

Abstract
Background
Drug interactions between antiretroviral drugs (ARVs) and anthelminthic drugs, ivermectin (IVM) and praziquantel (PZQ) were assessed by investigating their permeation through the Caco-2 cell monolayers in a transwell. The impact of anthelminthics on the transport of ARVs was determined by assessing the apical to basolateral (AP → BL) [passive] and basolateral to apical (BL → AP) [efflux] directions alone, and in presence of an anthelminthic. The reverse was conducted for the assessment of the influence of ARVs on anthelminthics.
Methods
Samples from the AP and BL compartments were taken at 60, 120, 180 and 240 min and quantified either by HPLC or radiolabeled assay using a liquid scintillating counter for the respective drugs. Transepithelial resistance (TEER) was used to assess the integrity of the monolayers. The amount of compound transported per second (apparent permeability, Papp) was calculated for both AP to BL (PappAtoB), and BL to AP (PappBtoA) movements. Samples collected after 60 min were used to determine the efflux ratio (ER), quotient of secretory permeability and absorptive permeability (PappBL-AP/PappAP-BL). The reverse, (PappAP-BL/PappBL-AP) constituted the uptake ratio. The impact of SQV, EFV and NVP on the transport of both IVM and PZQ were investigated. The effect of LPV on the transport of IVM was also determined. The influence of IVM on the transport of SQV, NVP, LPV and EFV; as well as the effect PZQ on the transport of SQV of was also investigated, and a two-tailed p value of <0.05 was considered significant.
Results
IVM significantly inhibited the efflux transport (BL → AP movement) of LPV (ER; 6.7 vs. 0.8, p = 0.0038) and SQV (ER; 3.1 vs. 1.2 p = 0.00328); and increased the efflux transport of EFV (ER; 0.7 vs. 0.9, p = 0.031) suggesting the possibility of drug transporter mediated interactions between the two drugs. NVP increased the efflux transport of IVM (ER; 0.8 vs. 1.8, p = 0.0094).
Conclusions
The study provides in vitro evidence of potential interactions between IVM, an anthelminthic drug with antiretroviral drugs; LPV, SQV, NVP and EFV. Further investigations should be conducted to investigate the possibility of in vivo interactions.
Electronic supplementary material
The online version of this article (doi:10.1186/s40360-017-0129-6) contains supplementary material, which is available to authorized users.
Keywords: Antiretroviral, Antiparasitic, Drug interactions, Caco-2 cell monolayers, Drug transport, Intestinal epithelium, TEER
Background
The sub-Saharan Africa still leads in the prevalence of Human immunodeficiency virus infection and acquired immune deficiency syndrome (HIV/AIDs), malaria, tuberculosis and helminthic infections. HIV positive patients are thus likely to be co-infected with any of the diseases, and the co-administration of anthelminthics and ARVs is not uncommon [1]. This can give rise to drug-drug interactions (DDIs) which are likely to alter the therapeutic outcome of each of the drugs. These interactions may result in an increase or decrease in the plasma concentrations of the drugs thereby increasing the risk of toxicity or development of resistance amongst other adverse effects. This may in some instances require dosage adjustments [2–4]. The knowledge of any potential interactions between these drugs is therefore important in optimization of HIV therapy [5, 6]. The potential mechanisms of the drug interactions include modulation by drug transporters (both efflux and influx), and inhibition or induction of drug metabolizing enzymes [6–14].
Several drugs are substrates and/or inhibitors of these efflux transporters and metabolic enzymes, especially CYP 3A4 [15, 16]. Among the ARVs, protease inhibitors (PIs) are known to be substrates of P-gp, ABCC 1 and ABCC 2 [3, 12, 17, 18]. Saquinavir (SQV) and lopinavir (LPV) are substrates and inhibitors of drug transporters [19, 20]. Nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) have also been characterized as substrates for drug transporters [21–24]. Newer NNRTIs such as etravirine have been reported to induce ABC transporters especially BCRP/ABCG2, and therefore potential for drug interactions with co-administered drugs that are substrates for these transporters [25]. Rilpivirine has also been reported to induce and inhibit several relevant drug-metabolizing enzymes and drug transporters albeit with less potential for drug interactions owing to low plasma concentrations [26].
PZQ and IVM are some of the most widely used anthelminthic drugs. PZQ is mainly used to treat schistosomiasis, whereas IVM is used in the treatment of lymphatic filariasis [27, 28]. Both diseases are endemic in developing countries with schistosomiasis afflicting over 200 million people [29, 30], and lymphatic filariasis having a global prevalence of over 120 million people with an estimated 1.3 billion at risk [31, 32]. IVM has been characterized as a substrate and inhibitor of P-gp [33–35]. IVM interacts with P-gp modulators [36, 37], and the inhibition of P-gp has been described as a potential strategy to counter the emerging resistance to IVM [38]. PZQ has not been conclusively characterized with regards to drug transporter specificity [39, 40], but from the available data, it is known to be metabolized by cytochrome P450 isoenzymes (CYP), mainly CYP2B1, CYP3A4, CYP2C9 and CYP2C19. It therefore has potential to interact with drugs which are inhibitors or inducers of these enzymes [41, 42]. Enzyme inducers such as carbamazepine, phenytoin and rifampicin reduce PZQ plasma levels, while ketoconazole, an enzyme inhibitor significantly increases its concentration [41, 43]. Previous researchers on transport of antiparasitic drugs along the Caco-2 cell monolayers (CCM) have also reported that PZQ is an inhibitor of P-gp without being a substrate [40]. However, it is not clear how the investigators concluded that this was specifically mediated by P-gp using the CCM model since CCM expresses several other transporters [44]. From the research conducted earlier in our laboratories we have demonstrated that PZQ is neither a substrate nor an inhibitor of P-gp in CEM T-lymphoblastoid cells [45].
The majority of drugs in use are orally administered, and their absorption from the gastrointestinal tract is pivotal for their therapeutic success [46]. The ability of a drug to cross the intestinal wall in order to reach portal circulation is to a large extent dependent on its permeability coefficient [47]. The Caco-2 cell model provides a simple and reliable method to assay in vitro permeability of drugs [48–50]. The permeability of drugs through the CCM correlates well with in vivo absorption in humans thus making the CCM an invaluable analytical tool in the screening of orally administered drugs [51–54]. CCM are derived from human colonic adenocarcinoma and have morphological as well as functional similarities to intestinal (absorptive) enterocytes [44, 55–57]. They have adherent properties and therefore form a monolayer with tight junctions which prevent paracellular diffusion so that drugs or other solutes can only pass through the cell, as illustrated in the cartoon (Fig. 1). This in addition results in development of cell polarity, and the efflux transporter P-glycoprotein (P-gp) has been shown to be localized on the apical brush border, approximately 20 microns above the base of the cells [58], while certain efflux transporters such as multidrug resistance-associated proteins (MRPs) are expressed on the basolateral side of the monolayer [59, 60]. Apart from P-gp and MRPs, CCM express a wide array of transporters (efflux and influx) as well as metabolic enzymes, thus making them suitable for the study of drug-drug interactions based on the permeability of drugs through the monolayers [20, 44, 55, 56, 61, 62].
Fig. 1.
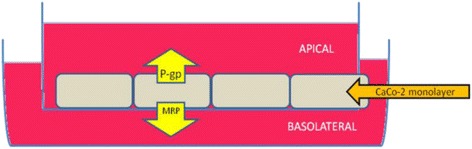
An illustration of a transwell depicting the growing Caco-2 cells including the compartments and the positions of the transporters
The permeation of drugs through the monolayers allows the study of the major absorptive mechanisms for drugs, such as passive transcellular transport and carrier-mediated influx as well as efflux mechanisms [63]. In this system, the passage of drugs from apical (AP) to basolateral (BL) compartment is attributable to passive diffusion and occurs at a lower rate, whereas the BL to AP passage occurs by active transport, presumed to be mediated by transporters [20, 44, 64]. The carrier-mediated transport is a saturable process, which raises a possibility that when two drugs are co-administered they may compete for a transporter (influx or efflux) which would lead to drug interactions leading to lower or higher exposure than when dosed alone [20, 65]. The cells are grown on a porous membrane and form differentiated monolayers after about 20 days. The membranes are polarized and evaluation of the monolayers can be performed by measuring the transepithelial resistance (TEER), using a volt-ohm meter equipped with electrodes placed in the upper and lower chambers of the insert. TEER increases with culture reaching a maximum in about 10-15 days [66–68], and depends on the number of cells seeded, plus the surface area of the filter. TEER values range from 150 to 1600 ohm.cm2 as compared to human ileum which is about 50 ohm [69].
To date there is scant information from the literature regarding the interactions between ARVs and anthelminthics despite the likelihood of their co-administration in tropical regions owing to their geographic overlaps. The aim of the study was therefore to assess the potential DDIs between ARVs and the anthelminthic drugs; PZQ and IVM. In the study, CCM was used to evaluate potential interactions between ARVs and anthelminthics. The impact of the anthelminthics on the ARVs transport was determined by assessing the AP → BL and BL → AP directions alone, and in presence of an anthelminthic, and the reverse was conducted for the assessment of the influence of ARVs on anthelminthics. Quantification was performed using an HPLC method described earlier [45], or radiolabeled assay using a liquid scintillating counter. PZQ and IVM were used as prototypes for the anthelminthics, and PIs, saquinavir (SQV) and lopinavir (LPV) as well as NNRTIs, efavirenz (EFV) and nevirapine (NVP) as prototypes of ARVs. The impact of SQV, EFV and NVP on the transport of PZQ as well as the impact of PZQ on the transport of SQV was determined. The influence of SQV, NVP, LPV and EFV on the transport of IVM, and IVM on the transport of SQV, NVP, LPV and EFV were also investigated.
Methods
Equipment
The HPLC consisted of a Dionex (Dionex Softron GmbH, Germany) HPLC system with a P 680 pump, an ASI-100 automated sample injector and a UVD 1704 detector. A 250μl injector with a 20μl loop was used. Reversed-phase-liquid chromatography was carried out using a Hypurity™ C18 analytical column, 5μm x 4.6mm (Thermo Electron Corporation, Runcorn, UK 22105-154630). A column guard (Thermo electron 60140-412) was used to protect the analytical column. The ultraviolet detector was set to monitor the 215 nm wavelength. A Packard Tri-Carb Liquid Scintillation Analyzer model 1900 TR (Packard instrument Co.) was used for radioactivity counting. A Millicell Electrical Resistance System (Fisher Scientific, Leicestershire, UK) was used for measuring the transepithelial electric resistance (TEER). Transwells (six-well transwell polycarbonate tissue culture treated plates, 4.67 cm2, 24 mm diameter; 0.4 μm pore size) were purchased from Corning life Sciences (Costar High Wycombe, Bucks; UK).
Materials
The human colon adenocarcinoma cell line, Caco-2 was purchased from European collection of cell cultures (ECACC No. 286010202), and the cells were counted using a Nucleo Counter (ChemoMetec, Denmark) cell counter. SQV was provided by Roche Discovery (Welwyn Garden City, UK), LPV by Abbott Laboratories (Chicago, USA), EFV by Dupont Bristol Myers Squibb (New Brunswick, NJ, USA), and NVP by Boehringer Ingelheim (Berkshire, UK). Radiolabelled SQV (3H SQV), LPV (3H LPV), EFV (14C EFV) and NVP (3H NVP) were purchased from Moravek Biochemicals (Brea California, USA). 3H IVM was kindly donated by Dr Iain Gardner of Pfizer (Sandwich, Kent, UK). PZQ, CLZ, DMEM, HBSS, FBS, DMSO and Trypsin-EDTA solution were purchased from Sigma Aldrich (Poole, UK). ACN and MeOH were purchased from VWR Laboratory Supplies (Poole, UK) whereas diethyl ether was purchased from Fisher Scientific, (Loughborough, UK). Ultima Gold liquid scintillation cocktail was obtained from Packard (Groningen, Netherlands). All the other chemicals used were of analytical or HPLC grade. Deionised water used to prepare the solutions or mobile phase, and was purified in an Elga DV 25 pure lab option system (Elga, High Wycombe, Bucks, and UK).
Caco-2 cell lines
Cell culture
Caco-2 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with fetal bovine serum (FBS) [15%v/v]. The cells were grown and routinely seeded in tissue cultured treated 162 cm2 flasks in a humidified chamber (37°C, 10% CO2 incubator) and harvested by regular trypsinization. The medium was changed every 2 to 3 days until the confluence of the cell monolayer was achieved. Trypsinization involved decanting the media, followed by washing twice with 6 ml of Hanks' Balanced Salt Solution (HBSS), and the detachment of the monolayer by addition of 4 ml of trypsin EDTA. The cells were then incubated for 10 min. The resulting suspension was then centrifuged (2000 g × 5 min, 4°C) and supernatant removed. The resulting pellet was then re-suspended in 20 ml of fresh DMEM (+15% FBS), 10 ml transferred two new flasks; and each made to 20 ml.
Storage of Caco-2 cells
The cells were trypsinised as described earlier after attaining confluence. The pellets were then re-suspended in DMEM (15%v/v), counted using a nucleocounter and centrifuged (2000 g × 5 min, 4°C). The cells were then re-suspended in warm FBS (FBS + 10% DMSO), mixed thoroughly and made up to a concentration of 5 × 106 cells per ml. 1 ml of the cell suspension was then transferred to pre-labelled 1.5 ml cryovials and frozen at -80°C for use as and when required. The viable semi-frozen cells were thawed by placing the cryovials rapidly in a waterbath (37°C), or by simply holding in the vials in the hands for a few minutes and re-suspending the pellets in 9 ml DMEM (+15%FBS) followed by culturing.
Determination of drug transport in Caco-2 cells
Cell seeding
The monolayers used were of between passage 20 and 65 after 15 days of growth. Each experiment was performed in duplicate using two six well-transwell culture plates. The cells were trypsinized as described earlier, and after centrifugation the pellet re-suspended in fresh DMEM (+15%), and the cells counted using a cell counter. A volume of DMEM was added enough to give a cell count of 2 × 106 cells per ml, and cells were seeded on the transwell culture plates at a density of 2 × 104 cells/cm2 (~ 100,000 cells per well, since insert membrane growth area = 4.67 cm2). The plates were then incubated at 37°C and 10% CO2 in a humidified chamber and the media changed every 2-3 days, by aspirating using a suction pump and replacing with an equal volume of DMEM. Transport experiments were conducted 15 to 20 days after seeding. The TEER across the cell monolayers was monitored using a Millicell-ERS in order to assess cell monolayer integrity and the monolayers considered appropriate for the experiment when the TEER values were typically above 500 cm2 [49, 70].
Transport experiments
Prior to transport studies, each monolayer was washed and equilibrated with the transport medium (DMEM without FBS). The medium was removed from all AP and BL compartments of the transwells and replaced with 2 ml of the transport medium (DMEM alone), to both compartments and equilibrated for 1 h (37°C, 10% CO2 incubator), after which the TEER was re-assessed and labeled. The medium was then removed from both compartments and replaced with an equal volume of pre-warmed medium containing the compound of interest at the appropriate concentration. For the AP → BL transport, 2 ml of medium containing the desired drug was placed in the AP chamber and 2 ml of the medium alone in the BL chamber, whereas 2 ml of medium containing the drug was placed in the BL and 2 ml of medium on the AP chamber for the BL → AP transport [71]. The effect of the second drug was then assessed by adding the medium containing the original drug and the drug under study to the AP side for AP → BL transport with the medium containing the original drug alone in the BL chamber, and vice-versa for the BL → AP transport. Transport in each direction was done in triplicate. The transwell plates were then incubated (37°C, 10% CO2 incubator), and 100μl of the samples from the AP and BL compartments were taken at 60, 120, 180 and 240 min and quantified either by HPLC method described earlier [45], or by the use of a liquid scintillating counter depending on the drug under study. The HPLC method involved liquid - liquid extraction followed by ultra-high performance liquid chromatography using a Hypurity C18 column and ultraviolet detection set at a wavelength of 215 nm. The mobile phase consisted of ammonium formate, acetonitrile and methanol (57:38:5 v/v). Separation was facilitated via isocratic elution at a flow rate of 1.5 ml/min. For HPLC assays, the concentration used for each drug was 20μg/ml. The concentrations that offered the best detection were selected for radiolabeled assay. Table 1 summarizes the type of assay and the various concentrations used in each experiment. HPLC was used in the assay of interactions between SQV/PZQ, IVM/SQV, EFV/PZQ and NVP/PZQ; while radiolabeled assay was utilized in the investigations of the interactions between PZQ/SQV, SQV/IVM, NVP/IVM, IVM/NVP, LPV/IVM, IVM/LPV, EFV/IVM and IVM/EFV. The integrity of the CCM during the experiment was monitored by measuring the TEER at the beginning (0 min) and the end of the experiment (240min).
Table 1.
Concentration and type of assay used for each experiment
| Concentrations used | Quantification method | |||||
|---|---|---|---|---|---|---|
| A | B | Drug | μg/ml | μM | ||
| 1 | PZQ | SQV | PZQ | 20 | 64 | HPLC |
| SQV | 20 | 29.8 | ||||
| 2 | SQV | PZQ | [3H] SQV | 1.07 | Radiolabeled assay | |
| PZQ | 20 | 64 | ||||
| 3 | IVM | SQV | [3H] IVM | 0.01 | Radiolabeled assay | |
| SQV | 20 | 29.8 | ||||
| 4 | SQV | IVM | SQV | 20 | 29.8 | HPLC |
| IVM | 20 | 22.75 | ||||
| 5 | PZQ | EFV | PZQ | 20 | 64 | HPLC |
| EFV | 20 | 63.4 | ||||
| 6 | PZQ | NVP | PZQ | 20 | 64 | HPLC |
| NVP | 20 | 75.1 | ||||
| 7 | IVM | NVP | [3H] IVM | 0.01 | Radiolabeled assay | |
| NVP | 20 | 75.1 | ||||
| 8 | NVP | IVM | [3H] NVP | 1.5 | Radiolabeled assay | |
| IVM | 20 | 22.75 | ||||
| 9 | IVM | LPV | [3H] IVM | 0.02 | Radiolabeled assay | |
| LPV | 20 | 31.81 | ||||
| 10 | LPV | IVM | [3H] LPV | 1.5 | Radiolabeled assay | |
| IVM | 20 | 22.75 | ||||
| 11 | IVM | EFV | [3H] IVM | 0.01 | Radiolabeled assay | |
| EFV | 20 | 63.34 | ||||
| 12 | EFV | IVM | [14C] EFV | 0.01 | Radiolabeled assay | |
| IVM | 20 | 22.75 | ||||
Drug A Drug whose transport is under investigation
Drug B Interacting drug
Apparent permeability
The results were expressed as apparent permeability coefficient (Papp, unit: cms-1), the amount of compound transported per second. Papp values were calculated for both AP to BL (PappAtoB), and BL to AP (PappBtoA) movement of the compound. Papp was calculated using the following equation [52, 72]:
Papp (cm/s) = (dQ/dt) x (1/(ACO)
dQ/dt = Steady-state flux (dpm s-1 or μmol s-1)
A = Surface area of the filter (cm2)
CO = Initial concentration in the donor chamber (dpm litre -1 or μM)
The quotient of secretory permeability and absorptive permeability (PappBL-AP/PappAP-BL) constitutes the efflux ratio, while the reverse (PappAP-BL/PappBL-AP) is the uptake ratio [71]. This calculation requires that the receiver concentration should not exceed 10% of the donor concentration, and therefore was only applied for the samples taken at 60 min. The permeability is a saturable process and depends on several physiological conditions such as accumulation, pH, and lipophilicity (sink conditions), which have an effect on Papp values with incubations over longer periods of time [71, 73]. In order to assess the potential interactions, ER of a respective drug alone was compared to that in presence of a second drug under investigation. Samples of both drugs collected after 60 min were used to investigate the trends over a period of 4 h.
Statistical analysis
The results were presented as mean ± standard deviation (SD) of three experiments with 95% confidence intervals for differences between the means where appropriate. The analysis of the transport results obtained after 60 min was performed using a two-way analysis of variance (ANOVA). A two-tailed p value of <0.05 was accepted as being significant.
Results
The results of the transport experiments are summarized in Table 2. IVM significantly inhibited the efflux transport (BL → AP movement) of LPV (ER; 6.7 vs. 0.8, p = 0.0038) and SQV (ER; 3.1 vs. 1.2 p = 0.0328). It also increased the efflux transport of EFV (ER; 0.7 vs. 0.9, p = 0.031) suggesting the possibility of drug transporter mediated interactions between the two drugs. NVP increased the efflux transport of IVM (ER; 0.8 vs. 1.8, p = 0.0094). Figs. 2, 3, 4, 5, 6 and 7 illustrate the trends of interactions between the respective drugs over a period of 4 h. Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 show the details of how the ER values of each experiment were computed from the obtained results.
Table 2.
Summary of the cumulative transport results showing the mean ER values after 60 min
| Papp (106 cm/s) | Mean ER value | SD | p value | Comments | ||||
|---|---|---|---|---|---|---|---|---|
| AP → BL | BL → AP | |||||||
| 1 | A | PZQ | 7.6 | 8.6 | 1.14 | 0.17 | 0.5008 | No significant influence on PZQ transport |
| B | PZQ + SQV | 5.0 | 6.6 | 1.34 | 0.27 | |||
| A | SQV | 6.6 | 10.8 | 3.55 | 1.72 | 0.6796 | No significant influence on SQV transport | |
| B | SQV + PZQ | 18.3 | 27.0 | 2.65 | 1.57 | |||
| 2 | A | PZQ | 28.1 | 28.6 | 1.03 | 0.19 | 0.0964 | No significant influence on PZQ transport |
| B | PZQ + NVP | 37.2 | 30.5 | 0.82 | 0.08 | |||
| A | PZQ | 35.0 | 30.3 | 0.87 | 0.17 | 0.4676 | No significant influence on PZQ transport | |
| B | PZQ + EFV | 35.1 | 39.2 | 1.19 | 0.18 | |||
| 3 | A | IVM | 7.4 | 5.9 | 0.86 | 0.18 | 0.2007 | No significant influence on IVM transport |
| B | IVM + SQV | 6.4 | 6.3 | 0.99 | 0.16 | |||
| A | SQV | 6.9 | 21.1 | 3.05 | 0.46 | 0.0328 | Potential for interaction; efflux transport of SQV inhibited | |
| B | SQV + IVM | 6.2 | 7.7 | 1.24 | 0.14 | |||
| 4 | A | IVM | 14.1 | 11.3 | 0.80 | 0.07 | 0.0094 | Potential for interaction; efflux transport of IVM increased |
| B | IVM + NVP | 4.8 | 8.6 | 1.76 | 0.13 | |||
| A | NVP | 42.7 | 39.5 | 0.91 | 0.12 | 0.6891 | No significant influence on IVM transport | |
| B | NVP + IVM | 39.4 | 36.6 | 0.98 | 0.17 | |||
| 5 | A | IVM | 4.5 | 13.2 | 2.89 | 0.50 | 0.1822 | No significant influence on IVM transport |
| B | IVM + LPV | 6.7 | 9.5 | 1.58 | 0.62 | |||
| A | LPV | 4.8 | 32.1 | 6.72 | 0.54 | 0.0038 | Potential for interaction; efflux transport of LPV inhibited | |
| B | LPV + IVM | 15.9 | 12.3 | 0.78 | 0.10 | |||
| 6 | A | IVM | 7.6 | 8.9 | 1.19 | 0.15 | 0.4280 | No significant influence on IVM transport |
| B | IVM + EFV | 4.7 | 7.3 | 1.67 | 0.69 | |||
| A | EFV | 16.5 | 10.8 | 0.66 | 0.07 | 0.0310 | Potential for interaction; efflux transport of EFV increased | |
| B | EFV + IVM | 8.6 | 7.7 | 0.90 | 0.03 | |||
Drug A Drug whose transport is under investigation
Drug B Drug A plus the interacting drug
ER Efflux ratio
Fig. 2.
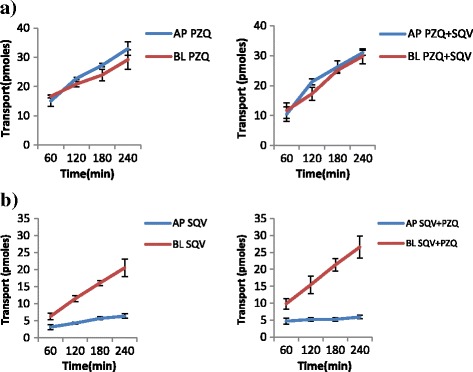
Influence of SQV on the transport of PZQ (a), and PZQ on the transport of SQV (b) across the Caco2 cell monolayers over a 4h period. The results are expressed as mean ± S.D (n = 3). a Cumulative transport of PZQ alone, and in presence of SQV. b Cumulative transport of SQV alone and in presence of PZQ
Fig. 3.
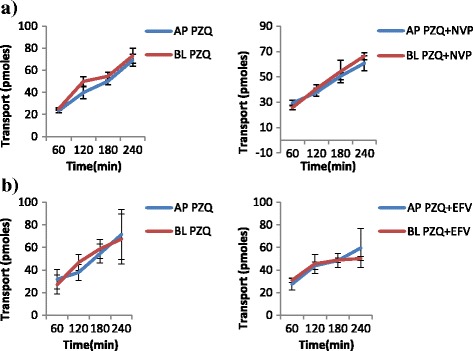
Influence of NVP (a) and EFV (b) on the transport of PZQ. a Cumulative transport of PZQ alone, and in presence of NVP. b Cumulative transport of PZQ alone, and in presence of EFV
Fig. 4.
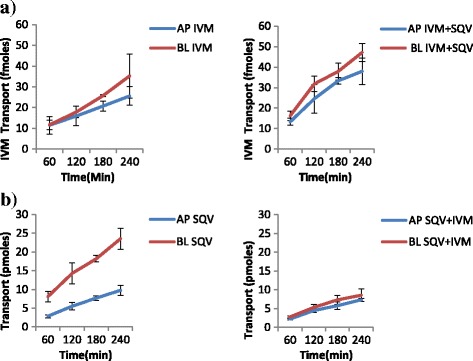
Influence of SQV on the transport of IVM a, and IVM on SQV transport b. a Cumulative transport of [3H] IVM alone, and in presence of SQV. b Cumulative transport of SQV alone, and in presence of IVM
Fig. 5.
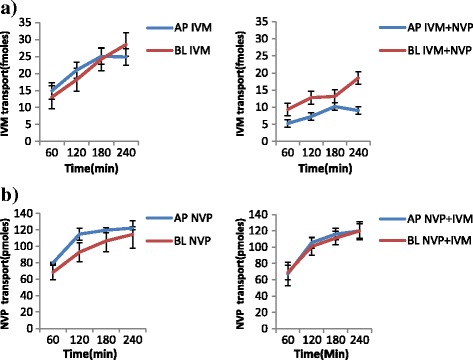
Influence of NVP on the transport of IVM a, and IVM on NVP transport b. a Cumulative transport of [3H] IVM alone, and in presence of NVP. b Cumulative transport of [3H] IVM alone, and in presence of NVP
Fig. 6.
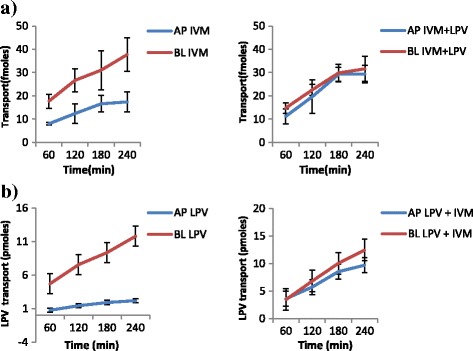
Influence of LVP on the transport of IVM a, and IVM on LVP transport b. a Cumulative transport of [3H] IVM alone, and in presence of LPV. b Cumulative transport of [3H] LPV alone, and in presence of IVM
Fig. 7.
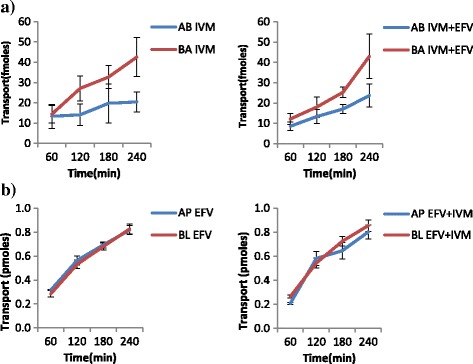
Influence of EFV on the transport of IVM a, and IVM on EFV transport b. a Cumulative transport of [3H] IVM alone, and in presence of EFV. b Cumulative transport of [14C] EFV alone, and in presence IVM
Discussion
The main aim of this study was to establish the potential interactions between the anthelminthic drugs, IVM and PZQ with ARVs by investigating their transport through CCM. PIs and NNRTIs were selected as they are widely used in management of HIV and may be co-administered with anthelminthics in the mass treatment of helminthic infections and HIV in third world countries because of the geographic overlap of the two diseases. In addition PIs and NNRTIs have both been characterized with regards to substrate specificity of both CYP 450 enzymes and drug transporters [19, 24, 74, 75]. SQV and LPV were selected as prototypes of PIs, while EFV and NVP were prototypes of NNRTIs. The CCM express a wide range of transporters making them suitable for the study of drug-drug interactions, since drug transporters play an integral role in the disposition of drugs and corresponding susceptibility to drug interactions [25, 44, 54–56].
The main findings from our study provide evidence that IVM influences the transport of SQV, LPV and EFV; whereas NVP influences the transport of IVM as illustrated by their transport characteristics along the CCM. IVM significantly inhibited the efflux transport of LPV and SQV; and increased the efflux of EFV. NVP increased the efflux transport of IVM. This raises the possibility of interactions between the drugs involving drug transporters. Drug interactions between ARVs and co-administered drugs may lead to treatment failures and adverse reactions, and understanding of the mechanism of interaction is pivotal for the optimal choice of the highly active antiretroviral therapy (HAART) regimens [76]. Increased efflux of a drug from the cells may cause resistance as the levels become sub-therapeutic [77–79], whereas an inhibition of its efflux may cause enhanced plasma toxicity and subsequent toxicity [80–82].
PZQ did not appear to significantly influence the transport of ARVs and likewise ARVs did not affect the transport of PZQ. SQV and other PIs have been demonstrated to be substrates of efflux transporters P-gp, MRP1 and MRP2 that are expressed by Caco-2 cells [17, 19]. IVM has also been characterized as a substrate of P-gp and has been shown to inhibit P-gp, MRP 1, 2 and 3 [34]. Altered expression of P-gp has been attributed to the neurotoxicity associated with IVM [83]. In an experiment involving collie dogs, the dogs that had a deletion of ABCB-1 gene displayed neurotoxicity when dosed with IVM, whereas normal dogs do not. The authors in this study concluded that P-gp plays a role in effluxing the IVM from the CNS [84]. In a related study on beagle dogs, co-administration of IVM with spinosad, a P-gp inhibitor, has been demonstrated to increase IVM neurotoxicity through the inhibition of P-gp at the blood brain barrier [85].
IVM has been reported to interact with other drugs including doxycycline and albendazole which improve its antiparasitic efficacy [86]. In the control of onchoerciasis, doxycycline was reported to enhance the ivermectin-induced suppression of microfiladermia [87]. Levamisole has been shown to increase the plasma bioavailability of IVM though without necessarily increasing its antiparasitory effects [88]. Previous studies have also reported that ketoconazole substantially increases IVM plasma concentrations in sheep upon co-administration. The authors attributed this to the reversal of P-gp effects [36]. P-gp modulators itraconazole and valspodar have also been shown to increase the concentration of IVM in plasma and gastrointestinal tissues of rats [37].
There was an eightfold inhibition of the efflux transport of LPV in the presence of IVM which could have possibly involved P-gp or other transporters. The transport of LPV and other PIs has been shown to be modulated by efflux transporters P-gp and MRP [12, 89]. Whereas studies have demonstrated that these efflux transporters limit the uptake of ARVs, influence of influx transporters such as Organic anion-transporting polypeptide (OATP) on their modulation and indeed most drugs has not been fully described. Authors from previous studies concluded that an interplay of influx transporter (OATP), efflux transporters (P-gp and MRP) and lipophilicity had implications on the cellular uptake and retention of SQV and LPV in some T-cell lines, CEM, CEMVBL and CEM1000 as well as peripheral blood mononuclear cells [90]. In this study, pre-treatment of cells with P-gp and MRP inhibitors, tariquidar (XR9576) for P-gp, and MK571 with frusemide for MRP respectively; followed by subsequent co-incubation with a human OATP substrate, estrone-3-sulphate (E-3-S) resulted in a reduction of the cellular accumulation of SQV and LPV. This may suggest involvement of OATP in the effect of IVM on the disposition of LPV. It is worth noting that LPV (Log Kow =5.94) is more lipophilic than SQV (Log Kow = 2.5), which may contribute to the difference in response to IVM between the two PIs [91–93]. In a related study the authors concluded that IVM may influence the absorption of fexofenadine by interfering with influx and efflux pumps OATP and P-gp [94]. In our study, LPV inhibited the influx transport of IVM, though to a lesser margin (twofold decrease in the efflux ratio, 2.9 to 1.4 [Table 2]). It is therefore evident from these results that there is likelihood of interactions between LPV and IVM, and that these interactions could most likely be influenced by drug transporters (influx and efflux). Further investigations should be carried out to determine the specific transporters responsible for the interactions, and the dosage range that would exhibit these interactions.
NVP increased the efflux transport of IVM, but IVM did not appear to significantly influence the transport of NVP. In a study to investigate the influence of NNRTIs on P-gp activity, NVP significantly reduced the uptake of rhodamine 123, a P-gp substrate into LS180V cells signifying decreased efflux as a result of inhibition of transport [24]. In a related study, the authors concluded that NNRTIs induced P-gp in LS 180 cells [95, 96]. The observed interactions between NVP and IVM may therefore be attributed to the activities of influx and efflux drug transporters. With regards to the interactions between IVM and EFV, there was a marginally significant increase in the efflux ratio of EFV in presence of IVM. EFV has been characterized as a substrate of P-gp and has also been reported to decrease plasma concentrations of co-administered drugs that are metabolized by CYP 450 enzymes without modifying intestinal absorption of co-administered substrates of P-gp [22, 97].
PZQ did not significantly influence the transport of ARVs and likewise ARVs did not significantly affect the transport of PZQ. The presence of PZQ did not alter the transport of SQV, whereas SQV, EFV and NVP did not affect the transport of PZQ. This is consistent with our earlier studies whereby we established that PZQ is neither a substrate nor an inhibitor of P-gp in accumulation experiments involving CEM parental and CEMVBL cells [45]. PZQ has not been conclusively characterized with respect to drug transporters and metabolic enzymes. In a study involving the transport of PZQ and other antiparasitic drugs along Caco-2 cell monolayers, PZQ appeared to be an inhibitor of P-gp without being a substrate, based on inhibition of P-gp mediated [3H]-taxol transport in Caco-2 cells [40]. It is however noteworthy that Caco-2 cell lines express several drug transporters, both influx as well as efflux; including metabolic enzymes, and interplay of several factors is therefore possible. Careful interpretation of the results may therefore be necessary before arriving at any conclusions [44, 98, 99]. Ketoconazole, a CYP-450 inhibitor has been reported to double the plasma concentration of PZQ in humans, while rifampicin; an inducer has been reported to dramatically reduce its concentration, and the authors recommended dose adjustment upon co-administration [41, 43]. An increase in Schistosome P-gp levels has also been postulated to confer resistance to PZQ [100].
Conclusions
This study provides in vitro evidence of potential interactions between IVM, an antihelminthic drug with the ARVs; LPV, SQV and NVP and EFV. Further investigations should be conducted to investigate the possibility of in vivo interactions. From a clinical perspective, the co-administration of IVM with these drugs may require dosage adjustments in order to minimize the incidences of drug-drug interactions. However, this requires corroboration from clinical studies.
Additional files
Summary of the methodology. (DOCX 15 kb)
Transport results. (DOCX 167 kb)
Apparent permeability (Papp) calculations. (DOCX 23 kb)
Efflux ratios. (DOCX 13 kb)
a Impact of SQV on the transport of PZQ along the CCM. b Impact of PZQ on the transport of SQV along the CCM. (ZIP 32 kb)
a Impact of NVP on the transport of PZQ along the CCM. 2b Impact of EFV on the transport of PZQ along the CCM. (ZIP 30 kb)
a Impact of SQV on the transport of IVM along the CCM. b Impact of IVM on the transport of SQV along the CCM. (ZIP 29 kb)
a Impact of NPV on the transport of IVM along the CCM. b Impact of IVM on the transport of NVP along the CCM. (ZIP 29 kb)
a Impact of LPV on the transport of IVM along the CCM. b Impact of IVM on the transport of LPV along the CCM. (ZIP 29 kb)
a Impact of EFV on the transport of IVM along the CCM. b Impact of IVM on the transport of EFV along the CCM. (ZIP 28 kb)
Acknowledgements
The authors would like to thank the staff from the Department of Molecular and Clinical Pharmacology, University of Liverpool for their assistance in carrying out the laboratory work.
Funding
This work was part of Gabriel Kigen’s PhD study at the University of Liverpool which was funded by the Government of the Republic of Kenya.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.
Authors’ contributions
GK and GE designed the study. GK and GE extracted and analyzed the data. GK carried out the HPLC and radioassays and drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that there are no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AIDS
Acquired Immune Deficiency Syndrome
- AP
Apical
- ARV
Antiretroviral
- BL
Basolateral
- CCM
Caco-2 cell monolayers
- DDIs
Drug-drug interactions
- DMEM
Dulbecco's Modified Eagle Medium
- DMSO
Dimethyl sulfoxide
- EFV
Efavirenz
- ER
Efflux ratio
- FBS
Fetal bovine serum
- HBSS
Hanks' Balanced Salt Solution
- HIV
Human Immunodeficiency Virus
- HPLC
High Pressure Liquid Chromatography
- IVM
Ivermectin
- LPV
Lopinavir
- MRP
Multidrug resistance-associated protein
- NNRTIs
Non-Nucleoside Reverse Transcriptase Inhibitors
- NVP
Nevirapine
- OATP
Organic anion-transporting polypeptide
- P-gp
P-glycoprotein
- PIs
Protease inhibitors
- PZQ
Praziquantel
- SQV
Saquinavir
- TEER
Transepithelial resistance
Contributor Information
Gabriel Kigen, Email: kigengfk@gmail.com.
Geoffrey Edwards, Email: ge1000@liverpool.ac.uk.
References
- 1.Steinmann P, Utzinger J, Du ZW, Zhou XN. Multiparasitism a neglected reality on global, regional and local scale. Adv Parasitol. 2010;73:21–50. doi: 10.1016/S0065-308X(10)73002-5. [DOI] [PubMed] [Google Scholar]
- 2.Young B. Review: mixing new cocktails: drug interactions in antiretroviral regimens. AIDS Patient Care STDS. 2005;19:286–297. doi: 10.1089/apc.2005.19.286. [DOI] [PubMed] [Google Scholar]
- 3.Perloff ES, Duan SX, Skolnik PR, Greenblatt DJ, von Moltke LL. Atazanavir: effects on P-glycoprotein transport and CYP3A metabolism in vitro. Drug Metab Dispos. 2005;33:764–770. doi: 10.1124/dmd.104.002931. [DOI] [PubMed] [Google Scholar]
- 4.Pontali E. Interactions of antiretroviral drugs with anti-infectives and other antiretrovirals. Chemotherapy. 2007;53:26–29. doi: 10.1159/000098247. [DOI] [PubMed] [Google Scholar]
- 5.Seden K, Khoo S, Back D, Prevatt N, Lamorde M, Byakika-Kibwika P, Mayito J, Ryan M, Merry C. Drug-drug interactions between antiretrovirals and drugs used in the management of neglected tropical diseases: important considerations in the WHO 2020 Roadmap and London Declaration on Neglected Tropical Diseases. AIDS (London, England) 2013;27:675–-686. doi: 10.1097/QAD.0b013e32835ca9b4. [DOI] [PubMed] [Google Scholar]
- 6.Zha W, Wang G, Xu W, Liu X, Wang Y, Zha BS, Shi J, Zhao Q, Gerk PM, Studer E, et al. Inhibition of P-glycoprotein by HIV protease inhibitors increases intracellular accumulation of berberine in murine and human macrophages. PLoS One. 2013;8:e54349. doi: 10.1371/journal.pone.0054349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel J, Mitra AK. Strategies to overcome simultaneous P-glycoprotein mediated efflux and CYP3A4 mediated metabolism of drugs. Pharmacogenomics. 2001;2:401–415. doi: 10.1517/14622416.2.4.401. [DOI] [PubMed] [Google Scholar]
- 8.Dupuy J, Alvinerie M, Menez C, Lespine A. Interaction of anthelmintic drugs with P-glycoprotein in recombinant LLC-PK1-mdr1a cells. Chem Biol Interact. 2010;186:280–286. doi: 10.1016/j.cbi.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Bünning P. Drug–Drug Interaction: Enzyme Induction. In: Vogel HG, Maas J, Hock FJ, Mayer D, editors. Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays. Berlin: Springer; 2013. pp. 975–987. [Google Scholar]
- 10.Shi S, Li Y. Interplay of drug-metabolizing enzymes and transporters in drug absorption and disposition. Curr Drug Metab. 2014;15:915–941. doi: 10.2174/1389200216666150401110610. [DOI] [PubMed] [Google Scholar]
- 11.Konig J, Muller F, Fromm MF. Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65:944–966. doi: 10.1124/pr.113.007518. [DOI] [PubMed] [Google Scholar]
- 12.Gimenez F, Fernandez C, Mabondzo A. Transport of HIV protease inhibitors through the blood-brain barrier and interactions with the efflux proteins, P-glycoprotein and multidrug resistance proteins. J Acquir Immune Defic Syndr. 2004;36:649–658. doi: 10.1097/00126334-200406010-00001. [DOI] [PubMed] [Google Scholar]
- 13.Muller F, Fromm MF. Transporter-mediated drug-drug interactions. Pharmacogenomics. 2011;12:1017–1037. doi: 10.2217/pgs.11.44. [DOI] [PubMed] [Google Scholar]
- 14.Ogu CC, Maxa JL. Drug interactions due to cytochrome P450. Proc (Baylor Univ Med Cent) 2000;13:421–423. doi: 10.1080/08998280.2000.11927719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murakami T, Takano M. Intestinal efflux transporters and drug absorption. Expert Opin Drug Metab Toxicol. 2008;4:923–939. doi: 10.1517/17425255.4.7.923. [DOI] [PubMed] [Google Scholar]
- 16.Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9:310–322. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 17.Profit L, Eagling VA, Back DJ. Modulation of P-glycoprotein function in human lymphocytes and Caco-2 cell monolayers by HIV-1 protease inhibitors. AIDS. 1999;13:1623–1627. doi: 10.1097/00002030-199909100-00004. [DOI] [PubMed] [Google Scholar]
- 18.Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101:289–294. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim AE, Dintaman JM, Waddell DS, Silverman JA. Saquinavir, an HIV protease inhibitor, is transported by P-glycoprotein. J Pharmacol Exp Ther. 1998;286:1439–1445. [PubMed] [Google Scholar]
- 20.Alsenz J, Steffen H, Alex R. Active apical secretory efflux of the HIV protease inhibitors saquinavir and ritonavir in Caco-2 cell monolayers. Pharm Res. 1998;15:423–428. doi: 10.1023/A:1011924314899. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Baba M. The role of breast cancer resistance protein (BCRP/ABCG2) in cellular resistance to HIV-1 nucleoside reverse transcriptase inhibitors. Antivir Chem Chemother. 2005;16:213–216. doi: 10.1177/095632020501600401. [DOI] [PubMed] [Google Scholar]
- 22.Peroni RN, Di Gennaro SS, Hocht C, Chiappetta DA, Rubio MC, Sosnik A, Bramuglia GF. Efavirenz is a substrate and in turn modulates the expression of the efflux transporter ABCG2/BCRP in the gastrointestinal tract of the rat. Biochem Pharmacol. 2011;82:1227–1233. doi: 10.1016/j.bcp.2011.07.081. [DOI] [PubMed] [Google Scholar]
- 23.Storch CH, Theile D, Lindenmaier H, Haefeli WE, Weiss J. Comparison of the inhibitory activity of anti-HIV drugs on P-glycoprotein. Biochem Pharmacol. 2007;73:1573–1581. doi: 10.1016/j.bcp.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 24.Stormer E, von Moltke LL, Perloff MD, Greenblatt DJ. Differential modulation of P-glycoprotein expression and activity by non-nucleoside HIV-1 reverse transcriptase inhibitors in cell culture. Pharm Res. 2002;19:1038–1045. doi: 10.1023/A:1016430825740. [DOI] [PubMed] [Google Scholar]
- 25.Zembruski NC, Haefeli WE, Weiss J. Interaction potential of etravirine with drug transporters assessed in vitro. Antimicrob Agents Chemother. 2011;55:1282–1284. doi: 10.1128/AAC.01527-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss J, Haefeli WE. Potential of the novel antiretroviral drug rilpivirine to modulate the expression and function of drug transporters and drug-metabolising enzymes in vitro. Int J Antimicrob Agents. 2013;41:484–487. doi: 10.1016/j.ijantimicag.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Hotez PJ, Molyneux DH, Fenwick A, Kumaresan J, Sachs SE, Sachs JD, Savioli L. Control of neglected tropical diseases. New Engl J Med. 2007;357:1018–1027. doi: 10.1056/NEJMra064142. [DOI] [PubMed] [Google Scholar]
- 28.Liu L, Li-Li J, Qiong C, Xiao-Lin F. "Recent Advances in Antischistosomal Drugs and Agents." Mini reviews in medicinal chemistry. 2017;17(5):467-84. [DOI] [PubMed]
- 29.Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6:411–425. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]
- 30.WHO Schistosomiasis: number of people treated worldwide in 2014. Releve epidemiologique hebdomadaire/Section d'hygiene du Secretariat de la Societe des Nations = Weekly epidemiological record/Health Section of the Secretariat of the League of Nations. 2016;91:53–60. [PubMed] [Google Scholar]
- 31.Ottesen EA. Lymphatic filariasis: treatment, control and elimination. Adv Parasitol. 2006;61:395–441. doi: 10.1016/S0065-308X(05)61010-X. [DOI] [PubMed] [Google Scholar]
- 32.WHO Global programme to eliminate lymphatic filariasis: progress report, 2011. Releve epidemiologique hebdomadaire/Section d'hygiene du Secretariat de la Societe des Nations = Weekly epidemiological record/Health Section of the Secretariat of the League of Nations. 2012;87:346–356. [PubMed] [Google Scholar]
- 33.Pouliot JF, L'Heureux F, Liu Z, Prichard RK, Georges E. Reversal of P-glycoprotein-associated multidrug resistance by ivermectin. Biochem Pharmacol. 1997;53:17–25. doi: 10.1016/S0006-2952(96)00656-9. [DOI] [PubMed] [Google Scholar]
- 34.Lespine A, Dupuy J, Orlowski S, Nagy T, Glavinas H, Krajcsi P, Alvinerie M. Interaction of ivermectin with multidrug resistance proteins (MRP1, 2 and 3) Chem Biol Interact. 2006;159:169–179. doi: 10.1016/j.cbi.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Brayden DJ, Griffin J. Avermectin transepithelial transport in MDR1- and MRP-transfected canine kidney monolayers. Vet Res Commun. 2008;32:93–106. doi: 10.1007/s11259-007-9007-9. [DOI] [PubMed] [Google Scholar]
- 36.Alvinerie M, Dupuy J, Kiki-Mvouaka S, Sutra JF, Lespine A. Ketoconazole increases the plasma levels of ivermectin in sheep. Vet Parasitol. 2008;157:117–122. doi: 10.1016/j.vetpar.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 37.Ballent M, Lifschitz A, Virkel G, Sallovitz J, Lanusse C. Modulation of the P-glycoprotein-mediated intestinal secretion of ivermectin: in vitro and in vivo assessments. Drug Metab Dispos. 2006;34:457–463. doi: 10.1124/dmd.105.007757. [DOI] [PubMed] [Google Scholar]
- 38.Lespine A, Alvinerie M, Vercruysse J, Prichard RK, Geldhof P. ABC transporter modulation: a strategy to enhance the activity of macrocyclic lactone anthelmintics. Trends Parasitol. 2008;24:293–298. doi: 10.1016/j.pt.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez-Esquivel D, Rivera J, Castro N, Yepez-Mulia L, Jung Cook H. In vitro characterization of some biopharmaceutical properties of praziquantel. Int J Pharm. 2005;295:93–99. doi: 10.1016/j.ijpharm.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 40.Hayeshi R, Masimirembwa C, Mukanganyama S, Ungell AL. The potential inhibitory effect of antiparasitic drugs and natural products on P-glycoprotein mediated efflux. Eur J Pharm Sci. 2006;29:70–81. doi: 10.1016/j.ejps.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 41.Ridtitid W, Wongnawa M, Mahatthanatrakul W, Punyo J, Sunbhanich M. Rifampin markedly decreases plasma concentrations of praziquantel in healthy volunteers. Clin Pharmacol Ther. 2002;72:505–513. doi: 10.1067/mcp.2002.129319. [DOI] [PubMed] [Google Scholar]
- 42.Godawska-Matysik A, Kiec-Kononowicz K. Biotransformation of praziquantel by human cytochrome p450 3A4 (CYP 3A4) Acta Pol Pharm. 2006;63:381–385. [PubMed] [Google Scholar]
- 43.Ridtitid W, Ratsamemonthon K, Mahatthanatrakul W, Wongnawa M. Pharmacokinetic interaction between ketoconazole and praziquantel in healthy volunteers. J Clin Pharm Ther. 2007;32:585–593. doi: 10.1111/j.1365-2710.2007.00862.x. [DOI] [PubMed] [Google Scholar]
- 44.van Breemen RB, Li Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin Drug Metab Toxicol. 2005;1:175–185. doi: 10.1517/17425255.1.2.175. [DOI] [PubMed] [Google Scholar]
- 45.Kigen G, Edwards G. Intracellular accumulation of Praziquantel in T lymphoblastoid cell lines, CEM (parental) and CEMVBL(P-gp-overexpressing) BMC Pharmacol Toxicol. 2016;17:37. doi: 10.1186/s40360-016-0079-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lennernas H. Human intestinal permeability. J Pharm Sci. 1998;87:403–410. doi: 10.1021/js970332a. [DOI] [PubMed] [Google Scholar]
- 47.Masaoka Y, Tanaka Y, Kataoka M, Sakuma S, Yamashita S. Site of drug absorption after oral administration: assessment of membrane permeability and luminal concentration of drugs in each segment of gastrointestinal tract. Eur J Pharm Sci. 2006;29:240–250. doi: 10.1016/j.ejps.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Adson A, Burton PS, Raub TJ, Barsuhn CL, Audus KL, Ho NF. Passive diffusion of weak organic electrolytes across Caco-2 cell monolayers: uncoupling the contributions of hydrodynamic, transcellular, and paracellular barriers. J Pharm Sci. 1995;84:1197–1204. doi: 10.1002/jps.2600841011. [DOI] [PubMed] [Google Scholar]
- 49.Artursson P. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorptive (Caco-2) cells. J Pharm Sci. 1990;79:476–482. doi: 10.1002/jps.2600790604. [DOI] [PubMed] [Google Scholar]
- 50.Shah P, Jogani V, Bagchi T, Misra A. Role of Caco-2 cell monolayers in prediction of intestinal drug absorption. Biotechnol Prog. 2006;22:186–198. doi: 10.1021/bp050208u. [DOI] [PubMed] [Google Scholar]
- 51.Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun. 1991;175:880–885. doi: 10.1016/0006-291X(91)91647-U. [DOI] [PubMed] [Google Scholar]
- 52.Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev. 2001;46:27–43. doi: 10.1016/S0169-409X(00)00128-9. [DOI] [PubMed] [Google Scholar]
- 53.Gres MC, Julian B, Bourrie M, Meunier V, Roques C, Berger M, Boulenc X, Berger Y, Fabre G. Correlation between oral drug absorption in humans, and apparent drug permeability in TC-7 cells, a human epithelial intestinal cell line: comparison with the parental Caco-2 cell line. Pharm Res. 1998;15:726–733. doi: 10.1023/A:1011919003030. [DOI] [PubMed] [Google Scholar]
- 54.Sulzbacher A, Jarosch A, Schuler R, Acerbi D, Ventura P, Puccini P, Woodcock BG. Validation of a Caco-2 cell monolayer culture for drug transport studies. Int J Clin Pharmacol Ther. 1998;36:86–89. [PubMed] [Google Scholar]
- 55.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–749. doi: 10.1016/S0016-5085(89)80072-1. [DOI] [PubMed] [Google Scholar]
- 56.Hilgers AR, Conradi RA, Burton PS. Caco-2 cell monolayers as a model for drug transport across the intestinal mucosa. Pharm Res. 1990;7:902–910. doi: 10.1023/A:1015937605100. [DOI] [PubMed] [Google Scholar]
- 57.Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- 58.Hunter J, Jepson MA, Tsuruo T, Simmons NL, Hirst BH. Functional expression of P-glycoprotein in apical membranes of human intestinal Caco-2 cells. Kinetics of vinblastine secretion and interaction with modulators. J Biol Chem. 1993;268:14991–14997. [PubMed] [Google Scholar]
- 59.Hirohashi T, Suzuki H, Chu XY, Tamai I, Tsuji A, Sugiyama Y. Function and expression of multidrug resistance-associated protein family in human colon adenocarcinoma cells (Caco-2) J Pharmacol Exp Ther. 2000;292:265–270. [PubMed] [Google Scholar]
- 60.Fossati L, Dechaume R, Hardillier E, Chevillon D, Prevost C, Bolze S, Maubon N. Use of simulated intestinal fluid for Caco-2 permeability assay of lipophilic drugs. Int J Pharm. 2008;360:148–155. doi: 10.1016/j.ijpharm.2008.04.034. [DOI] [PubMed] [Google Scholar]
- 61.Peters WH, Roelofs HM. Time-dependent activity and expression of glutathione S-transferases in the human colon adenocarcinoma cell line Caco-2. Biochem J. 1989;264:613–616. doi: 10.1042/bj2640613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hidalgo IJ. Assessing the absorption of new pharmaceuticals. Curr Top Med Chem. 2001;1:385–401. doi: 10.2174/1568026013395010. [DOI] [PubMed] [Google Scholar]
- 63.Alsenz J, Haenel E. Development of a 7-day, 96-well Caco-2 permeability assay with high-throughput direct UV compound analysis. Pharm Res. 2003;20:1961–1969. doi: 10.1023/B:PHAM.0000008043.71001.43. [DOI] [PubMed] [Google Scholar]
- 64.Hidalgo IJ, Borchardt RT. Transport of bile acids in a human intestinal epithelial cell line, Caco-2. Biochim Biophys Acta. 1990;1035:97–103. doi: 10.1016/0304-4165(90)90179-Z. [DOI] [PubMed] [Google Scholar]
- 65.Okamura N, Hirai M, Tanigawara Y, Tanaka K, Yasuhara M, Ueda K, Komano T, Hori R. Digoxin-cyclosporin A interaction: modulation of the multidrug transporter P-glycoprotein in the kidney. J Pharmacol Exp Ther. 1993;266:1614–1619. [PubMed] [Google Scholar]
- 66.D'Souza VM, Shertzer HG, Menon AG, Pauletti GM. High glucose concentration in isotonic media alters caco-2 cell permeability. AAPS PharmSci. 2003;5 doi: 10.1208/ps050324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bravo SA, Nielsen CU, Amstrup J, Frokjaer S, Brodin B. In-depth evaluation of Gly-Sar transport parameters as a function of culture time in the Caco-2 cell model. Eur J Pharm Sci. 2004;21:77–86. doi: 10.1016/S0928-0987(03)00205-7. [DOI] [PubMed] [Google Scholar]
- 68.Anderle P, Niederer E, Rubas W, Hilgendorf C, Spahn-Langguth H, Wunderli-Allenspach H, Merkle HP, Langguth P. P-Glycoprotein (P-gp) mediated efflux in Caco-2 cell monolayers: the influence of culturing conditions and drug exposure on P-gp expression levels. J Pharm Sci. 1998;87:757–762. doi: 10.1021/js970372e. [DOI] [PubMed] [Google Scholar]
- 69.Legen I, Salobir M, Kerc J. Comparison of different intestinal epithelia as models for absorption enhancement studies. Int J Pharm. 2005;291:183–188. doi: 10.1016/j.ijpharm.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 70.Delezay O, Yahi N, Tamalet C, Baghdiguian S, Boudier JA, Fantini J. Direct effect of type 1 human immunodeficiency virus (HIV-1) on intestinal epithelial cell differentiation: relationship to HIV-1 enteropathy. Virology. 1997;238:231–242. doi: 10.1006/viro.1997.8829. [DOI] [PubMed] [Google Scholar]
- 71.Hubatsch I, Ragnarsson EG, Artursson P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc. 2007;2:2111–2119. doi: 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- 72.Meaney C, O'Driscoll C. Mucus as a barrier to the permeability of hydrophilic and lipophilic compounds in the absence and presence of sodium taurocholate micellar systems using cell culture models. Eur J Pharm Sci. 1999;8:167–175. doi: 10.1016/S0928-0987(99)00007-X. [DOI] [PubMed] [Google Scholar]
- 73.Youdim KA, Avdeef A, Abbott NJ. In vitro trans-monolayer permeability calculations: often forgotten assumptions. Drug Discovery Today. 2003;8:997–1003. doi: 10.1016/S1359-6446(03)02873-3. [DOI] [PubMed] [Google Scholar]
- 74.von Moltke LL, Greenblatt DJ, Granda BW, Giancarlo GM, Duan SX, Daily JP, Harmatz JS, Shader RI. Inhibition of human cytochrome P450 isoforms by nonnucleoside reverse transcriptase inhibitors. J Clin Pharmacol. 2001;41:85–91. doi: 10.1177/00912700122009728. [DOI] [PubMed] [Google Scholar]
- 75.Erickson DA, Mather G, Trager WF, Levy RH, Keirns JJ. Characterization of the in vitro biotransformation of the HIV-1 reverse transcriptase inhibitor nevirapine by human hepatic cytochromes P-450. Drug Metab Dispos. 1999;27:1488–1495. [PubMed] [Google Scholar]
- 76.Pal D, Kwatra D, Minocha M, Paturi DK, Budda B, Mitra AK. Efflux transporters- and cytochrome P-450-mediated interactions between drugs of abuse and antiretrovirals. Life Sci. 2011;88:959–971. doi: 10.1016/j.lfs.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amaral L, Spengler G, Martins A, Armada A, Handzlik J, Kiec-Kononowicz K, Molnar J. Inhibitors of bacterial efflux pumps that also inhibit efflux pumps of cancer cells. Anticancer Res. 2012;32:2947–2957. [PubMed] [Google Scholar]
- 78.Ughachukwu P, Unekwe P. Efflux pump-mediated resistance in chemotherapy. Ann Med Health Sci Res. 2012;2:191–198. doi: 10.4103/2141-9248.105671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holmes AR, Cardno TS, Strouse JJ, Ivnitski-Steele I, Keniya MV, Lackovic K, Monk BC, Sklar LA, Cannon RD. Targeting efflux pumps to overcome antifungal drug resistance. Future Med Chem. 2016;8:1485–1501. doi: 10.4155/fmc-2016-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pfeifer ND, Hardwick RN, Brouwer KL. Role of hepatic efflux transporters in regulating systemic and hepatocyte exposure to xenobiotics. Ann Rev Pharmacol Toxicol. 2014;54:509–535. doi: 10.1146/annurev-pharmtox-011613-140021. [DOI] [PubMed] [Google Scholar]
- 81.Pal D, Mitra AK. MDR- and CYP3A4-mediated drug-drug interactions. J Neuroimmune Pharmacol. 2006;1:323–339. doi: 10.1007/s11481-006-9034-2. [DOI] [PubMed] [Google Scholar]
- 82.Cheng Y, El-Kattan A, Zhang Y, Ray AS, Lai Y. Involvement of drug transporters in organ toxicity: the fundamental basis of drug discovery and development. Chem Res Toxicol. 2016;29:545–563. doi: 10.1021/acs.chemrestox.5b00511. [DOI] [PubMed] [Google Scholar]
- 83.Edwards G. Ivermectin: does P-glycoprotein play a role in neurotoxicity? Filaria J. 2003;2 Suppl 1:S8. doi: 10.1186/1475-2883-2-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mealey KL, Bentjen SA, Gay JM, Cantor GH. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics. 2001;11:727–733. doi: 10.1097/00008571-200111000-00012. [DOI] [PubMed] [Google Scholar]
- 85.Dunn ST, Hedges L, Sampson KE, Lai Y, Mahabir S, Balogh L, Locuson CW. Pharmacokinetic interaction of the antiparasitic agents ivermectin and spinosad in dogs. Drug Metab Dispos. 2011;39:789–795. doi: 10.1124/dmd.110.034827. [DOI] [PubMed] [Google Scholar]
- 86.Gonzalez Canga A, Sahagun Prieto AM, Diez Liebana MJ, Fernandez Martinez N, Sierra Vega M, Garcia Vieitez JJ. The pharmacokinetics and interactions of ivermectin in humans--a mini-review. AAPS J. 2008;10:42–46. doi: 10.1208/s12248-007-9000-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hoerauf A, Mand S, Adjei O, Fleischer B, Buttner DW. Depletion of wolbachia endobacteria in Onchocerca volvulus by doxycycline and microfilaridermia after ivermectin treatment. Lancet. 2001;357:1415–1416. doi: 10.1016/S0140-6736(00)04581-5. [DOI] [PubMed] [Google Scholar]
- 88.Awadzi K, Edwards G, Opoku NO, Ardrey AE, Favager S, Addy ET, Attah SK, Yamuah LK, Quartey BT. The safety, tolerability and pharmacokinetics of levamisole alone, levamisole plus ivermectin, and levamisole plus albendazole, and their efficacy against Onchocerca volvulus. Ann Trop Med Parasitol. 2004;98:595–614. doi: 10.1179/000349804225021370. [DOI] [PubMed] [Google Scholar]
- 89.Agarwal S, Pal D, Mitra AK. Both P-gp and MRP2 mediate transport of Lopinavir, a protease inhibitor. Int J Pharm. 2007;339:139–147. doi: 10.1016/j.ijpharm.2007.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Janneh O, Hartkoorn RC, Jones E, Owen A, Ward SA, Davey R, Back DJ, Khoo SH. Cultured CD4T cells and primary human lymphocytes express hOATPs: intracellular accumulation of saquinavir and lopinavir. Br J Pharmacol. 2008;155:875–883. doi: 10.1038/bjp.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.PubChem. 2016. SAQUINAVIR | C38H50N6O5 - https://pubchem.ncbi.nlm.nih.gov/compound/441243. Accessed 16 Dec 2016.
- 92.PubChem. 2016. Lopinavir | C37H48N4O5 - https://pubchem.ncbi.nlm.nih.gov/compound/92727. Accessed 16 Dec 2016.
- 93.Diker B, Janneh O, van Heeswijk RP, Copeland KF. Comparative efflux of saquinavir, ritonavir and lopinavir from primary human cells. Drug MetabLett. 2010;4:241–245. doi: 10.2174/187231210792928297. [DOI] [PubMed] [Google Scholar]
- 94.Olsen L, Ingvast-Larsson C, Larsson P, Brostrom H, Bondesson U, Sundqvist M, Tjalve H. Fexofenadine in horses: pharmacokinetics, pharmacodynamics and effect of ivermectin pretreatment. J Vet Pharmacol Ther. 2006;29:129–135. doi: 10.1111/j.1365-2885.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 95.Weiss J, Weis N, Ketabi-Kiyanvash N, Storch CH, Haefeli WE. Comparison of the induction of P-glycoprotein activity by nucleotide, nucleoside, and non-nucleoside reverse transcriptase inhibitors. Eur J Pharmacol. 2008;579:104–109. doi: 10.1016/j.ejphar.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 96.Weiss J, Theile D, Ketabi-Kiyanvash N, Lindenmaier H, Haefeli WE. Inhibition of MRP1/ABCC1, MRP2/ABCC2, and MRP3/ABCC3 by nucleoside, nucleotide, and non-nucleoside reverse transcriptase inhibitors. Drug Metab Dispos. 2007;35:340–344. doi: 10.1124/dmd.106.012765. [DOI] [PubMed] [Google Scholar]
- 97.Berruet N, Sentenac S, Auchere D, Gimenez F, Farinotti R, Fernandez C. Effect of efavirenz on intestinal p-glycoprotein and hepatic p450 function in rats. J Pharm Pharm Sci. 2005;8:226–234. [PubMed] [Google Scholar]
- 98.Prieto P, Hoffmann S, Tirelli V, Tancredi F, Gonzalez I, Bermejo M, De Angelis I. An exploratory study of two Caco-2 cell models for oral absorption: a report on their within-laboratory and between-laboratory variability, and their predictive capacity. Altern Lab Anim. 2010;38:367–386. doi: 10.1177/026119291003800510. [DOI] [PubMed] [Google Scholar]
- 99.Turco L, Catone T, Caloni F, Di Consiglio E, Testai E, Stammati A. Caco-2/TC7 cell line characterization for intestinal absorption: how reliable is this in vitro model for the prediction of the oral dose fraction absorbed in human? Toxicol in vitro. 2011;25:13–20. doi: 10.1016/j.tiv.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 100.Messerli SM, Kasinathan RS, Morgan W, Spranger S, Greenberg RM. Schistosoma mansoni P-glycoprotein levels increase in response to praziquantel exposure and correlate with reduced praziquantel susceptibility. Mol Biochem Parasitol. 2009;167:54–59. doi: 10.1016/j.molbiopara.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the methodology. (DOCX 15 kb)
Transport results. (DOCX 167 kb)
Apparent permeability (Papp) calculations. (DOCX 23 kb)
Efflux ratios. (DOCX 13 kb)
a Impact of SQV on the transport of PZQ along the CCM. b Impact of PZQ on the transport of SQV along the CCM. (ZIP 32 kb)
a Impact of NVP on the transport of PZQ along the CCM. 2b Impact of EFV on the transport of PZQ along the CCM. (ZIP 30 kb)
a Impact of SQV on the transport of IVM along the CCM. b Impact of IVM on the transport of SQV along the CCM. (ZIP 29 kb)
a Impact of NPV on the transport of IVM along the CCM. b Impact of IVM on the transport of NVP along the CCM. (ZIP 29 kb)
a Impact of LPV on the transport of IVM along the CCM. b Impact of IVM on the transport of LPV along the CCM. (ZIP 29 kb)
a Impact of EFV on the transport of IVM along the CCM. b Impact of IVM on the transport of EFV along the CCM. (ZIP 28 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10.