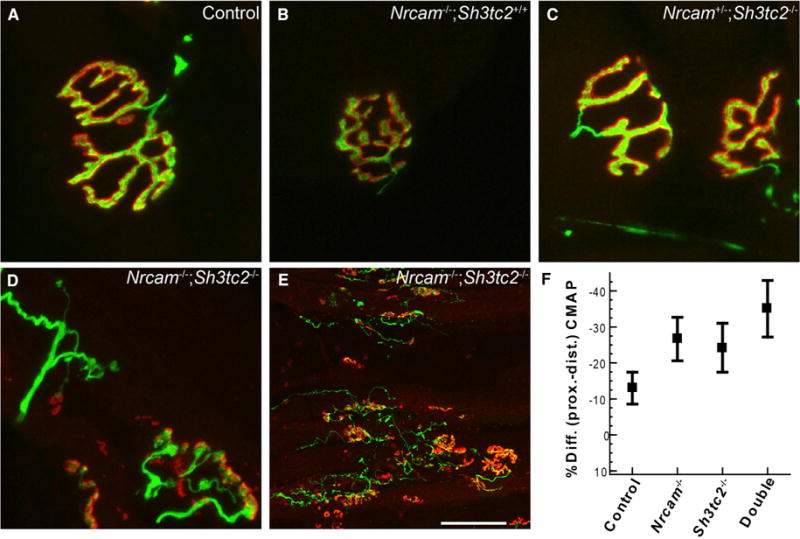
(A) Normally, the motor nerve terminal (green) completely overlays the postsynaptic acetylcholine receptors (red).
(B and C) Mice with mutations in Nrcam (B) or Sh3tc2 (C) did not show abnormal NMJ morphology or nerve occupancy.
(D and E) Double-mutant mice showed profound NMJ defects after 3.5 months of age, with sprouting nerve terminals, sites of partial and complete denervation, aneural puncta of AChRs, and fragmentation of NMJs. Few if any normal NMJs persist, as shown at a lower magnification (E). Sprouting and NMJ dysmorphology (D and E) were observed in all double-mutant mice >3 months of age (n = 5) and were never observed in control or single-mutant mice.
(F) The iCMAP evoked from distal stimulation was larger than when evoked by proximal stimulation in double-mutant mice (mean ± SD). The mice in (F) are the same as those analyzed in Figure 2H.
Scale bar in (E) represents 14 μm in (A)–(D) and 84 μm in (E).
Nerve terminal sprouting and extrasynaptic acetylcholine receptor (AChR) expression are also seen when peripheral nerves are silenced by chronic application of tetrodotoxin to the axon or botulinum toxin to the nerve terminal (Duchen and Strich, 1968; Pestronk and Drachman, 1978; Brown and Ironton, 1977). Consistent with motor neuron action potentials not reaching presynaptic terminals in the double-mutant mice, we saw evidence of conduction block (Mallik and Weir, 2005) in our NCV studies (Figures 2H and 3F). Normally, the integrated compound muscle action potential (iCMAP) amplitudes of the distally (ankle) and proximally (hip) evoked electromyogram (EMG) are equal, but with conduction block, the proximally evoked EMG is reduced. Clinically, decrements of at least 20% are classified as conduction block (Mallik and Weir, 2005). Conduction block was present in the double-mutant mice, with the proximal iCMAP reduced compared to distal (−35%) (Figure 3F). Interestingly, Nrcam and Sh3tc2 mutant mice had intermediate effects (~–25%), and conduction block was not present in control nerves (−12%). Statistically, the variability inherent in CMAP amplitude with transcutaneous stimulation resulted in no significant changes by ANOVA (p = 0.07), but double-mutant mice had significant conduction block when compared to control in pairwise comparisons (t test p = 0.01 double-mutant versus control). The duration of the proximal iCMAPs was increased by less than 10% compared to the distal in all genotypes. Thus, temporal dispersion is not pronounced. Taken together, the immunofluorescence and conduction block data are consistent with failures in action potential propagation with distance in the double mutants.
To further investigate this, one double-mutant mouse was examined for synaptic transmission at the NMJ by two-electrode voltage clamp. In this muscle, five of the ten fibers recorded produced normal postsynaptic responses following nerve stimulation (excitatory postsynaptic current [EPC]) (Figure S4). However, no evoked response was produced by nerve stimulation in the other five fibers despite the presence of spontaneous mini excitatory postsynaptic currents (mEPCs). Recently, we examined 184 NMJs in wild-type mice, only four (2%) had mEPCs without a corresponding EPC, and within single experiments where six to 11 NMJs were examined, an mEPC without an EPC never occurred more than once (Spaulding et al., 2016). Thus, the finding that 50% of double-mutant terminals showed spontaneous release without an evoked response is consistent with neuromuscular transmission being relatively normal at active synapses (Figure S4) but that action potentials fail to reach some nerve terminals.
These effects are completely recessive. The progressive paralysis phenotype was observed only in double-homozygous mice. Mice heterozygous for either or both the Nrcam and Sh3tc2 mutations were normal (Figure S5). Furthermore, if mice were homozygous for one mutation, being heterozygous for the second did not make the phenotype worse (Figure S5). Therefore, many of the control values reported are pooled data from heterozygous and homozygous wild-type mice.
Together, these results led us to hypothesize that mutations that compromise the axonal length constant will synergize with mutations that compromise depolarization, and specifically Na+ currents at nodes. However, since Nrcam mutations potentially affect more than Na+ currents, and since Sh3tc2 mutations also impact nodes as well as myelination, we performed additional experiments compromising the length constant by reducing axon diameter, and directly compromising NaV1.6.
Gars Dominant Axonal Neuropathy Is Exacerbated by Loss of Nrcam
We used a dominant mutation in glycyl tRNA synthetase (Gars) to specifically decrease axon diameter (Achilli et al., 2009; Motley et al., 2011; Spaulding et al., 2016). The GarsC201R/+ mice have smaller diameter axons than wild-type mice, but no axon loss. Thus, in GarsC201R/+ mice, the axonal length constant is compromised, and we would predict that diminished function at nodes would worsen the phenotype.
We bred GarsC201R/+ mice into an Nrcam−/− background and repeated our analysis of nerve and muscle histology and function. Cross-sections of the motor branch of the femoral nerve confirmed the reduced axon size in the GarsC201R/+ mice (p < 0.0001), which was not exacerbated by the additional loss of Nrcam (p < 0.0001 versus control and p = 0.12 versus GarsC201R/+) (Figures 4A–4D). Axon number was unchanged in any genotype (Figure 4E). Nrcam−/− muscles were of normal size, but GarsC201R/+ muscles were smaller, and muscles from Nrcam−/−; GarsC201R/+ mice were smaller still (p < 0.001), as assessed by the ratio of the muscle weights to total body weight (Figure 4F). NCV was further reduced in double-mutant mice compared to either single mutant (p < 0.001) (Figure 4G). In examining NMJs, no partial innervation or denervation was observed in Nrcam−/− mice alone (p = 0.56), but significant innervation defects were seen in GarsC201R/+ (p < 0.05) and were worsened by the loss of Nrcam (p < 0.01 control versus double and p < 0.01 GarsC210R/+ versus double) (Figure 4H). The double-mutant mice were also overtly more affected based on size, tremor, and poor motor performance. However, the mice did not progress to paralysis and premature death. These studies support the hypothesis that the introduction of defects at nodes caused by the Nrcam mutation synergize with the GarsC201R/+ mutation to cause a more severe phenotype.
Figure 4. Axonal Neuropathy Is Exacerbated by Loss of Nrcam.
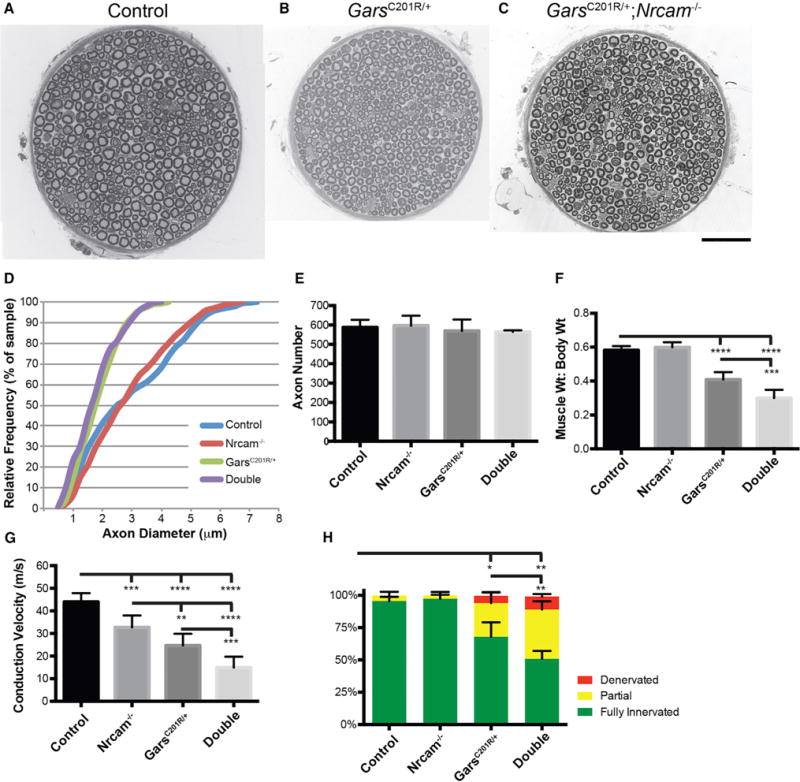
(A–C) Cross-sections of the motor branch of the femoral nerve were examined for 3-month-old wild-type (A, shown as a montage), GarsC201R/+ (B), and GarsC201R/+; Nrcam−/− (C) mice.
(D) Axon diameters were reduced in GarsC201R/+ compared to wild-type and Nrcam−/− mice, with no further reduction in double mutants.
(E) Axon number was not reduced in any genotype.
(F) Muscle size was assessed by the ratio of muscle weight (g) to total body weight (g) (MW/BW*100) for the triceps surae. No change was observed in Nrcam−/− mice, but GarsC201R/+ mice showed reduced MW:BW. Double-mutant mice showed the largest reduction in MW:BW ratio.
(G) NCV was more severely reduced in double-mutant mice than in either single mutant.
(H) The GarsC201R/+ mice have reduced NMJ occupancy, with more partial innervation of NMJs, and some instances of complete denervation. NMJ occupancy was more severely reduced in double-mutant mice, whereas Nrcam−/− mice had no change in NMJ occupancy compared to controls.
(D) and (E) are based on eight mice per genotype, (F) on seven control, seven Nrcam−/−, nine GarsC201R/+, and eight double-mutant mice, (G) on eight control, seven Nrcam−/−, nine GarsC201R/+, and nine double-mutant mice, and (H) on eight control, six Nrcam−/−, seven GarsC201R/+, and four double-mutant mice. Values for (E)–(H) are mean ± SD. Animals were 3 months of age, and an approximately equal mix of male and females were used. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar in (C), 50 μm for (A)–(C).
Heterozygosity for Scn8a, Encoding NaV1.6, Also Exacerbates Gars-Associated Axonal Neuropathy
Next, we directly compromised NaV1.6 to assess their possible contribution. Since homozygous mutations in Scn8a cause a variety of neurological phenotypes in mice and humans (O’Brien and Meisler, 2013; Burgess et al., 1995), we used mice heterozygous for a spontaneous point mutation in Scn8a, R914S, which produces a pronounced med-like phenotype as a homozygote (Duchen, 1970; https://www.jax.org/strain/023609) but no overt symptoms as a heterozygote. GarsC201R/+ mice were bred to Scn8a+/− mice, and offspring of all subsequent genotypes (wild-type, GarsC201R/+, Scn8a+/−, double heterozygotes) were analyzed. The Scn8a+/− mice did not show a reduction in axon size or axon diameter, and the reduced axon size observed in GarsC201R/+ was not exacerbated by heterozygosity for Scn8a (p = 0.11 GarsC201R/+ versus double heterozygotes) (Figures 5A–5F). NCV was reduced in the Scn8a+/− mice (p = 0.05), and the NCV in Gars;Scn8a double-heterozygous mice was lower than in either single mutant alone (p = 0.05 GarsC201R/+ versus double heterozygotes) (Figure 5G). Although additional muscle atrophy was not seen in the double-heterozygous mice (p = 0.63, GarsC201R/+ versus double heterozygotes), NMJ innervation status was worse than GarsC201R/+ alone (p < 0.01), whereas Scn8a+/− mice did not have NMJ defects (Figures 5H and 5I). Thus, Scn8a heterozygosity produced no phenotype on its own beyond a modest reduction in NCV but made the GarsC201R/+ phenotype more severe both for NCV and for NMJ innervation. The GarsC201R/+;Scn8a+/− double-heterozygous mice were also overtly more severe. The fact the Scn8a heterozygous mutation recapitulates much of what we observed for the Nrcam mutation supports our conclusion that the Nrcam effects are largely through NaV1.6 mislocalization or decreased NaV1.6 channel function.
Figure 5. Axonal Neuropathy Is Exacerbated by Partial Impairment of NaV1.6/Scn8a+/−.
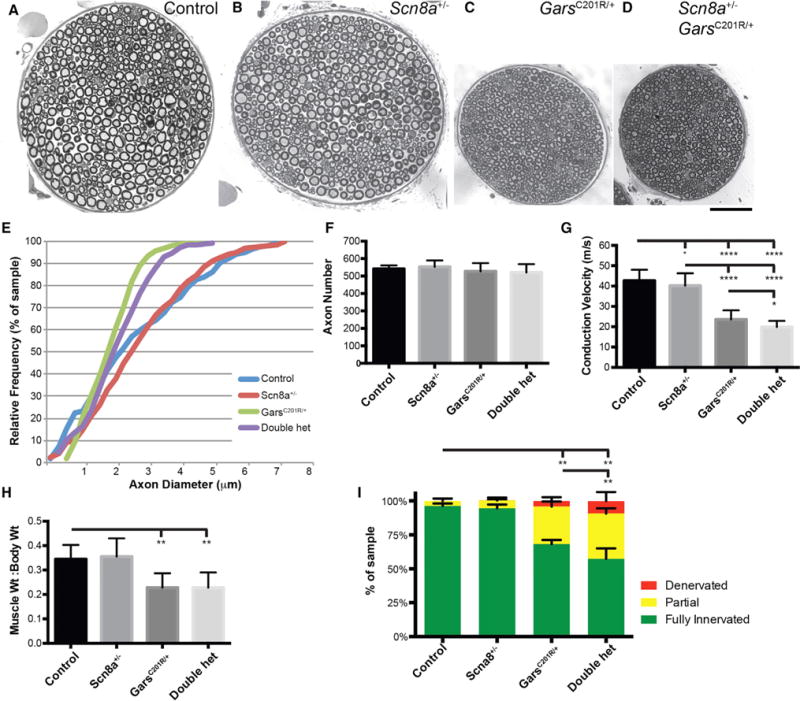
(A–D) Cross-sections from the motor branch of the femoral nerve for control (A), Scn8a+/− (B), GarsC201R/+ (C), and double-heterozygous mice (D); note (A) and (B) are montages to capture the entire nerve.
(E) Axon diameters were not changed in Scn8a+/−, and double-heterozygous mice did not show a greater reduction in axon diameters than GarsC201R/+ alone.
(F) Axon number in the motor branch of the femoral nerve was unchanged in any genotype.
(G) NCV was mildly reduced in Scn8a+/− mice, and double heterozygotes had conduction velocities reduced below either single mutant.
(H) No muscle atrophy was observed in Scn8a+/− mice, and no additional muscle atrophy beyond the effects of GarsC201R/+ was observed in double heterozygotes.
(I) Scn8a+/− mice did not show changes in innervation at the NMJ, but double-heterozygous mice had more partial innervation and denervation at NMJs than GarsC201R/+ mice.
Values in (F)–(I) are mean ± SD. Analyses were performed on eight mice per genotype at 3 months of age with an approximately equal mix of males and females.
*p < 0.05, **p < 0.01. Scale bar in (D), 50 μm for (A)–(D).
The GarsC201R/+;Scn8a+/− double-heterozygous mice also did not progress to paralysis and death, consistent with the slow progression of the GarsC201R/+ phenotype. However, we also mated Scn8a+/− mice to mice with a more severe allele of Gars, P278KY/+. These mice develop a similar, but more severe, motor and sensory neuropathy, and also lose 20% to 30% of their peripheral axons by a few months of age (Seburn et al., 2006; Motley et al., 2011). We saw a similar increase in the severity of the double-heterozygous phenotype over GarsP278KY/+ alone by 2.5–3.5 months of age (Figure 6). The GarsP278KY/+ mice did not have more severe axon atrophy or axon loss when heterozygous for Scn8a (Figures 6A and 6B) but did have more severe reductions in NCV (p < 0.01), muscle atrophy (p < 0.01), and NMJ occupancy (p < 0.0001) (Figures 6C and 6E). Eventually, these mice did become severely impaired in motor ability by 6 months of age and were not maintained longer.
Figure 6. Scn8a Heterozygosity Enhances a More Severe Axonal Neuropathy Model, GarsP278KY/+.
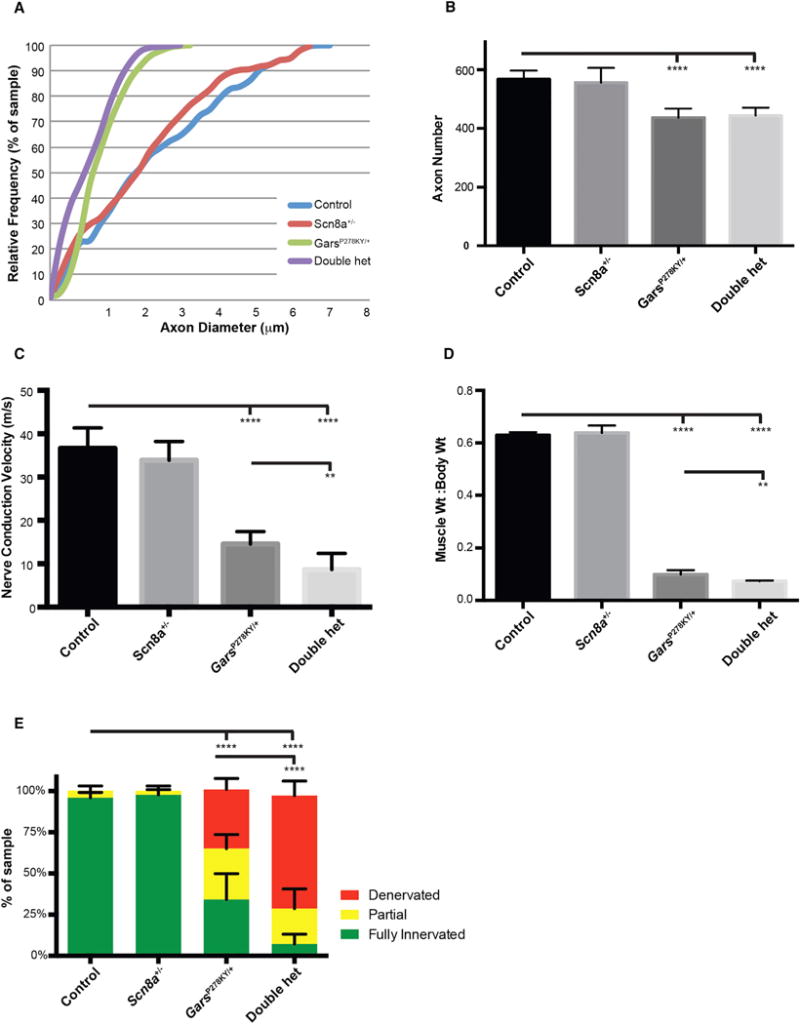
(A) There was no additional reduction in axon diameter in the motor branch of the femoral nerve over GarsP278KY/+ in mice also heterozygous for Scn8a.
(B) The GarsP278KY/+ mice have fewer myelinated axons, but this reduction was not exacerbated by Scn8a heterozygosity.
(C) NCV was further reduced in double heterozygotes, beyond the effects of GarsP278KY/+ alone.
(D) Additional muscle atrophy was present in double-heterozygous mice.
(E) Additional defects were present at NMJs, with increased denervation and partial innervation, and virtually no fully innervated junctions.
Analyses were performed on eight mice of each genotype between 2.5 and 3.5 months of age, except (C), in which eight control and nine Scn8a+/− mice were used, and results from one GarsP278KY/+ and two double-heterozygous mice were not included because a reliable EMG signal and conduction velocity could not be ascertained. Values in (B)–(E) are mean ± SD. An approximately equal mix of male and female mice were used. *p < 0.05, **p < 0.01 ***p < 0.001, ****p < 0.0001.
DISCUSSION
Our results indicate that mutations that subtly compromise nodes of Ranvier and sodium channel function in peripheral axons can be relatively innocuous on their own but synergize with mutations that cause thinner myelin or smaller diameter axons in the peripheral nervous system to cause a much more severe phenotype. Homozygous mutations in Nrcam or heterozygous mutations in Scn8a result in a minor reduction in NCV and occasional mis-localization and reduced numbers of sodium channels in peripheral nerves (Nrcam homozygotes). These mutations do not cause overt neuromuscular phenotypes. Furthermore, Nrcam−/− or Scn8a+/− mutations do not result in muscle atrophy or histological changes in nerve, muscle, or neuromuscular junctions. However, in combination with either demyelinating (Sh3tc2 homozygous) or axonal (Gars heterozygous) models of CMT, the Nrcam homozygous and Scn8a heterozygous mutations exacerbate the phenotype. With Gars mutations, this results in an additional reduction in NCV, additional muscle atrophy (in GarsP278KY/+), and additional denervation at the NMJ. With Sh3tc2, the effect is particularly severe, resulting in progressive paralysis and muscle wasting, leading to death by 5 months of age.
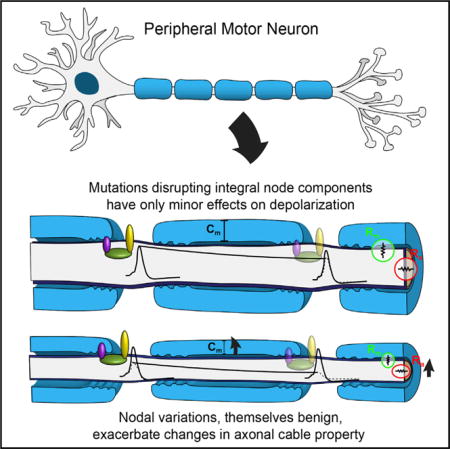
These results are consistent with peripheral nerve biophysics. Saltatory conduction results from depolarizing action potentials opening voltage-gated sodium channels localized to nodes. This local depolarization is propagated very rapidly from node to node by the passive “cable properties” of the axon. The depolarization decays over distance, but as long as it is supra-threshold and opens sodium channels at the next node, saltatory conduction continues. The “length constant” of the axon describes the passive decay in potential with distance and depends on the resistance of the membrane (Rm), the axial resistance along the axon (Ra), and the capacitance of the membrane (Cm), largely determined by myelin thickness (Figure 7). The peripheral nerve normally has a “safety factor,” such that the length constant is approximately five times longer than necessary to sustain saltatory conduction (Li, 2015). Thus, as predicted, our Nrcam homozygous or Scn8a heterozygous mutations that only slightly compromise sodium currents produce a very mild phenotype.
Figure 7. Compromised Axonal Length Constants Synergize with Compromised Depolarization at Nodes.
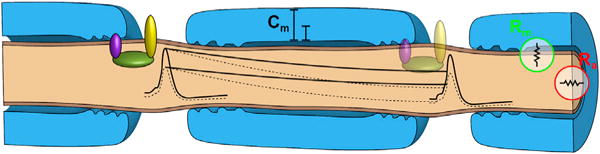
Changes in either the length constant or the amplitude of depolarization (dotted lines) are tolerated, but, when both are changed, depolarization may be subthreshold and salatory conduction may fail.
The modest reduction in NCV observed in both Nrcam homozygous and Scn8a heterozygous nerves is also consistent with a reduction in sodium currents at the nodes. The rapid passive propagation of depolarization has to overcome the capacitance current. If Cm is increased, or if current is decreased, the rate of change in potential will decrease. The reduced sodium influx may also slow the time to reach threshold depolarization and initiate an action potential.
To explore the feasibility of our hypothesized interaction, we used a simple mathematical model of myelinated axons (Neurons in Action 2, http://neuronsinaction.com/home/main), with default settings for Cm and Na+ current density based on Hodgkin and Huxley parameters for a standard myelinated axon. We used 8 μm for a large diameter axon (see Figures 2E, 4D, and 5E, as well as Seburn et al., 2006; Motley et al., 2011) and obtained a NCV of 40 m/s, closely matching our experimental results. The 17% decrease in NCV in the Nrcam−/− mice requires only a 33% decrease in Na+ current density, less than our estimated loss measured by western blotting (Figures 1K and 1L), although we did not detect a sodium channel decrease specifically at nodes by immunofluorescence. The 60% reduction in myelin thickness in Sh3tc2−/− axons produces an NCV of 28.6 m/s in this model, closely matching our experimental value. The combined effect of the Sh3tc2 and Nrcam mutations using the model is an NCV of 22 m/s, which underestimates the effects we see in vivo. A number of additional factors not considered by this model may also be involved. First, the axon initial segment may also be affected by the loss of NRCAM (Xu and Shrager, 2005). Our distal sciatic nerve stimulation circumvents this, but for normal physiological function, it is clearly a consideration. Second, in addition to thin myelin, the retraction of the Schwann cells from nodes in Sh3tc2 mutant mice may also be increasing K+ conductances and shunt current (Arnaud et al., 2009). Nrcam mutant axons do not have disrupted paranodes (Custer et al., 2003; Amor et al., 2014; data not shown), but loss of Nrcam interacts with loss of paranodal CASPR to create a more severe phenotype (Feinberg et al., 2010). Therefore, we cannot rule out that Nrcam;Sh3tc2 double-mutant mice have even more severely disrupted paranodes. However, whether the effect is decreased Na+ currents or increased K+ currents, our hypothesis and the effect on saltatory conduction are fundamentally unchanged. Third, peripheral axons typically conduct action potential trains, and propagation failures may become more widespread at higher frequency. Finally, demyelination or aberrant channel function may alter the resting potential of the axon (Kuwabara and Misawa, 2004).
Nonetheless, the schematic in Figure 7 illustrates the point that a change in the length constant that is still within the safety factor, and a change in the amplitude of depolarization that is still within the safety factor, could additively result in subthreshold depolarization and action potential failure, as seen in double-mutant mice. This reflects the functional interdependence of these processes and does not require physical interaction between the proteins.
These results can also be considered in the context of CMT genetics. Since virtually all CMTs compromise the axonal length constant, through demyelination or axon atrophy, we would anticipate any mutations that compromise nodes to be synergistic. Both Gars/CMT2D and Sh3tc2/CMT4C are reported to be influenced by genetic modifiers (Sivakumar et al., 2005; Colomer et al., 2006), but identifying modifiers is difficult in rare diseases. The SCN8A gene in humans is associated with cognitive impairment with and without cerebellar ataxia, and early infantile epileptic encephalopathy, and in mice it causes the motor endplate degeneration (med) phenotype (Burgess et al., 1995; Trudeau et al., 2006; Veeramah et al., 2012; O’Brien and Meisler, 2013). Nrcam mutations are associated with neurodevelopmental disorders and addictive behaviors in humans (Bonora et al., 2005; Sakurai et al., 2006; Ishiguro et al., 2006; Kim et al., 2009; Marui et al., 2009; Yoo et al., 2012). However, our point is not to make a direct case for NRCAM or SCN8A as human disease modifiers for CMT (indeed, their clinical associations are more severe than the “subclinical” mouse mutations we are describing) but instead to use them as a demonstration of the principle that variants that impact nodes will lead to more severe CMT phenotypes. Such variants may include any of the key components of the node of Ranvier (see Rasband and Peles, 2015 for review). Although the frequency of such modifiers is currently unknown, as genetic diagnostic sequencing improves, variants in such genes in patients diagnosed with CMT can be tested for correlation with the disease severity, which could improve the prognostic accuracy for disease severity and progression.
Finally, these results may also suggest a treatment strategy. Sodium channels are generally antagonized by more slowly gated potassium channels, which attenuate and truncate the sodium-mediated depolarization. Thus, if decreased sodium currents are detrimental, then perhaps reduced potassium currents would be beneficial. This is readily testable using genetic or pharmacological approaches in these mouse models.
In summary, genetic analyses in mice have led to the general hypothesis that mutations compromising the axonal length constant will synergize with mutations that alter nodes and/or reduce Na+-channel-mediated depolarization in peripheral axons. These findings are consistent with our understanding of axonal action potential propagation and have implications for the diagnosis and treatment of diseases resulting in neuropathy, such as CMT.
EXPERIMENTAL PROCEDURES
Mice
Mice were housed in pressurized individually ventilated (PIV) racks in the research animal facility at The Jackson Laboratory and provided food and water ad libitum. All mouse husbandry and experimental procedures were conducted according to the NIH Guide for Care and Use of Laboratory Animals and were reviewed and approved by The Animal Care and Use Committee of The Jackson Laboratory. Mice carrying mutations in Gars (CAST;B6-GarsNmf249/Rwb and C3.C(CBA)-GarsC201R/HRwb) have been previously described (Seburn et al., 2006; Achilli et al., 2009). The newly identified mutations in Sh3tc2 and Nrcam are B6(129P2)-Sh3tc2m1J/GrsrRwb and B6(129P2)-Nrcamm1J/GrsrRwb. For validation, we also examined mice carrying null alleles of each gene (B6N(Cg)-Nrcamtm2e.1(KOMP) Wtsi /J and B6N(Cg)– Sh3tc2tm1b(KOMP)Wtsi/J) obtained from the NIH repository for knockout mice (KOMP; https://www.komp.org/). The Scn8am10J mice carry a C to A transversion, changing arginine at amino acid 914 to serine (R914S, strain #23609, https://www.jax.org/strain/023609). Unless otherwise noted, all experimental cohorts used for direct comparisons consisted of littermate animals to match strain and age.
Mapping
The nm4302 mice were ascertained in a mixed B6.129 background (Jackson strain 5786, Cnr2tm1Dgen). Two spontaneous mutations were mapped by out crossing an affected mouse to BALB/cByJ mice to generate obligate heterozygous F1 animals and affected F2 offspring. Mapping with 129 SNPs spanning the entire genome in 11 affected mice and 16 unaffected littermates revealed two peaks of association. Testing additional polymorphic markers narrowed these intervals to between D12Mit222 (12:39503,868) and rs3724069 (12:79,259,640) on Chr12 and between D18Mit124 (18:57,617,140) and D18Mit183 18:64,987,826) on Chr18 in mm9 NCBI Build 37.
Exome Sequencing
Genomic DNA from an affected mouse was isolated and fragmented. Pre-capture libraries were constructed (Hodges et al., 2009) with nine cycles of PCR. The product was hybridized to the alpha version of the Roche NimbleGen Mouse Exome capture probe set (Roche NimbleGen). The sequencing library was quantified by qPCR and run on a single lane of an Illumina HiSeq GAIIX (Illumina) using a 75-base single-end protocol. Sequences were aligned to the mouse reference genome (mm9, NCBI Build 37) using Burrows-Wheeler transformation (BWA) version 0.5.8a, and variants were identified using SAMtools version 1.0 (Li et al., 2009; Li and Durbin, 2009), including a C > T conversion on chr18 at 62,127,702 (18:61,968,048 in mm38). No candidate variants were identified in the chr12 mapping interval; however, Nrcam was a strong candidate. Analysis of Nrcam by RT-PCR and Sanger sequencing revealed abnormal splicing events between exons 25 and 26 and a B2 element insertion in exon 26. High-throughput sequencing reads confirming this event were subsequently identified but were initially filtered out because the B2 element does not map uniquely in the genome, and because the insertion results in a 15-bp duplication of the flanking sequence (12:44,576,700-44,576,714 in mm38), allowing exon 26 reads to align with apparently uninterrupted overlap.
Genotyping
Genomic DNA was prepared from tail-tip or toe-tip biopsy lysed with proteinase K incubation. Primers NM4302Nrcam-F (5′-AAATTCCTGCCAACAAGACC-3′) and NM4302Nrcam-R (5′-TCCTCTGTGATCTGACTGCC-3′), which anneal to exon 26 and span the B2 insertion, were used in a PCR to identify the NM4302 Nrcam mutation (wild-type PCR product of 115 bp and mutant 374 bp). NM4302Sh3tc2F (5′-GTG GAG GAC ACC AAT TCA GG-3′) and NM4302Sh3tc2R (5′-CAC CCT CTG TCA GCC AGT TG-3′), with subsequent Sanger sequencing with NM4302Sh3tc2F to detect the single base pair C to T conversion in exon 3 of Sh3tc2, were used. Mu-KOMPNrcam-F (5′-CGG TCG CTA CCA TTTA CCA GT-3′), Mu-KOMPNrcam-R (5′-TGC CAA TCA AGC GAA TTA CT-3′), WT-KOMPNrcam-F (5′-CTA CTA TCA GCT AGC ATA ATG G-3′), and WT-KOMPNrcam-R (5′-CTT GTA CTA TGC CAA TCA AGC G-3′) primer sets were used to detect the KOMP mutant and wild-type Nrcam allele, respectively. Mu-KOMPSh3tc2-F (5′-GGG GGA AAA GAT ACT TCT GAGG-3′), Mu-KOMPSh3tc2-R (5′-CGG TCG CTA CCA TTA CCA GT-3′), WT-KOMPSh3tc2-F (5′-GTG CCA CAC TGG GCT TGA-3′), and WT-KOMPSh3tc2-R (TTC CAA ACT GAA GGG AAT GTG-3′) primer sets were used to identify the mutant and wild-type Sh3tc2 alleles, respectively.
Tissue Lysate Preparation
Whole-brain samples were isolated from animals immediately after euthanasia by CO2 inhalation. The tissues were frozen in liquid nitrogen and stored at −80°C and then homogenized using a mortar and pestle followed by a Dounce homogenizer in 1% NP-40 in PBS supplemented with Protease Inhibitor Cocktail Tablets (Roche) and then centrifuged at 14,000 × g twice for 5 min at 4°C. Cleared homogenates were then sonicated at 4°C and centrifuged again at 14,000 × g for 5 min. Sciatic nerve lysates were homogenized in 3% Triton X-100, 25 mM Tris, 1 mM EDTA, 5% glycerol, 150 mM NaCl, and 1% NP-40 in PBS supplemented with Protease Inhibitor Cocktail Tablets. Protein concentrations were assessed using a Bradford assay (Bio-Rad). 20 μg of protein for brain and 10 μg for sciatic nerve were then analyzed by immunoblot.
Western Blot
Protein lysates were resolved on Mini-PROTEAN 4%–15% Tris-glycine gels (Bio-Rad) and transferred to an Invitrolon and Immobilon-P polyvinylidene fluoride (PVDF) membrane. Membranes were blocked with 5% skim milk in TBST (1×3 Tris-buffered saline, 0.1% Tween 20) and incubated overnight with anti-NRCAM (rabbit, Abcam, 1:1,000 dilution) and anti-GAPDH (mouse monoclonal, Cell Signaling Technology, 1:1,000) or anti-SCN8A (rabbit polyclonal, Abcam), followed by Anti-Actin (rabbit polyclonal, Sigma) diluted in blocking solution at 4°C. Following three 10-min washes in TBST, the blots were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (PerkinElmer) diluted in blocking solution. After three 10-min washes in TBST, the blots were developed using Western Lightening Plus-ECL, Enhanced Chemiluminescence Substrate (PerkinElmer).
Immunohistochemistry and Histology
The entire triceps surae (medial and lateral gastrocnemius, soleus, and plantaris) were dissected free from both hind legs and weighed. Detailed methods for immunolabeling and histology are described elsewhere (Seburn et al., 2006; Burgess et al., 2010; Motley et al., 2011). For basic histology, muscles were fixed in Bouin’s, paraffin-embedded, and sectioned for H&E staining.
Neuromuscular Junction Staining and Innervation Analysis
NMJ immunohistochemistry was performed as previously described (Burgess et al., 2010; Motley et al., 2011). Innervation status was scored blind to genotype. Full occupancy was defined as NMJs where the nerve completely overlapped the AChRs, partial occupancy as a portion of receptors was clearly vacated by the presynaptic nerve, and denervation as AChR plaques had no associated nerve.
Axons
The femoral nerves were dissected free and prepared for histology as previously described (Scherer et al., 2005; Motley et al., 2011). Images were collected at 40× magnification on a Nikon Eclipse 600 microscope with differential interference contrast (DIC)/Nomarski optics, and axons were counted using an automated method in Fiji/ImageJ with manual confirmation. Axon diameter and myelin thickness were measured in Fiji/ImageJ. Larger nerves could not be captured in a single photomicrograph at 403, and images in Figures 2A–2D, 4A, 5A, and 5B are montages to show the entire nerve.
Myelin Analysis
Femoral nerves prepared above were thin sectioned (75 nm) and post-stained on grids for transmission electron microscopy. Grids were viewed on a Jeol 1230 electron microscope and photographed at 50,000× magnification with a Hamamatsu digital camera. Spacing of myelin was determined by measuring the total thickness of the myelin using Fiji/ImageJ and counting the number of wraps.
Nodes of Ranvier
Nodes of Ranvier in sciatic nerves were labeled by immunofluorescence as previously described (Seburn et al., 2014). For determining internodal distances, axons were viewed under Nomarski/DIC optics at 200× magnification, and fiber diameters and internodal distances were determined using Fiji/ ImageJ. For immunofluorescence, primary antibodies were diluted in blocking buffer and applied overnight at 4°C. Following washes, secondary antibodies were also applied overnight at 4°C. Semiquantitative analysis of NaV1.6 density at nodes was performed by collecting confocal images of NaV1.6 and neurofilament labeling on teased sciatic nerve axons under standardized parameters. The labeling intensity in a region of interest including the NaV1.6 signal was compared to the labeling intensity of neurofilament in the adjacent axon in an equivalently sized region. Background intensity of neighboring blank space (no tissue) was subtracted from both values. The following primary antibodies were used: anti-Nrcam (rabbit, Abcam, 1:200 dilution), anti-NaV1.6 (rabbit, Alamone, 1:200 dilution), anti-NaV1.6 (mouse monoclonal IgG1, NeuroMab/Antibodies, 1:5 dilution), anti-Kv1.2 (mouse monoclonal IgG2bK, Millipore; 1:100 dilution), anti-AnkyrinG (rabbit, Santa Cruz Biotechnology, 1:200), and anti-neurofilament light chain (mouse IgG1 2H3, Developmental Studies Hybridoma Bank, 1:500). The following secondary antibodies (Invitrogen) were used for visualization: anti-mouse IgG1 594, anti-mouse IgG1 Alexa 488, anti-mouse 488, anti-mouse IgG2bK Alexa IgG2bK 488, and anti-rabbit Alexa 594.
Nerve Conduction
Mice were prepared and electrodes placed as previously described (Burgess et al., 2010; Motley et al., 2011). Sciatic NCV was determined by measuring CMAP latency and the distance between points of stimulation. The presence of conduction block was evaluated by measuring the integrated CMAP (iCMAP) in response to distal and proximal stimulation and expressing the difference as a percentage (proximal-distal)/distal)*100.
Voltage Clamp
Detailed methods have been previously described (Wang et al., 2005; Rich et al., 2002; Spaulding et al., 2016). Mini endplate currents (MEPCs) were recorded continuously for 1 min, and 20 evoked endplate currents (EPCs) (when present) were recorded in response to single stimuli delivered at 0.5 Hz.
Statistics
A one-way ANOVA followed by Tukey’s HSD post hoc comparison (R-Team, 2008) was used for evaluating differences between genotypes for axon counts, conduction velocity, body weight, and muscle-to-body-weight ratio (MW:BW). Axon diameters and myelin thickness were compared using the non-parametric Kolmogorov-Smirnov two-sample test. NMJ innervation status between genotypes and categories (fully innervated, partially innervated, and denervated) was evaluated with two-way ANOVA followed by Tukey’s HSD and Roy post hoc comparisons (R-Team, 2008). Probability of p < 0.05 was used as a cutoff for declaring statistical significance in all comparisons. Unless otherwise stated, values are reported as mean ± SD.
Supplementary Material
Highlights.
Mutations in Nrcam subtly alter structure of nodes of Ranvier and Nav1.6 levels
Loss of Nrcam increases the severity of both demyelinating and axonal neuropathy
Heterozygosity for Scn8a (Nav1.6) largely recapitulates loss of Nrcam
Decreasing axonal depolarization synergizes with poor myelination and axon atrophy
Acknowledgments
We would like to thank the scientific services at The Jackson Laboratory for their assistance in these studies, including Mr. Pete Finger in the electron microscopy service and the genome technologies and computational sciences services for assistance in whole-exome capture, sequencing, and analysis. We would also like to thank Dr. Laura Reinholdt and the Mouse Mutant Resource for facilitating the identification of the nm4302 strain, the honors biology class from Bangor Christian High School (Dylan Merchant, Cody Collins, and Zach Palmeter) for assistance in quantifying electron micrographs, and Dr. Rosalinda Doty for consultation in muscle histology and neurogenic atrophy. This work was supported by grants from the NIH (R21 NS072675 to R.W.B., G.A.C., and K.L.S.; F31 NS100328 to E.L.S.; F31 NS098540 to K.H.M.; RO1 AR054170 to G.A.C.; RO1 NS054154 and U54 OD020351 to R.W.B.; and R24 NS098523 to R.W.B. and K.L.S.). The scientific services at The Jackson Laboratory are supported in part by CA34196.
Footnotes
ACCESSION NUMBERS
The accession number for the exome sequencing data reported in this paper is BioSample: SAMN06546078.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.03.009.
AUTHOR CONTRIBUTIONS
The original nm4302 strain was ascertained by L.A.D. D.G.S. and G.A.C. oversaw mapping and exome sequencing efforts to identify the spontaneous mutations. E.L.S. analyzed myelination. K.H.M., K.L.S., and R.W.B. performed phenotyping experiments, analyzed data, and designed and executed genetic crosses with neuropathy models. R.W.B. wrote the manuscript with input from all authors.
References
- Achilli F, Bros-Facer V, Williams HP, Banks GT, AlQatari M, Chia R, Tucci V, Groves M, Nickols CD, Seburn KL, et al. An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis Model Mech. 2009;2:359–373. doi: 10.1242/dmm.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor V, Feinberg K, Eshed-Eisenbach Y, Vainshtein A, Frechter S, Grumet M, Rosenbluth J, Peles E. Long-term maintenance of Na+ channels at nodes of Ranvier depends on glial contact mediated by gliomedin and NrCAM. J Neurosci. 2014;34:5089–5098. doi: 10.1523/JNEUROSCI.4752-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud E, Zenker J, de Preux Charles AS, Stendel C, Roos A, Médard JJ, Tricaud N, Kleine H, Luscher B, Weis J, et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc Natl Acad Sci USA. 2009;106:17528–17533. doi: 10.1073/pnas.0905523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora E, Lamb JA, Barnby G, Sykes N, Moberly T, Beyer KS, Klauck SM, Poustka F, Bacchelli E, Blasi F, et al. Mutation screening and association analysis of six candidate genes for autism on chromosome 7q. Eur J Hum Genet. 2005;13:198–207. doi: 10.1038/sj.ejhg.5201315. [DOI] [PubMed] [Google Scholar]
- Brown MC, Ironton R. Motor neurone sprouting induced by prolonged tetrodotoxin block of nerve action potentials. Nature. 1977;265:459–461. doi: 10.1038/265459a0. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- Burgess RW, Cox GA, Seburn KL. Neuromuscular disease models and analysis. Methods Mol Biol. 2010;602:347–393. doi: 10.1007/978-1-60761-058-8_20. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci USA. 2000;97:5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer J, Gooding R, Angelicheva D, King RH, Guillén-Navarro E, Parman Y, Nascimento A, Conill J, Kalaydjieva L. Clinical spectrum of CMT4C disease in patients homozygous for the p.Arg1109X mutation in SH3TC2. Neuromuscul Disord. 2006;16:449–453. doi: 10.1016/j.nmd.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Custer AW, Kazarinova-Noyes K, Sakurai T, Xu X, Simon W, Grumet M, Shrager P. The role of the ankyrin-binding protein NrCAM in node of Ranvier formation. J Neurosci. 2003;23:10032–10039. doi: 10.1523/JNEUROSCI.23-31-10032.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269:27163–27166. [PubMed] [Google Scholar]
- Douglas DS, Moran JL, Bermingham JR, Jr, Chen XJ, Brindley DN, Soliven B, Beier DR, Popko B. Concurrent Lpin1 and Nrcam mouse mutations result in severe peripheral neuropathy with transitory hindlimb paralysis. J Neurosci. 2009;29:12089–12100. doi: 10.1523/JNEUROSCI.2029-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen LW. Hereditary motor end-plate disease in the mouse: Light and electron microscopic studies. J Neurol Neurosurg Psychiatry. 1970;33:238–250. doi: 10.1136/jnnp.33.2.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen LW, Strich SJ. The effects of botulinum toxin on the pattern of innervation of skeletal muscle in the mouse. Q J Exp Physiol Cogn Med Sci. 1968;53:84–89. doi: 10.1113/expphysiol.1968.sp001948. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Thomas PK. Peripheral Neuropathy. Elsevier Saunders; 2005. [Google Scholar]
- Eshed-Eisenbach Y, Peles E. The making of a node: A co-production of neurons and glia. Curr Opin Neurobiol. 2013;23:1049–1056. doi: 10.1016/j.conb.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg K, Eshed-Eisenbach Y, Frechter S, Amor V, Salomon D, Sabanay H, Dupree JL, Grumet M, Brophy PJ, Shrager P, Peles E. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron. 2010;65:490–502. doi: 10.1016/j.neuron.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Harel T, Gambin T, Kousi M, Griffin LB, Francescatto L, Ozes B, Karaca E, Jhangiani SN, Bainbridge MN, et al. Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep. 2015;12:1169–1183. doi: 10.1016/j.celrep.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges E, Rooks M, Xuan Z, Bhattacharjee A, Benjamin Gordon D, Brizuela L, Richard McCombie W, Hannon GJ. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat Protoc. 2009;4:960–974. doi: 10.1038/nprot.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Liu QR, Gong JP, Hall FS, Ujike H, Morales M, Sakurai T, Grumet M, Uhl GR. NrCAM in addiction vulnerability: Positional cloning, drug-regulation, haplotype-specific expression, and altered drug reward in knockout mice. Neuropsychopharmacology. 2006;31:572–584. doi: 10.1038/sj.npp.1300855. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim HG, Kim MH, Kwack KB, Park JK, Kim T, Kim JW, Ban JY, Chung JH. Association between neuronal cell adhesion molecule (NRCAM) single nucleotide polymorphisms and schizophrenia in a Korean population. Psychiatry Clin Neurosci. 2009;63:123–124. doi: 10.1111/j.1440-1819.2008.01893.x. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Misawa S. Axonal ionic pathophysiology in human peripheral neuropathy and motor neuron disease. Curr Neurovasc Res. 2004;1:373–379. doi: 10.2174/1567202043362162. [DOI] [PubMed] [Google Scholar]
- Li J. Molecular regulators of nerve conduction—lessons from inherited neuropathies and rodent genetic models. Exp Neurol. 2015;267:209–218. doi: 10.1016/j.expneurol.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallik A, Weir AI. Nerve conduction studies: Essentials and pitfalls in practice. J Neurol Neurosurg Psychiatry. 2005;76(Suppl 2):ii23–ii31. doi: 10.1136/jnnp.2005.069138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marui T, Funatogawa I, Koishi S, Yamamoto K, Matsumoto H, Hashimoto O, Nanba E, Nishida H, Sugiyama T, Kasai K, Watanabe K, Kano Y, Sasaki T, Kato N. Association of the neuronal cell adhesion molecule (NRCAM) gene variants with autism. Int J Neuropsychopharmacol. 2009;12:1–10. doi: 10.1017/S1461145708009127. [DOI] [PubMed] [Google Scholar]
- Motley WW, Seburn KL, Nawaz MH, Miers KE, Cheng J, Antonellis A, Green ED, Talbot K, Yang XL, Fischbeck KH, Burgess RW. Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet. 2011;7:e1002399. doi: 10.1371/journal.pgen.1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): Properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet. 2013;4:213. doi: 10.3389/fgene.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8:654–667. doi: 10.1016/S1474-4422(09)70110-3. [DOI] [PubMed] [Google Scholar]
- Pestronk A, Drachman DB. Motor nerve sprouting and acetylcholine receptors. Science. 1978;199:1223–1225. doi: 10.1126/science.204007. [DOI] [PubMed] [Google Scholar]
- R-TEAM. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2008. [Google Scholar]
- Rasband MN, Peles E. The nodes of Ranvier: Molecular assembly and maintenance. Cold Spring Harb Perspect Biol. 2015;8:a020495. doi: 10.1101/cshperspect.a020495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich MM, Waldeck RF, Cork LC, Balice-Gordon RJ, Fyffe RE, Wang X, Cope TC, Pinter MJ. Reduced endplate currents underlie motor unit dysfunction in canine motor neuron disease. J Neurophysiol. 2002;88:3293–3304. doi: 10.1152/jn.00270.2002. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Ramoz N, Reichert JG, Corwin TE, Kryzak L, Smith CJ, Silverman JM, Hollander E, Buxbaum JD. Association analysis of the NrCAM gene in autism and in subsets of families with severe obsessive-compulsive or self-stimulatory behaviors. Psychiatr Genet. 2006;16:251–257. doi: 10.1097/01.ypg.0000242196.81891.c9. [DOI] [PubMed] [Google Scholar]
- Saporta MA, Shy ME. Inherited peripheral neuropathies. Neurol Clin. 2013;31:597–619. doi: 10.1016/j.ncl.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Messing A, Willecke K, Fischbeck KH, Jeng LJ. Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32-null mice. J Neurosci. 2005;25:1550–1559. doi: 10.1523/JNEUROSCI.3082-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 2006;51:715–726. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Seburn KL, Morelli KH, Jordanova A, Burgess RW. Lack of neuropathy-related phenotypes in hint1 knockout mice. J Neuropathol Exp Neurol. 2014;73:693–701. doi: 10.1097/NEN.0000000000000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senderek J, Bergmann C, Stendel C, Kirfel J, Verpoorten N, De Jonghe P, Timmerman V, Chrast R, Verheijen MH, Lemke G, et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am J Hum Genet. 2003;73:1106–1119. doi: 10.1086/379525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrager P. Sodium channels in single demyelinated mammalian axons. Brain Res. 1989;483:149–154. doi: 10.1016/0006-8993(89)90046-2. [DOI] [PubMed] [Google Scholar]
- Sivakumar K, Kyriakides T, Puls I, Nicholson GA, Funalot B, Antonellis A, Sambuughin N, Christodoulou K, Beggs JL, Zamba-Papanicolaou E, et al. Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain. 2005;128:2304–2314. doi: 10.1093/brain/awh590. [DOI] [PubMed] [Google Scholar]
- Spaulding EL, Sleigh JN, Morelli KH, Pinter MJ, Burgess RW, Seburn KL. Synaptic Deficits at Neuromuscular Junctions in Two Mouse Models of Charcot-Marie-Tooth Type 2d. J Neurosci. 2016;36:3254–3267. doi: 10.1523/JNEUROSCI.1762-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman V, Strickland AV, Züchner S. Genetics of Charcot-Marie-Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel) 2014;5:13–32. doi: 10.3390/genes5010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet. 2006;43:527–530. doi: 10.1136/jmg.2005.035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschöp J, Kasten KR, Nogueiras R, Goetzman HS, Cave CM, England LG, Dattilo J, Lentsch AB, Tschöp MH, Caldwell CC. The cannabinoid receptor 2 is critical for the host response to sepsis. J Immunol. 2009;183:499–505. doi: 10.4049/jimmunol.0900203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90:502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Engisch KL, Nakanishi ST, Dodson SE, Miller GW, Cope TC, Pinter MJ, Rich MM. Activity-dependent presynaptic regulation of quantal size at the mammalian neuromuscular junction in vivo. J Neurosci. 2005;25:343–351. doi: 10.1523/JNEUROSCI.3252-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Shrager P. Dependence of axon initial segment formation on Na+ channel expression. J Neurosci Res. 2005;79:428–441. doi: 10.1002/jnr.20378. [DOI] [PubMed] [Google Scholar]
- Yoo BK, Shim JC, Lee BD, Kim C, Chung YI, Park JM, Kim SG, Kim JH, Lee YM, Moon ES, Kwon DH. Association of the neuronal cell adhesion molecule (NrCAM) gene variants with personality traits and addictive symptoms in methamphetamine use disorder. Psychiatry Investig. 2012;9:400–407. doi: 10.4306/pi.2012.9.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.