

Abstract
Diabetic kidney disease (DKD) is a worldwide public health problem. The definition of DKD is under discussion. Although the term DKD was originally defined as ‘kidney disease specific to diabetes,’ DKD frequently means chronic kidney disease with diabetes mellitus and includes not only classical diabetic nephropathy, but also kidney dysfunction as a result of nephrosclerosis and other causes. Metabolic memory plays a crucial role in the progression of various complications of diabetes, including DKD. The mechanisms of metabolic memory in DKD are supposed to include advanced glycation end‐products, deoxyribonucleic acid methylation, histone modifications and non‐coding ribonucleic acid including micro ribonucleic acid. Regardless of the presence of diabetes mellitus, the final common pathway in chronic kidney disease is chronic kidney hypoxia, which influences epigenetic processes, including deoxyribonucleic acid methylation, histone modification, and conformational changes in micro ribonucleic acid and chromatin. Therefore, hypoxia and oxidative stress are appropriate targets of therapies against DKD. Prolyl hydroxylase domain inhibitor enhances the defensive mechanisms against hypoxia. Bardoxolone methyl protects against oxidative stress, and can even reverse impaired renal function; a phase 2 trial with considerable attention to heart complications is currently ongoing in Japan.
Keywords: Diabetic kidney disease, Metabolic memory, Renal hypoxia
Introduction
Diabetic kidney disease (DKD) is one of the complications of diabetes mellitus, and is the major cause of end‐stage kidney disease in developed countries1, 2. In past years, diabetic patients suffered from classical diabetic nephropathy (DN), which starts with glomerular hyperfiltration, followed by microalbuminuria, macroalbuminuria and nephrotic‐range proteinuria with a decrease in glomerular filtration and eventual development of end‐stage kidney disease. However, improvements of blood glucose control, utilization of renin–angiotensin blockers and an increase in the aging population have changed the phenotypes of DKD. A recent comparison of type 1 diabetes patients with a control population in Finland revealed the absence of ‘glomerular hyperfiltration’ these days3. Among adults with diabetes in the USA, the overall prevalence of kidney disease did not change significantly, but that of albuminuria declined and that of reduced estimated glomerular filtration rate (eGFR) increased4. These show that the role of classical DN in the pathogenesis of DKD is decreasing, whereas that of glomerulosclerosis as a result of aging, atherosclerosis and so on is increasing. The definition of DKD is still under discussion. The term DKD was originally used as a reference to ‘kidney disease specific to diabetes’5. Although DKD is still used as its original meaning,2 and sometimes clearly separated from glomerulosclerosis6, nowadays the term DKD frequently covers classical DN and other types of kidney dysfunction in diabetic patients, and is currently the term being used to reflect the changes in kidney disease phenotypes of diabetes patients. DN was originally a pathological definition that was characterized by mesangial expansion, nodular glomerular sclerosis and tubulointerstitial fibrosis7, 8. Now, DN is frequently diagnosed without renal biopsy, usually in patients with long‐standing diabetes mellitus, absence of hematuria and existence of diabetic retinopathy. Therefore, the definition of DN is a part of DKD, which has a wide heterogeneity (Figure 1).
Figure 1.
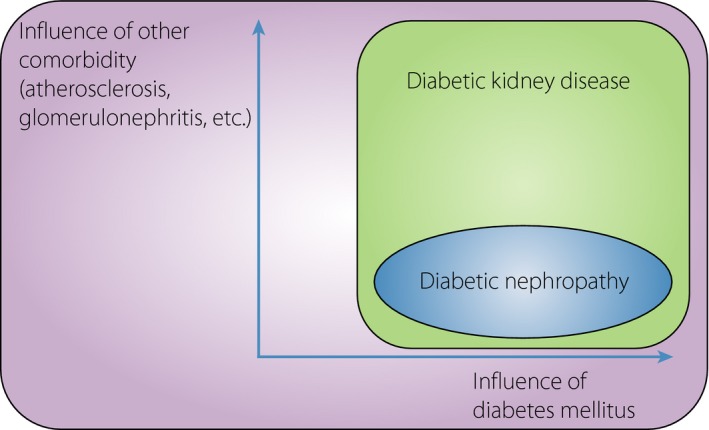
A Venn diagram of the notions of diabetic kidney disease and diabetic nephropathy. Diabetic nephropathy, which was originally a pathological diagnosis, includes only patients or animals whose kidney damage was obviously from diabetes mellitus. In contrast, diabetic kidney disease means chronic kidney disease with diabetes, regardless of other comorbidities, such as atherosclerosis and glomerulonephritis. Therefore, diabetic kidney disease encompasses diabetic nephropathy.
Recent large clinical trials on DKD included patients with kidney diseases other than the classical DN; the inclusion criteria in these trials did not require proof of DN by renal biopsy, absence of hematuria, duration of diabetes mellitus or existence of diabetic retinopathy in type 2 diabetes mellitus patients9, 10, 11, 12, 13. Diabetic animal models have been utilized, but these have rarely shown the classic pathological changes in DN14, 15, 16, 17, 18. These differences must be taken into consideration when comparing clinical studies and animal experiments.
Metabolic Memory
Accumulating evidence clarifies the involvement of various factors in the pathogenesis of DKD; these factors include high blood glucose, activation of the renin–angiotensin system, oxidative stress, increase in advanced glycation end‐products (AGEs) and glomerular hyperfiltration19, 20. In the clinical course of DKD, there is a famous phenomenon called ‘metabolic memory’21, 22, which indicates the long‐term effect of glycemic control on diabetic complications including DKD. Further understanding and clarification of this mechanism have drawn increasing attention. AGEs and epigenetic changes are candidates for the underlying mechanism of metabolic memory23, 24, as both AGE accumulation and epigenetic changes are prolonged phenomena.
Advanced Glycation End‐products and their Detoxifier, Glyoxalase 1
AGE is a collective term for compounds formed through non‐enzymatic glycation of proteins and nucleic acids25. The rate of AGE formation is affected by inflammation and oxidative stress, in addition to plasma glucose level26. AGEs are chemically stable and are toxic to living organs, either by direct means or through receptors for AGEs (RAGE). AGEs can directly bind and produce degenerative changes in extracellular matrix protein, such as collagen. AGEs can also bind to RAGE to increase oxidative stress and activate nuclear factor‐κΒ and the subsequent inflammatory pathways27, 28. Therefore, accumulation of AGE is considered to be the cause of diabetic complications.
Indeed, skin collagen glycation, which is correlated with complications of type 1 diabetes mellitus29, is reduced under strict glycemic control. Surprisingly, this difference in skin AGEs affects the long‐term risk for diabetic complications for as long as 10 years30. Although an invasive procedure, AGEs on skin biopsy were used in past research to estimate whole‐body AGEs. Later on, non‐invasive techniques to assess skin AGE accumulation were developed, an example of which is the measurement of skin autofluorescence that is unique to AGEs, which emit strong fluorescence at a wavelength of 440 nm when excited at 370 nm31, 32. Recently, the measurement of skin autofluorescence has attracted major concern, and has been shown to be related to the risk of developing complications in diabetes patients33, 34. Skin autofluorescence was reported to correlate with all‐cause mortality and vascular diseases in CKD patients35, 36, 37, suggesting that whole‐body AGE accumulation is closely related to the prognosis of DKD.
In rats, AGEs were shown to cause direct kidney injury, and induce albuminuria, glomerular sclerosis, basement membrane widening and mesangial matrix expansion38. Furthermore, as the kidney is the major site of AGE clearance, renal dysfunction results in AGE accumulation39. Therefore, renal dysfunction and AGE accumulation potentiate each other, and reduction of AGEs is a promising strategy against renal dysfunction, including DKD.
Glyoxalase 1 (GLO‐1) is a detoxifier of AGEs. Overexpression of GLO‐1 gene was shown to ameliorate diabetic kidney changes in streptozotocin‐induced type 1 diabetes mellitus in rats40. In contrast, in vivo knockdown of the GLO‐1 gene in non‐diabetic mice resulted in DN‐like phenotypes, such as albuminuria, glomerular enlargement, mesangial matrix expansion and basement membrane thickening without diabetes41. Taken together, changes in GLO‐1 activity and subsequent changes in AGE content might be closely related with the development of DN.
Epigenetics in Diabetic Kidney Disease
Epigenetics indicates persistent changes in gene expression and the resulting phenotypic changes, which occur without changes in deoxyribonucleic acid (DNA) sequence. Epigenetic changes include DNA methylation, histone modification, non‐coding ribonucleic acids (ncRNAs) and chromosome conformational changes42, 43. As these epigenetic changes have long‐term effects, they are likely to play a critical role in metabolic memory.
DNA methylation
DNA methylation occurs mainly at the 5′ cytosines of CpG dinucleotides. In general, DNA methylation at the promoter region results in gene repression44. Although the status can differ among various organs, DNA methylation in whole blood or circulating monocytes was estimated to reflect whole‐body DNA methylation status in diabetes patients, although DNA methylation status can differ among various organs45, 46, 47. Therefore, investigation of DNA methylation, at least in circulating cells, can be done in DKD patients. Recently, Chen et al.46 have shown that changes in DNA methylation status in circulating cells of diabetic patients persisted for over 15 years. They investigated whole blood and blood monocytes, from which 12 annotated and differentially methylated loci, including hypomethylation of thioredoxin‐interacting protein (TXNIP), were extracted. As systemic knockout of the TXNIP gene is known to ameliorate streptozotocin‐induced DN48, persistent hypomethylation in this gene could render the patients vulnerable to DKD. In contrast, Ko et al.49 examined the DNA methylation status in the tubules of surgically resected human kidneys. As DNA methylation, like other epigenomes, is cell‐specific, they microdissected tubules from an entire kidney of DKD patients, and discovered the presence of hypomethylation of the RUNX1 gene and upregulation of RUNX1.
Several interesting changes in DNA methylation have been shown in DKD model mice. Hasegawa et al.50 reported that DN phenotypes of podocytopathy were aggravated in tubular‐specific Sirtuin 1 (Sirt1) knockout diabetic mice, and ameliorated in SIRT1 transgenic diabetic mice. One interesting point of this research was the proof that proximal tubules affect glomeruli. Another point was that the SIRT1 transgene might protect the kidney, especially the podocytes, from diabetic insult through claudin‐1 hypermethylation. They showed that the SIRT1 transgene suppressed claudin‐1 expression and increased methylated CpG, and that claudin‐1 aggravated diabetic podocytopathy in microdissected tubules of diabetic mice. The SIRT1 is a histone deacetylase that is well known to have a role against histones, not DNA. Although there was a report showing that SIRT1 activated DNA methyltransferase 1, which binds to hemi‐methylated CpG sites and methylates CpG51, the mechanism of DNA hypomethylation resulting from SIRT1 overexpression remains obscure. Another report focused on the temporal profile of DNA methylation changes in purified proximal tubules of diabetic mice kidneys, which were prepared using a cell sorter52. They identified several aberrant hypomethylation and hypermethylation sites, and highlighted the hypomethylation of the Agt gene, which is known to persist after pioglitazone antidiabetic therapy. As the Agt protein, or angiotensinogen, in proximal tubular cells is closely related to the progression of DN53, changes in its methylation status must have a role in metabolic memory, at least in mice.
Histone modifications
DNA is packaged in the chromatin, which is composed of the nucleosome as its fundamental unit. The nucleosome comprises an octamer of four core histones (H3, H4, H2A and H2B), which is wrapped by DNA. The histones can undergo modifications, such as acetylation and methylation on specific lysines54. In general, histone acetylation at gene promoters is linked to active transcription. H3K9ac, H3K14ac and H4K5ac are representative histone acetylations. In contrast, whether histone methylation results in gene activation or gene suppression depends on the amino acid residue and extent of methylation (i.e., mono‐, di‐ or tri‐methylation). H3K4me1/2/3 and H3K36me2/3 are representative transcriptionally active marks, whereas H3K9me3 and H3K27me3 are repressive ones24. Miao et al.55 have elucidated the histone modifications of peripheral blood lymphocytes and monocytes in type 1 diabetes patients. They checked H3K9ac, H3K4me3 and H3K9me2, and found that monocytes from the high glycated hemoglobin group displayed greater enrichment of H3K9ac at promoter regions than those from the low glycated hemoglobin group. Interestingly, many genes with high concentrations of H3K9ac in the promoter region are related to the nuclear factor‐κΒ pathway, which is closely related to the development of diabetic complications19, 56. This result might partly explain systemic metabolic memory.
How about kidney‐specific histone modification? From this viewpoint, several experimental studies have been carried out on diabetic animal models. Chen et al.57 focused on monocyte chemotactic protein‐1 (MCP‐1), a chemokine that plays an important role in DKD progression58, and investigated the histone modifications in the promoter region of its gene. They clarified that SET7/9, a histone lysine methyltransferase that monomethylates H3K4, accumulated in the promoter region of the MCP‐1 gene, and that the H3K4me1 level was higher in the same region in diabetic mice than in control mice. Cai et al.59 focused on osteopontin, which is recognized as an important gene for mesangial matrix expansion in DN60. They showed that the kidneys of diabetic mice upregulated the osteopontin gene; increased H3K9ac, H3K4me1 and H3K4me3 levels; and decreased H3K27me3 levels in the promoter region of the osteopontin gene59. However, these results need to be carefully interpreted, because they analyzed whole kidney lysate; epigenetics are dependent on cell types, and solation of single or low‐cell types must yield more interpretable results.
To overcome this limitation, glomerular fractions have been used to determine more specific and reliable changes in histone modification, although it is important to note that glomeruli contain various types of cells, such as podocytes, endothelial cells and mesangial cells. De Marinis et al.61 examined histone modifications on the promoter region of the Txnip gene in a diabetic mouse model and showed increased Txnip expression; increased levels of H3K9ac, H3K4me1 and H3K4me3; and decreased level of H3K27me3. They also showed that an increase in glomerular Txnip gene expression depended on fasting blood glucose, which is consistent with hypomethylation of the TXNIP gene in the peripheral blood cells of diabetic patients46. Reddy et al.62 examined the effect of losartan, an angiotensin II type 1 receptor blocker, on epigenetics in diabetic mice. They found that the elevated glomerular fractions of the Mcp‐1, plasminogen activator inhibitor‐1 (Pai‐1) and Rage genes in diabetic mice decreased after losartan administration. Similar to Mcp‐1 and Rage, Pai‐1 upregulation is supposed to worsen DN, as Pai‐1 knockout alleviates DN in mice63, 64. They also analyzed glomerular fractions from diabetic kidneys, with and without losartan, and showed an increase of H3K9/14Ac in the promoter regions of Pai‐1 and Rage genes; after losartan treatment, the elevated H3K36me3 in the promoter region of the Rage gene was diminished. They showed that several histone acetyltransferases, histone deacetylases and histone methyltransferases were upregulated in the glomerular fraction of diabetic mice, and were diminished by angiotensin receptor blocker administration. Although it is still obscure whether these epigenetic changes were directly induced or indirectly caused, such as by blood pressure lowering, these results gave new insights into the renoprotective effect of angiotensin receptor blockers. Furthermore, the epigenetic changes in the promoter region of the RAGE gene implied a cross‐talk between epigenetics and AGEs, which are two candidate factors related to metabolic memory.
These results showed that histone modifications play a role in metabolic memory, and that targeting histone modification seems a promising strategy against DKD. Indeed, two inhibitors of EZH2, which trimethylates H3K27, were shown to ameliorate renal fibrosis induced by unilateral ureteral obstruction65. One limitation of these studies on histone modification in the context of DKD was the lack of data obtained from diseased kidneys, as histone modification must depend on differences between species, as well between clinical DKD and experimental DN.
Non‐coding RNA and MicroRNA
NcRNAs are ubiquitous and endogenous RNAs that are transcribed from DNA, but do not code protein; small ncRNAs that are approximately 22‐nucleotide long are called microRNA (miR)24, 66. These ncRNAs, including miRs, can work as epigenetic factors, because they can regulate gene expressions through transcriptional and post‐transcriptional mechanisms. Among numerous ncRNAs, miRs are eagerly researched partly because detection of miR is generally easier than that of long ncRNA.
One milestone of miRNA research in DN is about miR‐192 and its relationship with transforming growth factor‐β1 (TGF‐β), which is an important mediator of DN67. Kato et al.68, 69 showed that miR‐192, miR‐200b and miR‐200c were upregulated in the glomeruli of two independent diabetic model mice. The miR‐192 targets smad‐interacting protein 1, and is upregulated by TGF‐β in mouse mesangial cells. Silencing of miR‐192 has been shown to increase Col1a2 promoter activation, probably through smad‐interacting protein 168. The expression of miR‐200b and miR‐200c were downregulated by in vivo inhibition of miR‐192, showing that miR‐200b and miR‐200c are downstream genes of miR‐192 in mouse mesangial cells, although miR‐192 does not directly bind to either of them. They also showed that both miR‐192 and miR‐200b increased the expression of TGF‐β and Col1a2 in mouse mesangial cells69. Given that mesangial matrix expansion is a hallmark of DN, they concluded that miR‐192 and miR‐200b/c must play important roles in the pathogenesis of DN. Indeed, in vivo silencing of miR‐192 reversed mesangial matrix expansion, and decreased urinary protein in streptozotocin‐induced diabetic mice70.
How about microRNAs in human DKD? From this viewpoint, Krupa et al.71 examined miR‐array of renal biopsy samples of DKD, and found that miR‐192 expression positively correlated with eGFR and negatively correlated with fibrosis score. Although this research had a limitation of using whole lysate only, it provided evidence of changes in miR concentration in human DKD. Another approach to miR changes in DKD is examination of plasma or urine where RNAse is abundant, because carrier proteins, such as argonaute2, protect these from degradation72, 73. Plasma miR‐126 concentration was shown to be relatively low in type 2 diabetes patients, and was speculated to reflect the loss of systemic endothelial miR‐126, not from a single organ74. In contrast, urinary miR profile must reflect that in the kidney, especially in podocytes and tubular cells. Using urine miR microarray, Delic et al. showed significant changes in 16 miR concentrations in type 2 diabetes patients with reduced eGFR75. Among these 16 miRs, they focused on miR‐320c, which positively correlates with the urinary albumin‐to‐creatinine ratio and negatively correlates with eGFR, meaning that miR‐320c increases with the progression of DKD. Evidence of the relationship between miR320c and DKD progression was based on the fact that thrompospondine 1 was a predicted target of miR320c, and that antagonizing thrompospondine 1‐mediated latent TGF‐β activation resulted in amelioration of DN in mice76. Another group also showed that urinary miR profiles differed according to the different stages of DKD in type 1 diabetes patients, including downregulation of miR‐221 in several conditions77. Interestingly, miR‐221 is known to be downregulated by AGEs in human endothelial cells78. Taken together, urinary miRs must be useful biomarkers, but their additional pathogenic role has not been elucidated.
Renal Hypoxia and its Link to Metabolic Memory
The progression of renal damage in CKD becomes consistent and irreversible above a certain threshold, independent of the cause of CKD. Renal hypoxia, especially tubulointerstitial hypoxia, is the ‘final common pathway’ in CKD progression79, 80. Although the kidneys receive 20% of the cardiac output, oxygen tension in the kidney is innately low compared with that in other organs. This innately low oxygen tension in the kidney has been proven by needle electrodes and visualization of hypoxia‐inducible factor (HIF) activity using luciferase in mice81, 82. One reason is the existence of an oxygen shunt between the intrarenal arteries and veins83. With the progression of CKD, the kidneys suffer from much lower oxygen tension or hypoxia. This pathophysiology of renal hypoxia in CKD is multifactorial, and includes loss of peritubular capillaries84, 85, renal anemia86 and increased oxygen demand in the renal tubules87, 88. Of these, increased oxygen demand in the renal tubules has been shown in a mouse model of DN. Several clinical studies implicated the effect of renal hypoxia on renal prognosis. Increased hemoglobin level, which must alleviate renal hypoxia, correlates with better renal prognosis. In contrast, increased CKD risk has been proven with the existence of intermittent or persistent hypoxemia in patients with sleep apnea syndrome or chronic obstructive pulmonary disease, respectively89, 90.
Another factor is oxidative stress. Generally, hypoxia is linked to increased oxidative stress, which is closely involved in the development of diabetic complications56, 91. Therefore, tubulointerstitial hypoxia is supposed to be an additional aggravating factor in DKD. However, determination of renal oxygenation on blood oxygen level‐dependent magnetic resonance imaging in DKD patients yielded conflicting results on the correlation between renal hypoxia and renal function in DKD patients92, 93, 94, 95. These findings are considered the basis for the heterogeneity of DKD.
A master regulator of cellular response against hypoxia is HIF, which comprises two subunits, HIF‐α and HIF‐β96. HIF‐α is an oxygen‐dependent subunit, and is hydroxylated through prolyl hydroxylase domain (PHD). Hydroxylated HIF‐α is recognized by von Hippel–Lindau tumor suppressor, resulting in proteasomal degradation. Because the rate‐limiting step of HIF degradation is hydroxylation of HIF by PHD, the concentration of oxygen determines the HIF‐α concentration. Under hypoxic conditions or pharmaceutical PHD inhibition, HIF‐α is stabilized in the cytosol and forms a heterodimer with HIF‐1β, which is an oxygen‐insensitive heterodimer that can translocate into the nucleus and act as a transcriptional factor by binding with the consensus enhancer through hypoxia‐responsive elements (Figure 2a). Erythropoietin and vascular endothelial growth factor are well‐known HIF downstream genes that counteract hypoxia through erythropoiesis and neovascularization, respectively.
Figure 2.
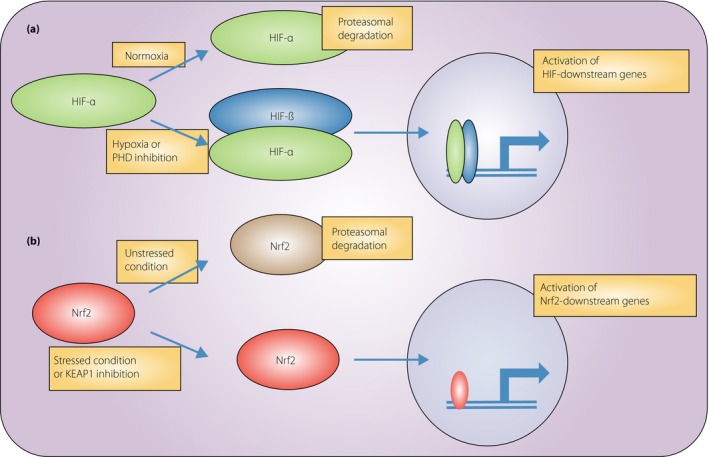
Regulation of hypoxia‐inducible factor (HIF) and NF‐E2‐related factor 2 (Nrf2). (a) Regulation of HIF. HIF‐α undergoes hydroxylation by prolyl hydroxylase domain (PHD) in a normoxic condition, resulting in proteasomal degradation. Under hypoxia or PHD inhibition, HIF‐α is not hydroxylated, but is stabilized in cytosol and forms a heterodimer with HIF‐β. This heterodimer translocates into the nucleus, binds to the consensus enhancer through hypoxia‐responsive elements and activates downstream genes. (b) Regulation of Nrf2. Nrf2 is recognized by Kelch‐like ECH‐associated protein 1 (KEAP1) under non‐stressful conditions, followed by proteasomal degradation. Under a stressful condition of pharmaceutical KEAP1 inhibition, Nrf2 cannot be recognized by KEAP1, and is stabilized in the cytosol. Increased concentration of Nrf2 results in nuclear translocation, binding to the consensus enhancer through anti‐oxidant‐responsive elements, and activation of downstream genes.
Hypoxia also induces epigenetic changes. The oxygen molecule is required for the activity of the ten‐eleven translocation enzyme, an oxidation enzyme against 5‐methylcytosine that is similar to PHDs with regard to being dependent on Fe2+ and α‐ketoglutarate97. Oxidized 5‐methylcytosine results in replacement to unmodified cytosine; therefore, the ten‐eleven translocation enzyme has power to inhibit methylation98, 99. This confirms the importance of hypoxia in DNA methylation and that the promoters of tumor suppressor genes are more methylated in hypoxic tumor tissues100. Another mechanism of hypoxia‐induced changes in epigenetics is through Dicer, which is a key endoribonuclease that processes precursor miR to mature miR, and whose expression and activity are impaired in hypoxia101. This downregulation of Dicer expression is through hypoxia‐induced increase in the H3K27me3 level in the Dicer promoter region in tumor cells102. The expression of miRs in renal tubular cells also changes after hypoxia–reoxygenation in human cultured tubular cells and in human kidney 2 cells103. Among these, the expression of miR‐205, which binds to the 3′‐untranslated region of the PHD1, was most decreased in hypoxia–reoxygenation. MiR‐205 inhibition leads to decreased expression of anti‐oxidant enzymes, such as hemeoxygenase 1, copper/zinc superoxide dismutase and manganese superoxide dismutase, all of which are downstream genes of HIF.
Chromatin conformational change, which is another mechanism of epigenetics, has been reported to occur in hypoxia. Mimura et al.104 showed that chromatin conformational change occurred in the promoter region of the SLC2A3 (GLUT3) gene in human umbilical vein endothelial cells. In normoxic conditions, HIF binds to the transcriptional starting sites (TSS) and enhancer 1 (−35 kbp from TSS), resulting in conformational proximity between these two regions. Under hypoxic conditions, the levels of H3K27ac increase in the TSS, enhancer 1 and enhancer 2 (−24 kbp from TSS), resulting in HIF binding with enhancer 2. This process changes the chromatin conformation and brings the TSS, enhancer 1 and enhancer 2 closer to form lysine (K)‐specific demethylase 3A, an H3K9me2 demethylase; therefore, H3K9me2 is recruited and is subsequently demethylated in these three regions. Removal of H3K9me2, a repressive histone mark, activates SLC2A3 gene transcription; this chromatin conformational change is the mechanism of robust upregulation of the SLC2A3 gene in hypoxic conditions. Inoue et al.105 reported a similar chromatin conformational change in the angiopoietin‐like 4 (ANGPTL4) gene. They elucidated that hypoxia and peroxisome proliferator‐activated receptors‐β/δ agonists synergistically activated ANGPLT4. When peroxisome proliferator‐activated receptor‐β/δ agonists and HIF1α coexist, the peroxisome proliferator‐activated receptor‐response elements come closer to and result in increased H3K27ac levels in the HIF1α binding site.
Therapeutics
Existing therapeutics against DKD
Accumulating clinical evidence showed that strict glycemic control106, 107, 108 and blood pressure control109, 110 improves the prognosis of diabetic patients, including those with DKD. Pharmaceutical inhibition of the renin–angiotensin–aldosterone system by angiotensin‐converting enzyme inhibitors or angiotensin receptor blockers were also shown to protect the DKD kidneys of both type 1111, 112, 113 and type 211, 12, 13 diabetes patients; these agents are now used as first‐line therapy against hypertension in DKD. The effectiveness of direct renin inhibitors, another renin–angiotensin–aldosterone system blocker, is still controversial10, 114. Dietary protein restriction was found to be ineffective based on the results of a meta‐analysis. According to the standards of medical care in diabetes, reducing the amount of dietary protein below the recommended daily allowance of 0.8 g/kg ideal bodyweight was not advisable for people with DKD, because it does not alter glycemic measures, cardiovascular risk measures or the course of GFR decline (grade A evidence)115, 116.
Sodium glucose co‐transporter 2 (SGLT2) inhibitors improve glycemic control by enhancing excretion of glucose into urine. The Randomized, Placebo‐Controlled Cardiovascular Outcome Trial of Empagliflozin (EMPA‐REG OUTCOME trial) showed that the use of empagliflozin, an SLGT2 inhibitor, in addition to standard care prevented composite cardiovascular outcomes and death from any cause117. A subanalysis of this trial showed that empagliflozin slowed the progression of DKD and lowered the rates of clinically relevant renal events compared with a placebo118. The mechanism of renoprotection by an SGLT2 inhibitor is believed to be mediated by amelioration of glomerular hyperfiltration and, possibly, renal hypoxia. An SGLT2 inhibitor improves anemia in CKD by enhancing the expression of erythropoietin119. Reabsorption of glucose from the glomerular filtrate also requires simultaneous reabsorption of sodium, which is driven by the concentration gradient between the glomerular filtrate and tubular cells. Maintenance of this gradient requires adenosine triphosphate; therefore, glucose reabsorption through SGLT increases energy demand and oxygen consumption88. A recent report showed that the low cortical oxygen tension in diabetic rats was normalized by administration of phlorizin, another SGLT2 inhibitor120. However, phlorizin reduces medullary oxygen tension independent of the presence of diabetes mellitus. Therefore, it remains to be fully elucidated whether SGLT2 inhibitor counteracts renal hypoxia.
Hypoxia‐oriented therapies against DKD
As hypoxia serves as the final common pathway, it is expected to be an effective target of treatment strategies against CKD, including DKD. Cobalt chloride, a classic PHD inhibitor, has been shown to have protective effects on experimental CKD models without diabetes, and to reduce proteinuria and interstitial fibrosis in DN in mice after amelioration of renal hypoxia121, 122, 123. However, cobalt chloride cannot be used for human patients because of its side‐effects. Therefore, synthesis of new and safe PHD inhibitors has been encouraged; at least six PHD inhibitors of renal anemia are now under clinical trials124. FG‐4497, which is one of these PHD inhibitors, has been shown to ameliorate lipid metabolism and insulin resistance in mice that were fed a high‐fat diet125. Taken together, PHD inhibitors might protect the kidneys from DKD by targeting the kidney itself and/or by metabolic dysfunction; therefore, these are promising drugs not only for renal anemia, but also for DKD progression.
Another approach is targeting oxidative stress. Activation of NF‐E2‐related factor 2 (Nrf2) results in powerful anti‐oxidative effects. Clinical trials on bardoxolone methyl, an Nrf2 activator, showed encouraging results in DKD patients, although there is a problem that requires attention. Nrf2 is a transcriptional factor that activates the expression of anti‐oxidant genes126. Interestingly, Nrf2 also activates GLO‐1 gene expression and reduces glycative stress; therefore, Nrf2 activation might intervene with metabolic memory through AGEs127. Nrf2 is recognized by Kelch‐like ECH‐associated protein 1 and is subsequently ubiquitinated, resulting in proteasomal degradation under non‐stressful conditions. Once cellular stress, such as oxidative stress, is induced, Nrf2 recognition by Kelch‐like ECH‐associated protein 1 is inhibited and Nrf2 is stabilized in cytosol, followed by nuclear translocation (Figure 2b). Bardoxolone methyl is a Kelch‐like ECH‐associated protein 1 inhibitor that was found to have a renoprotective effect in CKD patients when used as an anticancer drug128. This renoprotective effect of bardoxolone methyl against DKD has attracted attention, as oxidative stress was the main pathogenic factor of DKD56. Two phase 2 trials showed that bardoxolone methyl increased the eGFR of patients with type 2 diabetes mellitus and those in stage 3b–4 CKD9, 128. However, a subsequent phase 3 trial (Bardoxolone Methyl in Type 2 Diabetes and Stage 4 Chronic Kidney Disease [BEACON] trial) was terminated because of a relatively high rate of cardiovascular events, especially heart failure, in the first month6. In that phase 3 trial, an increase in eGFR was also documented in the bardoxolone methyl group. Secondary analysis of the BEACON study showed that elevated B‐type natriuretic peptide and a history of hospitalization for heart failure were risk factors for cardiovascular events in the bardoxolone group129. After the first month, the risks for cardiovascular events were similar between the bardoxolone group and the placebo groups. Considering that bardoxolone methyl might be a revolutionary drug that can reverse eGFR decline in DKD, a the Phase II Study of Bardoxolone Methyl in Patients with Chronic Kidney Disease and Type 2 Diabetes (TSUBAKI study) on bardoxolone methyl for type 2 diabetes patients with CKD is currently underway in Japan, with considerable caution regarding the occurrence of cardiovascular events.
Conclusion
The present review focused on the mechanism of metabolic memory, its relationship with renal hypoxia and on treatment, especially hypoxia‐oriented pharmaceutical therapies, of DKD. It is now widely recognized that experimental animal DN and human DKD differ widely; therefore, careful analysis of results is required to overcome the worldwide public health problem of DKD. Hypoxia‐oriented therapies seem to be promising for this disease.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the Grant‐in‐Aid for Scientific Research on Innovative Areas (26111003 to MN).
J Diabetes Investig 2017; 8: 261–271
References
- 1. Thomas MC, Cooper ME, Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat Rev Nephrol 2016; 12: 73–81. [DOI] [PubMed] [Google Scholar]
- 2. Tuttle KR, Bakris GL, Bilous RW, et al Diabetic kidney disease: a report from an ADA Consensus Conference. Diabetes Care 2014; 37: 2864–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thomas MC, Moran JL, Harjutsalo V, et al Hyperfiltration in type 1 diabetes: does it exist and does it matter for nephropathy? Diabetologia 2012; 55: 1505–1513. [DOI] [PubMed] [Google Scholar]
- 4. Afkarian M, Zelnick LR, Hall YN, et al Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988‐2014. JAMA 2016; 316: 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis 2007; 49: S12–S154. [DOI] [PubMed] [Google Scholar]
- 6. de Zeeuw D, Akizawa T, Audhya P, et al Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 2013; 369: 2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maezawa Y, Takemoto M, Yokote K. Cell biology of diabetic nephropathy: roles of endothelial cells, tubulointerstitial cells and podocytes. J Diabetes Investig 2015; 6: 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Breyer MD, Bottinger E, Brosius FC, et al Diabetic nephropathy: of mice and men. Adv Chronic Kidney Dis 2005; 12: 128–145. [DOI] [PubMed] [Google Scholar]
- 9. Pergola PE, Raskin P, Toto RD, et al Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 2011; 365: 327–336. [DOI] [PubMed] [Google Scholar]
- 10. Parving HH, Brenner BM, McMurray JJ, et al Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med 2012; 367: 2204–2213. [DOI] [PubMed] [Google Scholar]
- 11. Lewis EJ, Hunsicker LG, Clarke WR, et al Renoprotective effect of the angiotensin‐receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345: 851–860. [DOI] [PubMed] [Google Scholar]
- 12. Brenner BM, Cooper ME, de Zeeuw D, et al Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345: 861–869. [DOI] [PubMed] [Google Scholar]
- 13. Barnett AH, Bain SC, Bouter P, et al Angiotensin‐receptor blockade versus converting‐enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med 2004; 351: 1952–1961. [DOI] [PubMed] [Google Scholar]
- 14. Betz B, Conway BR. An Update on the Use of Animal Models in Diabetic Nephropathy Research. Curr Diab Rep 2016; 16: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuzawa Y, Niki I, Kosugi T, et al Overexpression of calmodulin in pancreatic beta cells induces diabetic nephropathy. J Am Soc Nephrol 2008; 19: 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hudkins KL, Pichaiwong W, Wietecha T, et al BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol 2010; 21: 1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kawano K, Hirashima T, Mori S, et al OLETF (Otsuka Long‐Evans Tokushima Fatty) rat: a new NIDDM rat strain. Diabetes Res Clin Pract 1994; 24(Suppl): S317–S320. [DOI] [PubMed] [Google Scholar]
- 18. Brosius FC 3rd, Alpers CE, Bottinger EP, et al Mouse models of diabetic nephropathy. J Am Soc Nephrol 2009; 20: 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev 2013; 93: 137–188. [DOI] [PubMed] [Google Scholar]
- 20. Rask‐Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab 2013; 17: 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nathan DM, Cleary PA, Backlund JY, et al Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353: 2643–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chalmers J, Cooper ME. UKPDS and the legacy effect. N Engl J Med 2008; 359: 1618–1620. [DOI] [PubMed] [Google Scholar]
- 23. Ceriello A. Hypothesis: the “metabolic memory”, the new challenge of diabetes. Diabetes Res Clin Pract 2009; 86(Suppl 1): S2–S6. [DOI] [PubMed] [Google Scholar]
- 24. Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015; 58: 443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brownlee M, Vlassara H, Cerami A. Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Ann Intern Med 1984; 101: 527–537. [DOI] [PubMed] [Google Scholar]
- 26. Uribarri J, Cai W, Peppa M, et al Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci 2007; 62: 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fleming TH, Humpert PM, Nawroth PP, et al Reactive metabolites and AGE/RAGE‐mediated cellular dysfunction affect the aging process: a mini‐review. Gerontology 2011; 57: 435–443. [DOI] [PubMed] [Google Scholar]
- 28. Coughlan MT, Thorburn DR, Penfold SA, et al RAGE‐induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol 2009; 20: 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Monnier VM, Bautista O, Kenny D, et al Skin collagen glycation, glycoxidation, and crosslinking are lower in subjects with long‐term intensive versus conventional therapy of type 1 diabetes: relevance of glycated collagen products versus HbA1c as markers of diabetic complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes 1999; 48: 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Genuth S, Sun W, Cleary P, et al Glycation and carboxymethyllysine levels in skin collagen predict the risk of future 10‐year progression of diabetic retinopathy and nephropathy in the diabetes control and complications trial and epidemiology of diabetes interventions and complications participants with type 1 diabetes. Diabetes 2005; 54: 3103–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Monnier VM, Vishwanath V, Frank KE, et al Relation between complications of type I diabetes mellitus and collagen‐linked fluorescence. N Engl J Med 1986; 314: 403–408. [DOI] [PubMed] [Google Scholar]
- 32. Meerwaldt R, Graaff R, Oomen PH, et al Simple non‐invasive assessment of advanced glycation endproduct accumulation. Diabetologia 2004; 47: 1324–1330. [DOI] [PubMed] [Google Scholar]
- 33. Gerrits EG, Lutgers HL, Kleefstra N, et al Skin autofluorescence: a tool to identify type 2 diabetic patients at risk for developing microvascular complications. Diabetes Care 2008; 31: 517–521. [DOI] [PubMed] [Google Scholar]
- 34. Meerwaldt R, Lutgers HL, Links TP, et al Skin autofluorescence is a strong predictor of cardiac mortality in diabetes. Diabetes Care 2007; 30: 107–112. [DOI] [PubMed] [Google Scholar]
- 35. Fraser SD, Roderick PJ, McIntyre NJ, et al Skin autofluorescence and all‐cause mortality in stage 3 CKD. Clin J Am Soc Nephrol 2014; 9: 1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Vos LC, Noordzij MJ, Mulder DJ, et al Skin autofluorescence as a measure of advanced glycation end products deposition is elevated in peripheral artery disease. Arterioscler Thromb Vasc Biol 2013; 33: 131–138. [DOI] [PubMed] [Google Scholar]
- 37. Wang AY, Wong CK, Yau YY, et al Skin autofluorescence associates with vascular calcification in chronic kidney disease. Arterioscler Thromb Vasc Biol 2014; 34: 1784–1790. [DOI] [PubMed] [Google Scholar]
- 38. Vlassara H, Striker LJ, Teichberg S, et al Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc Natl Acad Sci USA 1994; 91: 11704–11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miyata T, Ueda Y, Horie K, et al Renal catabolism of advanced glycation end products: the fate of pentosidine. Kidney Int 1998; 53: 416–422. [DOI] [PubMed] [Google Scholar]
- 40. Brouwers O, Niessen PM, Miyata T, et al Glyoxalase‐1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014; 57: 224–235. [DOI] [PubMed] [Google Scholar]
- 41. Giacco F, Du X, D'Agati VD, et al Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 2014; 63: 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kato M, Natarajan R. Diabetic nephropathy–emerging epigenetic mechanisms. Nat Rev Nephrol 2014; 10: 517–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mimura I, Tanaka T, Nangaku M. New insights into molecular mechanisms of epigenetic regulation in kidney disease. Clin Exp Pharmacol Physiol 2016; 43: 1159–1167. [DOI] [PubMed] [Google Scholar]
- 44. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13: 484–492. [DOI] [PubMed] [Google Scholar]
- 45. Agardh E, Lundstig A, Perfilyev A, et al Genome‐wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med 2015; 13: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen Z, Miao F, Paterson AD, et al Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci USA 2016; 113: E3002–E3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swan EJ, Maxwell AP, McKnight AJ. Distinct methylation patterns in genes that affect mitochondrial function are associated with kidney disease in blood‐derived DNA from individuals with Type 1 diabetes. Diabet Med 2015; 32: 1110–1115. [DOI] [PubMed] [Google Scholar]
- 48. Shah A, Xia L, Masson EA, et al Thioredoxin‐Interacting Protein Deficiency Protects against Diabetic Nephropathy. J Am Soc Nephrol 2015; 26: 2963–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ko YA, Mohtat D, Suzuki M, et al Cytosine methylation changes in enhancer regions of core pro‐fibrotic genes characterize kidney fibrosis development. Genome Biol 2013; 14: R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hasegawa K, Wakino S, Simic P, et al Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin‐1 overexpression in podocytes. Nat Med 2013; 19: 1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peng L, Yuan Z, Ling H, et al SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol Cell Biol 2011; 31: 4720–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marumo T, Yagi S, Kawarazaki W, et al Diabetes Induces Aberrant DNA Methylation in the Proximal Tubules of the Kidney. J Am Soc Nephrol 2015; 26: 2388–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kobori H, Nangaku M, Navar LG, et al The intrarenal renin‐angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007; 59: 251–287. [DOI] [PubMed] [Google Scholar]
- 54. Kouzarides T. Chromatin modifications and their function. Cell 2007; 128: 693–705. [DOI] [PubMed] [Google Scholar]
- 55. Miao F, Chen Z, Genuth S, et al Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014; 63: 1748–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001; 414: 813–820. [DOI] [PubMed] [Google Scholar]
- 57. Chen J, Guo Y, Zeng W, et al ER stress triggers MCP‐1 expression through SET7/9‐induced histone methylation in the kidneys of db/db mice. Am J Physiol Renal Physiol 2014; 306: F916–F925. [DOI] [PubMed] [Google Scholar]
- 58. Tesch GH. MCP‐1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol 2008; 294: F697–F701. [DOI] [PubMed] [Google Scholar]
- 59. Cai M, Bompada P, Atac D, et al Epigenetic regulation of glucose‐stimulated osteopontin (OPN) expression in diabetic kidney. Biochem Biophys Res Commun 2016; 469: 108–113. [DOI] [PubMed] [Google Scholar]
- 60. Susztak K, Bottinger E, Novetsky A, et al Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes 2004; 53: 784–794. [DOI] [PubMed] [Google Scholar]
- 61. De Marinis Y, Cai M, Bompada P, et al Epigenetic regulation of the thioredoxin‐interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int 2016; 89: 342–353. [DOI] [PubMed] [Google Scholar]
- 62. Reddy MA, Sumanth P, Lanting L, et al Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int 2014; 85: 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee HB, Ha H. Plasminogen activator inhibitor‐1 and diabetic nephropathy. Nephrology 2005; 10(Suppl): S11–S13. [DOI] [PubMed] [Google Scholar]
- 64. Lassila M, Fukami K, Jandeleit‐Dahm K, et al Plasminogen activator inhibitor‐1 production is pathogenetic in experimental murine diabetic renal disease. Diabetologia 2007; 50: 1315–1326. [DOI] [PubMed] [Google Scholar]
- 65. Zhou X, Zang X, Ponnusamy M, et al Enhancer of Zeste Homolog 2 Inhibition Attenuates Renal Fibrosis by Maintaining Smad7 and Phosphatase and Tensin Homolog Expression. J Am Soc Nephrol 2016; 27: 2092–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Simpson K, Wonnacott A, Fraser DJ, et al MicroRNAs in Diabetic Nephropathy: from Biomarkers to Therapy. Curr Diab Rep 2016; 16: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ziyadeh FN, Hoffman BB, Han DC, et al Long‐term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor‐beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA 2000; 97: 8015–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kato M, Zhang J, Wang M, et al MicroRNA‐192 in diabetic kidney glomeruli and its function in TGF‐β‐induced collagen expression via inhibition of E‐box repressors. Proc Natl Acad Sci USA 2007; 104: 3432–3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kato M, Arce L, Wang M, et al A microRNA circuit mediates transforming growth factor‐beta1 autoregulation in renal glomerular mesangial cells. Kidney Int 2011; 80: 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Putta S, Lanting L, Sun G, et al Inhibiting microRNA‐192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol 2012; 23: 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Krupa A, Jenkins R, Luo DD, et al Loss of MicroRNA‐192 promotes fibrogenesis in diabetic nephropathy. J Am Soc Nephrol 2010; 21: 438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Arroyo JD, Chevillet JR, Kroh EM, et al Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA 2011; 108: 5003–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Beltrami C, Clayton A, Newbury L, et al Stabilization of Urinary MicroRNAs by Association with Exosomes and Argonaute 2 Protein. Non‐Coding RNA 2015; 1: 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zampetaki A, Kiechl S, Drozdov I, et al Plasma microRNA profiling reveals loss of endothelial miR‐126 and other microRNAs in type 2 diabetes. Circ Res 2010; 107: 810–817. [DOI] [PubMed] [Google Scholar]
- 75. Delic D, Eisele C, Schmid R, et al Urinary Exosomal miRNA Signature in Type II Diabetic Nephropathy Patients. PLoS One 2016; 11: e0150154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lu A, Miao M, Schoeb TR, et al Blockade of TSP1‐dependent TGF‐beta activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am J Pathol 2011; 178: 2573–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Argyropoulos C, Wang K, McClarty S, et al Urinary microRNA profiling in the nephropathy of type 1 diabetes. PLoS One 2013; 8: e54662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Togliatto G, Trombetta A, Dentelli P, et al MIR221/MIR222‐driven post‐transcriptional regulation of P27KIP1 and P57KIP2 is crucial for high‐glucose‐ and AGE‐mediated vascular cell damage. Diabetologia 2011; 54: 1930–1940. [DOI] [PubMed] [Google Scholar]
- 79. Fine LG, Bandyopadhay D, Norman JT. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia. Kidney Int Suppl 2000; 75: S22–S26. [PubMed] [Google Scholar]
- 80. Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end‐stage renal failure. J Am Soc Nephrol 2006; 17: 17–25. [DOI] [PubMed] [Google Scholar]
- 81. Safran M, Kim WY, O'Connell F, et al Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci USA 2006; 103: 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. van Bommel J, Siegemund M, Henny ChP, et al Heart, kidney, and intestine have different tolerances for anemia. Transl Res 2008; 151: 110–117. [DOI] [PubMed] [Google Scholar]
- 83. Schurek HJ, Jost U, Baumgartl H, et al Evidence for a preglomerular oxygen diffusion shunt in rat renal cortex. Am J Physiol 1990; 259: F910–F915. [DOI] [PubMed] [Google Scholar]
- 84. Kang DH, Hughes J, Mazzali M, et al Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. J Am Soc Nephrol 2001; 12: 1448–1457. [DOI] [PubMed] [Google Scholar]
- 85. Ohashi R, Shimizu A, Masuda Y, et al Peritubular capillary regression during the progression of experimental obstructive nephropathy. J Am Soc Nephrol 2002; 13: 1795–1805. [DOI] [PubMed] [Google Scholar]
- 86. Astor BC, Muntner P, Levin A, et al Association of kidney function with anemia: the Third National Health and Nutrition Examination Survey (1988‐1994). Arch Intern Med 2002; 162: 1401–1408. [DOI] [PubMed] [Google Scholar]
- 87. Adler S, Huang H. Impaired regulation of renal oxygen consumption in spontaneously hypertensive rats. J Am Soc Nephrol 2002; 13: 1788–1794. [DOI] [PubMed] [Google Scholar]
- 88. Korner A, Eklof AC, Celsi G, et al Increased renal metabolism in diabetes. Mechanism and functional implications. Diabetes 1994; 43: 629–633. [DOI] [PubMed] [Google Scholar]
- 89. Chen CY, Liao KM. Chronic Obstructive Pulmonary Disease is associated with risk of Chronic Kidney Disease: a Nationwide Case‐Cohort Study. Sci Rep 2016; 6: 25855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Iseki K, Tohyama K, Matsumoto T, et al High Prevalence of chronic kidney disease among patients with sleep related breathing disorder (SRBD). Hypertens Res 2008; 31: 249–255. [DOI] [PubMed] [Google Scholar]
- 91. Siervo M, Riley HL, Fernandez BO, et al Effects of Prolonged Exposure to Hypobaric Hypoxia on Oxidative Stress, Inflammation and Gluco‐Insular Regulation: the Not‐So‐Sweet Price for Good Regulation. PLoS One 2014; 9: e94915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Michaely HJ, Metzger L, Haneder S, et al Renal BOLD‐MRI does not reflect renal function in chronic kidney disease. Kidney Int 2012; 81: 684–689. [DOI] [PubMed] [Google Scholar]
- 93. Inoue T, Kozawa E, Okada H, et al Noninvasive evaluation of kidney hypoxia and fibrosis using magnetic resonance imaging. J Am Soc Nephrol 2011; 22: 1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wang ZJ, Kumar R, Banerjee S, et al Blood oxygen level‐dependent (BOLD) MRI of diabetic nephropathy: preliminary experience. J Magn Reson Imaging 2011; 33: 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yin WJ, Liu F, Li XM, et al Noninvasive evaluation of renal oxygenation in diabetic nephropathy by BOLD‐MRI. Eur J Radiol 2012; 81: 1426–1431. [DOI] [PubMed] [Google Scholar]
- 96. Semenza GL. Oxygen Sensing, Hypoxia‐Inducible Factors, and Disease Pathophysiology. Annu Rev Pathol 2014; 9: 47–71. [DOI] [PubMed] [Google Scholar]
- 97. Ploumakis A, Coleman ML. OH, the Places You'll Go! Hydroxylation, Gene Expression, and Cancer. Mol Cell 2015; 58: 729–741. [DOI] [PubMed] [Google Scholar]
- 98. Tahiliani M, Koh KP, Shen Y, et al Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shen L, Wu H, Diep D, et al Genome‐wide analysis reveals TET‐ and TDG‐dependent 5‐methylcytosine oxidation dynamics. Cell 2013; 153: 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Thienpont B, Steinbacher J, Zhao H, et al Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016; 537: 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ho JJ, Metcalf JL, Yan MS, et al Functional importance of Dicer protein in the adaptive cellular response to hypoxia. J Biol Chem 2012; 287: 29003–29020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. van den Beucken T, Koch E, Chu K, et al Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat Commun 2014; 5: 5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Muratsu‐Ikeda S, Nangaku M, Ikeda Y, et al Downregulation of miR‐205 modulates cell susceptibility to oxidative and endoplasmic reticulum stresses in renal tubular cells. PLoS One 2012; 7: e41462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mimura I, Nangaku M, Kanki Y, et al Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia‐inducible factor 1 and KDM3A. Mol Cell Biol 2012; 32: 3018–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Inoue T, Kohro T, Tanaka T, et al Cross‐enhancement of ANGPTL4 transcription by HIF1 alpha and PPAR beta/delta is the result of the conformational proximity of two response elements. Genome Biol 2014; 15: R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329: 977–986. [DOI] [PubMed] [Google Scholar]
- 107. Reichard P, Nilsson BY, Rosenqvist U. The effect of long‐term intensified insulin treatment on the development of microvascular complications of diabetes mellitus. N Engl J Med 1993; 329: 304–309. [DOI] [PubMed] [Google Scholar]
- 108. Patel A, MacMahon S, Chalmers J, et al Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358: 2560–2572. [DOI] [PubMed] [Google Scholar]
- 109. Pohl MA, Blumenthal S, Cordonnier DJ, et al Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the irbesartan diabetic nephropathy trial: clinical implications and limitations. J Am Soc Nephrol 2005; 16: 3027–3037. [DOI] [PubMed] [Google Scholar]
- 110. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective Diabetes Study Group. BMJ 1998; 317: 713–720. [PMC free article] [PubMed] [Google Scholar]
- 111. Lewis EJ, Hunsicker LG, Bain RP, et al The effect of angiotensin‐converting‐enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329: 1456–1462. [DOI] [PubMed] [Google Scholar]
- 112. Lewis JB, Berl T, Bain RP, et al Effect of intensive blood pressure control on the course of type 1 diabetic nephropathy. Collaborative Study Group. Am J Kidney Dis 1999; 34: 809–817. [DOI] [PubMed] [Google Scholar]
- 113. Mauer M, Zinman B, Gardiner R, et al Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med 2009; 361: 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Heerspink HJ, Persson F, Brenner BM, et al Renal outcomes with aliskiren in patients with type 2 diabetes: a prespecified secondary analysis of the ALTITUDE randomised controlled trial. Lancet Diabetes Endocrinol 2016; 4: 309–317. [DOI] [PubMed] [Google Scholar]
- 115. Pan Y, Guo LL, Jin HM. Low‐protein diet for diabetic nephropathy: a meta‐analysis of randomized controlled trials. Am J Clin Nutr 2008; 88: 660–666. [DOI] [PubMed] [Google Scholar]
- 116. Foundations of care . education, nutrition, physical activity, smoking cessation, psychosocial care, and immunization. Diabetes Care 2015; 38(Suppl): S20–S30. [DOI] [PubMed] [Google Scholar]
- 117. Zinman B, Wanner C, Lachin JM, et al Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med 2015; 373: 2117–2128. [DOI] [PubMed] [Google Scholar]
- 118. Wanner C, Inzucchi SE, Lachin JM, et al Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J Med 2016; 375: 323–334. [DOI] [PubMed] [Google Scholar]
- 119. Lambers Heerspink HJ, de Zeeuw D, Wie L, et al Dapagliflozin a glucose‐regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab 2013; 15: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. O'Neill J, Fasching A, Pihl L, et al Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am J Physiol Renal Physiol 2015; 309: F227–F234. [DOI] [PubMed] [Google Scholar]
- 121. Nordquist L, Friederich‐Persson M, Fasching A, et al Activation of hypoxia‐inducible factors prevents diabetic nephropathy. J Am Soc Nephrol 2015; 26: 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tanaka T, Kojima I, Ohse T, et al Cobalt promotes angiogenesis via hypoxia‐inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest 2005; 85: 1292–1307. [DOI] [PubMed] [Google Scholar]
- 123. Tanaka T, Matsumoto M, Inagi R, et al Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int 2005; 68: 2714–2725. [DOI] [PubMed] [Google Scholar]
- 124. Maxwell PH, Eckardt KU. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat Rev Nephrol 2016; 12: 157–168. [DOI] [PubMed] [Google Scholar]
- 125. Rahtu‐Korpela L, Karsikas S, Horkko S, et al HIF prolyl 4‐hydroxylase‐2 inhibition improves glucose and lipid metabolism and protects against obesity and metabolic dysfunction. Diabetes 2014; 63: 3324–3333. [DOI] [PubMed] [Google Scholar]
- 126. Bryan HK, Olayanju A, Goldring CE, et al The Nrf2 cell defence pathway: Keap1‐dependent and ‐independent mechanisms of regulation. Biochem Pharmacol 2013; 85: 705–717. [DOI] [PubMed] [Google Scholar]
- 127. Xue M, Rabbani N, Momiji H, et al Transcriptional control of glyoxalase 1 by Nrf2 provides a stress‐responsive defence against dicarbonyl glycation. Biochem J 2012; 443: 213–222. [DOI] [PubMed] [Google Scholar]
- 128. Pergola PE, Krauth M, Huff JW, et al Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b‐4 CKD. Am J Nephrol 2011; 33: 469–476. [DOI] [PubMed] [Google Scholar]
- 129. Chin MP, Wrolstad D, Bakris GL, et al Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J Card Fail 2014; 20: 953–958. [DOI] [PubMed] [Google Scholar]