

Abstract
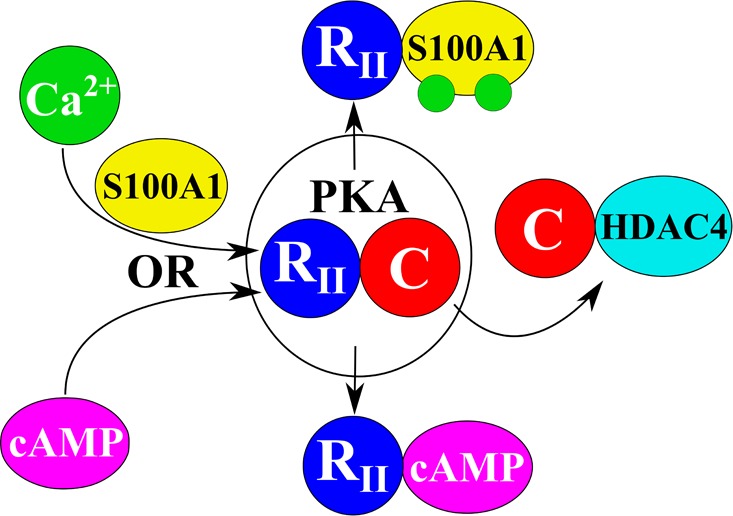
Biochemical and structural studies demonstrate that S100A1 is involved in a Ca2+-dependent interaction with the type 2α and type 2β regulatory subunits of protein kinase A (PKA) (RIIα and RIIβ) to activate holo-PKA. The interaction was specific for S100A1 because other calcium-binding proteins (i.e., S100B and calmodulin) had no effect. Likewise, a role for S100A1 in PKA-dependent signaling was established because the PKA-dependent subcellular redistribution of HDAC4 was abolished in cells derived from S100A1 knockout mice. Thus, the Ca2+-dependent interaction between S100A1 and the type 2 regulatory subunits represents a novel mechanism that provides a link between Ca2+ and PKA signaling, which is important for the regulation of gene expression in skeletal muscle via HDAC4 cytosolic–nuclear trafficking.
Skeletal muscle, the most abundant tissue of the human body, constituting >40% by mass, controls vital functions such as locomotion and breathing.1 These processes are regulated by phosphorylation and by Ca2+ signaling. Ca2+ signaling is a vital process for signal transduction, homeostasis, protein–protein interactions, muscle contraction, and a host of other biochemical processes.2
Eukaryotic protein kinases (EPKs) are critical for the regulation of numerous cellular processes and signaling pathways.3 From the time the first EPK structure was determined and the superfamily was determined to consist of highly regulated molecular switches rather than efficient catalysts, EPKs have become the second-most targeted enzyme class for drug development.4 Protein phosphorylation plays a vital role in innumerable biochemical processes, including protein–protein interactions, muscle contraction, and regulation of the foremost drug target, G-protein-coupled receptors.2 Irregularities in EPK regulation have been implicated in diseases and disorders ranging from Alzheimer’s disease and cancer to diabetes and heart disease.5,6 EPKs have evolved to initiate signal cascades often under single-turnover conditions; as such, precise regulation of EPKs is critical for appropriate cell and tissue function. The most well-known and studied of this superfamily, PKA, has long been considered to be regulated exclusively by the presence of cAMP and directed throughout the cell by A kinase anchoring proteins.7 On rare occasions, however, evidence has been reported suggesting activation of PKA that is independent of cAMP, though the precise mechanism for cAMP-independent activation of PKA has remained elusive.8 For example, in peripheral ganglion neurons and cardiac cells, S100A1 increases the Cav1 channel current amplitude. This effect was blocked by the inhibition of PKA, suggesting it is the result of a PKA-dependent process. However, the PKA-dependent effect on Cav1 current did not require cAMP, so its mechanism of activation has long remained elusive.9 Herein, we describe a mechanism of cAMP-independent activation of PKA in skeletal muscle that involves the calcium-binding protein S100A1.
S100A1, an 11 kDa dimeric Ca2+-binding protein, plays an important role in Ca2+ signaling and homeostasis in heart and skeletal muscle.10 In cardiac and skeletal muscle, S100A1 binds to the RyR to promote the release of Ca2+ from the sarcoplasmic reticulum, a process necessary for muscle contraction.11,12 Here, we demonstrate for the first time that S100A1 also binds to full-length regulatory subunits type II α and β (RIIα and RIIβ, respectively) of the PKA heterotetramer. In parallel with cAMP, this interaction activates the catalytic subunit, PKA-CA (Cα), and PKA-dependent signal cascades in muscle.
To explore the physiological relevance of the S100A1–PKA interaction in skeletal muscle function, we also examined the effects of activating PKA in regulating nucleocytoplasmic movement of HDAC4 in wild-type and S100A1 knockout (S100A1KO) mice. Class IIa histone deacetylases (HDACs), including HDAC isoforms 4, 5, 7, and 9, move between skeletal muscle fiber cytoplasm and nuclei in response to diverse cellular cues, suppressing activity of the nuclear transcription factor, myocyte enhancer factor 2.13 PKA phosphorylates HDAC4 in skeletal muscle, resulting in HDAC nuclear accumulation.14,15 Using HDAC4-green fluorescent protein (HDAC4-GFP) expressed in isolated skeletal muscle fibers and time-lapse confocal microscopy, we show that activation of PKA, by the β-receptor agonist isoproterenol, caused a steady HDAC4-GFP nuclear influx. The PKA inhibitor H-89 blocked the effects of isoproterenol on the nuclear influx of HDAC4-GFP. Interestingly, the effect of isoproterenol on HDAC4 nuclear influx was also attenuated in muscle fibers from S100A1KO mice. Collectively, our results demonstrate a novel interaction between S100A1 and PKA and suggest that this interaction could play an important role in the regulation of gene expression in skeletal muscle by modulating HDAC4 nuclear–cytoplasmic movement. Other physiological roles resulting from formation of the Ca2+-dependent S100A1–PKA complex and cAMP-independent activation of PKA are certainly possible and worthy of investigation.
Materials and Methods
Materials
M-280 tosyl-activated Dynabeads were obtained from Invitrogen and protease inhibitor cocktails from Sigma-Aldrich. Column resins were obtained from GE Healthcare Life Sciences. All buffers were passed through Chelex-100 resin (Bio-Rad) to remove trace metals and divalent ions. All proteins used were recombinant and purified (>99%) and were dialyzed using Chelex-100 resin for the same purpose. All other chemical reagents used were ACS grade or higher and were purchased from Sigma-Aldrich unless otherwise stated.
Expression and Purification of S100A1 and PKA Subunits
All purification steps were performed on ice or at 4 °C unless otherwise stated. Recombinant human S100A1 was expressed in Escherichia coli strain BL21(DE3) cells transformed with an expression plasmid (pET-11b, Novagen) containing the gene for human S100A1. Protein expression and bacterial lysis were performed as recommended by the manufacturer (Novagen). S100A1-containing bacterial lysates were precipitated with ammonium sulfate at 65% saturation. The supernatant was dialyzed against buffer A [50 mM Tris (pH 7.50) and 0.5 mM DTT] to remove ammonium sulfate, and the sample was loaded onto a DE52 diethylaminoethyl-Sepharose (DEAE) column (Whatman, Inc.). The column was equilibrated, and unbound proteins were washed away with buffer A. S100A1 was eluted stepwise with increasing NaCl concentrations. Fractions were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and S100A1 protein-containing fractions were pooled. Pooled DEAE fractions were concentrated to 1 mL and further purified by gel filtration on a Sephadex G-25 column equilibrated in buffer B [0.25 mM Tris (pH 7.50), 50 mM NaCl, and 0.25 mM DTT]. S100A1 aliquots were stored at −20 °C.
Recombinant full-length rat PKA subunits were individually expressed in E. coli strain BL21(DE3) cells transformed with an expression plasmid (pET-15b) containing the gene for rat RIα, RIIα, RIβ, or RIIβ. RIIβ was expressed with a 10-histidine tag and an enterokinase cleavage site. All other PKA regulatory subunits were expressed with a six-histidine tag and a tobacco etch virus (TEV) protease cleavage site. Protein expression was performed as recommended by the manufacturer (Novagen). The cell pellet was resuspended in buffer C [50 mM Tris (pH 7.50), 5 mM BME, 20 mM imidazole, 0.5 mM AEBSF, 0.1% Triton X-100, and 0.5 mg/mL lysozyme] with 0.1% protease inhibitor cocktail (Sigma-Aldrich). The cell suspension was sonicated to ensure sufficient lysing, and the debris was pelleted by centrifugation. The supernatant was filtered and loaded over a HisTrap IMAC FF column (GE Healthcare Life Sciences). The column was equilibrated, and the unbound proteins were washed away with buffer D [50 mM Tris (pH 7.50), 300 mM NaCl, 5 mM BME, and 20 mM imidazole]. Each PKA subunit was eluted with a stepwise gradient (0 to 100%) with buffer D containing 0.5 M imidazole. Fractions were analyzed by SDS–PAGE, and PKA subunit-containing fractions were pooled. Purified subunits were dialyzed against buffer C. His tags were removed using 2 mg of enterokinase or TEV protease via overnight treatment (8 h). The protease mixture was loaded over a HisTrap HP column (GE Healthcare Life Sciences), and pure PKA subunits were eluted with buffer D containing no imidazole. Fractions were analyzed by SDS–PAGE, and PKA subunit-containing fractions were pooled. Pooled fractions were concentrated to 1–2 mg/mL and stored at −4 °C to prevent aggregation and degradation.
Enzymatic Characterization of PKA
The enzymatic activity for PKA was measured as the change in fluorescence intensity at 485 nm (excitation at 360 nm) upon PKA-dependent phosphorylation of the commercially available Sox peptide (i.e., Omnia assay, from Thermo Fisher Scientific).16,17 This assay was optimized for fluorescence measurements in black, 384-well, flat-bottom plates (Corning) using a PHERAstarPlus plate reader (BMG Labtech) in total volumes of 20 μL/well. For enzymatic assays, a kinase buffer solution that contained 1.0 mM ATP, 10.0 mM DTT, 100 μg/mL BSA, and a buffer solution provided by the vendor (pH 7.5) was prepared. In all studies in which Ca2+ was a dependent variable, solutions were chelexed prior to the addition of 1.0 mM Mg2+. For PKA assays with other subunits (RIα, RIIα, or RIβ), the same kinase buffer solution required the regulatory subunit (5.0 μM) to inhibit the catalytic subunit. RIIα also required RNase treatment (5 mg/mL, Sigma-Aldrich) to inhibit the catalytic subunit. For cAMP-dependent enzymatic activation, kinase buffer, the catalytic subunit (PKACat, 3 nM), RIIβ (1.0 μM) or RIIα (5.0 μM), and cAMP (0–5.0 μM) were incubated 12 h prior to the addition of substrate (10.0 μM Sox peptide). The plate was then read every 30 s for 1 h at 25 °C.16,17 Addition of CaCl2 (0–2 mM) in the absence of S100A1 was tested under these conditions as a control. For studies with a varying S100A1 concentration (0–200 μM), Ca2+ concentrations were kept constant (2 mM). The most closely related family member, S100B, and calmodulin (CaM) were also tested under the same conditions to determine whether activation was specific for S100A1. To determine the level of free Ca2+ needed for S100A1-dependent activation of PKA, smaller amounts of RIIβ (0.1 μM) and S100A1 (1.0 μM) were used and the free Ca2+ concentrations (0–6 μM) were prepared using EGTA-containing calcium buffers (Thermo Fisher Scientific). To test whether S100A1 directly affects the activity of PKACat (3 nM), S100A1 (0–200 μM) was added to the enzyme in the presence of Ca2+ (1.0 mM) but with no RIIβ or cAMP present. Similar experiments with calmodulin or another S100 protein (i.e., S100B) were completed in the same manner, but they had no measurable effect on PKA activity (not shown). Enzymatic data were processed using Origin 6.1 (OriginLab) and fit using a dose–response curve.
Fluorescence Polarization Competition Assay (FPCA)
The ability of RIIα or RIIβ to compete with a peptide probe, TAMRA-labeled Hdm4, bound to Ca2+-S100A1 was evaluated as detailed by Wilder et al.18 FPCA titrations of RII subunits into a solution of S100A1 bound to TAMRA-Hdm4 were performed in triplicate with three biological replicates in a 384-well black polypropylene microplate with a final volume of 20 μL. Conditions were as follows: 10 nM TAMRA-Hdm4, 6 μM S100A1, 50 mM HEPES (pH 7.4), 50 mM NaCl, 5 mM CaCl2, 5 mM DTT, and 0.1% Triton X-100. Fluorescence polarization was measured at room temperature in a BMG PHERAstar Plus multimode microplate reader equipped with dual-detection PMTs with excitation at 544 nm and emission at 590 nm. FPCA data were processed using Origin 6.1 and fit using the Hill function. Binding affinities were calculated using the Cheng–Prusoff equation.19
Isothermal Titration Calorimetry (ITC)
Heat changes during the titration of Ca2+-S100A1 into RIIβ were measured using a VP-ITC titration microcalorimeter (MicroCal) as performed previously.20 All proteins involved were dialyzed into ITC buffer [20 mM HEPES, 50 mM NaCl, 10 mM MgCl2, 10 mM CaCl2, and 0.5 mM TCEP (pH 7.40)] prior to use. All solutions were degassed under vacuum and equilibrated to 37 °C prior to titration. The sample cell (1.4 mL) contained ITC buffer with 40 μM RIIβ, or no protein, while the reference cell contained water. A 700 μM S100A1 solution in the same buffer without RIIβ was injected in 30× 10 μL aliquots using the default injection rate with a 180 s interval between each injection to allow the sample to return to baseline. The resulting titration curves were corrected with a buffer control in the absence of RIIβ in the cell and analyzed using Origin for ITC (MicroCal).
Nuclear Magnetic Resonance Spectroscopy (NMR)
NMR spectra were collected at 37 °C with a Bruker AVANCE 800 NMR spectrometer (800.27 MHz for protons), equipped with four frequency channels and a 5 mm triple-resonance z-axis gradient cryogenic probe head. Chemical shift perturbations were obtained by comparing 15N TROSY-HSQC data of 15N-labeled Ca2+-S100A1 assigned previously to those of titrations with individual, unlabeled PKA regulatory subunits up to a 1:1 ratio.21 The sample conditions during these titrations included 10 mM HEPES (pH 7.40), 15 mM NaCl, 10 mM CaCl2, 2 mM DTT, 200 μM S100A1, and 0.34 mM NaN3 at 37 °C. Perturbations were determined as previously described.22,23
S100A1–RIIβ Pull-Down Assay Using Magnetic Beads
M-280 tosyl-activated magnetic beads (i.e., Dynabeads) were washed with buffer A [0.1 M sodium borate (pH 9.50)]; 100 μg of full-length, purified, rat RIIβ (>99%) in 150 μL of buffer B [3 M ammonium sulfate in buffer A (pH 9.50)] was directly conjugated to 5 mg of washed magnetic beads according to the manufacturer’s instructions (Invitrogen), and 100 μg of purified human S100A1 (>99%) in 150 μL of buffer C [10 mM HEPES and 150 mM NaCl (pH 7.40)] was used to interact with conjugated RIIβ. The RIIβ conjugation and S100A1 binding experiments were performed at 4 °C and included [Ca2+]total values of 1000, 500, and 100 nM. Buffer D (5 mM EDTA in buffer C) was used for the control experiment in the absence of Ca2+. After each binding experiment, unbound S100A1 was washed with buffer C while [Ca2+]total was maintained; S100A1 was eluted with buffer D for Western blot analysis.
Western Blots
For the analysis of recombinant PKA regulatory subunits, the protein concentration was determined with the Bio-Rad Protein Assay. Protein was resolved on a 4 to 12% Nu-PAGE gel (Invitrogen) and transferred to a PVDF membrane (EMD Millipore). The amounts of protein were 22, 22, and 13 μg for RIα, RIIα, and RIIβ, respectively. The RIIβ monoclonal rabbit antibody (1:50000; 75993, Abcam, Cambridge, MA) was used. Blots were incubated with the primary antibody followed by horseradish peroxidase-conjugated goat anti-rabbit IgG (KPL laboratories) and developed with enhanced chemiluminescence (EMD Millipore) and autoradiography film (Denville).
For the analysis of the pull-down assay, the protein concentration was determined with the Bio-Rad Protein Assay. Protein (8.33 μg) was resolved on 4 to 12% Nu-PAGE gels (Invitrogen) and transferred to PVDF membranes (EMD Millipore). The S100A1 polyclonal rabbit antibody (1:1000; 5066, Cell Signaling) was used. Blots were incubated with the primary antibody followed by horseradish peroxidase-conjugated goat anti-rabbit IgG (KPL laboratories) and developed with enhanced chemiluminescence (EMD Millipore) and autoradiography film (Denville).
For the analysis of RIIβ expression in muscle tissue, protein extraction and Western blotting techniques were performed as previously described with slight modifications.24 Muscle tissue was dissected and immediately frozen in liquid nitrogen to minimize tissue proteolytic damage. Frozen muscle tissue was ground with a pestle in T-PER lysis buffer (Thermo Scientific, Rockford, IL) supplemented with protease inhibitors (Complete-Mini EDTA free, Roche Diagnostics, Indianapolis, IN) and kept on ice with periodic trituration for up to 2 h. Insoluble debris was removed by centrifugation. The supernatant was removed, and the concentration was estimated with a Nanodrop-1000 spectrophotometer (Thermo Scientific, Wilmington, DE). Purified RIIβ was used as a control. Thirty micrograms of sample per lane was denatured at 74 °C, resolved on a precast 4 to 12% SDS–PAGE gel, and transferred to a PVDF membrane. Blots were then processed and probed with the primary rabbit antibody against RIIβ (1:5000) and the mouse antibody against Hsp90 (1:5000; 610418, BD Biosciences, San Jose, CA). Blots were incubated with the secondary antibodies, Alexa-647 goat anti-rabbit and Alexa-488 goat anti-mouse (1:1000; A21244 and A11029, respectively, Thermo-Fisher, Rockford, IL). Membranes were imaged on a Typhoon FLA 9500 biomolecular imager (GE Healthcare Bio-Sciences, Pittsburgh, PA). Bands were visualized using ImageJ (National Institutes of Health, Bethesda, MD), following automated background subtraction.
Immunofluorescence
Immunostaining was performed according to previously published methods.25 Muscle fibers were fixed in phosphate-buffered saline [PBS (pH 7.4)] containing 4% (w/v) paraformaldehyde for 20 min, permeabilized in PBS containing 0.1% (v/v) Triton X-100 (Sigma) for 15 min, and then incubated in PBS containing 8% (v/v) goat serum for 1 h at room temperature to block nonspecific labeling. Fibers were incubated overnight in primary antibodies. Primary antibodies were then washed out, and secondary fluorescent antibodies were applied for 24 h and washed out. Sequential incubation (first primary antibody and then first secondary followed by second primary and then second secondary) was used for α-actinin and dystrophin as both were developed in mouse. All antibodies used are commercially available as follows: RIIβ (1:100; ab75993), α-actinin (1:250; A7811, Sigma, St. Louis, MO), dystrophin (1:100; MANDRA1, Developmental Studies Hybridoma Bank, Iowa City, IA), Alexa-488 goat anti-mouse, Alexa-568 goat anti-mouse, and Alexa-647 goat anti-rabbit (1:1000; A21244, A11004, and A11029, respectively, Thermo-Fisher, Rockford, IL). POPO-1 (1:1000; P3580, Thermo Fisher) was used to stain the nuclei for 30 min and applied before the last washout. For each primary antibody-treated dish, another dish was treated with the secondary antibody only and used as a control. Antibody fluorescence and POPO-1-labeled muscle fibers were imaged on a Fluoview 500 Olympus LSM system, based on an IX/71 inverted microscope using a 60× NA 1.2 water immersion objective lens. Sequential excitation for POPO-1, Alexa-488, Alexa-568, and Alexa-647 was provided by using 440, 488, 533, and 633 nm lasers, respectively; the emitted light was collected with a 460–500 nm band-pass filter (BPF), a 510–530 nm BPF, a 560–600 nm BPF, and a >640 nm long-pass filter, respectively. Confocal images were collected using the same image acquisition settings and enhancing parameters so that all images could be directly compared. Images were background corrected and processed using ImageJ.
Animals
All experiments, protocols, and mice care guidelines were approved by the Institutional Animal Care and Use Committee of the University of Maryland (Baltimore, MD), in compliance with the National Institutes of Health guidelines. Young adult, male S100A1KO and wild-type mice on a hybrid C57/129 background were used in this study. The generation and genotyping of these animals have been previously reported.11 S100A1KO transgenic founders were obtained from D. Zimmer (University of Maryland, Baltimore, MD). Mice were housed in groups in a pathogen-free area at the University of Maryland (Baltimore, MD). Mice were killed by regulated delivery of a compressed CO2 overdose followed by cervical dislocation. The flexor digitorum brevis (FDB) muscles were dissected for further evaluation.
Infection of Recombinant Adenoviruses in Muscle Fibers
Single muscle fibers were enzymatically dissociated from FDB muscles of 4–6-week-old S100A1KO and wild-type C57/129 mice and cultured as previously described.14 Isolated fibers were cultured on laminin-coated glass-bottom Petri dishes. Fibers were cultured in minimal essential medium (MEM) containing 10% fetal bovine serum and 50 μg/mL gentamicin sulfate in 5% CO2 (37 °C). Recombinant adenovirus (Ad5) containing HDAC4-GFP was produced as described previously.14,15 Viral infections were performed with approximately 108 particles per muscle fiber. The recombinant adenoviruses were added to the culture dishes with MEM without serum. One hour after infection, the medium was changed to virus-free MEM with serum for continued culture.
Microscopy, Image Acquisition, and Analysis
To study the intracellular localization of HDAC4-GFP, 2 days after infection the culture medium was changed to Ringer’s solution [135 mM NaCl, 4 mM KCl, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, and 1.8 mM CaCl2 (pH 7.40)]. The culture dish was mounted on an Olympus IX70 inverted microscope equipped with an Olympus FluoView 500 laser scanning confocal imaging system. Fibers were viewed with an Olympus 60× 1.2 NA water immersion objective and scanned at 2.0× zoom with a constant laser power and gain. These imaging experiments were performed at room temperature.
The average fluorescence values of pixels within user-specified areas of interest in each image were quantified using ImageJ (NIH). The nuclear fluorescence values at each time point were normalized by the nuclear fluorescence value of 0 min of that specific muscle fiber to obtain the N/N0 ratio. Results are expressed as means ± the standard error of the mean (SEM). If an image of a fiber had more than one nucleus in focus, then all the nuclei in good focus were analyzed and multiple nuclei were treated equally.
Data Analysis
All data processing and statistical analysis were performed using OriginPro 8.0. All data are presented as means ± SEM unless otherwise noted. Statistical significance was assessed using either a parametric two-sample t test or the nonparametric Mann–Whitney rank-sum test. Significance was set at p < 0.05.
Results
The PKA holoenzyme comprises different isoforms of regulatory (R) (RIα, RIβ, RIIα, and RIIβ) and catalytic (C) (Cα, Cβ, and Cγ) subunits that are encoded by different genes and splice variants. Previous reports have shown that in skeletal muscle, the PKA subunits are concentrated at the neuromuscular junction but are also expressed outside of the end-plate region.26,27 While the cytoplasmic expression of RIα and RIIα subunits tends to predominate, the RIIβ subunit is also present in the cytosolic fraction.28 To characterize the expression and localization of RIIβ, we conducted Western blot experiments in flexor digitorum brevis (FDB), extensor digitorum longus (EDL), and soleus (SOL) muscle homogenates. As a control, the specificity of the antibody used in our Western blot assays was tested (Figure 1A). The blot shows that the RIIβ antibody does not detect RIα and RIIα. Similar to previous studies in skeletal muscle, a band for RIIβ was detected in FDB, EDL, and SOL muscles (Figure 1B).28 The cellular distribution of RIIβ was examined using indirect immunofluorescence and confocal microscopy on isolated FDB muscle fibers that were also labeled with antibodies against dystrophin, to delineate the surface membrane, α-actinin, to track the z-line, which forms sarcomere boundaries in striated muscles, and labeled with POPO-1, to stain the nuclei (Figure 1C,D). As shown in Figure 1D, RIIβ is localized in the myoplasm in a double-band pattern between the z-lines. Transversely oriented and regularly spaced bands of RIIβ fluorescence were observed as two fluorescent lines separated by a thin unlabeled region. The fluorescence signal in the no-primary control was negligible (Figure 1E,F). These in vitro and in cellulo studies demonstrate the presence of RIIβ in skeletal muscle tissue.
Figure 1.
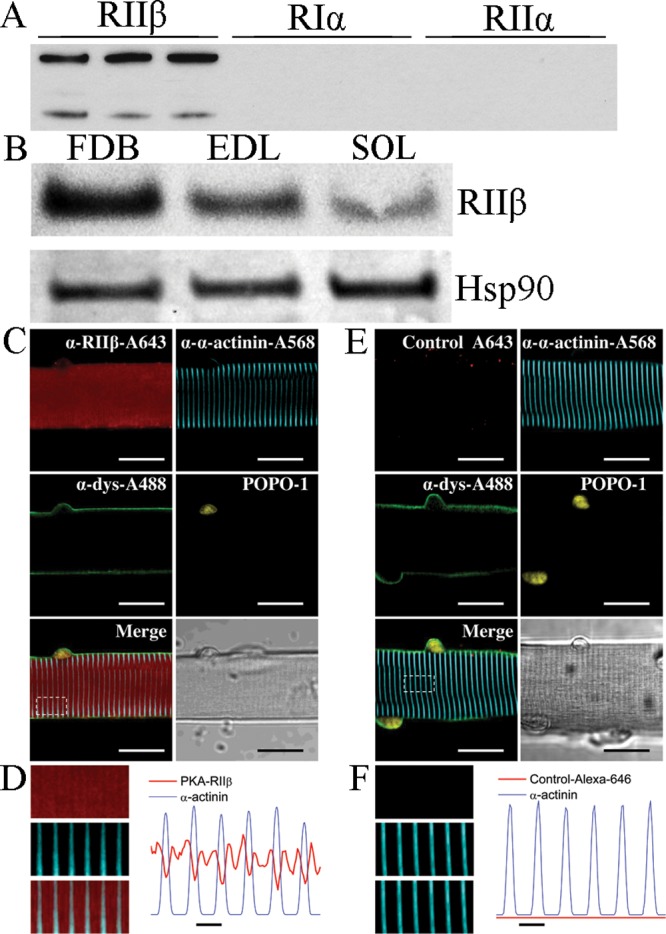
Western blot and immunofluorescence analysis of PKA. (A) Western blot for RIIβ showing antibody specificity. Lanes are 1000-, 2000-, and 5000-fold dilutions for RIIβ (lanes 1–3, respectively), RIα,4−6 and RIIα.7−9 (B) Western blot showing the presence of RIIβ in FDB, EDB, and soleus muscle tissue. (C) Representative confocal images of a segment of FDB fiber indirectly immunolabeled with antibodies against RIIβ (red), α-actinin (cyan), dystrophin (green), and POPO-1, to define the nuclei. (D) Close-ups (left) of the boxed region indicated in panel C for RIIβ (top), α-actinin (middle), and merged images (bottom) and averaged fluorescence profiles (right) of RIIβ (red trace) and α-actinin (blue trace) signals across the box. (E and F) Same labeling as in panels C and D, respectively, except that anti-RIIβ was not included. Scale bars in panels C and E are 20 μm and in panels D and F are 2 μm.
The Activation of PKA by S100A1 Is Ca2+-Dependent
To gain further biochemical information about the activation of holo PKA, a tetramer that contains two catalytic (PKACat) and two regulatory subunits (RIIβ) was studied in titrations with cAMP and S100A1 (Figure 2A,B). While S100A1 and cAMP were both found to activate holo PKA [cAMPEC50 = 236 ± 11 nM; S100A1EC50 = 5.2 ± 0.2 μM (Figure 2 and Table 1)], S100A1-dependent activation required calcium. The Ca2+ dependence of PKA activation by S100A1 was determined next [CaEC50 = 341 ± 90 nM (Figure 2D and Table 1)] and demonstrates that S100A1-dependent PKA activation occurs at physiologically relevant calcium-free concentrations.29 Activation of holo PKA by S100A1 was also assessed using regulatory subunit RIIα in place of RIIβ [S100A1EC50 = 149 ± 13 μM (Figure 2C and Table 1)]. Notably, it required 5-fold more RIIα (5.0 μM) to inhibit PKAcatin vitro as compared to RIIβ (1.0 μM). No activation by S100A1 was observed with RIα or RIβ. The addition of CaCl2 alone was tested as a control (0–2 mM) and had no effect on PKA activity in the absence of S100A1 under the conditions tested. In addition, S100A1 was found to have no measurable effect on PKA activity in the absence of Ca2+ or when it is was added to the PKA catalytic subunit alone (not shown).
Figure 2.

Enzymatic activation of PKA by S100A1, measured as the increase in fluorescence intensity (ΔF). (A) Increasing PKA activity in response to cAMP, in the absence of S100A1 (1.0 μM RIIβ). (B) Increasing PKA activity in response to Ca2+-S100A1, in the absence of cAMP (1.0 μM RIIβ). (C) Increasing PKA activity in response to Ca2+-S100A1, in the absence of cAMP (5.0 μM RIIα). Error bars represent one standard deviation from the mean. (D) Increasing PKA activity in response to addition of Ca2+-EGTA in the absence of cAMP (100 nM RIIβ and 10 μM S100A1). Error bars represent the standard error of the mean (α = 0.01). Each experiment was performed in triplicate with at least two biological replicates.
Table 1. PKA Binding and Activation Constants.
| Binding (μM) | |
|---|---|
| S100A1–RIIα | 0.99 ± 0.08 |
| S100A1–RIIβ | 2.16 ± 1.3 |
| Activation (μM)a | |
|---|---|
| RIIα-S100A1EC50 | 149 ± 13 |
| RIIβ-S100A1EC50 | 5.2 ± 0.2 |
| RIIβ-S100BEC50 | >130 |
| RIIβ-CaEC50 | 0.34 ± 0.10 |
| RIIβ-cAMPEC50 | 0.24 ± 0.01 |
Activation constants were determined with 1.0 μM RIIβ or 5.0 μM RIIα.
PKA activation was shown next to be highly specific for S100A1 as titrations with other EF-hand Ca2+-binding proteins (i.e., S100B and CaM) showed little or no PKA activation. For example, the activation constant for S100B (S100BEC50 > 130 μM, with 1.0 μM RIIβ present) was well above the relevant cellular concentrations, and no activation was detected with CaM with either RIIα or RIIβ (Table 1) under conditions identical to those used in the S100A1-dependent activation experiments. These studies demonstrate that S100A1 specifically activates PKA via the RII subunits, in a Ca2+-dependent manner, in the absence of cAMP, and represents a novel Ca2+-dependent activation mechanism for this important protein kinase.
Direct Interaction of S100A1 with Full-Length RIIβ at Physiological Ca2+ Levels
To confirm that S100A1-dependent PKA activation was the result of a calcium-dependent interaction with RIIβ, magnetic-bead pull-down studies (i.e., with Dynabeads) were completed in the absence or presence of calcium (100–1000 nM). In these experiments, S100A1 was found to interact with RIIβ only in the presence of Ca2+, and the complex was detected at Ca2+ concentrations as low as 100 nM; no interaction was observed in the absence of Ca2+ (Figure 3). Additionally, the binding affinity of this interaction in the presence of a saturating (5 mM) Ca2+ concentration was determined to be 2.16 ± 1.3 μM by FPCA, with secondary confirmation by ITC; no interaction was observed in the absence of Ca2+. The binding affinity for the interaction with RIIα was determined to be 990 ± 80 nM by FPCA (Table 1). NMR titrations were completed next and confirmed that formation of the CaS100A1–RIIβ complex was calcium-dependent because a large number of chemical shift perturbations were observed upon addition of RIIβ to 15N-labeled Ca2+-S100A1, but no perturbations were observed upon titrations of RIIβ into apo-S100A1.
Figure 3.
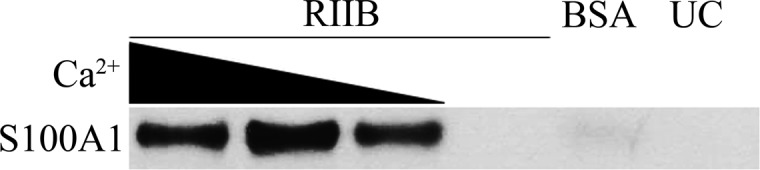
Western blot of Ca2+-S100A1 interacting with RIIβ-conjugated (lanes 1–4) or BSA-conjugated (lane 5) Dynabeads. Lane 6 (UC) was eluted from unconjugated beads. RIIβ (100 μg) or BSA (5 mg) was used for conjugation. S100A1 (100 μg) was used for binding. Binding was performed in the presence of 1000, 500, and 100 nM total Ca2+, or 5 mM EGTA with 0 nM total Ca2+ (lanes 1–4); 1000 nM Ca2+ was used for BSA-conjugated and unconjugated beads.
Characterization of the S100A1–RIIβ Binding Interface
NMR was next used to characterize the binding site of full-length RIIβ on Ca2+-S100A1 (Figure 4). To do this, unlabeled RIIβ was titrated slowly into a sample of 15N-labeled Ca2+-S100A1, and a series of TROSY-HSQC spectra were collected upon formation of the S100A1–RIIβ complex (Figure 4A). Upon saturation, most of the backbone N–H amide correlations in the titration could be assigned without difficulty (75 of 94, 80%) because they could be monitored unambiguously throughout the titration. Perturbation values assigned for other residues were ambiguous under these conditions due to resonance overlap (11 residues) and/or because of broadening due to exchange characteristics (6 residues). Despite peak broadening, it was still possible to assign perturbations for residues G44 and F45 in the hinge region, but these values have larger associated errors. The amide and/or amide proton chemical shift perturbations that could be assigned unambiguously upon formation of the CaS100A1–RIIβ complex were for residues in helix 1 (E10 and N14), the hinge region (Q49), helix 3 (A54 and K60), helix 4 (V76, V77, L68, L78, A80, T83, F90, and E92), and the Ca2+-binding EF hands (G24, E64, and V70) of S100A1 (Figure 4B). The average chemical shift perturbation for these perturbed residues was quantified by Euclidean weighting (Figure 4C)22,23 and consistent with RIIβ binding in a region of Ca2+-S100A1 similar to that observed for PKA-derived peptides.22,23,30
Figure 4.

Chemical shift perturbations of Ca2+-S100A1 from RIIβ binding. (A) Representative region of the 15N TROSY-HSQC experiment for Ca2+-S100A1 (black) and the 1:1 Ca2+-S100A1–RIIβ complex (red), showing peak shifts of Ala-8, Ala-18, Leu-37, and Glu-64. (B) Quantification of chemical shift perturbations calculated using Euclidean weighting and using 2 times the σ0 cutoff of 0.051 ppm for greater stringency in perturbations. (C) Ribbon diagram highlighting significantly perturbed residues. Modified from Protein Data Bank entry 2LP3.44 These data were collected using a sample of [15N]Ca2+-S100A1 with unlabeled FL-RIIβ.
S100A1 Modulates the PKA-Dependent Nuclear–Cytoplasmic Distribution of HDAC4 in Skeletal Muscle
We next examined whether S100A1 modulated PKA action in skeletal muscle. S100A1 is an S100 family member most strongly expressed in cardiac and skeletal muscle.31,32 PKA phosphorylates HDAC4 in skeletal muscle, resulting in HDAC nuclear accumulation.15 To examine the physiological roles for S100A1-dependent activation of PKA in skeletal muscle, we examined the kinetics of PKA-dependent nuclear fluxes of HDAC4-GFP expressed in living skeletal muscle fibers. For these studies, we monitored the time course of mean pixel fluorescence due to HDAC4-GFP in nuclear [Nuc (Figure 5A)] or cytoplasmic [Cyto (Figure 5A)] areas of interest in muscle fibers from wild-type and S100A1KO mice.
Figure 5.

HDAC4 nuclear localization is dependent on PKA and S100A1. (A) Confocal microscope image of a live resting skeletal muscle fiber expressing HDAC4-GFP. Nuc1 and Nuc2 are the areas of interest monitored in two different nuclei in the same muscle fiber. Cyt is a cytoplasmic area of interest. (B) Time course of nuclear HDAC4-GFP mean pixel fluorescence in muscle fibers from wild-type (empty black squares) or S100A1KO (empty red circles) mice, before and during application of isoproterenol (5 μM). Filled blue symbols give the time course of nuclear HDAC4-GFP in wild-type (filled squares) or S100A1KO (filled circles) muscle fibers exposed to isoproterenol after pre-exposure to PKA inhibitor H89 (5 μM), which completely blocks the increase in the leve of nuclear HDAC4-GFP. Error bars are SEM and are smaller than the size of the symbol when not shown. (C) The difference in nuclear HDAC due to β-adrenergic stimulation is significantly larger in wild-type than in S100A1KO muscle fibers. An asterisk indicates p < 0.05 from a two-sample t test. (D and E) Confocal images of exemplar nuclei expressing HDAC4-GFP before and after isoproterenol treatment for 60 min in a muscle fiber from the S100A1KO group and from a corresponding WT counterpart.
We previously found that both isoproterenol (Figure 5B,C) and the membrane permeable cAMP analogue, dibutyryl cAMP (not shown), cause net nuclear accumulation of HDAC4-GFP in skeletal muscle, whereas the cytoplasmic concentration of HDAC4-GFP remains constant in all cases examined (not shown) because the cytoplasmic volume is much larger than the nuclear volume.15 Prior to isoproterenol application, nuclear HDAC4-GFP remains constant (Figure 5B, □, t < 0).
During the application of isoproterenol to muscle fibers from wild-type mice (Figure 5B, □, t > 0), the nuclear concentration of HDAC4-GFP increases with time, indicating activation of net nuclear influx of HDAC4 by isoproterenol.
We have also shown that the HDAC4-GFP net nuclear influx during isoproterenol application is mediated by PKA activation and that the resulting PKA phosphorylation of HDAC4 at specific PKA sites is important for muscle function.15Figure 5B presents evidence that the effect of β-adrenergic activation of PKA is decreased in S100A1KO muscle fibers. As seen in panels B and C of Figure 5, muscle fibers from mice lacking S100A1 exhibit a suppressed net nuclear import of HDAC4-GFP compared to muscle fibers from wild-type littermates. Likewise, pre-application of the PKA kinase inhibitor (H89) eliminated the isoproterenol-induced HDAC4-GFP nuclear influx in wild-type and S100A1KO muscle (Figure 5B,C). Panels D and E of Figure 5 present representative nuclei from WT and S100A1KO muscle fibers, respectively, before (left) and after (right) application of isoproterenol. Our results indicate that Ca2+-S100A1, via PKA regulation, constitutes an additional molecular switch in the modulation of the HDAC4 nuclear–cytoplasmic distribution by β-adrenergic activation in skeletal muscle.
Discussion
S100A1 is a well-known enhancer of cardiac and skeletal muscle contractility and exhibits strong potential as a gene therapeutic agent for the treatment of cardiomyopathy. PKA is an extremely well-characterized molecular effector involved in a number of biological processes, including activation of RyR1 in skeletal muscle.33 In addition to the involvement of S100A1 with the ryanodine receptor in heart and skeletal muscle, we now present evidence of a novel mechanism of cAMP-independent PKA activation by S100A1. This interaction may serve as an additional and important means of PKA regulation.
As with most S100 protein interactions, S100A1 binds to PKA in a Ca2+-dependent manner (Figures 2 and 3). Interestingly, like cAMP, this interaction was shown to activate PKA even in the absence of cAMP, but only when calcium ions are present (Figure 2). It was also determined that S100A1 has no effect on the catalytic domain of PKA in the absence of the regulatory domain (not shown).
While the S100A1–RIIβ interaction was observed at very low Ca2+ levels (100 nM), significant PKA activation was not observed until the Ca2+ level was elevated somewhat above this basal level [CaEC50 = 341 ± 90 nM (Table 1)]. The implication of this CaEC50 value is that S100A1-dependent PKA activation may be in response to intracellular Ca2+ release mechanisms. In skeletal muscle, this includes Ca2+ release via a mechanical coupling of the dihydropyridine receptor (DHPR) and RyR1, rather than through calcium-induced calcium release (CICR).34−36 As previously established, Ca2+ release in skeletal and cardiac muscle is finely tuned through the interaction between S100A1 and RyR.11,12 As such, activation of PKA by S100A1 may be a result of Ca2+ release in response to activation of RyR by DHPR, PKA, or even S100A1 itself, in a feed-forward manner.
Upon Ca2+ binding, S100A1 undergoes a significant conformational change, rotating helix 3 (the entering helix) by 90° to expose a hydrophobic pocket in helix 3, the hinge region, and helix 4 (the exiting helix) of each subunit for binding of target protein.21,37 These residues were the most dramatically perturbed as a result of binding of S100A1 to RIIβ. Substantial perturbations were observed primarily in the EF-hands, responsible for Ca2+ binding, and in the hinge region and helices 3 and 4 of S100A1, and make up the binding interface between S100A1 and the target protein, RIIβ (Figure 4). Significant broadening effects were also observed for residues G44 and F45, which reside in the hinge region and may be broadened as a result of direct interaction with RIIβ.
The perturbations in Ca2+-S100A1 resonances upon binding of RIIβ were found to be relatively similar to those observed for binding of S100A1 to the CapZ peptide, TRTK,21,38,39 and the RyR peptide, RyRP-12.21,38,39 In each case, the largest perturbations were localized in the hinge region (loop 2), helix 3, and helix 4. Perturbations were observed for known S100 target-binding site residues for binding sites 1 (F45, Q49, A54, V58, L78, and A85), 2 (G44, A85, C86, and F90), and 3 (E10, N14, F45, A85, C86, and N88) of S100A1. A significant number of perturbations were also observed for adjacent residues, hence the need for greater stringency in the cutoff threshold to focus on the most significantly perturbed residues. These data suggest a typical interaction of the S100 protein with Ca2+ tightening and binding of the target protein within the exposed hydrophobic pocket. S100A1 was also found to bind identically to a truncated form of RIIβ [Δ1–100 (not shown)] as determined by NMR. Experiments are underway to monitor the perturbations of S100A1–RIIβ binding from the side of truncated RIIβ as the full-length protein proved to be too large and too dynamic to observe by NMR. S100A1 was also found to bind full-length RIIα by NMR; however, no binding was observed with S100A1 in the presence of RIα or RIβ (not shown). Taken together with previously published data using a peptide derived from RIIβ, our results indicate that S100A1 forms a specific interaction with the second cyclic nucleotide-binding domain (CNB-B) of RIIβ and shows selectivity for the type 2 regulatory subunits.30,40 However, further characterization of this complex at atomic resolution will be necessary for a more complete understanding of the mechanistic and functional significance of this interaction and the cAMP-independent activation of PKA via S100A1 in skeletal muscle.
The expression of RIIβ in skeletal muscle tissue has been reported previously. The presence of the RIIβ mRNA in EDL and SOL muscle was reported by Hoover et al.26 Western blot assays demonstrated expression of RIIβ in SOL and EDL muscle.28 While the localization of RIIβ was reported to be punctate and negligible in intercostal muscles, here we found that RIIβ is expressed in FDB, EDL, and SOL skeletal muscle.28 Our immunofluorescence assays provide further evidence that RIIβ is localized within the FDB muscle fiber displaying a sarcomeric pattern between z-lines. S100A1 was found to localize in a sarcomeric pattern by Prosser et al.11 A previous report by Weiss et al. showing no RIIβ expression in fetal muscle tissue also suggested developmental differences in the expression of PKA regulatory subunits.41
Previous work from our group has shown that β-adrenergic agonists or membrane permeable cAMP analogues modulate HDAC4 localization in skeletal muscle, enhancing the nuclear influx of HDAC4 via activation of PKA and the resulting PKA-dependent phosphorylation of HDAC4.15 Mutation of Ser265 and Ser266, the PKA phosphorylation sites of HDAC4, blocks the effects of PKA activation on HDAC4 nuclear influx, thus confirming that these effects are indeed mediated by PKA.15 Previous results demonstrated that nuclear efflux of HDAC4 enhances transcriptional activity of MEF2, thereby promoting slow fiber gene expression.14 Here, we monitored intracellular fluorescence changes of PKA-dependent HDAC4-GFP nuclear translocation in living single-muscle fibers using time series confocal microscopy. The nuclear traffic of HDAC4-GFP was used as a biosensor for PKA activation. We present evidence that the β-adrenergic-induced and PKA-dependent HDAC4 nuclear influx is suppressed on muscle fibers lacking the expression of S100A1. This effect suggests the contribution of S100A1 in the activation of the PKA-dependent HDAC4 nuclear influx initiated by β-adrenergic activation. Judging by the effects of S100A1 on HDAC4 traffic described above and considering the function of S100A1 in other systems, it seems that S100A1 “tunes” the effects of PKA activation on the HDAC4 nuclear influx response mediated by β-adrenergic activation.42,43 These observations directly demonstrate the synergistic effects of β-adrenergic signaling and S100A1 on HDAC4 nuclear influx. The β-adrenergic signaling pathway to HDAC4 can include the following: β-adrenergic receptor → cAMP → PKA → HDAC4 phosphorylation, which causes HDAC4 nuclear influx (Figure 6, Scheme 1). The β-adrenergic signaling pathway can also include, in parallel, an S100A1-dependent route to HDAC4 as follows: β-adrenergic receptor → cAMP and/or S100A1 → PKA → HDAC4 phosphorylation, which causes enhanced HDAC4 nuclear influx (Figure 6, Schemes 2 and 3). Thus, an S100A1-regulated PKA-dependent nuclear increase in the level of HDAC4 should suppress the transcriptional activity of MEF2, which would otherwise promote slow fiber gene expression.
Figure 6.

Reaction schemes for activation of PKA by cAMP or Ca2+-S100A1 in skeletal muscle fibers during β-adrenergic activation (Scheme 1), Ca2+-S100A1-mediated activation (Scheme 2), or parallel activation by adrenergic signaling and Ca2+-S100A1 (Scheme 3). S, R, and C represent S100A1 and the regulatory and catalytic subunits of PKA, respectively. PKA moves from left to right from the inactive to the activated due to β-adrenergic activation cAMP-dependent activation (Scheme 1) or in parallel with Ca2+-S100A1 dependent activation (Scheme 3). Scheme 2 illustrates the case for Ca2+-S100A1-dependent PKA activation with no cAMP present.
Conclusions
These data describe a novel interaction between S100A1 and the type 2 regulatory subunits of PKA. This interaction is sufficient for the cAMP-independent activation of PKA. This pathway utilizes S100A1 in a Ca2+-dependent manner, and data from muscle cells suggest it may act in parallel with activation by cAMP. The interaction between S100A1 and PKA appears to play an important role in the regulation of gene expression in skeletal muscle by modulation of HDAC4 nuclear–cytoplasmic translocation. This may represent a major advance for pharmacological PKA regulation as a therapeutic topic. Studies are ongoing to further characterize this interaction by NMR and X-ray crystallography.
In isolated sympathetic ganglion neurons, S100A1 enhances Ca2+ channel currents in a PKA-dependent manner and amplifies action potential-induced Ca2+ transients.30 By NMR chemical shift perturbations, S100A1 was shown to interact with a peptide derived from RIIβ.30 These data suggested a direct interaction between S100A1 and PKA, which may have significant biological implications. To fully characterize Ca2+-dependent S100A1–PKA complex formation, full-length PKA regulatory subunits were used here. First, the binding interface of Ca2+-S100A1 that interacts with full-length RIIβ was identified via NMR, and the S100A1 interaction with PKA was shown to be calcium-dependent. Next, we showed that S100A1 was able to activate PKA enzymatic activity in vitro in a cAMP-independent manner in the presence of either RIIα or RIIβ. Lastly, PKA-dependent effects via S100A1 were demonstrated in muscle via HDAC translocation, which were suppressed in a side-by-side comparison with studies of muscle fibers isolated from S100A1KO mice.
Acknowledgments
We thank Dana Zimmer for providing S100A1KO mice.
Glossary
Abbreviations
- EPK
eukaryotic protein kinase
- PKA
protein kinase A
- cAMP
cyclic adenosine monophosphate
- RyR
ryanodine receptor
- RIIα
type 2α regulatory subunit of PKA
- RIIβ
type 2β regulatory subunit of PKA
- S100A1KO
S100A1 knockout
- HDAC
histone deacetylase
- DEAE
diethylaminoethyl
- FDB
flexor digitorum brevis
- EFL
extensor digitorum longus
- DHPR
dihydropyridine receptor.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00117.
Figures S1 and S2 (PDF)
Author Contributions
Z.M. and E.O.H.-O. contributed equally to this work. Z.M. and E.O.H.-O. conducted most of these experiments, analyzed the results, and wrote most of the paper. E.O.H.-O., S.J.P.P., and Y.L. conducted immunofluorescence and skeletal muscle fiber experiments. Z.M., P.T.W., and D.H.B. conducted enzymatic assays. Z.M., A.D.P., and K.A.A. conducted pull-down and Western blotting experiments. Z.M. and K.M.V. conducted NMR experiments. M.F.S. and D.J.W. conceived the idea for the project and wrote the paper with Z.M. and E.O.H.-O. The abstract graphic was created by Z.M.
This work was supported by research grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R37-AR055099 to M.F.S.) and the National Institutes of Health (NIH) (R01-GM58888 and R01-CA107331 to D.J.W.). The NMR spectrometer used in these studies was purchased, in part, with funds from shared instrumentation grants from the NIH (S10-RR015741 to D.J.W.). Z.M. was supported by the Interdisciplinary Training Program in Muscle Biology (NIH Grant T32 AR007592).
The authors declare no competing financial interest.
Supplementary Material
References
- Janssen I.; Heymsfield S. B.; Baumgartner R. N.; Ross R. (2000) Estimation of skeletal muscle mass by bioelectrical impedance analysis. J. Appl. Physiol. 89, 465–471. [DOI] [PubMed] [Google Scholar]
- Berchtold M. W.; Brinkmeier H.; Müntener M. (2000) Calcium Ion in Skeletal Muscle: Its Crucial Role for Muscle Function, Plasticity, and Disease. Physiol. Rev. 80, 1215–1265. [DOI] [PubMed] [Google Scholar]
- Endicott J. A.; Noble M. E. M.; Johnson L. N. (2012) The Structural Basis for Control of Eukaryotic Protein Kinases. Annu. Rev. Biochem. 81, 587–613. 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Taylor S. S.; Keshwani M. M.; Steichen J. M.; Kornev A. P. (2012) Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc., B 367, 2517–2528. 10.1098/rstb.2012.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo G. L.; Russo M.; Ungaro P. (2013) AMP-activated protein kinase: A target for old drugs against diabetes and cancer. Biochem. Pharmacol. 86, 339–350. 10.1016/j.bcp.2013.05.023. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Lee H. G.; Raina A. K.; Perry G.; Smith M. A. (2002) The Role of Mitogen-Activated Protein Kinase Pathways in Alzheimer’s Disease. Neurosignals 11, 270–281. 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- Taylor S. S.; Zhang P.; Steichen J. M.; Keshwani M. M.; Kornev A. P. (2013) PKA: Lessons Learned after Twenty Years. Biochim. Biophys. Acta, Proteins Proteomics 1834, 1271–1278. 10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L. B.; Moran J. M.; Scarim A. L.; Ford D. A.; Yoon J.-W.; McHowat J.; Buller R. M. L.; Corbett J. A. (2002) Novel Role for Calcium-independent Phospholipase A(2) in the Macrophage Antiviral Response of Inducible Nitric-oxide Synthase Expression. J. Biol. Chem. 277, 38449–38455. 10.1074/jbc.M206247200. [DOI] [PubMed] [Google Scholar]
- Weiss S.; Oz S.; Benmocha A.; Dascal N. (2013) Regulation of cardiac L-type Ca2+ channel CaV1.2 via the β-adrenergic-cAMP-protein kinase A pathway: old dogmas, advances, and new uncertainties. Circ. Res. 113, 617. 10.1161/CIRCRESAHA.113.301781. [DOI] [PubMed] [Google Scholar]
- Zimmer D. B.; Eubanks J. O.; Ramakrishnan D.; Criscitiello M. F. (2013) Evolution of the S100 family of calcium sensor proteins. Cell Calcium 53, 170–179. 10.1016/j.ceca.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Prosser B. L.; Wright N. T.; Hernãndez-Ochoa E. O.; Varney K. M.; Liu Y.; Olojo R. O.; Zimmer D. B.; Weber D. J.; Schneider M. F. (2008) S100A1 Binds to the Calmodulin-binding Site of Ryanodine Receptor and Modulates Skeletal Muscle Excitation-Contraction Coupling. J. Biol. Chem. 283, 5046–5057. 10.1074/jbc.M709231200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright N. T.; Prosser B. L.; Varney K. M.; Zimmer D. B.; Schneider M. F.; Weber D. J. (2008) S100A1 and Calmodulin Compete for the Same Binding Site on Ryanodine Receptor. J. Biol. Chem. 283, 26676–26683. 10.1074/jbc.M804432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel-Duby R.; Olson E. N. (2006) Signaling pathways in skeletal muscle remodeling. Annu. Rev. Biochem. 75, 19–37. 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Randall W. R.; Schneider M. F. (2005) Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J. Cell Biol. 168, 887–897. 10.1083/jcb.200408128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Schneider M. F. (2013) Opposing HDAC4 nuclear fluxes due to phosphorylation by β-adrenergic activated protein kinase A or by activity or Epac activated CaMKII in skeletal muscle fibres. J. Physiol. 591, 3605–3623. 10.1113/jphysiol.2013.256263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shults M. D.; Imperiali B. (2003) Versatile Fluorescence Probes of Protein Kinase Activity. J. Am. Chem. Soc. 125, 14248–14249. 10.1021/ja0380502. [DOI] [PubMed] [Google Scholar]
- Shults M. D.; Janes K. A.; Lauffenburger D. A.; Imperiali B. (2005) A multiplexed homogeneous fluorescence-based assay for protein kinase activity in cell lysates. Nat. Methods 2, 277–284. 10.1038/nmeth747. [DOI] [PubMed] [Google Scholar]
- Wilder P. T.; Charpentier T. H.; Liriano M. A.; Gianni K.; Varney K. M.; Pozharski E.; Coop A.; Toth E. A.; MacKerell A. D.; Weber D. J. (2010) In vitro screening and structural characterization of inhibitors of the S100B-p53 interaction. Int. J. High Throughput Screening 2010, 109–126. 10.2147/IJHTS.S8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Wilder P. T.; Baldisseri D. M.; Udan R.; Vallely K. M.; Weber D. J. (2003) Location of the Zn2+-Binding Site on S100B As Determined by NMR Spectroscopy and Site-Directed Mutagenesis. Biochemistry 42, 13410–13421. 10.1021/bi035334q. [DOI] [PubMed] [Google Scholar]
- Wright N. T.; Varney K. M.; Ellis K. C.; Markowitz J.; Gitti R. K.; Zimmer D. B.; Weber D. J. (2005) The Three-dimensional Solution Structure of Ca2+-bound S100A1 as Determined by NMR Spectroscopy. J. Mol. Biol. 353, 410–426. 10.1016/j.jmb.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Schumann F.; Riepl H.; Maurer T.; Gronwald W.; Neidig K.-P.; Kalbitzer H. (2007) Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR 39, 275–289. 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- Williamson M. P. (2013) Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16. 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Hernández-Ochoa E. O.; Robison P.; Contreras M.; Shen T.; Zhao Z.; Schneider M. F. (2012) Elevated extracellular glucose and uncontrolled type 1 diabetes enhance NFAT5 signaling and disrupt the transverse tubular network in mouse skeletal muscle. Exp. Biol. Med. (London, U. K.) 237, 1068–1083. 10.1258/ebm.2012.012052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ochoa E. O.; Vanegas C.; Iyer S. R.; Lovering R. M.; Schneider M. F. (2015) Alternating bipolar field stimulation identifies muscle fibers with defective excitability but maintained local Ca(2+) signals and contraction. Skeletal Muscle 6, 6. 10.1186/s13395-016-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover F.; Kalhovde J. M.; Dahle M. K.; Skålhegg B.; Taskén K.; Lømo T. (2002) Electrical Muscle Activity Pattern and Transcriptional and Posttranscriptional Mechanisms Regulate PKA Subunit Expression in Rat Skeletal Muscle. Mol. Cell. Neurosci. 19, 125–137. 10.1006/mcne.2001.1053. [DOI] [PubMed] [Google Scholar]
- Hoover F.; Mathiesen I.; Skålhegg B.; Lømo T.; Taskén K. (2001) Differential expression and regulation of the PKA signalling pathway in fast and slow skeletal muscle. Anat. Embryol. 203, 193–201. 10.1007/s004290000155. [DOI] [PubMed] [Google Scholar]
- Perkins G. A.; Wang L.; Huang L. J.-s.; Humphries K.; Yao V. J.; Martone M.; Deerinck T. J.; Barraclough D. M.; Violin J. D.; Smith D.; Newton A.; Scott J. D.; Taylor S. S.; Ellisman M. H. (2001) PKA, PKC, and AKAP localization in and around the neuromuscular junction. BMC Neurosci. 2, 17–17. 10.1186/1471-2202-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudier J.; Glasser N.; Gerard D. (1986) Ions binding to S100 proteins. I. Calcium- and zinc-binding properties of bovine brain S100 alpha alpha, S100a (alpha beta), and S100b (beta beta) protein: Zn2+ regulates Ca2+ binding on S100b protein. J. Biol. Chem. 261, 8192–8203. [PubMed] [Google Scholar]
- Hernández-Ochoa E. O.; Prosser B. L.; Wright N. T.; Contreras M.; Weber D. J.; Schneider M. F. (2009) Augmentation of Ca(v)1 channel current and action potential duration after uptake of S100A1 in sympathetic ganglion neurons. Am. J. Physiol. Cell. Physiol 297, C955–C970. 10.1152/ajpcell.00140.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimoto H.; Hosoda S.; Kato K. (1987) Differential distribution of immunoreactive S100-alpha and S100-beta proteins in normal nonnervous human tissues. Lab. Invest. 57, 489–498. [PubMed] [Google Scholar]
- Haimoto H.; Kato K. (1988) S100a0 (alpha alpha) protein in cardiac muscle. Isolation from human cardiac muscle and ultrastructural localization. Eur. J. Biochem. 171, 409–415. 10.1111/j.1432-1033.1988.tb13805.x. [DOI] [PubMed] [Google Scholar]
- Reiken S.; Lacampagne A.; Zhou H.; Kherani A.; Lehnart S. E.; Ward C.; Huang F.; Gaburjakova M.; Gaburjakova J.; Rosemblit N.; Warren M. S.; He K.-l.; Yi G.-h.; Wang J.; Burkhoff D.; Vassort G.; Marks A. R. (2003) PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J. Cell Biol. 160, 919–928. 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M. (2009) Calcium-Induced Calcium Release in Skeletal Muscle. Physiol. Rev. 89, 1153–1176. 10.1152/physrev.00040.2008. [DOI] [PubMed] [Google Scholar]
- Fabiato A. (1983) Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245, C1–C14. [DOI] [PubMed] [Google Scholar]
- Schneider M. F. (1994) Control of Calcium Release in Functioning Skeletal Muscle Fibers. Annu. Rev. Physiol. 56, 463–484. 10.1146/annurev.ph.56.030194.002335. [DOI] [PubMed] [Google Scholar]
- Wright N. T.; Cannon B. R.; Zimmer D. B.; Weber D. J. (2009) S100A1: Structure, Function, and Therapeutic Potential. Curr. Chem. Biol. 3, 138–145. 10.2174/187231309788166460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman K. G.; Yang R.; Rustandi R. R.; Miller K. E.; Baldisseri D. M.; Weber D. J. (2002) Solution NMR Structure of S100B Bound to the High-affinity Target Peptide TRTK-12. J. Mol. Biol. 324, 1003–1014. 10.1016/S0022-2836(02)01152-X. [DOI] [PubMed] [Google Scholar]
- Wright N. T.; Cannon B. R.; Wilder P. T.; Morgan M. T.; Varney K. M.; Zimmer D. B.; Weber D. J. (2009) Solution structure of S100A1 bound to the CapZ peptide (TRTK12). J. Mol. Biol. 386, 1265–1277. 10.1016/j.jmb.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil D.; Blumenthal D. K.; Heller W. T.; Brown S.; Canaves J. M.; Taylor S. S.; Trewhella J. (2004) Conformational Differences Among Solution Structures of the Type Iα, IIα and IIβ Protein Kinase A Regulatory Subunit Homodimers: Role of the Linker Regions. J. Mol. Biol. 337, 1183–1194. 10.1016/j.jmb.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Imaizumi-Scherrer T.; Faust D. M.; Benichou J.-C.; Hellio R.; Weiss M. C. (1996) Accumulation in fetal muscle and localization to the neuromuscular junction of cAMP-dependent protein kinase A regulatory and catalytic subunits RI alpha and C alpha. J. Cell Biol. 134, 1241–1254. 10.1083/jcb.134.5.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R.; Cannon B. R.; Sorci G.; Riuzzi F.; Hsu K.; Weber D. J.; Geczy C. L. (2013) Functions of S100 Proteins. Curr. Mol. Med. 13, 24–57. 10.2174/156652413804486214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub M. C.; Heizmann C. W. (2008) Calcium, troponin, calmodulin, S100 proteins: From myocardial basics to new therapeutic strategies. Biochem. Biophys. Res. Commun. 369, 247–264. 10.1016/j.bbrc.2007.10.082. [DOI] [PubMed] [Google Scholar]
- Nowakowski M.; Ruszczyńska-Bartnik K.; Budzińska M.; Jaremko L.; Jaremko M.; Zdanowski K.; Bierzyński A.; Ejchart A. (2013) Impact of Calcium Binding and Thionylation of S100A1 Protein on Its Nuclear Magnetic Resonance-Derived Structure and Backbone Dynamics. Biochemistry 52, 1149–1159. 10.1021/bi3015407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.