

Abstract
Traumatic brain injuries (TBI) often involve vascular dysfunction that leads to longterm alterations in physiological and cognitive functions of the brain. Indeed, all the cells that form blood vessels and that are involved in maintaining their proper function can be altered by TBI. In this review, we focus on the different types of cerebrovascular dysfunction that occur after TBI, ranging from cerebral blood flow (CBF) alterations, autoregulation impairments, subarachnoid hemorrhage (SAH), vasospasms, blood-brain barrier (BBB) disruption and edema formation. We also discuss the mechanisms that mediate these dysfunctions, focusing on the cellular components of cerebral blood vessels (endothelial cells, smooth muscle cells, astrocytes, pericytes, perivascular nerves) and their known and potential roles in the secondary injury cascade.
Graphical abstract
Traumatic brain injuries lead to alteration of cerebrovascular function by inducing decreased cerebral blood flow, impaired autoregulation, subarachnoid hemorrhage, vasospasms, edema, blood-brain barrier disruption and long-term neurodegeneration. The roles of endothelial cells, smooth muscle cells, astrocytes, and perivascular innervation in these injury cascades are discussed in this review.
I) Introduction
During the last decades, researchers have focused on neurocentric dysfunctions after acute brain injuries with an emphasis on the molecular mechanisms involved in early cell death. Traumatic brain injury (TBI) research has moved from acute vascular dysfunction to an increasing focus on neuronal death. As noted in various review articles (Jullienne and Badaut 2013; Zhang et al. 2012), this strategy has failed to transfer new therapeutic compounds from research tools to clinical therapies. It is within this context that in 2002 the NIH (National Institutes of Health) and NINDS (National Institute of Neurological Disorders and Stroke) hosted a workshop that suggested that the neurovascular unit (NVU) should be considered, as well as a larger micro-system comprised of vessels and the associated glial cells, as the functional target of injury (Grotta et al. 2002; Zhang et al. 2012). A dysfunctional NVU has been recently proposed to be involved in the mechanisms underlying neurodegenerative diseases, including Alzheimer’s disease (AD) (Iadecola 2004; Zlokovic 2011). Interestingly, TBI is frequently associated with higher long-term risk for AD, and vascular dysfunction has recently been suggested to be involved in the development of AD (Johnson et al. 2010). Therefore, the NVU and vascular malfunction could be involved in poor cognitive outcomes after TBI. This review emphasizes the importance of the cerebrovascular dysfunction and related molecular mechanisms post-TBI.
II) Traumatic brain injuries: clinical definition
TBI contributes to more than 30% of all injury-related deaths in the United States (Faul et al. 2010) and represents in excess of 75,000 deaths each year in Europe (Maas et al. 2015). TBI is defined as a brain lesion caused by a direct or indirect external mechanical impact like the penetration of a projectile, or by blast waves, inducing the disruption of the normal structure and function of the brain. The most frequent causes of TBI in the general population are falls (35%), motor vehicle and traffic related accidents (17%), being struck by or against an object (16%), and assaults (10%) (Faul et al. 2010). While TBI affects all ages, pediatric and elderly populations are more vulnerable than adults (Faul et al. 2010). Some populations including military personnel and rescue workers are more likely to endure blast waves, resulting in a higher risk for TBI. Finally, sports-related TBI is a public health concern because of the repetitive nature of these injuries and the fact that they are often unreported.
Three main levels of TBI severity can be defined clinically, based on the Glasgow coma scale (3-to 15-point scale used to assess the patient’s level of consciousness and neurologic function) or on the duration of the loss of consciousness. The majority of TBI (75%) is mild, and is also known as concussion. There is no skull fracture and little or minor changes observed on computed tomography scan (CT-scan) and/or standard MRI (Ashwal et al. 2014). For moderate and severe TBI, the presence of bleeding is frequently observed, especially in the case of blast injuries (Maas et al. 2008; Taber et al. 2006).
There are two distinct injury phases in TBI: the primary acute event is often followed by a secondary injury cascade that includes glutamatergic excitotoxicity, calcium overload and vascular dysfunction. These secondary injuries usually last for several months or even years, resulting in a pattern of “chronic brain disease” (Johnson et al. 2010; Pop and Badaut 2011; Smith et al. 2013). Several clinical studies have shown that after TBI and the acute period, there are long lasting behavioral dysfunctions including cognitive decline or emergence of psychiatric disorders remote from the initial injury (Smith et al. 2013). For a more complete review of these deficits see Obenaus (2015). For example, even if patients recover well from physical problems, 30% of adults report altered cognitive function, such as memory and concentration deficits, up to 3 months after a mild TBI (Ponsford et al. 2011). It has also been shown that a TBI occurring early in life could lead to a higher risk of mortality, independently of its severity (Himanen et al. 2011; McMillan and Teasdale 2007). Repeated TBIs are a known as a risk factor for dementia, particularly when related to sport injuries. Indeed, repeated TBI is associated with long-term cognitive impairment, as reported in retired athletes (Guskiewicz et al. 2005; Lakhan and Kirchgessner 2012).
TBI is a massive health burden worldwide, not only because of the cognitive and psychological impairments but also because of the economic costs that include medical expenses, as well as indirect expenses such as losses of productivity (Corso et al. 2006; Gustavsson et al. 2011). Despite recent advances, a comprehensive research on TBI pathophysiology is marginal compared to other acute injuries such as stroke, or other neurodegenerative diseases. The numerous veterans from the Iraq and Afghanistan wars and the increased public awareness regarding sport-related concussion injuries, has led to an increase in research to understand the pathophysiology of this heterogeneous disorder to develop better treatments. Various animal models, from rodents to larger animals, have been developed in order to obtain a better understanding of the cellular and molecular mechanisms (Obenaus 2012; Petraglia et al. 2014; Prins and Hovda 2003). The variety of pre-clinical models reflects the variety of severity and heterogeneity of clinical TBI. However, the definition of degree of TBI severity in different animal models is still controversial and remains poorly defined. This is a critical point because the cellular and molecular responses to the injury depend upon not only its severity, but also on the location of the impact. Moreover, the age of the animals is important because the outcome is more severe for younger patients (Himanen et al. 2011; McMillan and Teasdale 2007; Pop and Badaut 2011). However, it is not the focus of our review and we refer the reader to other reviews addressing this question (Obenaus 2012; Petraglia et al. 2014; Prins and Hovda 2003). It is crucial to keep in mind that most pre-clinical TBI models exhibit vascular dysfunction ranging from blood-brain barrier (BBB) disruption to hemorrhage (DeWitt and Prough 2003; Golding 2002; Pop and Badaut 2011). A recent review article illustrates the importance of the vascular responses and changes in morphology and perfusion after TBI (Kenney et al. 2015). The early vascular dysfunction has been known for almost two decades and the long-term changes could be associated with premature aging of the brain or emergence of brain dysfunction after TBI.
III) TBI as a cerebrovascular injury: clinical evidence
1) Characteristics of the brain vasculature
Cerebral vessel morphology is composed of three distinct layers, each one having unique roles. The innermost layer is called tunica intima, and is composed of a single layer of endothelial cells surrounded by a basement membrane. In the case of capillaries, the basement membrane encloses pericytes (Figure 1). Around this first layer is the tunica media, which is the muscular portion of the vessel. This medial layer is composed of vascular smooth muscle cells (SMC), surrounded by a basement membrane. The thickness of the smooth muscle layer is dependent on the size of the vessel. Pial arteries generally have two or three SMC layers, whereas penetrating arteries have only one or two SMC per circumference. The penetrating arteries give rise to arterioles that are composed of a single layer of SMC. Then, in the capillaries, pericytes can replace SMC, even though their density and distribution remains controversial. Finally, the outermost layer of the cerebral blood vessels, the tunica adventitia, is a strong layer composed of connective tissue allowing the blood vessel to resist forces acting on the vessel wall. The tunica adventitia contains mostly collagen fibers, fibroblasts, and associated cells including terminal nerve fibers in pial arteries. However, after the end of the Virchow-Robin space, the tunica adventitia is almost absent and the intraparenchymal arterioles are in direct contact with astrocyte endfeet (Cipolla et al. 2004; Golding 2002) (Figure 1).
FIGURE 1. Morphology and characteristics of cerebral blood vessels.
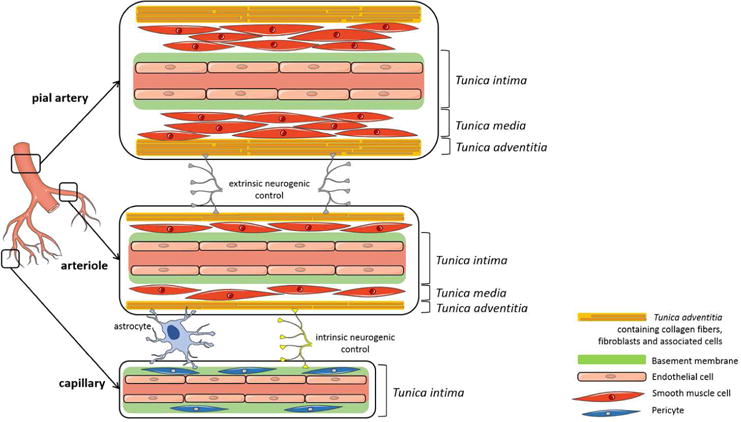
Cerebral blood vessels are formed by 3 distinct layers: the tunica intima with endothelial cells and a basement membrane; the tunica media with smooth muscle cells; and the tunica adventitia with collagen fibers, fibroblasts and associated cells like nerve fibers and astrocytes. Capillaries are formed with only a tunica intima with a basement membrane enclosing pericytes. (This figure was produced using Servier Medical Art; www.servier.com).
2) Extra-and intraparenchymal blood vessels
Pial vessels are located on the surface of the brain and give rise to smaller arteries that penetrate into the brain parenchyma with the Virchow-Robin space filled by cerebrospinal fluid (CSF). These penetrating arteries become intraparenchymal arterioles once the Virchow-Robin space disappears and are almost completely surrounded by astrocyte endfeet (Cipolla et al. 2004). Pial and penetrating arteries present mostly an “extrinsic” neurogenic control of the perfusion via a perivascular innervation from the peripheral nervous system (Rennels and Nelson 1975).
Even though the myogenic regulation and the role of vascular SMCs and endothelial cells in maintaining cerebral perfusion are involved in the vascular tone, the control of perfusion in intraparenchymal arterioles is also influenced by “intrinsic” innervation coming from the surrounding brain neuropil including interneurons, as well as astrocytes (Cohen et al. 1996) (Figure 1). In fact, intraparenchymal arterioles have only one layer of vascular SMCs and are completely covered by astrocytes endfeet on their abluminal side. It has been recently well documented that astrocytes have an important role in the regulation of cerebral blood flow (CBF) (Attwell et al. 2010; Filosa et al. 2015; Howarth 2014). Indeed, astrocytes have a unique position within the NVU where they contact synapses with their processes as well as 99% of the blood vessels surface (Filosa et al. 2015) (Figure 1). Therefore, by responding to neuronal activity (buffering neurotransmitters and ions, e.g. potassium), astrocytes can subsequently relay information to blood vessels to ensure adequate blood supply to areas with increased neuronal activity: blood flow-metabolism coupling, a physiological response also termed neurovascular coupling. Several studies, both in vitro (Blanco et al. 2008; Gordon et al. 2008; Mulligan and MacVicar 2004) and in vivo (Lind et al. 2013; Otsu et al. 2015; Winship et al. 2007) have shown that in response to neuronal activation there is an increase in the intracellular calcium concentration in astrocytes and a subsequent release of vasoactive substances such as prostaglandin E2 and epoxyeicosatrienoic acids that induce vasodilation of the blood vessels. Moreover, increases in astrocytic calcium contribute to the production of other vasoactive substances such as nitric oxide (NO), glutamate, adenosine, and adenosine tri-phosphate (ATP) (Filosa et al. 2015). In addition to the vasoactive substances, arachidonic acid is produced and released by astrocytes and can also diffuse to the arteriole’s SMC where it is metabolized to the vasoconstrictor 20-HETE (20-hydroxyeicosatetraenoic acid) (Gebremedhin et al. 2000). Indeed, studies have shown that in addition to vasodilation, vasoconstriction of blood vessels can be induced by astrocytic activation (Blanco et al. 2008; Gordon et al. 2008; Metea and Newman 2006; Mulligan and MacVicar 2004).
The ability of astrocytes to induce vasoconstriction and vasodilation suggests that the balance of both vasodilatory and vasoconstrictive substances might contribute to the regulation of resting vascular tone and CBF, in addition to their role in neurovascular coupling (Filosa et al. 2015). Indeed, recent studies have shown that astrocytes are also involved in the regulation of parenchymal arteriole’s vascular tone and cerebral autoregulation (Kim et al. 2015; Rosenegger et al. 2015). It is well known that astrocytes respond to an injury in the central nervous system by changing their phenotype and transform into reactive astrocytes with a change in their morphology and the level of expression of different proteins, such as glial fibrillary acidic protein (GFAP) and extracellular matrix proteins (i.e. laminin, perlecan…). The differences in the evoked vascular responses (vasoconstriction versus vasodilation) after astrocyte activation might also contribute to the differences observed in experimental approaches (i.e. non-physiological stimulations, species of animals, different developmental stages) (reviewed in Filosa et al. (2015)). Regardless of the controversy surrounding the polarity of the evoked vascular response upon astrocyte stimulation, these studies firmly demonstrate an important role of astrocytes in the regulation of CBF under physiological conditions. This also suggests that astrocyte phenotype changes (quiescent versus reactive astrocyte) should not be overlooked when studying pathologies that involve vascular dysfunction.
3) Clinical observations
a. Decreased cerebral blood flow and impaired cerebrovascular autoregulation
CBF has been studied in humans with computed tomography using different compounds like 113Xe, stable Xe or 15O2. Investigations showed that early after a TBI (within the first 6 h), a significant number of patients present with depressed CBF values going as low as 22.5mL/100g/min when a normal CBF in adult humans should be around 45–50mL/100g/min (Bouma et al. 1991; DeWitt and Prough 2003). It has been shown that the values can even reach 15mL/100g/min in patients with severe TBI (Bouma et al. 1992). In children, it was shown that low CBF (<20mL/100g/min) and younger age (<5 years-old) were predictive of unfavorable outcomes, such as severe disability or vegetative state (Adelson et al. 2011).
This decreased CBF and its evolution during the first few weeks after a head injury is related to the recovery. Indeed, the reduction in CBF improves as recovery occurs (Langfitt et al. 1977). Moreover, when the CBF returns to normal within 3 weeks, the neurological outcome is significantly better than for the patients who still have a subnormal CBF after this time (Inoue et al. 2005).
Cerebrovascular autoregulation is an essential mechanism aimed at maintaining adequate blood perfusion in the brain by changes of cerebrovascular resistance. CBF is therefore maintained so that there is a constant local perfusion over a pressure range of 60 to 160 mmHg (Golding 2002). After a brain injury, cerebrovascular autoregulation is impaired or abolished in many patients (Enevoldsen and Jensen 1978; Rangel-Castilla et al. 2008). In summary, changes of CBF have been shown in patients after TBI, with a possible correlation with the outcomes.
b. Subarachnoid hemorrhage and vasospasms
Moderate and severe TBI may induce intracranial bleeding that can be associated with skull fracture or parenchymal contusions. Hematomas can increase intracranial pressure due to the distortion of the injured brain tissue, and ischemic lesions due to the compression of the surrounding vasculature. Several types of bleeding can occur between the skull and the brain: epidural hematoma (between the skull and the dura mater), subdural hematoma (between the dura and the arachnoid mater), and subarachnoid hemorrhage (between the arachnoid and the pia mater).
Subarachnoid hemorrhage (SAH) is a common feature of severe TBI. Approximately 40% of the patients with a severe head trauma present with SAH, which is associated with poor functional outcomes (Eisenberg et al. 1990; Servadei et al. 2002). Severe SAH can lead to death or severe disability even if it is detected and treated early, although treatment options are limited.
Cerebral vasospasms have been shown to correlate with severe SAH. They are characterized by contractions of the cerebral vasculature and usually predict a poor outcome. Indeed, 30 to 50% of the patients with SAH will develop vasospasms, increasing the risk of ischemic injuries (Armin et al. 2006). The onset occurs between days 2 to 15 after injury (Martin et al. 1997) and is accompanied by hypoperfusion in 50% of the patients. With regard to the type of injury, it has been shown that blast injury patients are more likely to develop vasospasms when compared to patients with other types of TBI (Zubkov et al. 2000). Moreover, vasospasms in blast-induced TBI have been shown to last up to 30 days (Armonda et al. 2006) while they tend to resolve completely after 14 days in closed head TBI without blast (Oertel et al. 2005).
Bleeding after severe and moderate TBI is a landmark of the acute vascular dysfunction. However, even without major bleeding, the BBB can still be injured in all TBI severities, from mild to severe, and be associated with edema formation.
c. Edema formation and BBB disruption
Decreased CBF induces hypoxic/ischemic conditions further contributing to the evolution of secondary injuries as well as the presence of micro-bleeding or hemorrhagic components. Following a brain injury, edema formation can have dramatic consequences on morbidity and mortality because it induces intracranial hypertension, and contributes to the vicious cycle of the secondary injury cascade (Unterberg et al. 2004). Classically, 2 types of brain edema are described: cytotoxic and vasogenic. Cytotoxic edema is defined by cell swelling, without an initial BBB alteration, whereas vasogenic edema is caused by an initial BBB disruption and an increased permeability of endothelial cells. However, this concept has recently been revised, suggesting that during the phase of edema build-up following an acute brain injury, anoxic and ionic edema precede vasogenic edema (Badaut et al. 2013). Anoxic edema occurs within a few minutes after the initial injury, it is characterized by the swelling of astrocytes and neuronal dendrites, due to the lack of energy induced by oxygen and nutrient deprivation. Without sufficient energy, control of the ionic gradients is affected, resulting in water entry in the cells. Anoxic edema is quickly followed by ionic edema, where the absence of oxygen and nutrients leads to the alteration of the ionic gradients within endothelial cells, with transcapillary flux of Na+ ions and tissue swelling (Badaut et al. 2013). Finally, vasogenic edema appears, due to the increased permeability of endothelial cells.
In patients, BBB disruption allows diffusion of molecules over 500kDa in the peripheral circulation and can therefore be measured by serum markers. Typically, the assessment of BBB status is done through the measurement of the cerebrospinal fluid-serum albumin quotient (Andersson et al. 1994). A study showed that serum levels of S100-β, an astrocytic protein, could also indicate BBB dysfunction 12 h after severe TBI, thereby using a less invasive technique (Blyth et al. 2009). However, a recent meta-analysis suggests that this measurement would be more useful in evaluating the TBI severity and in determining the long-term prognosis in patients with moderate to severe brain injury (Mercier et al. 2013). In a clinical study involving patients with severe non-penetrating brain injuries, 44% of the patients had significant BBB disruption which was associated with more severe TBI and worse outcomes after 18 months (Ho et al. 2014). Moreover, another study suggests that patients with severe TBI and BBB disruption had a higher risk for intracranial pressure as well as a trend towards increased mortality (Saw et al. 2014).
In general there is a better knowledge on molecular changes in BBB dysfunction in rodent models than in human (see below). After a TBI, the upregulation of different matrix metalloproteases (MMPs) is known to alter proteins of the extracellular matrix and participate in the degradation of the BBB. Despite limited information on MMP expression in the human brain after TBI, a recent clinical study involving 8 patients with severe TBI investigated the temporal response of several MMPs from cerebral microdialysates and in CSF over 6 days following the injury. MMP-8 and MMP-9 are increased in the brain and in the arterial plasma immediately following trauma. They progressively decrease while MMP-7 starts rising slowly after 4 days. Interestingly, MMP-8 levels are associated with mortality (Roberts et al. 2013). A very recent study involving 100 patients with severe TBI did not observe any difference in serum MMP-9 levels between survivors and non-surviving patients at 30 days. However they showed significantly higher serum TIMP-1 (tissue inhibitor of MMP-1) levels in non-surviving patients compared with survivors, suggesting TIMP-1 could be used as a prognostic biomarker of mortality in TBI patients (Lorente et al. 2014).
Vascular dysfunction is present after TBI, from BBB alteration (capillary level) to modification of the brain perfusion (large blood vessels). Altogether, these clinical findings strongly suggest alterations at different levels of the vascular tree.
IV) Preclinical observations
Preclinical TBI models have the advantage of advancing the knowledge of the cerebrovascular and NVU changes that may occur after TBI.
1) Cerebral blood flow and energy metabolism
As seen in clinical settings (Bouma et al. 1991; Bouma et al. 1992), changes in the CBF have been observed in animal models of TBI. Reduction of CBF after injury has been observed in animal models of severe controlled cortical impact (CCI) (Bryan et al. 1995; Kochanek et al. 1995; Plesnila et al. 2003). In a severe CCI model, and using MRI with iron as a contrast agent, a marked drop in the cerebral blood volume has been shown after injury in adult rats, which did not recover at the lesion site 2 weeks post-TBI (Immonen et al. 2010). In a lateral fluid percussion (LFP) injury model, which induces a more diffuse type of brain injury, decreased CBF in the perilesional cortex and hippocampus has been observed up to 8 months after injury in adult rats (Hayward et al. 2010). Importantly, a reduction of CBF has also been observed in mild TBI models (Long et al. 2015; Villapol et al. 2014). In a mild CCI model using adult rats, a decrease in CBF at the lesion site was observed acutely after the injury with recovery to near normal values after 2 weeks (Long et al. 2015). In a mild/moderate CCI model, the reduction of CBF was delayed with restoration at 30 days post-TBI in young adult mice (Villapol et al. 2015).
Altered CBF is likely to contribute to secondary injuries after TBI by decreasing glucose and oxygen delivery. The brain does not have sufficient energy stores and is therefore highly dependent on continuous CBF. Moreover, it has been shown that during the first 6 h after concussion in rats, cells in the injured brain show evidence of hypermetabolism (Yoshino et al. 1991). Consequently, the decreased CBF and the hypermetabolism occurring early after TBI result in a mismatch between demand and supply, known as uncoupling of CBF and glucose metabolism. After this hypermetabolic phase, glucose metabolism has been shown to be decreased in an age-dependent manner. In young adult rodent models (P35), it lasts for 5 to 10 days (Hovda et al. 1991; Yoshino et al. 1991), whereas it only lasts 3 days in juvenile rats (P17) (Thomas et al. 2000). In human studies, it can last up to 1 month (Bergsneider et al. 2001; Glenn et al. 2015). Aerobic metabolism is decreased and anaerobic glycolysis is increased, resulting in production of lactate (Verweij et al. 2007). Even though lactate has long been considered a waste product of impaired metabolism, it is now clear that it has a key role in energy metabolism after head injury. Not only can it be used by neurons to produce energy (Pellerin 2003), it is now suggested that it is a preferential fuel for the brain (Bartnik et al. 2007). In a study performed on TBI patients and using isotope tracers, Glenn and colleagues showed that around 70% of the carbohydrate consumed by the injured brain comes from the peripheral lactate production (Glenn et al. 2015). On the other hand, it has been shown that increased lactate in the cerebral parenchyma is associated with poor neurological outcomes in pediatric population (Ashwal et al. 2000). To explain this difference, Glenn and colleagues suggest that lactate accumulation in the brain is in the first case due to high production complicated by limited disposal, and in the second case due to a metabolic stasis leading to acidosis (Glenn et al. 2015).
Intravenous lactate therapy has been used in rats after fluid percussion injury and has been shown to have beneficial effects on cognition (Holloway et al. 2007). Lauritzen and colleagues suggest that these beneficial effects could be due not only to an increased metabolism of lactate but also to its binding to the receptor HCA1 (hydrocarboxylic acid receptor 1). Indeed, the authors suggest that, when lactate concentration rises, HCA1 inhibits cAMP generation, thereby slowing glycolysis rates (Lauritzen et al. 2014).
Several studies have investigated the expression of lactate transporters, MCTs (monocarboxylate transporters) after TBI. MCTs in brain lysates are increased during the first week post-CCI in young adult rats at P35 and P75 (Prins and Giza 2006). The same research group performed a study where they used a ketogenic diet on the rats for 1 week after the injury. This diet increased MCT levels, improved the behavioral outcomes and reduced the cortical lesion volumes, but only in younger rats (Appelberg et al. 2009).
It is also interesting to note that another study by Prins and collaborators showed that the metabolic depression occurring after a single head injury reflects the time-course of vulnerability of the brain. They suggest that glucose metabolism could serve as a biomarker to determine the duration of cerebral vulnerability. This concept would be very important to consider, especially for repeated brain injuries within the context of sports injuries (Prins et al. 2013).
2) Early BBB opening
The effect of TBI on the BBB differs according to the age, the type and the severity of the injury. The changes of the BBB have mainly been studied at early time points and they can range from simple alterations to complete rupture of the biochemical and functional properties. Opening of the BBB happens immediately after a TBI and contributes to vasogenic edema. In a concussion model in cats, the opening has been shown as early as 3 minutes after injury (Povlishock et al. 1978).
Several components of the BBB can be affected after a TBI. First, the endothelium itself has been shown to sustain morphological lesions. In a cat model of moderate to severe fluid percussion injury, electron microscopy revealed the presence of two types of lesions on the luminal surface of pial arterioles: cratershaped indentations and dome-shaped projections of the endothelial cell surface, typically corresponding to necrotic cells (Wei et al. 1980). In a cortical cold injury model in rats, microvessels in the lesion area (from the pial surface to the fourth cortical layer) are found necrotic early after the impact (Nag et al. 2007). Interestingly, the smooth muscle layer of the vessels was not morphologically altered by the impact (Wei et al. 1980).
Moreover, the tight junction complex has been shown to be affected after different experimental models of TBI. Early after the injury, pial and intracerebral vessels show decreased claudin-5 (at 2 days) and occludin (at 2 and 4 days) levels in a model of cortical cold injury in rats (Nag et al. 2007). Interestingly, in a model of mild closed head injury, tight junction complexes appear intact under electron microscopy during the first hours after injury (Rafols et al. 2007). However, it has been shown that in models of mild TBI induced by blast shock waves, there is a loss in tight junction proteins (occludin, claudin-5 and zonula occludens protein-1) at 6 and 24 h post-injury (Abdul-Muneer et al. 2013) and increased immunoglobulin G extravasation 5 minutes, 24 and 48 h post-injury (Yeoh et al. 2013), as reviewed in Shetty et al. (2014). In a juvenile CCI model, immunoglobulin G extravasation levels are high near the injury site and surrounding tissue at 1 and 3 days, and are lower by 7 days (Pop and Badaut 2011).
After a TBI, the upregulation of different MMPs is known to participate in the degradation of the BBB. MMP-9 and MMP-2 increase acutely after TBI in rodents (Wang et al. 2000; Zhang et al. 2010). MMP-3 activity, however, is increased chronically after TBI in rats and may play a role in synapse restoration (Zhang et al. 2010). After TBI in a P7 rat, MMP-2 and MMP-9 mRNA and protein levels are elevated in the injured tissue (Sifringer et al. 2007).
Finally, astrocytes, which are a key component of the BBB (Abbott et al. 2006), have been shown to be decreased in number after lateral fluid percussion injury in the adult rat hippocampus (Hill-Felberg et al. 1999; Zhao et al. 2003), contributing to BBB alteration. Astrocyte loss starts as early as 30 minutes post-injury and continues until 24 h (Zhao et al. 2003). Another study focused on later time points and showed that the astrocyte population in the injured hippocampus at 7 days was reduced by 64% of the total population compared to uninjured rats. After 1 month, the astrocyte population is restored to 85%, showing compensation for the earlier cell loss (Hill-Felberg et al. 1999).
3) Smooth muscle cell changes
As described earlier, the cerebral blood vessels contain a smooth muscle layer called tunica media in their vascular wall, in cortical arterioles consisting of one to three layers of smooth muscle cells (SMC; Figure 1) (Rafols et al. 2007). During vascular development, SMCs present mostly a synthetic phenotype, with high growth and synthetic rates, contributing to the secretion of extracellular matrix proteins like collagen and elastin, which are important for the stability of the blood vessels (Wagenseil and Mecham 2009). In adult blood vessels, SMCs acquire a contractile phenotype, with a low proliferation rate and a low synthetic activity. They express a number of contractile proteins such as alpha smooth muscle actin (αSMA), smooth muscle-myosin heavy chain (SM-MHC), smoothelin-A/B, calponin, as well as ion channels, and signaling molecules required for the contractile function of the cell (Owens et al. 2004; Rensen et al. 2007). Non-muscle MHC isoform-B and cellular retinol binding protein (CRBP)-1 are described as suitable markers for synthetic SMCs. They are upregulated when MHC is decreased in the proliferating SMCs (Rensen et al. 2007). Both synthetic and contractile phenotypes exist in the smooth muscle layer, but it should be noted that they represent two ends of a wide spectrum of SMCs with intermediate phenotypes. The ratio of synthetic and contractile markers changes depending on developmental age, the vascular environment and the physio/pathological situation (Rensen et al. 2007).
SMCs have been shown to exhibit long-term changes after a TBI and are therefore involved in different types of secondary injuries. Endothelin-1 is a peptide released mainly from the vascular endothelium and induced after TBI (Inoue et al. 1989). It contributes to increased αSMA in SMC and pericyte during the first hours post-injury, resulting in a decreased diameter of arterioles (Dore-Duffy et al. 2011). Molecular changes have been observed in other potential contractile proteins such as calponin in rodent models of TBI. Calponin expression is significantly increased during the first 48h, in association with enhanced vasoreactivity. This modification is also under the influence of the endothelin pathway (Kreipke and Rafols 2009). Inhibition of calponin phosphorylation reduces changes in vasoreactivity post-TBI and is associated with improved CBF (Kreipke and Rafols 2009). Similar phenotypic switching has been observed in an in vitro model of SMCs exposed to blast injury showing a mRNA decrease of the contractile protein smoothelin and an absence of SM-MHC in relation to vasospasm after blast-TBI (Alford et al. 2011).
Together with autoregulation, myogenic regulation is one of the mechanisms regulating the constancy of the blood flow during changes in perfusion pressure. The myogenic response is initiated by the vascular smooth muscle itself. For example, when the smooth muscle stretches, SMCs contract. After head trauma, the myogenic response can be impaired by responding abnormally to pressure changes due to various molecular mechanisms, including elevated Protein Kinase C activity and altered Transient Receptor Potential channels (Golding et al. 1998; Mathew et al. 1999).
Nitric oxide (NO) signaling has been shown to be involved in the regulation of cerebrovascular tone (Hamel 2006). The effects of NO are balanced between favorable, due to NO-mediated stimulation of cGMP and vasodilation at low concentrations, and unfavorable due to free-radical related pro-inflammatory effects at high concentrations. The activity of endothelial NOS (eNOS) exhibits a bimodal change after TBI with an initial increase spanning a few minutes followed by a ~50% decrease relative to baseline levels for 7 days before normalizing (Cherian et al. 2004; Wada et al. 1998). This decrease of constitutive NOS activity may contribute to altered CBF and cerebral autoregulation, in combination with changes in the myogenic response of the SMCs. Inducible NOS (iNOS) expression and activity have been shown to be increased not only in SMCs but also in neurons, macrophages, neutrophils, astrocytes, and oligodendrocytes, reaching peak levels between 4h and 48h after injury (Cherian et al. 2004; Clark et al. 1996; Steiner et al. 2004). Unfortunately, up-regulation of iNOS results in a harmful increase of tissue NO levels (Cherian et al. 2004) that are well known to contribute to the secondary injury cascade including neuroinflammation, apoptosis, excitotoxicity, energy depletion, and production of reactive oxygen species (Guix et al. 2005).
4) Changes in perivascular innervation
Along with the changes observed in endothelial and SMCs, it has been suggested that the perivascular innervation could be altered after TBI and thereby involved in the cerebrovascular dysfunction. The perivascular nerve plexus is part of the neurogenic regulation of the vascular tone of the pial and larger arteries and several studies have shown that the cerebrovascular response to vasoactive substances is impaired after TBI (Armstead 1997; Fujita et al. 2012; Wei et al. 1980). Significant changes are observed in the perivascular nerves of cerebral arteries during the first week after severe diffuse TBI induced by impact acceleration (Ueda et al. 2006). A decrease in the number of perivascular nerve fibers peaking at 24h after injury have been described, and in some instances a decrease in perivascular nerve fibers up to 7 days post-injury (Ueda et al. 2006). This decrease in the number of fibers could be due to erythrocyte toxicity after SAH since blood is known to cause the denervation of cerebral arteries 3 to 7 days after exposure (Duff et al. 1986; Sercombe et al. 2002). It is associated with a decrease in the concentration of vasoactive substances like acetylcholine, vasoactive intestinal peptide, substance P, and calcitonin gene-related peptide (Sercombe et al. 2002). Therefore, the neurogenic control of the vascular tone is affected by a TBI, due to the decreased number of nerve fibers in the perivascular area.
5) Astrogliosis and consequences on blood perfusion and BBB
In severe cases of injury, astrocytes become reactive and proliferate to form a glial scar (Burda and Sofroniew 2014). The role of astrogliosis is still debated as it can be both beneficial and detrimental to the surrounding tissues, depending on the timeline of the pathological processes (Sofroniew 2009). Regardless of this controversy, the process of astrogliosis can have a profound impact on the events following a brain injury as astrocytes have numerous functions in the healthy central nervous system such as providing energy for neurons, regulating ion and neurotransmitter homeostasis, participating in synapse development and function, and regulation of CBF (Sofroniew and Vinters 2010).
Astrocytes are among the first cells to respond to TBI and the mechanical forces of TBI trigger astrogliosis (Burda et al. 2015). It seems that astrocytes are especially vulnerable to mechanical injury and even though it is still not clear how this response is mediated, it could involve the activation of astrocytic mechanoreceptive channels (Burda et al. 2015). The activation of astrocytes with stretch injury or shear stress involves a rapid calcium influx (Maneshi et al. 2015; Rzigalinski et al. 1998). Moreover, TBI induces activation of signaling pathways in astrocytes including inositol triphosphate (IP3) signaling with IP3 concentration being increased up to 48 h post-injury (Floyd et al. 2001). The induction of IP3 signaling increases intracellular calcium concentration, which in turn activates phospholipase A2 (among many other calcium-sensitive enzymes such as Protein Kinase C and calpain) and can influence the release of vasoactive substances (Howarth 2014). Therefore, any changes in the intracellular calcium concentration within astrocytes post-injury could influence the release of vasoactive substances and as a consequence the regulation of CBF, perfusion, and contribute to the cerebrovascular dysfunction post-TBI.
It has been demonstrated that several vasoactive substances can be released from astrocytes after trauma. Stretch injury to primary astrocyte cultures induced a release of isoprostanes (Hoffman et al. 2000), shown to be vasoconstrictors of cerebral arterioles (Hoffman et al. 1997). Interestingly, increased levels of isoprostanes in CSF have been observed in adult and pediatric patients after moderate and severe TBI (Bayir et al. 2002; Varma et al. 2003; Yen et al. 2015). Therefore, increased secretion of isoprostanes from astrocytes could contribute to the decreased cerebral perfusion after TBI. Similarly, a stretch injury on primary astrocyte cultures provoked endothelin 1 release from astrocytes associated with calcium influx (Ostrow et al. 2000). Endothelin 1 is another powerful vasoconstrictor and its levels are increased after TBI in multiple animal models (Armstead and Kreipke 2011; Armstead and Raghupathi 2011; Petrov and Rafols 2001). Increases in endothelin 1 have been associated with unfavorable outcomes in children after severe TBI (Salonia et al. 2010). In addition, an increase in the expression of endothelin 1 receptors has also been observed after TBI in several animal models (Kallakuri et al. 2010).
In addition to the role of astrocytes in vascular tone, they play a key role in the integrity of the BBB (Abbott et al. 2006). In fact, they can release various factors such as cytokines and inflammatory mediators that can affect the BBB after brain injury (Chodobski et al. 2011; Sofroniew 2014). For example, astrocytic secretion of chemokines (Chodobski et al. 2011) and three isoforms of transforming growth factor-β (TGF-β) (Constam et al. 1992) have been shown to contribute to the BBB leakage after brain injury (Shen et al. 2011). In patients, increased TGF-β expression has been observed in patients with severe TBI where it parallels BBB function (Morganti-Kossmann et al. 1999). As already discussed, MMPs affect the structure of the BBB after TBI and they are extensively produced by reactive astrocytes (Chen and Swanson 2003).
The expression of the water channel aquaporin 4, expressed mainly on astrocyte endfeet in proximity of blood vessels in the cortex, are also altered after TBI, which is related to the formation of cerebral edema (Badaut et al. 2013). There is a correlation between the level of aquaporin 4 expression and disruption of BBB (Fukuda and Badaut 2012). In addition to the secretion of different molecules that can affect the BBB integrity and cerebral perfusion, the physical interaction between astrocytes and the vasculature can be also changed after TBI (Villapol et al. 2014), which all can contribute to the vascular dysfunction after TBI.
Altogether, astrocytes change their phenotype and can contribute to the vascular pathology observed after TBI. The neurodegeneration observed after TBI could be a result of chronic neuroinflammation and astrocyte activation (Faden and Loane 2015). Therefore, it may be important to consider the contribution of reactive astrocytes into the long-term consequences of vascular dysfunction.
V) Long-term pathological mechanisms behind vascular dysfunctions
1) Blood-brain barrier dysfunctions and long-term degeneration
As discussed above, there is a wide range of alterations in early time points after TBI to the brain vasculature. These changes support the hypothesis that TBI is a cerebrovascular injury with major dysfunction of the NVU after the primary impact. Most of the studies have so far focused on neuronal cell death and long-term recovery after TBI. However, little is known about the long-lasting changes in the brain vasculature and their involvement in functional outcome.
For a long time, the opening of the BBB was considered a short-term event that normalized within one week, as we observed in our rodent TBI model (Pop and Badaut 2011). However, the BBB still remains opened as late as 30 days after an insult in a stroke model (Strbian et al. 2008), suggesting long lasting changes of the endothelial properties. At 2 months post-injury in our juvenile TBI model, the BBB function seems to be restored since IgG staining was not detected around the lesion site (Pop et al. 2013). Moreover, claudin-5 expression in the penetrating arteries is significantly increased 2 months post-injury (Pop et al. 2013) as well as 2 weeks after moderate compression of rat somatosensory cortex in a different study (Lin et al. 2010). These observations suggest that the long-term phenotypic transformations of the endothelial cells compensate for the early BBB alteration (Pop et al. 2013).
TBI-induced long-term neurodegeneration is in part related to its effects on the BBB. Many studies showed that TBI can accelerate brain aging and promote accumulation of aberrant proteins such as amyloid-β (Aβ) (Johnson et al. 2010). Our recent studies suggest that when a TBI occurs early in life it can have long-term consequences. We previously reported in juvenile mild TBI an impairment of sensorimotor function and spatial memory deficits up to 6 months after the injury (Kamper et al. 2013). Some studies have suggested a link between vascular function and cognitive deficits (Alosco et al. 2013) and it is possible that the cognitive impairments we observed could be due to phenotypic changes of the BBB, including a decrease of the efflux pump P-glycoprotein (P-gp), and an increase of the perivascular matrix proteins perlecan and fibronectin. These changes are observed at 2 and 6 months after injury (Jullienne et al. 2014), and the matrix changes have also been observed in AD patients (Lepelletier et al. 2015). Changes in the matrix properties possibly participate in neurodegenerative processes by leading to the accumulation of Aβ, decreasing its clearance and its perivascular drainage (Jullienne et al. 2014; Pop et al. 2013).
P-gp has been suggested to be a key player in Aβ clearance from the brain parenchyma since its expression is decreased on endothelial cells in aged human and AD brains as well as in aged rodents (Silverberg et al. 2010). In addition, P-gp knock-out models have increased Aβ deposition after injection of Aβ in the brain (Cirrito et al. 2005).
The caveolin protein family takes part in the caveolae formation, which is known to be involved in endocytosis, transcytosis, and exocytosis in endothelial cells (Jodoin et al. 2003; Predescu et al. 2007). Caveolin-1 (cav-1), is one of 3 isoforms that is expressed by brain endothelial cells (Lisanti et al. 1994; Virgintino et al. 2002). Cav-1 also modulates the activity of signaling molecules, including inhibition of eNOS (Bauer et al. 2005; Bucci et al. 2000). Its expression is increased in the endothelium during the first week after a cold injury model of TBI (Nag et al. 2007). In support of the phenotypic transformation of the endothelium after juvenile TBI, we observed an increase in cav-1 immunostaining in brain cortical vessels at 2 months post-injury (Badaut et al. 2015; Badaut and Bix 2014). This observation also suggests that cav-1 could play a role in altered claudin-5 and P-gp expression that occur after TBI. Interestingly, cav-1 has been associated with stabilization of claudin-5 within the caveolae (McCaffrey et al. 2007) and a decrease of P-gp activity (McCaffrey et al. 2012).
We believe that the changes in the BBB properties at short and long-term after TBI will have direct and indirect consequences on the astrocytic properties and subsequently brain perfusion.
2) SMC changes long-term after TBI
It is known that phenotypic changes of SMCs play a critical role in various major human diseases, including cancer, hypertension, asthma, and atherosclerosis. SMCs exhibit a high phenotypic plasticity, which allows them to switch from a contractile function (to regulate blood vessel diameter) to a secretive function (to produce extracellular matrix proteins) (Alexander and Owens 2012).
The decrease of the CBF observed after TBI would generate chronic hypoxia throughout the vascular tree. In sheep, the SMCs of the carotid artery after chronic hypoxia differentiate from contractile to synthetic phenotype under a VEGF-driven mechanism, leading to an altered arterial contractility (Hubbell et al. 2012). SMCs lose their contractile properties via a decrease of SM-MHC and an increase of nonmuscle MHC (Hubbell et al. 2012). Moreover, hypoxia induces an increase in contractile protein myosin light chain (MLC)-20 and a decrease of MLC-kinase by up-regulating VEGF-receptors Flk-1 and Flt-1 (Adeoye et al. 2013). VEGF levels change in TBI (Mellergard et al. 2010; Morgan et al. 2007), but so do other growth factors and cytokines. SMC phenotypic modulation in the periphery is under the control of several molecular pathways that could be present after TBI, such as PDGF-BB, PDGF-DD, and IL-1β. All these pathways can induce rapid downregulation in expression of multiple SMC differentiation marker genes, including αSMA, SM-MHC, and calponin (Alexander and Owens 2012). A wide range of signaling pathways are involved in the responses to these molecular proteins and it is highly possible that the early changes in the NVU environment can contribute to long-term changes in the SMC phenotype.
VI) Conclusions and future therapeutic strategies
TBI is a massive health burden wherein basic science researchers and clinicians have shown that it is undeniably also a cerebrovascular injury. Each of the components of the NVU play key roles in the pathogenesis of injury, during the short and long-term time points after injury. Molecular and cellular mechanisms leading to long-term impairments are dependent upon the type of injury, the severity, as well as the age of the patient, and these parameters need to be considered in each case to improve recovery. This review highlights important targets regarding the management of secondary injuries as they relate to the cerebral vasculature. Indeed, endothelial cells, astrocytes, and smooth muscle cells are all involved in CBF regulation, edema formation, BBB disruption, vasospasms, and energy metabolism. The review pinpoints the involvement of multiple players within the NVU. Therefore, future therapeutic strategies should reflect this complexity and combine different treatment at different time points. This strategy has recently been discussed by Margulies and colleagues in a review entitled “Combination Therapies for Traumatic Brain Injury: Retrospective Considerations” (Margulies et al. 2016). This article provides an overview of six different projects aiming for multidrug testing with insights, difficulties and recommendations for future therapy development for TBI. Our review of the literature suggests that intravenous injection of lactate (Holloway et al. 2007), inhalation of NO (Terpolilli et al. 2013), injection of an inhibitor of c-Jun-N-terminal kinase (Borsello et al. 2003), and acting on extracellular matrix proteins such as perlecan domain V (Jullienne et al. 2014) are promising targets and could be considered to be combined in the aim of restoring vascular function. There is therefore a continuous need to study the mechanisms by which these cellular constituents respond to TBI and how they modulate the cerebrovascular tone.
Significance statement.
Traumatic brain injury (TBI) is a major health burden and researchers have been extensively focusing on the neurocentric aspect of the disorder. Early changes in cerebral blood vessels after TBI have been known for a long time. However, it is now becoming clear that the different types of neurovascular dysfunction play major roles in TBI pathogenesis. We review and outline the various cerebrovascular dysfunctions occurring after a TBI, such as cerebral blood flow and autoregulation impairment, subarachnoid hemorrhage, vasospasm, blood-brain barrier disruption and edema formation.
Acknowledgments
Preparation of this article was supported in part by funds from NIH NINDS (1 P01 NS082184-01A1), NIH NICHD R01HD061946, Fondation des gueules cassées, EraNet Neuron CNS-Aflame and TRAIL-ANR investissement d’avenir, France
Footnotes
Conflict of interest statement:
No author has a conflict of interest.
References
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature reviews Neuroscience. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, Zimmerman MC, Chandra N, Haorah J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free radical biology & medicine. 2013;60:282–291. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelson PD, Srinivas R, Chang Y, Bell M, Kochanek PM. Cerebrovascular response in children following severe traumatic brain injury. Child’s nervous system: ChNS: official journal of the International Society for Pediatric Neurosurgery. 2011;27(9):1465–1476. doi: 10.1007/s00381-011-1476-z. [DOI] [PubMed] [Google Scholar]
- Adeoye OO, Butler SM, Hubbell MC, Semotiuk A, Williams JM, Pearce WJ. Contribution of increased VEGF receptors to hypoxic changes in fetal ovine carotid artery contractile proteins. American journal of physiology Cell physiology. 2013;304(7):C656–665. doi: 10.1152/ajpcell.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annual review of physiology. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- Alford PW, Dabiri BE, Goss JA, Hemphill MA, Brigham MD, Parker KK. Blast-induced phenotypic switching in cerebral vasospasm. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(31):12705–12710. doi: 10.1073/pnas.1105860108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alosco ML, Spitznagel MB, Cohen R, Raz N, Sweet LH, Josephson R, Hughes J, Rosneck J, Gunstad J. Reduced cerebral perfusion predicts greater depressive symptoms and cognitive dysfunction at a 1-year follow-up in patients with heart failure. Int J Geriatr Psychiatry. 2013 doi: 10.1002/gps.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M, Alvarez-Cermeno J, Bernardi G, Cogato I, Fredman P, Frederiksen J, Fredrikson S, Gallo P, Grimaldi LM, Gronning M, et al. Cerebrospinal fluid in the diagnosis of multiple sclerosis: a consensus report. Journal of neurology, neurosurgery, and psychiatry. 1994;57(8):897–902. doi: 10.1136/jnnp.57.8.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelberg KS, Hovda DA, Prins ML. The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. Journal of neurotrauma. 2009;26(4):497–506. doi: 10.1089/neu.2008.0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armin SS, Colohan AR, Zhang JH. Traumatic subarachnoid hemorrhage: our current understanding and its evolution over the past half century. Neurological research. 2006;28(4):445–452. doi: 10.1179/016164106X115053. [DOI] [PubMed] [Google Scholar]
- Armonda RA, Bell RS, Vo AH, Ling G, DeGraba TJ, Crandall B, Ecklund J, Campbell WW. Wartime traumatic cerebral vasospasm: recent review of combat casualties. Neurosurgery. 2006;59(6):1215–1225. doi: 10.1227/01.NEU.0000249190.46033.94. discussion 1225. [DOI] [PubMed] [Google Scholar]
- Armstead WM. Brain injury impairs ATP-sensitive K+ channel function in piglet cerebral arteries. Stroke. 1997;28(11):2273–2279. doi: 10.1161/01.str.28.11.2273. discussion 2280. [DOI] [PubMed] [Google Scholar]
- Armstead WM, Kreipke CW. Endothelin-1 is upregulated after traumatic brain injury: a cross-species, cross-model analysis. Neurological research. 2011;33(2):133–136. doi: 10.1179/016164111X12881719352174. [DOI] [PubMed] [Google Scholar]
- Armstead WM, Raghupathi R. Endothelin and the neurovascular unit in pediatric traumatic brain injury. Neurological research. 2011;33(2):127–132. doi: 10.1179/016164111X12881719352138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal S, Holshouser BA, Shu SK, Simmons PL, Perkin RM, Tomasi LG, Knierim DS, Sheridan C, Craig K, Andrews GH, Hinshaw DB. Predictive value of proton magnetic resonance spectroscopy in pediatric closed head injury. Pediatric neurology. 2000;23(2):114–125. doi: 10.1016/s0887-8994(00)00176-4. [DOI] [PubMed] [Google Scholar]
- Ashwal S, Tong KA, Ghosh N, Bartnik-Olson B, Holshouser BA. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. Journal of child neurology. 2014;29(12):1704–1717. doi: 10.1177/0883073814538504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468(7321):232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Ajao DO, Sorensen DW, Fukuda AM, Pellerin L. Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: signs of blood-brain barrier healing? Neuroscience. 2015;285:215–226. doi: 10.1016/j.neuroscience.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Bix GJ. Vascular neural network phenotypic transformation after traumatic injury: potential role in long-term sequelae. Translational stroke research. 2014;5(3):394–406. doi: 10.1007/s12975-013-0304-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Fukuda AM, Jullienne A, Petry KG. Aquaporin and brain diseases. Biochimica et Biophysica Acta – General Subjects. 2013 doi: 10.1016/j.bbagen.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartnik BL, Lee SM, Hovda DA, Sutton RL. The fate of glucose during the period of decreased metabolism after fluid percussion injury: a 13C NMR study. Journal of neurotrauma. 2007;24(7):1079–1092. doi: 10.1089/neu.2006.0210. [DOI] [PubMed] [Google Scholar]
- Bauer PM, Yu J, Chen Y, Hickey R, Bernatchez PN, Looft-Wilson R, Huang Y, Giordano F, Stan RV, Sessa WC. Endothelial-specific expression of caveolin-1 impairs microvascular permeability and angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(1):204–209. doi: 10.1073/pnas.0406092102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatric research. 2002;51(5):571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Bergsneider M, Hovda DA, McArthur DL, Etchepare M, Huang SC, Sehati N, Satz P, Phelps ME, Becker DP. Metabolic recovery following human traumatic brain injury based on FDG-PET: time course and relationship to neurological disability. The Journal of head trauma rehabilitation. 2001;16(2):135–148. doi: 10.1097/00001199-200104000-00004. [DOI] [PubMed] [Google Scholar]
- Blanco VM, Stern JE, Filosa JA. Tone-dependent vascular responses to astrocyte-derived signals. Am J Physiol Heart Circ Physiol. 2008;294(6):H2855–2863. doi: 10.1152/ajpheart.91451.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyth BJ, Farhavar A, Gee C, Hawthorn B, He H, Nayak A, Stocklein V, Bazarian JJ. Validation of serum markers for blood-brain barrier disruption in traumatic brain injury. Journal of neurotrauma. 2009;26(9):1497–1507. doi: 10.1089/neu.2008.0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nature medicine. 2003;9(9):1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Bouma GJ, Muizelaar JP, Choi SC, Newlon PG, Young HF. Cerebral circulation and metabolism after severe traumatic brain injury: the elusive role of ischemia. Journal of neurosurgery. 1991;75(5):685–693. doi: 10.3171/jns.1991.75.5.0685. [DOI] [PubMed] [Google Scholar]
- Bouma GJ, Muizelaar JP, Stringer WA, Choi SC, Fatouros P, Young HF. Ultraearly evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. Journal of neurosurgery. 1992;77(3):360–368. doi: 10.3171/jns.1992.77.3.0360. [DOI] [PubMed] [Google Scholar]
- Bryan RM, Jr, Cherian L, Robertson C. Regional cerebral blood flow after controlled cortical impact injury in rats. Anesthesia and analgesia. 1995;80(4):687–695. doi: 10.1097/00000539-199504000-00007. [DOI] [PubMed] [Google Scholar]
- Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, Sessa WC. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nature medicine. 2000;6(12):1362–1367. doi: 10.1038/82176. [DOI] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Exp Neurol. 2015 doi: 10.1016/j.expneurol.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81(2):229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. Astrocytes and brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23(2):137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- Cherian L, Hlatky R, Robertson CS. Nitric oxide in traumatic brain injury. Brain Pathol. 2004;14(2):195–201. doi: 10.1111/j.1750-3639.2004.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Translational stroke research. 2011;2(4):492–516. doi: 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ, Li R, Vitullo L. Perivascular innervation of penetrating brain parenchymal arterioles. Journal of cardiovascular pharmacology. 2004;44(1):1–8. doi: 10.1097/00005344-200407000-00001. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. The Journal of clinical investigation. 2005;115(11):3285–3290. doi: 10.1172/JCI25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Schwarz MA, Schiding JK, Turner DS, Chen M, Carlos TM, Watkins SC. Inducible nitric oxide synthase expression in cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats. Pediatric research. 1996;39(5):784–790. doi: 10.1203/00006450-199605000-00007. [DOI] [PubMed] [Google Scholar]
- Cohen Z, Bonvento G, Lacombe P, Hamel E. Serotonin in the regulation of brain microcirculation. Progress in neurobiology. 1996;50(4):335–362. doi: 10.1016/s0301-0082(96)00033-0. [DOI] [PubMed] [Google Scholar]
- Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. Journal of immunology. 1992;148(5):1404–1410. [PubMed] [Google Scholar]
- Corso P, Finkelstein E, Miller T, Fiebelkorn I, Zaloshnja E. Incidence and lifetime costs of injuries in the United States. Injury prevention: journal of the International Society for Child and Adolescent Injury Prevention. 2006;12(4):212–218. doi: 10.1136/ip.2005.010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt DS, Prough DS. Traumatic cerebral vascular injury: the effects of concussive brain injury on the cerebral vasculature. Journal of neurotrauma. 2003;20(9):795–825. doi: 10.1089/089771503322385755. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, Wang S, Mehedi A, Katyshev V, Cleary K, Tapper A, Reynolds C, Ding Y, Zhan P, Rafols J, Kreipke CW. Pericyte-mediated vasoconstriction underlies TBI-induced hypoperfusion. Neurological research. 2011;33(2):176–186. doi: 10.1179/016164111X12881719352372. [DOI] [PubMed] [Google Scholar]
- Duff TA, Scott G, Feilbach JA. Ultrastructural evidence of arterial denervation following experimental subarachnoid hemorrhage. Journal of neurosurgery. 1986;64(2):292–297. doi: 10.3171/jns.1986.64.2.0292. [DOI] [PubMed] [Google Scholar]
- Eisenberg HM, Gary HE, Jr, Aldrich EF, Saydjari C, Turner B, Foulkes MA, Jane JA, Marmarou A, Marshall LF, Young HF. Initial CT findings in 753 patients with severe head injury. A report from the NIH Traumatic Coma Data Bank. Journal of neurosurgery. 1990;73(5):688–698. doi: 10.3171/jns.1990.73.5.0688. [DOI] [PubMed] [Google Scholar]
- Enevoldsen EM, Jensen FT. Autoregulation and CO2 responses of cerebral blood flow in patients with acute severe head injury. Journal of neurosurgery. 1978;48(5):689–703. doi: 10.3171/jns.1978.48.5.0689. [DOI] [PubMed] [Google Scholar]
- Faden AI, Loane DJ. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2015;12(1):143–150. doi: 10.1007/s13311-014-0319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, Coronado V. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths, 2002–2006. Atlanta, GA: CDC; 2010. (National Center for Injury Prevention and Control). [Google Scholar]
- Filosa JA, Morrison HW, Iddings JA, Du W, Kim KJ. Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. Neuroscience. 2015 doi: 10.1016/j.neuroscience.2015.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd CL, Rzigalinski BA, Weber JT, Sitterding HA, Willoughby KA, Ellis EF. Traumatic injury of cultured astrocytes alters inositol (1,4,5)-trisphosphate-mediated signaling. Glia. 2001;33(1):12–23. doi: 10.1002/1098-1136(20010101)33:1<12::aid-glia1002>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Fujita M, Wei EP, Povlishock JT. Intensity-and interval-specific repetitive traumatic brain injury can evoke both axonal and microvascular damage. Journal of neurotrauma. 2012;29(12):2172–2180. doi: 10.1089/neu.2012.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda AM, Badaut J. Aquaporin 4: a player in cerebral edema and neuroinflammation. Journal of neuroinflammation. 2012;9:279. doi: 10.1186/1742-2094-9-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Lange AR, Lowry TF, Taheri MR, Birks EK, Hudetz AG, Narayanan J, Falck JR, Okamoto H, Roman RJ, Nithipatikom K, Campbell WB, Harder DR. Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ Res. 2000;87(1):60–65. doi: 10.1161/01.res.87.1.60. [DOI] [PubMed] [Google Scholar]
- Glenn TC, Martin NA, Horning MA, McArthur DL, Hovda DA, Vespa P, Brooks GA. Lactate: brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. Journal of neurotrauma. 2015;32(11):820–832. doi: 10.1089/neu.2014.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding EM. Sequelae following traumatic brain injury. The cerebrovascular perspective. Brain research Brain research reviews. 2002;38(3):377–388. doi: 10.1016/s0165-0173(02)00141-8. [DOI] [PubMed] [Google Scholar]
- Golding EM, Contant CF, Jr, Robertson CS, Bryan RM., Jr Temporal effect of severe controlled cortical impact injury in the rat on the myogenic response of the middle cerebral artery. Journal of neurotrauma. 1998;15(11):973–984. doi: 10.1089/neu.1998.15.973. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456(7223):745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotta JC, Moskowitz MA, Jacobs TP, Marler J, Woodbury-Harris K, Radziszewska B, Scott PA. Report of the Stroke Progress Review Group. National Institute of Neurological Disorders and Stroke; 2002. [online], < http://www.ninds.nih.gov/findpeople/groups/strokeprg/StrokePRGreport-4-23-02.pdf>. [Google Scholar]
- Guix FX, Uribesalgo I, Coma M, Munoz FJ. The physiology and pathophysiology of nitric oxide in the brain. Progress in neurobiology. 2005;76(2):126–152. doi: 10.1016/j.pneurobio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Cantu RC, Randolph C, Jordan BD. Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery. 2005;57(4):719–726. doi: 10.1093/neurosurgery/57.4.719. discussion 719–726. [DOI] [PubMed] [Google Scholar]
- Gustavsson A, Svensson M, Jacobi F, Allgulander C, Alonso J, Beghi E, Dodel R, Ekman M, Faravelli C, Fratiglioni L, Gannon B, Jones DH, Jennum P, Jordanova A, Jonsson L, Karampampa K, Knapp M, Kobelt G, Kurth T, Lieb R, Linde M, Ljungcrantz C, Maercker A, Melin B, Moscarelli M, Musayev A, Norwood F, Preisig M, Pugliatti M, Rehm J, Salvador-Carulla L, Schlehofer B, Simon R, Steinhausen HC, Stovner LJ, Vallat JM, Van den Bergh P, van Os J, Vos P, Xu W, Wittchen HU, Jonsson B, Olesen J, Group CD Cost of disorders of the brain in Europe 2010. European neuropsychopharmacology: the journal of the European College of Neuropsychopharmacology. 2011;21(10):718–779. doi: 10.1016/j.euroneuro.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100(3):1059–1064. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- Hayward NM, Immonen R, Tuunanen PI, Ndode-Ekane XE, Grohn O, Pitkanen A. Association of chronic vascular changes with functional outcome after traumatic brain injury in rats. Journal of neurotrauma. 2010;27(12):2203–2219. doi: 10.1089/neu.2010.1448. [DOI] [PubMed] [Google Scholar]
- Hill-Felberg SJ, McIntosh TK, Oliver DL, Raghupathi R, Barbarese E. Concurrent loss and proliferation of astrocytes following lateral fluid percussion brain injury in the adult rat. Journal of neuroscience research. 1999;57(2):271–279. doi: 10.1002/(SICI)1097-4547(19990715)57:2<271::AID-JNR13>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Himanen L, Portin R, Hamalainen P, Hurme S, Hiekkanen H, Tenovuo O. Risk factors for reduced survival after traumatic brain injury: a 30-year follow-up study. Brain injury : [BI] 2011;25(5):443–452. doi: 10.3109/02699052.2011.556580. [DOI] [PubMed] [Google Scholar]
- Ho KM, Honeybul S, Yip CB, Silbert BI. Prognostic significance of blood-brain barrier disruption in patients with severe nonpenetrating traumatic brain injury requiring decompressive craniectomy. Journal of neurosurgery. 2014;121(3):674–679. doi: 10.3171/2014.6.JNS132838. [DOI] [PubMed] [Google Scholar]
- Hoffman SW, Moore S, Ellis EF. Isoprostanes: free radical-generated prostaglandins with constrictor effects on cerebral arterioles. Stroke. 1997;28(4):844–849. doi: 10.1161/01.str.28.4.844. [DOI] [PubMed] [Google Scholar]
- Hoffman SW, Rzigalinski BA, Willoughby KA, Ellis EF. Astrocytes generate isoprostanes in response to trauma or oxygen radicals. Journal of neurotrauma. 2000;17(5):415–420. doi: 10.1089/neu.2000.17.415. [DOI] [PubMed] [Google Scholar]
- Holloway R, Zhou Z, Harvey HB, Levasseur JE, Rice AC, Sun D, Hamm RJ, Bullock MR. Effect of lactate therapy upon cognitive deficits after traumatic brain injury in the rat. Acta neurochirurgica. 2007;149(9):919–927. doi: 10.1007/s00701-007-1241-y. discussion 927. [DOI] [PubMed] [Google Scholar]
- Hovda DA, Yoshino A, Kawamata T, Katayama Y, Becker DP. Diffuse prolonged depression of cerebral oxidative metabolism following concussive brain injury in the rat: a cytochrome oxidase histochemistry study. Brain research. 1991;567(1):1–10. doi: 10.1016/0006-8993(91)91429-5. [DOI] [PubMed] [Google Scholar]
- Howarth C. The contribution of astrocytes to the regulation of cerebral blood flow. Frontiers in neuroscience. 2014;8:103. doi: 10.3389/fnins.2014.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell MC, Semotiuk AJ, Thorpe RB, Adeoye OO, Butler SM, Williams JM, Khorram O, Pearce WJ. Chronic hypoxia and VEGF differentially modulate abundance and organization of myosin heavy chain isoforms in fetal and adult ovine arteries. American journal of physiology Cell physiology. 2012;303(10):C1090–1103. doi: 10.1152/ajpcell.00408.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nature reviews Neuroscience. 2004;5(5):347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Immonen R, Heikkinen T, Tahtivaara L, Nurmi A, Stenius TK, Puolivali J, Tuinstra T, Phinney AL, Van Vliet B, Yrjanheikki J, Grohn O. Cerebral blood volume alterations in the perilesional areas in the rat brain after traumatic brain injury–comparison with behavioral outcome. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30(7):1318–1328. doi: 10.1038/jcbfm.2010.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, Masaki T. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(8):2863–2867. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Shiozaki T, Tasaki O, Hayakata T, Ikegawa H, Yoshiya K, Fujinaka T, Tanaka H, Shimazu T, Sugimoto H. Changes in cerebral blood flow from the acute to the chronic phase of severe head injury. Journal of neurotrauma. 2005;22(12):1411–1418. doi: 10.1089/neu.2005.22.1411. [DOI] [PubMed] [Google Scholar]
- Jodoin J, Demeule M, Fenart L, Cecchelli R, Farmer S, Linton KJ, Higgins CF, Beliveau R. P-glycoprotein in blood-brain barrier endothelial cells: interaction and oligomerization with caveolins. Journal of neurochemistry. 2003;87(4):1010–1023. doi: 10.1046/j.1471-4159.2003.02081.x. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nature reviews Neuroscience. 2010;11(5):361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullienne A, Badaut J. Molecular contributions to neurovascular unit dysfunctions after brain injuries: lessons for target-specific drug development. Future neurology. 2013;8(6):677–689. doi: 10.2217/fnl.13.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullienne A, Roberts JM, Pop V, Paul Murphy M, Head E, Bix GJ, Badaut J. Juvenile traumatic brain injury induces long-term perivascular matrix changes alongside amyloid-beta accumulation. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014 doi: 10.1038/jcbfm.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallakuri S, Kreipke CW, Schafer PC, Schafer SM, Rafols JA. Brain cellular localization of endothelin receptors A and B in a rodent model of diffuse traumatic brain injury. Neuroscience. 2010;168(3):820–830. doi: 10.1016/j.neuroscience.2010.01.018. [DOI] [PubMed] [Google Scholar]
- Kamper JE, Pop V, Fukuda A, Ajao D, Hartman R, Badaut J. Juvenile traumatic brain injury evolves into a chronic brain disorder: Behavioral and histological changes over 6 months. Exp Neurol. 2013;250:8–19. doi: 10.1016/j.expneurol.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney K, Amyot F, Haber M, Pronger A, Bogoslovsky T, Moore C, Diaz-Arrastia R. Cerebral Vascular Injury in Traumatic Brain Injury. Exp Neurol. 2015 doi: 10.1016/j.expneurol.2015.05.019. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Iddings JA, Stern JE, Blanco VM, Croom D, Kirov SA, Filosa JA. Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35(21):8245–8257. doi: 10.1523/JNEUROSCI.4486-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek PM, Marion DW, Zhang W, Schiding JK, White M, Palmer AM, Clark RS, O’Malley ME, Styren SD, Ho C, et al. Severe controlled cortical impact in rats: assessment of cerebral edema, blood flow, and contusion volume. Journal of neurotrauma. 1995;12(6):1015–1025. doi: 10.1089/neu.1995.12.1015. [DOI] [PubMed] [Google Scholar]
- Kreipke CW, Rafols JA. Calponin control of cerebrovascular reactivity: therapeutic implications in brain trauma. Journal of cellular and molecular medicine. 2009;13(2):262–269. doi: 10.1111/j.1582-4934.2008.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhan SE, Kirchgessner A. Chronic traumatic encephalopathy: the dangers of getting “dinged”. SpringerPlus. 2012;1:2. doi: 10.1186/2193-1801-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfitt TW, Obrist WD, Gennarelli TA, O’Connor MJ, Weeme CA. Correlation of cerebral blood flow with outcome in head injured patients. Annals of surgery. 1977;186(4):411–414. doi: 10.1097/00000658-197710000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen KH, Morland C, Puchades M, Holm-Hansen S, Hagelin EM, Lauritzen F, Attramadal H, Storm-Mathisen J, Gjedde A, Bergersen LH. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cerebral cortex. 2014;24(10):2784–2795. doi: 10.1093/cercor/bht136. [DOI] [PubMed] [Google Scholar]
- Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathology and applied neurobiology. 2015 doi: 10.1111/nan.12295. [DOI] [PubMed] [Google Scholar]
- Lin JL, Huang YH, Shen YC, Huang HC, Liu PH. Ascorbic acid prevents blood-brain barrier disruption and sensory deficit caused by sustained compression of primary somatosensory cortex. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30(6):1121–1136. doi: 10.1038/jcbfm.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind BL, Brazhe AR, Jessen SB, Tan FC, Lauritzen MJ. Rapid stimulus-evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(48):E4678–4687. doi: 10.1073/pnas.1310065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski-Vosatka A, Tu YH, Cook RF, Sargiacomo M. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. The Journal of cell biology. 1994;126(1):111–126. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JA, Watts LT, Li W, Shen Q, Muir ER, Huang S, Boggs RC, Suri A, Duong TQ. The effects of perturbed cerebral blood flow and cerebrovascular reactivity on structural MRI and behavioral readouts in mild traumatic brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015 doi: 10.1038/jcbfm.2015.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente L, Martin MM, Lopez P, Ramos L, Blanquer J, Caceres JJ, Sole-Violan J, Solera J, Cabrera J, Argueso M, Ortiz R, Mora ML, Lubillo S, Jimenez A, Borreguero-Leon JM, Gonzalez A, Orbe J, Rodriguez JA, Paramo JA. Association between serum tissue inhibitor of matrix metalloproteinase-1 levels and mortality in patients with severe brain trauma injury. PloS one. 2014;9(4):e94370. doi: 10.1371/journal.pone.0094370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Menon DK, Steyerberg EW, Citerio G, Lecky F, Manley GT, Hill S, Legrand V, Sorgner A, Participants C-T, Investigators Collaborative European NeuroTrauma Effectiveness Research in Traumatic Brain Injury (CENTER-TBI): a prospective longitudinal observational study. Neurosurgery. 2015;76(1):67–80. doi: 10.1227/NEU.0000000000000575. [DOI] [PubMed] [Google Scholar]
- Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7(8):728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- Maneshi MM, Sachs F, Hua SZ. A Threshold Shear Force for Calcium Influx in an Astrocyte Model of Traumatic Brain Injury. Journal of neurotrauma. 2015;32(13):1020–1029. doi: 10.1089/neu.2014.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies S, Anderson G, Atif F, Badaut J, Clark R, Empey P, Guseva M, Hoane M, Huh J, Pauly J, Raghupathi R, Scheff S, Stein D, Tang H, Hicks M. Combination Therapies for Traumatic Brain Injury: Retrospective Considerations. Journal of neurotrauma. 2016;33(1):101–112. doi: 10.1089/neu.2014.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin NA, Patwardhan RV, Alexander MJ, Africk CZ, Lee JH, Shalmon E, Hovda DA, Becker DP. Characterization of cerebral hemodynamic phases following severe head trauma: hypoperfusion, hyperemia, and vasospasm. Journal of neurosurgery. 1997;87(1):9–19. doi: 10.3171/jns.1997.87.1.0009. [DOI] [PubMed] [Google Scholar]
- Mathew BP, DeWitt DS, Bryan RM, Jr, Bukoski RD, Prough DS. Traumatic brain injury reduces myogenic responses in pressurized rodent middle cerebral arteries. Journal of neurotrauma. 1999;16(12):1177–1186. doi: 10.1089/neu.1999.16.1177. [DOI] [PubMed] [Google Scholar]
- McCaffrey G, Staatz WD, Quigley CA, Nametz N, Seelbach MJ, Campos CR, Brooks TA, Egleton RD, Davis TP. Tight junctions contain oligomeric protein assembly critical for maintaining blood-brain barrier integrity in vivo. Journal of neurochemistry. 2007;103(6):2540–2555. doi: 10.1111/j.1471-4159.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- McCaffrey G, Staatz WD, Sanchez-Covarrubias L, Finch JD, Demarco K, Laracuente ML, Ronaldson PT, Davis TP. P-glycoprotein trafficking at the blood-brain barrier altered by peripheral inflammatory hyperalgesia. Journal of neurochemistry. 2012;122(5):962–975. doi: 10.1111/j.1471-4159.2012.07831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan TM, Teasdale GM. Death rate is increased for at least 7 years after head injury: a prospective study. Brain: a journal of neurology. 2007;130(Pt 10):2520–2527. doi: 10.1093/brain/awm185. [DOI] [PubMed] [Google Scholar]
- Mellergard P, Sjogren F, Hillman J. Release of VEGF and FGF in the extracellular space following severe subarachnoidal haemorrhage or traumatic head injury in humans. British journal of neurosurgery. 2010;24(3):261–267. doi: 10.3109/02688690903521605. [DOI] [PubMed] [Google Scholar]
- Mercier E, Boutin A, Lauzier F, Fergusson DA, Simard JF, Zarychanski R, Moore L, McIntyre LA, Archambault P, Lamontagne F, Legare F, Randell E, Nadeau L, Rousseau F, Turgeon AF. Predictive value of S-100beta protein for prognosis in patients with moderate and severe traumatic brain injury: systematic review and meta-analysis. Bmj. 2013;346:f1757. doi: 10.1136/bmj.f1757. [DOI] [PubMed] [Google Scholar]
- Metea MR, Newman EA. Calcium signaling in specialized glial cells. Glia. 2006;54(7):650–655. doi: 10.1002/glia.20352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R, Kreipke CW, Roberts G, Bagchi M, Rafols JA. Neovascularization following traumatic brain injury: possible evidence for both angiogenesis and vasculogenesis. Neurological research. 2007;29(4):375–381. doi: 10.1179/016164107X204693. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Hans VH, Lenzlinger PM, Dubs R, Ludwig E, Trentz O, Kossmann T. TGF-beta is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. Journal of neurotrauma. 1999;16(7):617–628. doi: 10.1089/neu.1999.16.617. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431(7005):195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta neuropathologica. 2007;114(5):459–469. doi: 10.1007/s00401-007-0274-x. [DOI] [PubMed] [Google Scholar]
- Obenaus A. Traumatic Brain Injury. In: Friedman HS, editor. Encyclopedia of Mental Health. 2nd. Academic Press; Waltham, MA: 2015. pp. 329–340. [Google Scholar]
- Obenaus AHL, Coats J, Hartman R, Badaut J, Ashwal S. Animal models of mild pediatric TBI. In: Kirkwood MW, Yeates KO, editors. Mild Traumatic Brain Injury in Children and Adolescents: From Basic Science to Clinical Management. 2012. pp. 53–76. [Google Scholar]
- Oertel M, Boscardin WJ, Obrist WD, Glenn TC, McArthur DL, Gravori T, Lee JH, Martin NA. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. Journal of neurosurgery. 2005;103(5):812–824. doi: 10.3171/jns.2005.103.5.0812. [DOI] [PubMed] [Google Scholar]
- Ostrow LW, Langan TJ, Sachs F. Stretch-induced endothelin-1 production by astrocytes. Journal of cardiovascular pharmacology. 2000;36(5 Suppl 1):S274–277. doi: 10.1097/00005344-200036051-00081. [DOI] [PubMed] [Google Scholar]
- Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, Pfrieger FW, Bergles DE, Charpak S. Calcium dynamics in astrocyte processes during neurovascular coupling. Nature neuroscience. 2015;18(2):210–218. doi: 10.1038/nn.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Pellerin L. Lactate as a pivotal element in neuron-glia metabolic cooperation. Neurochemistry international. 2003;43(4–5):331–338. doi: 10.1016/s0197-0186(03)00020-2. [DOI] [PubMed] [Google Scholar]
- Petraglia AL, Dashnaw ML, Turner RC, Bailes JE. Models of mild traumatic brain injury: translation of physiological and anatomic injury. Neurosurgery. 2014;75(Suppl 4):S34–49. doi: 10.1227/NEU.0000000000000472. [DOI] [PubMed] [Google Scholar]
- Petrov T, Rafols JA. Acute alterations of endothelin-1 and iNOS expression and control of the brain microcirculation after head trauma. Neurological research. 2001;23(2–3):139–143. doi: 10.1179/016164101101198479. [DOI] [PubMed] [Google Scholar]
- Plesnila N, Friedrich D, Eriskat J, Baethmann A, Stoffel M. Relative cerebral blood flow during the secondary expansion of a cortical lesion in rats. Neuroscience letters. 2003;345(2):85–88. doi: 10.1016/s0304-3940(03)00396-3. [DOI] [PubMed] [Google Scholar]
- Ponsford J, Cameron P, Fitzgerald M, Grant M, Mikocka-Walus A. Long-term outcomes after uncomplicated mild traumatic brain injury: a comparison with trauma controls. Journal of neurotrauma. 2011;28(6):937–946. doi: 10.1089/neu.2010.1516. [DOI] [PubMed] [Google Scholar]
- Pop V, Badaut J. A Neurovascular Perspective for Long-Term Changes After Brain Trauma. Translational stroke research. 2011;2(4):533–545. doi: 10.1007/s12975-011-0126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop V, Sorensen DW, Kamper JE, Ajao DO, Murphy MP, Head E, Hartman RE, Badaut J. Early brain injury alters the blood-brain barrier phenotype in parallel with beta-amyloid and cognitive changes in adulthood. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33(2):205–214. doi: 10.1038/jcbfm.2012.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povlishock JT, Becker DP, Sullivan HG, Miller JD. Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain research. 1978;153(2):223–239. doi: 10.1016/0006-8993(78)90404-3. [DOI] [PubMed] [Google Scholar]
- Predescu SA, Predescu DN, Malik AB. Molecular determinants of endothelial transcytosis and their role in endothelial permeability. American journal of physiology Lung cellular and molecular physiology. 2007;293(4):L823–842. doi: 10.1152/ajplung.00436.2006. [DOI] [PubMed] [Google Scholar]
- Prins ML, Alexander D, Giza CC, Hovda DA. Repeated mild traumatic brain injury: mechanisms of cerebral vulnerability. Journal of neurotrauma. 2013;30(1):30–38. doi: 10.1089/neu.2012.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Giza CC. Induction of monocarboxylate transporter 2 expression and ketone transport following traumatic brain injury in juvenile and adult rats. Developmental neuroscience. 2006;28(4–5):447–456. doi: 10.1159/000094170. [DOI] [PubMed] [Google Scholar]
- Prins ML, Hovda DA. Developing experimental models to address traumatic brain injury in children. Journal of neurotrauma. 2003;20(2):123–137. doi: 10.1089/08977150360547053. [DOI] [PubMed] [Google Scholar]
- Rafols JA, Kreipke CW, Petrov T. Alterations in cerebral cortex microvessels and the microcirculation in a rat model of traumatic brain injury: a correlative EM and laser Doppler flowmetry study. Neurological research. 2007;29(4):339–347. doi: 10.1179/016164107X204648. [DOI] [PubMed] [Google Scholar]
- Rangel-Castilla L, Gasco J, Nauta HJ, Okonkwo DO, Robertson CS. Cerebral pressure autoregulation in traumatic brain injury. Neurosurgical focus. 2008;25(4):E7. doi: 10.3171/FOC.2008.25.10.E7. [DOI] [PubMed] [Google Scholar]
- Rennels ML, Nelson E. Capillary innervation in the mammalian central nervous system: an electron microscopic demonstration. The American journal of anatomy. 1975;144(2):233–241. doi: 10.1002/aja.1001440208. [DOI] [PubMed] [Google Scholar]
- Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Netherlands heart journal: monthly journal of the Netherlands Society of Cardiology and the Netherlands Heart Foundation. 2007;15(3):100–108. doi: 10.1007/BF03085963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DJ, Jenne CN, Leger C, Kramer AH, Gallagher CN, Todd S, Parney IF, Doig CJ, Yong VW, Kubes P, Zygun DA. A prospective evaluation of the temporal matrix metalloproteinase response after severe traumatic brain injury in humans. Journal of neurotrauma. 2013;30(20):1717–1726. doi: 10.1089/neu.2012.2841. [DOI] [PubMed] [Google Scholar]
- Rosenegger DG, Tran CH, Wamsteeker Cusulin JI, Gordon GR. Tonic Local Brain Blood Flow Control by Astrocytes Independent of Phasic Neurovascular Coupling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35(39):13463–13474. doi: 10.1523/JNEUROSCI.1780-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzigalinski BA, Weber JT, Willoughby KA, Ellis EF. Intracellular free calcium dynamics in stretch-injured astrocytes. Journal of neurochemistry. 1998;70(6):2377–2385. doi: 10.1046/j.1471-4159.1998.70062377.x. [DOI] [PubMed] [Google Scholar]
- Salonia R, Empey PE, Poloyac SM, Wisniewski SR, Klamerus M, Ozawa H, Wagner AK, Ruppel R, Bell MJ, Feldman K, Adelson PD, Clark RS, Kochanek PM. Endothelin-1 is increased in cerebrospinal fluid and associated with unfavorable outcomes in children after severe traumatic brain injury. Journal of neurotrauma. 2010;27(10):1819–1825. doi: 10.1089/neu.2010.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saw MM, Chamberlain J, Barr M, Morgan MP, Burnett JR, Ho KM. Differential disruption of blood-brain barrier in severe traumatic brain injury. Neurocritical care. 2014;20(2):209–216. doi: 10.1007/s12028-013-9933-z. [DOI] [PubMed] [Google Scholar]
- Sercombe R, Dinh YR, Gomis P. Cerebrovascular inflammation following subarachnoid hemorrhage. Japanese journal of pharmacology. 2002;88(3):227–249. doi: 10.1254/jjp.88.227. [DOI] [PubMed] [Google Scholar]
- Servadei F, Murray GD, Teasdale GM, Dearden M, Iannotti F, Lapierre F, Maas AJ, Karimi A, Ohman J, Persson L, Stocchetti N, Trojanowski T, Unterberg A. Traumatic subarachnoid hemorrhage: demographic and clinical study of 750 patients from the European brain injury consortium survey of head injuries. Neurosurgery. 2002;50(2):261–267. doi: 10.1097/00006123-200202000-00006. discussion 267–269. [DOI] [PubMed] [Google Scholar]
- Shen W, Li S, Chung SH, Zhu L, Stayt J, Su T, Couraud PO, Romero IA, Weksler B, Gillies MC. Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-beta1-induced permeability of centrally derived vascular endothelium. European journal of cell biology. 2011;90(4):323–332. doi: 10.1016/j.ejcb.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Mishra V, Kodali M, Hattiangady B. Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Frontiers in cellular neuroscience. 2014;8:232. doi: 10.3389/fncel.2014.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifringer M, Stefovska V, Zentner I, Hansen B, Stepulak A, Knaute C, Marzahn J, Ikonomidou C. The role of matrix metalloproteinases in infant traumatic brain injury. Neurobiology of disease. 2007;25(3):526–535. doi: 10.1016/j.nbd.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Silverberg GD, Messier AA, Miller MC, Machan JT, Majmudar SS, Stopa EG, Donahue JE, Johanson CE. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. Journal of neuropathology and experimental neurology. 2010;69(10):1034–1043. doi: 10.1097/NEN.0b013e3181f46e25. [DOI] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nature reviews Neurology. 2013;9(4):211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends in neurosciences. 2009;32(12):638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2014;20(2):160–172. doi: 10.1177/1073858413504466. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta neuropathologica. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner J, Rafols D, Park HK, Katar MS, Rafols JA, Petrov T. Attenuation of iNOS mRNA exacerbates hypoperfusion and upregulates endothelin-1 expression in hippocampus and cortex after brain trauma. Nitric Oxide. 2004;10(3):162–169. doi: 10.1016/j.niox.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, Pedrono E, Abo-Ramadan U, Tatlisumak T. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. 2008;153(1):175–181. doi: 10.1016/j.neuroscience.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Taber KH, Warden DL, Hurley RA. Blast-related traumatic brain injury: what is known? The Journal of neuropsychiatry and clinical neurosciences. 2006;18(2):141–145. doi: 10.1176/jnp.2006.18.2.141. [DOI] [PubMed] [Google Scholar]
- Terpolilli NA, Kim SW, Thal SC, Kuebler WM, Plesnila N. Inhaled nitric oxide reduces secondary brain damage after traumatic brain injury in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33(2):311–318. doi: 10.1038/jcbfm.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Prins ML, Samii M, Hovda DA. Cerebral metabolic response to traumatic brain injury sustained early in development: a 2-deoxy-D-glucose autoradiographic study. Journal of neurotrauma. 2000;17(8):649–665. doi: 10.1089/089771500415409. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Walker SA, Povlishock JT. Perivascular nerve damage in the cerebral circulation following traumatic brain injury. Acta neuropathologica. 2006;112(1):85–94. doi: 10.1007/s00401-005-0029-5. [DOI] [PubMed] [Google Scholar]
- Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129(4):1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Varma S, Janesko KL, Wisniewski SR, Bayir H, Adelson PD, Thomas NJ, Kochanek PM. F2-isoprostane and neuron-specific enolase in cerebrospinal fluid after severe traumatic brain injury in infants and children. Journal of neurotrauma. 2003;20(8):781–786. doi: 10.1089/089771503767870005. [DOI] [PubMed] [Google Scholar]
- Verweij BH, Amelink GJ, Muizelaar JP. Current concepts of cerebral oxygen transport and energy metabolism after severe traumatic brain injury. Progress in brain research. 2007;161:111–124. doi: 10.1016/S0079-6123(06)61008-X. [DOI] [PubMed] [Google Scholar]
- Villapol S, Balarezo MG, Affram K, Saavedra JM, Symes AJ. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain: a journal of neurology. 2015 doi: 10.1093/brain/awv172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Byrnes KR, Symes AJ. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Frontiers in neurology. 2014;5:82. doi: 10.3389/fneur.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgintino D, Robertson D, Errede M, Benagiano V, Tauer U, Roncali L, Bertossi M. Expression of caveolin-1 in human brain microvessels. Neuroscience. 2002;115(1):145–152. doi: 10.1016/s0306-4522(02)00374-3. [DOI] [PubMed] [Google Scholar]
- Wada K, Chatzipanteli K, Busto R, Dietrich WD. Role of nitric oxide in traumatic brain injury in the rat. Journal of neurosurgery. 1998;89(5):807–818. doi: 10.3171/jns.1998.89.5.0807. [DOI] [PubMed] [Google Scholar]
- Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89(3):957–989. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20(18):7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei EP, Dietrich WD, Povlishock JT, Navari RM, Kontos HA. Functional, morphological, and metabolic abnormalities of the cerebral microcirculation after concussive brain injury in cats. Circ Res. 1980;46(1):37–47. doi: 10.1161/01.res.46.1.37. [DOI] [PubMed] [Google Scholar]
- Winship IR, Plaa N, Murphy TH. Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(23):6268–6272. doi: 10.1523/JNEUROSCI.4801-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Chen TW, Yang TC, Wei HJ, Hsu JC, Lin CL. Levels of F-isoprostanes, F-neuroprostanes, and total nitrate/nitrite in plasma and cerebrospinal fluid of patients with traumatic brain injury. Free radical research. 2015:1–12. doi: 10.3109/10715762.2015.1080363. [DOI] [PubMed] [Google Scholar]
- Yeoh S, Bell ED, Monson KL. Distribution of blood-brain barrier disruption in primary blast injury. Annals of biomedical engineering. 2013;41(10):2206–2214. doi: 10.1007/s10439-013-0805-7. [DOI] [PubMed] [Google Scholar]
- Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper-and subsequent hypometabolic state. Brain research. 1991;561(1):106–119. doi: 10.1016/0006-8993(91)90755-k. [DOI] [PubMed] [Google Scholar]
- Zhang H, Adwanikar H, Werb Z, Noble-Haeusslein LJ. Matrix metalloproteinases and neurotrauma: evolving roles in injury and reparative processes. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2010;16(2):156–170. doi: 10.1177/1073858409355830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Badaut J, Tang J, Obenaus A, Hartman R, Pearce WJ. The vascular neural network–a new paradigm in stroke pathophysiology. Nature reviews Neurology. 2012;8(12):711–716. doi: 10.1038/nrneurol.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ahram A, Berman RF, Muizelaar JP, Lyeth BG. Early loss of astrocytes after experimental traumatic brain injury. Glia. 2003;44(2):140–152. doi: 10.1002/glia.10283. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature reviews Neuroscience. 2011;12(12):723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubkov AY, Lewis AI, Raila FA, Zhang J, Parent AD. Risk factors for the development of post-traumatic cerebral vasospasm. Surgical neurology. 2000;53(2):126–130. doi: 10.1016/s0090-3019(99)00178-0. [DOI] [PubMed] [Google Scholar]