

Abstract
Congenital malformations of spine and spinal cord are collectively termed as spinal dysraphism. It includes a heterogeneous group of anomalies which result from faulty closure of midline structures during development. Magnetic resonance imaging (MRI) is now considered the imaging modality of choice for diagnosing these conditions. The purpose of this article is to review the normal development of spinal cord and spine and reviewing the MRI features of spinal dysraphism. Although imaging of spinal dysraphism is complicated, a systematic approach and correlation between neuro-radiological, clinical and developmental data helps in making the correct diagnosis.
Keywords: Spinal dysraphism, Magnetic resonance imaging, Open spinal dysraphism, Meningomyelocele, Closed spinal dysraphism
Core tip: Imaging of spinal dysraphism may appear complicated as it is a group of diverse conditions which can have variable imaging appearance. It includes a heterogeneous group of anomalies which result from faulty closure of midline structures during development. Magnetic resonance imaging is now considered the imaging modality of choice for diagnosing these conditions. A systematic approach and correlation with neuroradiological, clinical and developmental data helps in making the correct diagnosis.
INTRODUCTION
Congenital malformations of spine and spinal cord are collectively termed as spinal dysraphism. It includes a heterogeneous group of anomalies resulting from incomplete midline closure of osseous, mesenchymal and nervous tissue[1]. Most of these conditions are diagnosed at or soon after birth, but some are discovered late in childhood or in adulthood because of absence of clinical manifestations. Magnetic resonance imaging (MRI) is the imaging modality of choice for diagnosing spinal dysraphism because of its superior soft tissue characterisation and multiparametric imaging capabilities[2]. The purpose of this article is to review the normal development of spinal cord and MRI features of spinal dysraphism with clinico-embryologic and radiological correlation.
EMBRYOLOGY
Development of spinal cord occurs during early embryogenesis (between 2-6 wk of gestation) in three main stages - gastrulation, primary neurulation and secondary neurulation[3,4]. During the stage of gastrulation, the bilaminar embryonic disc composed of ectoderm and endoderm is converted to a trilaminar disc by the formation of mesoderm (Figure 1).
Figure 1.
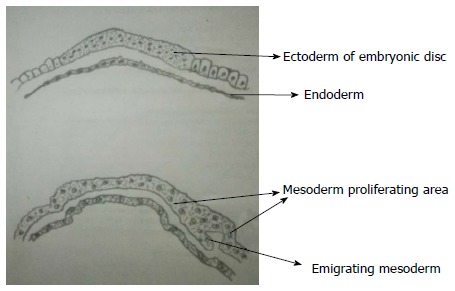
Gastrulation. During gastrulation, the bilaminar embryonic disc is converted to a trilaminar disc by the formation of mesoderm. There is rapid division of cells in embryonic disc which then detach and migrate between endoderm and ectoderm to form mesoderm. Notochord later develops from mesoderm.
Notochord which is formed from midline mesoderm interacts with overlying ectoderm resulting in the formation of neuroectoderm and neural plate[5]. Primary neurulation begins with the formation of neural plate and ends with the closure of caudal end of neural plate (Figure 2). A small depression develops along the central axis of neural plate to form neural groove and neural folds are formed on both sides of the neural groove. The neural plate then bends and neural folds fuse together converting the linear neural plate into a cylindrical neural tube. Closure of neural groove proceeds bidirectionally with the cephalic end (anterior or rostral neuropore) closing on day 25 and the caudal end (posterior or caudal neuropore) closing on day 27 or 28[6].
Figure 2.
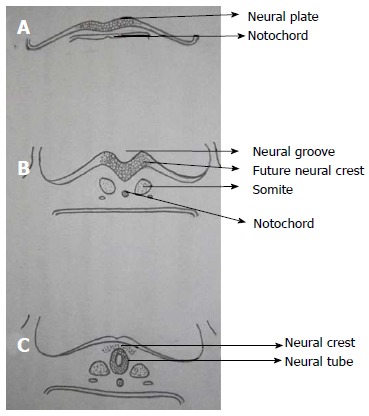
Primary neurulation. A: Thickening of embryonic ectoderm is seen dorsal to notochord to form neuroectoderm and neural plate; B: Neural plate invaginates along its central axis to form neural groove. Neural folds are formed on both sides of the neural groove; C: The neural plate then bends and neural folds fuse together to form neural tube and simultaneously separate from surface ectoderm (dysjunction).
During secondary neurulation there is formation of caudal cell mass composed of undifferentiated pluripotent cells from the caudal end of neural tube and notochord distal to caudal neuropore. Neurons and vacuoles develop within the caudal cell mass. Vacuoles then coalesce and eventually connect to the central canal by the process called cavitation. Finally the cells of caudal cell mass undergo retrograde differentiation during which cells undergo programmed cell death or apoptosis to form conus medullaris, filum terminale and ventriculus terminalis (Figure 3)[7,8].
Figure 3.

Secondary neurulation. Schematic diagram of caudal end of embryo showing secondary neurulation which forms the distal part of spinal cord. There is formation of solid caudal cell mass distal to the caudal neuropore which then develops a lumen by a process called “cavitation”. This then becomes continuous with the central cavity of neural tube.
Various disorders due to defective primary neurulation include open spinal dysraphism (OSD), closed spinal dysraphism (CSD) and dorsal dermal sinus while defects during secondary neurulation results in filar lipoma, tight filum terminale, caudal agenesis and sacrococcygeal teratoma. Defective development of notochord results in diastematomyelia, neurenteric cyst, caudal agenesis and segmental spinal dysgenesis[9].
CLASSIFICATION
On the basis of presence or absence of overlying skin covering, spinal dysraphism is divided into open and closed types[10-12] (Table 1). In OSD overlying skin covering is absent and the neural elements are exposed to the external environment whereas, in closed type the neural elements have a skin covering (Figure 4). CSD can be further divided based on the presence or absence of associated subcutaneous mass.
Table 1.
Classification of spinal dysraphisms
| Open spinal dysraphisms |
| Myelomeningocele |
| Myelocele |
| Hemimyelomeningocele |
| Hemimyelocele |
| Closed spinal dysraphisms |
| With subcutaneous mass |
| Lipomyelomeningocele |
| Lipomyelocele |
| Terminal myelocystocele |
| Meningocele |
| Myelocystocele |
| Without subcutaneous mass |
| Simple dysraphic states |
| Intradural lipoma |
| Filar lipoma |
| Tight filum terminale |
| Persistent terminal ventricle |
| Dermal sinus |
| Complex dysraphic states |
| Dorsal enteric fistula |
| Neuroenteric cyst |
| Diastematomyelia |
| Caudal agenesis |
| Segmental spinal dysgenesis |
Figure 4.
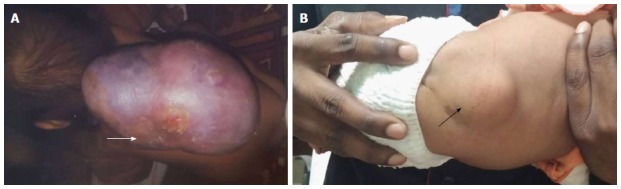
Open and closed spinal dysraphism. Clinical pictures of open (A) and closed (B) spinal dysraphism. In open spinal dysraphism, a case of cervical myelomeningocele the neural placode is directly exposed to the environment and is surrounded by partially epithelized skin (arrow in A). In closed spinal dysraphism with subcutaneous mass there is continuous skin coverage over the abnormality (arrow in B).
In our review, we will follow the clinic-radiological classification as it is easier to understand and is more prevalent in use.
OSDS
Myelomeningocele and myelocele
OSDs result from faulty primary neurulation due to defective closure of the neural tube. About 98.8% of all OSDs are constituted by Myelomeningocele (MMC) where both the neural placode and meningeal lining protrude through the bony and cutaneous defect in the midline[3]. OSDs are commonly diagnosed clinically as the neonate presents with a midline reddish exposed neural placode and immediate surgical repair is usually done, so imaging studies are not always performed. It usually involves the lower lumbar and sacral regions (98%) and is rare in cervical and upper thoracic spine, probably because the lesion in these areas are more severe leading to fetal demise[4,13].
In MMC, the protruding neural placode extends beyond the skin surface as there is enlargement of the adjacent subarachnoid space (Figure 5)[5]. This help to distinguish MMC from the far rarer myelocele, where the placode is flush with the skin surface (Figures 6 and 7). About 80% of myelocele and MMC have associated hydrocephalus and 100% patients have Chiari II malformation which involve cerebellum, brain stem, skull base, spine and spinal column (Figure 8)[3,4]. Studies have shown that defective neural tube closure resulting in abnormal drainage of CSF and hence decompression of the primitive ventricular system resulting in various manifestations of Chiari II malformation[14,15].
Figure 5.
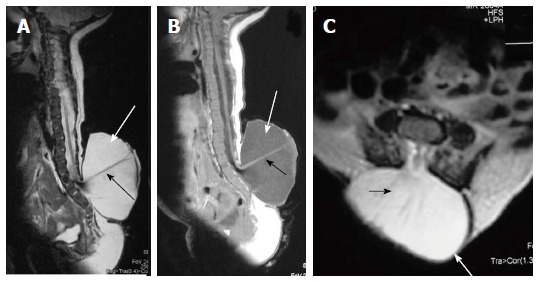
Lumbar myelomeningocele. T2 weighted sagittal (A), T1 weighted sagittal (B) and T2 weighted axial (C) images of lumbar spine showing myelomeningocele. There is posterior herniation of a CSF filled sac (white arrows) containing cord and nerve fibers (black arrows). There is absence of skin covering with interruption of subcutaneous fat.
Figure 6.
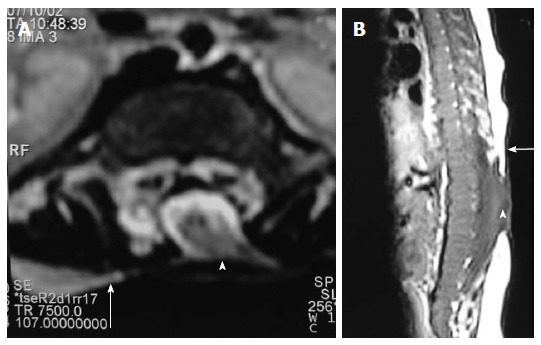
Myelocele. Axial T2 weighted (A) and sagittal T1 weighted (B) images of dorsolumbar spine in a case of myelocele showing the neural placode (arrowhead) flush with the skin surface (arrow). Skin covering is absent over the myelocele.
Figure 7.
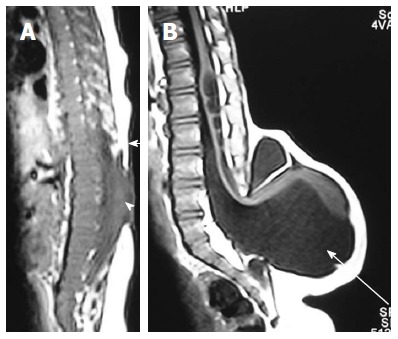
Myelocele and myelomeningocele. Sagittal T1 weighted images of spine in cases of myelocele (A) and myelomeningocele (B). In myelocele the placode (arrowhead) is flush with the skin surface (arrow). In myelomeningocele (B), the protruding neural placode extends beyond the skin surface as there is enlargement of the underlying subarachnoid space (arrow).
Figure 8.
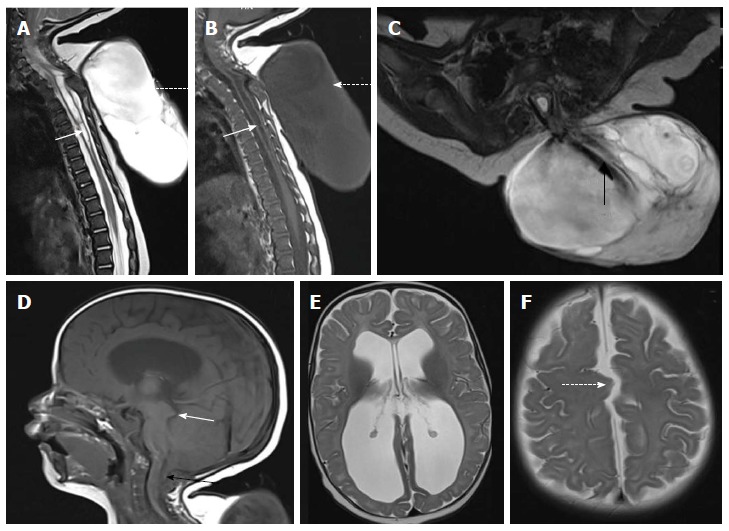
Cervico-dorsal myelomeningocele with Chiari II malformation. T2 weighted sagittal (A), T1 weighted sagittal (B) and T2 weighted axial (C) images of cervicodorsal spine showing myelomeningocele. There is posterior herniation of a CSF filled sac (dashed arrows) containing nerve fibers (black arrow). Spinal cord show kinking at the same level and dilated central canal (white arrow) suggestive of syringohydromyelia. There is no overlying skin cover. Sagittal T1 weighted (D) and axial T2 weighted (E and F) images of brain shows herniation of cerebellar tonsils (black arrow) with tectal beaking (white arrow). Fourth ventricle is elongated and tubular shaped. There is colpocephaly suggestive of corpus callosal agenesis (in E) and interdigitating gyri (dashed arrow in F) suggestive of fenestrated falx.
Advancements in prenatal diagnosis permit diagnosis of neural tube defects in fetus as early as first trimester, and now most of the cases of MMC are diagnosed prenatally during screening sonography. Although sonography is the modality of choice for screening fetus for any gross congenital anomalies, MRI is being increasingly used for prenatal evaluation of CNS anomalies with the advent of faster MR sequences (Figure 9)[16,17].
Figure 9.
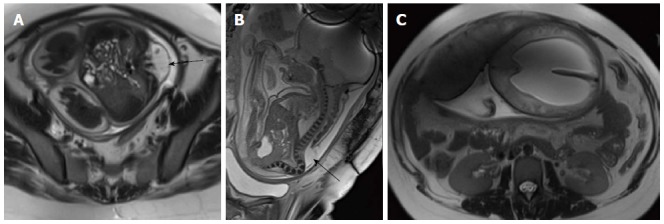
Fetal Myelomeningocele with Chiari II malformation. Axial (A) and sagittal (B) T2 weighted images of lumbar spine of fetus showing splaying of posterior elements of lumbar vertebrae with herniation of spinal canal contents (black arrow) suggestive of MMC. Axial T2 weighted image of brain (C) shows associated hydrocephalus. Hydrocephalus in the setting of MMC in a fetus is considered highly suggestive of Chiari II malformation. MMC: Myelomeningocele.
Prenatal diagnosis and developments in fetal surgery has made possible inutero repair of the neural tube defects which can arrest the development of other malformations developing secondary to abnormal tube closure. The Management of Myelomeningocele Study trial, a prospective randomized study done in United States has shown that fetal surgery for MMC in second trimester preserves neurologic function, reverses the changes of Chiari II malformation and reduces the need for postnatal ventriculoperitoneal shunt[18-21].
Hemimyelomeningocele and hemimyelocele
These conditions are extremely rare and are caused by defective gastrulation and primary neurulation[22]. Diastematomyelia is a common association with OSDs but only when one of the hemicords shows defective neurulation, the malformation is labelled hemimyelo(meningo)cele[3]. Here one of the two hemicords exhibits a myelomeningocele or myelocele while the other hemicord can be normal or is tethered.
CSDS WITH SUBCUTANEOUS MASS
Lipomas with dural defect - lipomyelocele and lipomyelomeningocele
Lipomyelocele and Lipomyelomeningocele result from defective primary neurulation where there is premature focal disjunction of cutaneous ectoderm and neuroectoderm allowing mesenchyme to enter the neural tube. This mesenchyme later forms the lipomatous tissue for unknown reasons[23,24]. Clinically they are characterized by the presence of subcutaneous fatty mass lesion above the intergluteal line which may extend to buttocks. Sagittal T1 WI images show high intensity fat on the dorsal aspect of the placode which is continuous with the adjacent subcutaneous fat. T1 weighted fat saturated images show suppression of fat signal. Lipomyelocele and Lipomyelomeningocele are differentiated based on the position of neural placode - lipoma interface (Figure 10). It lies within or at the edge of the spinal canal in lipomyelocele and outside the spinal canal in lipomyelomeningocele (Figure 11)[11]. In lipomyelomeningocele there is expansion of subarachnoid space anterior to the cord pushing the neural placode - lipoma interface posteriorly to lie outside the boundaries of spinal canal. In hemilipomyelocele or hemilipomyelomeningocele there is associated diastematomyelia with one of the hemicord showing lipomyelocele or lipomyelomeningocele respectively (Figure 12).
Figure 10.
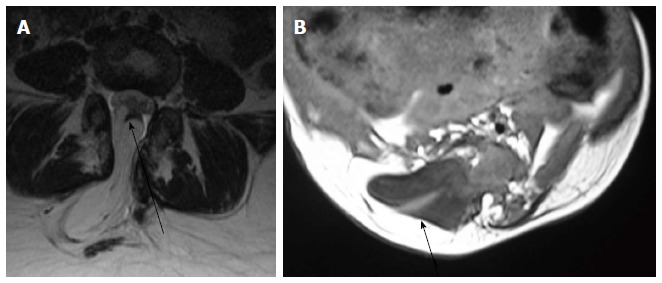
Lipoma placode interface in lipomyelocele and lipomyelomeningocele. Axial T2 weighted images in cases of lipomyelocele (A) and axial T1 weighted images in case of lipomyelomeningocele (B) showing high intensity fat on the dorsal aspect of the neural placode which is continuous with the adjacent subcutaneous fat. The lipoma placode interface (black arrow) is within the spinal canal in lipomyelocele and outside the spinal canal in lipomyelomeningocele. Both these conditions have intact skin covering.
Figure 11.
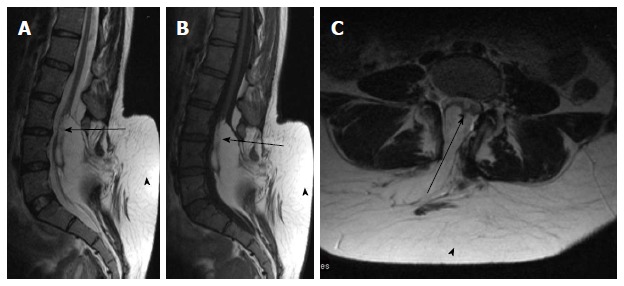
Lipomyelocele. T2 weighted sagittal (A), T1 weighted sagittal (B) and T2 weighted axial (C) images of lumbosacral spine in a case of lumbar lipomyelocele. High intensity fat is seen on the dorsal aspect of the neural placode which is continuous with the adjacent subcutaneous fat (arrowhead) through an open defect in the posterior aspect of spinal canal. The placode lipoma interface is within the spinal canal (arrow).
Figure 12.
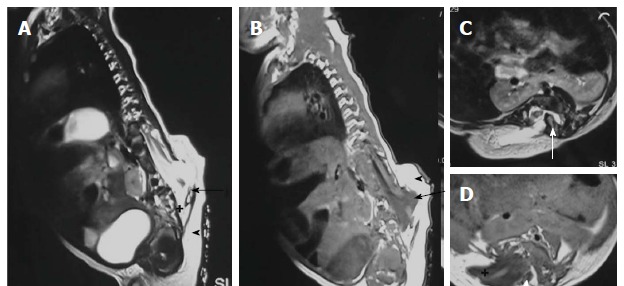
Hemilipomyelomeningocele. T2 weighted sagittal (A), T1 weighted sagittal (B), T2 weighted axial (C) and T1 weighted axial (D) images in a case of hemilipomyelomeningocele. There is diastematomyelia with bony septum (white arrow) suggestive of type I diastematomyelia. There is defect in the posterior spinal canal on the right side through which the spinal canal contents herniate with intact overlying skin and subcutaneous tissue (arrowhead). There is expansion of subarachnoid space (+) anterior to the cord pushing the neural placode - lipoma interface posteriorly to lie outside the boundaries of spinal canal (arrow).
Meningocele
Meningocele refers to herniation of CSF filled sac lined by dura and arahnoid mater. The exact embryogenesis is unknown but is thought to be caused by ballooning of meninges due to CSF pulsation. By definition, spinal cord should not be seen within the meningocele but may be seen tethered to its neck. Meningocele may contain nerve roots and or filum terminale which usually appear hypertrophied.
Posterior meningocele is due to herniation of meningial lining through posterior spina bifida (Figure 13). It is usually lumbosacral in location but can be seen in other locations also. Anterior meningocele is almost always presacral in location[25].
Figure 13.
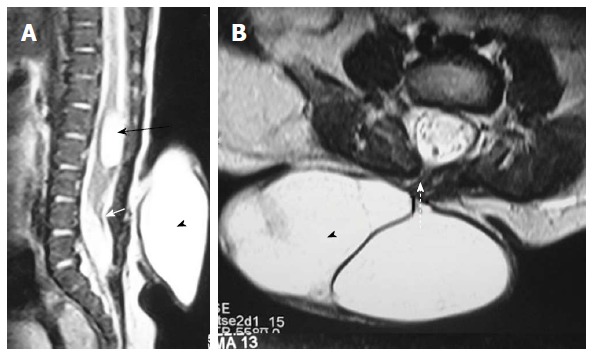
Posterior meningocele with persistent terminal ventricle. T2 weighted sagittal (A) and axial (B) images of lumbosacral spine showing posterior meningocele and persistent terminal ventricle. There is herniation of a CSF filled sac (arrowhead) through a posterior spina bifida (dashed arrow) consistent with posterior meningocele. No neural tissue is noted within the CSF sac. There is another CSF attenuation lesion in conus medullaris (black arrow) suggestive of persistent terminal ventricle. The filum terminale is short and thick (white arrow) with low lying spinal cord suggestive of tight filum terminale. CSF: Cerebrospinal fluid.
Terminal myelocystocele
Terminal myelocystocele involves herniation of a dilated terminal central canal forming terminal syringohydromyelia (syringocele) through a posterior vertebral defect into an expanded CSF filled dural sheath (meningocele). It results from defective secondary neurulation which affects the CSF flow dynamics. The inner terminal syrinx communicates with the central canal of the spinal cord and the outer meningocele is continuous with the spinal subarachnoid space. The syringocele and meningocele usually do not communicate with each other[26-28].
CSDS WITHOUT SUBCUTANEOUS MASS
Simple dysraphic states
These are a heterogeneous group of conditions which arise due to abnormalities of primary and secondary neurulation and are the most common type of spinal dysraphism seen in older children[29]. Simple dysraphic states include intradural lipoma, filar lipoma, tight filum terminale, dermal sinus and persistent terminal ventricle.
Intradural lipoma
It is a midline lipoma located in the groove of unopposed neural placode in its dorsal surface within an intact dural sac. The intact dura help to differentiate this from lipomyelocele and lipomyelomeningocele. They are usually seen in lumbosacral region and are associated with tethered cord syndrome. Large lipomas may cause cord displacement. On MRI lipomas follow the signal intensity of subcutaneous fat on all sequences (Figure 14)[10].
Figure 14.
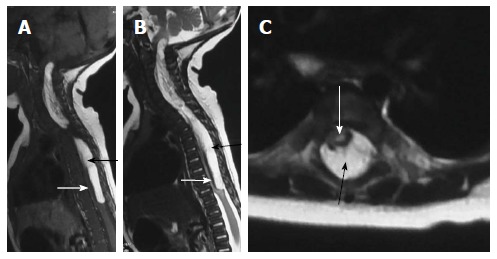
Dural lipoma. T2 weighted sagittal (A), T1 weighted sagittal (B) and T2 weighted axial (C) images of cervico-dorsal spine showing a T1 and T2 hyperintense lesion in the spinal canal (black arrow) causing compression and anterior displacement of the spinal cord (white arrow). The lesion is isointense to subcutaneous fat on all sequences.
Filar lipoma
Filar lipoma is an abnormality of secondary neurulation which shows fibrolipomatous thickening of the filum terminale. On imaging filar lipoma appears hyperintense on T1 and T2 weighted images within a thickened filum terminale (Figure 15). One point five percent to 5% of normal adult population may show fat within filum terminale on MRI and hence the finding is considered a normal variant (Figure 16) unless it is associated with tethered cord syndrome[30,31]. Tethered cord syndrome is characterized clinically by progressive neurological deficit and on imaging, there is low lying conus medullaris with a short thick filum terminale which is tethered to dural sac[32].
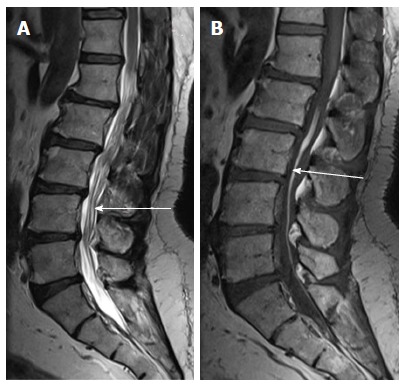
Figure 15.
Filarlipoma. T2 weighted sagittal (A) and T1 weighted axial images of lumbosacral spine in a case of filar lipoma. The lesion (black arrow) appears hyperintense on both T1 and T2 weighted images and follows the signal of subcutaneous fat on all sequences. Filum terminale is thickenined and is tethered (white arrow).
Figure 16.
Fat in filumterminale. T2 weighted (A) and T1 weighted (B) sagittal images of lumbosacral spine in a 60-year-old male showing linear T1 and T2 hyperintense focus in filumterminale (arrow) noted incidentally. Spinal cord is not low lying and there is no tethering of cauda equina fibres. Note is made of degenerative changes in lumbar spine.
Tight filum terminale
It is characterized by shortening and hypertrophy offilum terminale which cause tethering of cord and impairs the ascent of conus medullaris. Embryologically the defect lies in retrogressive differentiation during secondary neurulation.
On imaging it is characterized by a thick filum terminale (thickness measuring > 2 mm) and a low lying conus medullaris - below L2 vertebral body (Figure 17)[8]. It is usually seen in association with other malformations and isolated cases are rare.
Figure 17.
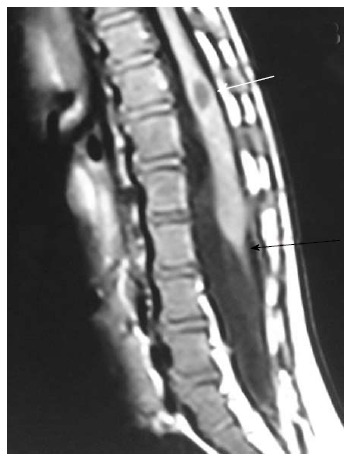
Tight filumterminale. T1 weighted sagittal image of the lumbosacral spine showing thick and short filum terminale (black arrow) with low lying spinal cord. There is a CSF attenuation lesion in conus medullaris (white arrow) suggestive of syrinx. CSF: Cerebrospinal fluid.
Dermal sinus
It is an epithelial lined fistulous communication between CNS or its meningeal covering and skin. It results from focal incomplete disjunction between neuroectoderm and cutaneous ectoderm.
Clinically a midline dimple or ostium is found on the cutaneous surface and is commonly associated with cutaneous stigmata of underlying occult spinal dysraphism like hairy nevus, hemangioma or hyperpigmentation. The tract then ascends and opens into spinal canal (Figure 18). Dermal sinus may be associated with intraspinal dermoids or epidermoids which show variable imaging findings depending on their contents[33,34]. Dermoids usually appear hyperintense on both T1 and T2 weighted images while epidermoids are hypointense on T1 weighted and hyperintense on T2 weighted images. CNS infection is a common complication because of fistulous communication and hence these cases require early surgical repair (Figure 19).
Figure 18.
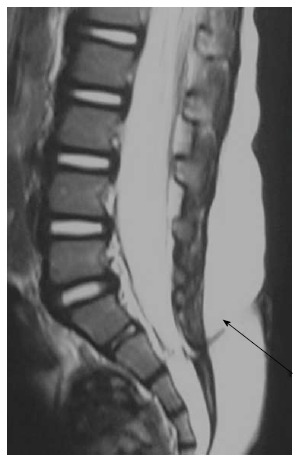
Dermal sinus. T2 weighted sagittal images of lumbosacral spine showing a T2 hypointense tract extending from the posterior skin surface to the spinal canal (black arrow). There is associated tethered cord.
Figure 19.
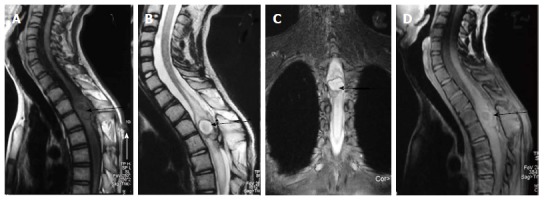
Infected dermal sinus with intraspinal dermoid. T1 weighted sagittal image (A) showing a dermal sinus (white arrow) with an intraspinal lesion (black arrow) showing mildly hyperintense signal with expansion of spinal cord. On T2 weighted sagittal (B) and STIR coronal (C) images the intraspinal lesion appears hyperintense with well defined hypointense rim. On post contrast image (D) there is peripheral enhancement of the intraspinal lesion and the tract.
Persistent terminal ventricle
Terminal ventricle is a small, ependyma lined cavity within conus medullaris (Figures 13 and 17). Embroyologically incomplete regression of terminal ventricle during the stage of secondary neurulation is responsible for the condition. Location just above filum terminale helps to differentiate it from hydromyelia and lack of enhancement is the differentiating feature from intramedullary tumors[35].
COMPLEX DYRAPHIC STATES
Any abnormality occurring at the time of gastrulation affects the spinal cord and various other structures which are derived from notochord resulting in complex anomalies[36]. Most of these abnormalities are covered by skin and subcutaneous masses are absent. On the basis of their embryogenesis complex dysraphic states are divided into two subtypes - disorders of midline notochordal integration and disorders of notochordal formation.
Disorders of midline notochordal integration
The process of fusion of paired notochordal anlagen to form a single midline notochordal process is called midline notochordal integration[3]. Any abnormality at this stage results in longitudinal splitting of spinal cord. Most important entities in this group are neurenteric cyst and diastematomyelia.
Neurenteric cyst: Most severe form of disorder of midline notochordal integration is dorsal enteric fistula - a fistulous communication between skin surface and bowel - which is an extremely rare condition. Neurenteric cyst is a localized form of dorsal enteric fistula and is seen anterior to spinal cord with adjacent vertebral anomalies. These cysts are typically seen in extramedullary intradural compartment of cervicothoracic spine, however may be seen in other locations too[37]. On MRI, neurenteric cysts usually appear iso- to hyper-intense to CSF on both T1 and T2 weighted images due to high protein content and show absent contrast enahncment (Figure 20)[38,39].
Figure 20.
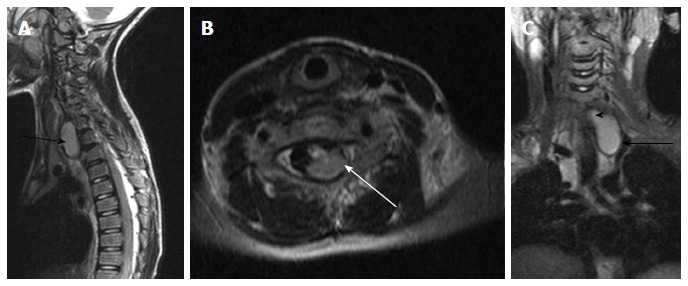
Neuroenteric cyst. T2 weighted images of cervicodorsal spine in sagittal (A), axial (B) and coronal (C) planes show a bilobed lesion with both extraspinal (black arrow) and intraspinal extramedullary component (white arrow). The communication between them (arrowhead) is better appreciated in the coronal plane (C).
Diastematomyelia: It is the most common form of defective midline notochordal integration. Due to defective midline integration there are two notochordal processes each of which induces formation of separate neural plate with intervening primitive streak tissue. The development of the primitive streak tissue decides the type of diastematomyelia. In type I diastematomyelia the intervening primitive streak develops into bone or cartilage, resulting in two hemicords in different dural sacs separated by an osteocartilaginous septum (Figure 12). In type II diastematomyelia, the primitive streak is reabsorbed or forms a fibrous septum with the hemicords lying within the same dural sac (Figure 21)[40]. Diastematomyelia is commonly associated with vertebral anomalies and hydromyelia. A high lying hairy tuft over a child’s back is a reliable indicator for underlying diastematomyelia[41].
Figure 21.
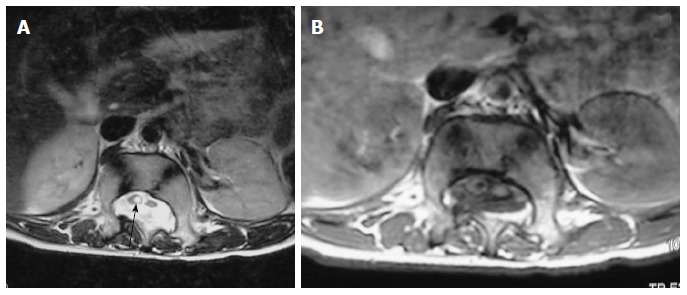
Diastematomyelia II. Axial T2 weighted (A) and T1 weighted (B) scan of lumbar spine showing two hemicords. No intervening bony septum was noted. The hemicord on the right shows dilated central canal or syringohydromyelia (arrow).
Disorders of notochordal formation
Apoptosis or programmed cell death is an important process occurring during different steps of embryogenesis. Abnormal apoptosis results in disorders of notochord formation and these disorders include caudal agenesis and segmental spinal dysgenesis[29].
Caudal agenesis: It is characterized by partial or total agenesis of spinal column and is commonly associated with genital anomalies, anal imperforation, pulmonary hypoplasia, renal aplasia or dysplasia and limb abnormalities. Caudal agenesis (CA) is broadly divided into two types.
In type I CA both caudal cell mass and notochord formation is affected, resulting in high position (most commonly at the level of D12 vertebra) and abnormal termination of conus medullaris. There is accompanying varying degree of vertebral aplasia, with the last vertebra as L5 through S2 in majority of patients.
In type II CA there is abnormal development of only caudal cell mass with unaffected true notochord formation.Hence, there is defective secondary neurulation with normal primary neurulation. As a result only the most caudal part of conus medullaris is absent in type II CA (Figure 22). Vertebral dysgenesis is less severe in these cases and these patients present with tethered cord syndrome as the conus in these cases is stretched and tethered[42,43].
Figure 22.
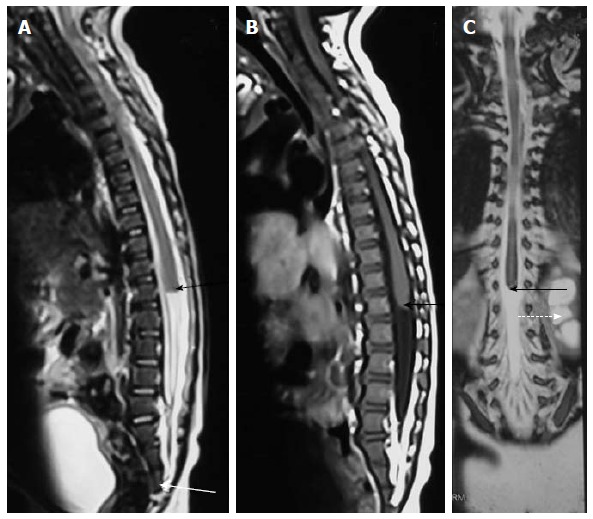
Caudal agenesis. T2 weighted sagittal (A), T1 weighted sagittal (B) and T2 weighted coronal (C) MR images showing type II caudal agenesis. There is non-development of distal sacral vertebra (white arrow in A) with abnormal termination of conus medullaris (black arrow). The child also has associated left hydronephrosis (dashed arrow in C).
Segmental spinal dysgenesis: This is an extremely rare condition characterized by: (1) segmental agenesis or dysgenesis of lumbar or thoacolumbar spine; (2) segmental abnormality of spinal cord or nerve roots; (3) congenital pareparesis or paraplegia; and (4) congenital lower limb deformities. This occurs due to notochordal abnormality occurring during gastrulation which involves an intermediate segment of notochord[29,44].
SACROCOCCYGEAL TERATOMA
Sacrococcygeal teratoma, although a tumor, needs special mentioning as it develops from the pleuripotent cells of caudal cell mass. It is the most common tumor of fetus and new born and commonly present as a large complex solid cystic mass caudal to coccyx. Most of the teratomas are benign and contains derivatives from all three germ layers. Based on the presence of external and internal components they are divided into four types - type I: Primarily external, type II: Equal external and internal portions, type III: Primarily internal and type IV: Entirely internal.
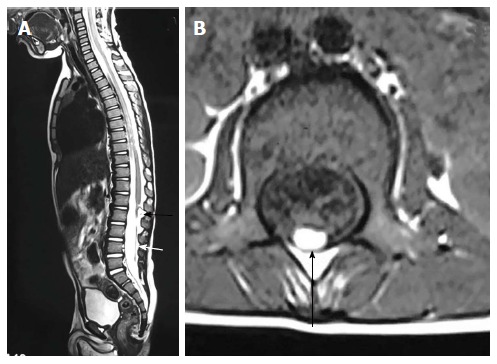
On MRI, sacrococcygeal teratoma has variable signal on T1 and T2 weighted images depending on the internal contents (fat, soft tissue, fluid, calcium). On post contrast images, there is heterogeneous enhancement of the solid portion (Figure 23)[45,46]. This may also be picked up on antenatal scan where MRI can accurately depict its extension into the pelvis and its mass effect on pelvic organs (Figure 24).
Figure 23.
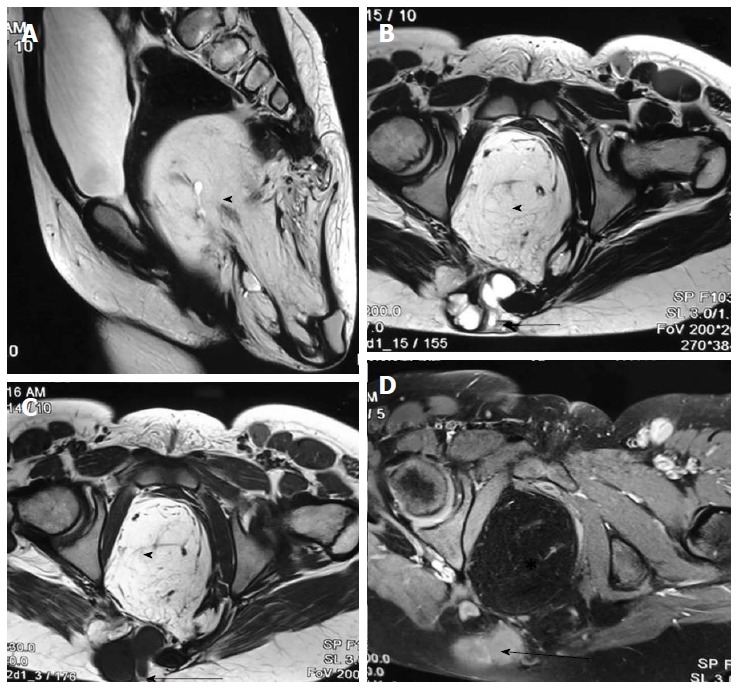
Sacrococcygeal teratoma (type III). T2 weighted sagittal (A), T2 weighted axial (B), T1 weighted axial (C) and T1 weighted fat suppressed axial post contrast (D) images show a large heterogeneous presacral lesion. It has a large internal component which is predominately fat (arrowhead) appearing hyperintense on both T1 and T2 weighted images with signal suppression on post contrast fat saturated image. It also has a small complex solid cystic external component (arrow) appearing hyperintense on T2 wighted and hypointense on T1 weighted with enhancement on post contrast images.
Figure 24.
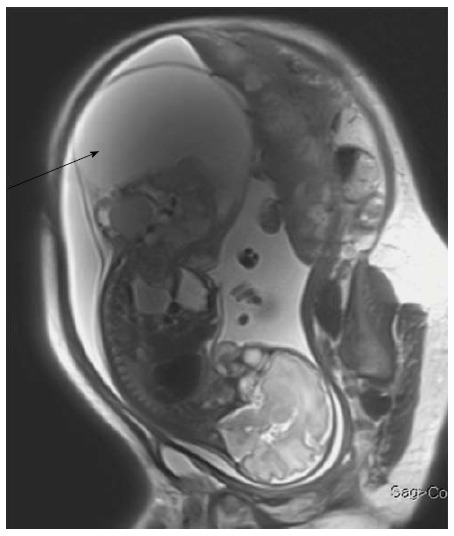
Fetal sacrococcygeal teratoma. T2 weighted sagittal scans through the fetus show a large heterogeneous lesion in the lumbosacral region (arrow) with extension into the pelvis. The lesion shows both solid and cystic components consistent with sacrococcygeal teratoma.
POST-OPERATIVE IMAGING
Surgery is the treatment of choice for spinal dysraphism and surgery includes closure of the neural tube defect and detethering followed by lifelong supportive care and follow up. During follow-up, worsening of symptoms should be looked for and MRI should be done at the earliest suspicion. During post-operative imaging, various immediate and late complications of spinal surgery like wound infection, shunt infection, wound dehiscence, cerebrospinal fluid leak, problems related to kyphectomy, adhesions with retethering or dermoid formation should be looked for (Figure 25). Retethering is due to post-surgical fibrosis resulting in tethering of cauda equine fibres. MRI may be done in prone position to look for CSF dorsal to the conus for ruling out retethering[47].
Figure 25.
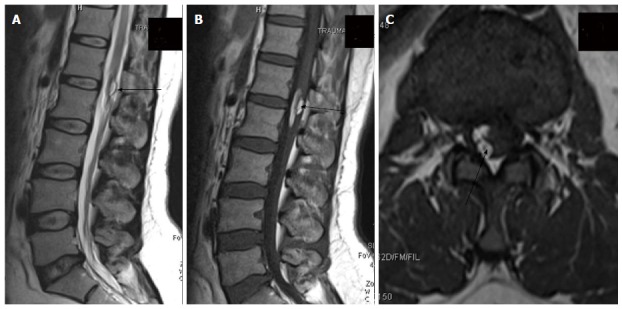
Post-operative dermoid. In a post-operative case of spinal dyraphysm, T2 weighted sagittal, T1 weighted sagittal and T1 weighted axial images of lumbosacral spine showing a T1 and T2 hyperintense lesion in the conusmedullaris and filumterminale consistent with dermoid formation (black arrow).
CONCLUSION
Imaging of spinal dysraphism may appear complicated as it is a group of diverse conditions which can have variable imaging appearance. A systematic approach and correlation with neuroradiological, clinical and developmental data helps in making the correct diagnosis.
Footnotes
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Manuscript source: Invited manuscript
Specialty type: Radiology, nuclear medicine and medical imaging
Country of origin: India
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: October 17, 2016
First decision: January 16, 2017
Article in press: March 2, 2017
P- Reviewer: Battal B, Doglietto F S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.French BN. The embryology of spinal dysraphism. Clin Neurosurg. 1983;30:295–340. doi: 10.1093/neurosurgery/30.cn_suppl_1.295. [DOI] [PubMed] [Google Scholar]
- 2.Rossi A, Cama A, Piatelli G, Ravegnani M, Biancheri R, Tortori-Donati P. Spinal dysraphism: MR imaging rationale. J Neuroradiol. 2004;31:3–24. doi: 10.1016/s0150-9861(04)96875-7. [DOI] [PubMed] [Google Scholar]
- 3.Tortori-Donati P, Rossi A, Cama A. Spinal dysraphism: a review of neuroradiological features with embryological correlations and proposal for a new classification. Neuroradiology. 2000;42:471–491. doi: 10.1007/s002340000325. [DOI] [PubMed] [Google Scholar]
- 4.Barkovich AJ. Pediatric neuroradiology, 4th ed. Philadelphia, PA: Lippincott Willims & Wilkins; 2011. pp. 857–916. [Google Scholar]
- 5.Naidich TP, Blaser SI, Delman BN. Congenital Anomalies of the Spine and Spinal Cord: Embryology and Malformations. In: Atlas SW eds., editor. Magnetic Resonance Imaging of the Brain and Spine, 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2009. pp. 1364–1447. [Google Scholar]
- 6.Müller F, O’Rahilly R. The first appearance of the neural tube and optic primordium in the human embryo at stage 10. Anat Embryol (Berl) 1985;172:157–169. doi: 10.1007/BF00319598. [DOI] [PubMed] [Google Scholar]
- 7.Catala M. Genetic control of caudal development. Clin Genet. 2002;61:89–96. doi: 10.1034/j.1399-0004.2002.610202.x. [DOI] [PubMed] [Google Scholar]
- 8.Warder DE. Tethered cord syndrome and occult spinal dysraphism. Neurosurg Focus. 2001;10:e1. doi: 10.3171/foc.2001.10.1.2. [DOI] [PubMed] [Google Scholar]
- 9.Huisman TA, Rossi A, Tortori-Donati P. MR imaging of neonatal spinal dysraphia: what to consider? Magn Reson Imaging Clin N Am. 2012;20:45–61. doi: 10.1016/j.mric.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Tortori-Donati P, Rossi A, Biancheri R, Cama A. Magnetic resonance imaging of spinal dysraphism. Top Magn Reson Imaging. 2001;12:375–409. doi: 10.1097/00002142-200112000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Rossi A, Biancheri R, Cama A, Piatelli G, Ravegnani M, Tortori-Donati P. Imaging in spine and spinal cord malformations. Eur J Radiol. 2004;50:177–200. doi: 10.1016/j.ejrad.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Thompson D. Spinal dysraphic anomalies; classification, presentation and management. Paediatrics and Child Health. 2010;20:397–403. [Google Scholar]
- 13.Osaka K, Matsumoto S, Tanimura T. Myeloschisis in early human embryos. Childs Brain. 1978;4:347–359. doi: 10.1159/000119791. [DOI] [PubMed] [Google Scholar]
- 14.McLone DG, Naidich TP. Developmental morphology of the subarachnoid space, brain vasculature, and contiguous structures, and the cause of the Chiari II malformation. AJNR Am J Neuroradiol. 1992;13:463–482. [PMC free article] [PubMed] [Google Scholar]
- 15.McLone DG, Knepper PA. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15:1–12. doi: 10.1159/000120432. [DOI] [PubMed] [Google Scholar]
- 16.von Koch CS, Glenn OA, Goldstein RB, Barkovich AJ. Fetal magnetic resonance imaging enhances detection of spinal cord anomalies in patients with sonographically detected bony anomalies of the spine. J Ultrasound Med. 2005;24:781–789. doi: 10.7863/jum.2005.24.6.781. [DOI] [PubMed] [Google Scholar]
- 17.Reddy UM, Filly RA, Copel JA. Prenatal imaging: ultrasonography and magnetic resonance imaging. Obstet Gynecol. 2008;112:145–157. doi: 10.1097/01.AOG.0000318871.95090.d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adzick NS, Thom EA, Spong CY, Brock JW, Burrows PK, Johnson MP, Howell LJ, Farrell JA, Dabrowiak ME, Sutton LN, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364:993–1004. doi: 10.1056/NEJMoa1014379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzick NS. Fetal surgery for spina bifida: past, present, future. Semin Pediatr Surg. 2013;22:10–17. doi: 10.1053/j.sempedsurg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adzick NS. Fetal surgery for myelomeningocele: trials and tribulations. Isabella Forshall Lecture. J Pediatr Surg. 2012;47:273–281. doi: 10.1016/j.jpedsurg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grivell RM, Andersen C, Dodd JM. Prenatal versus postnatal repair procedures for spina bifida for improving infant and maternal outcomes. Cochrane Database Syst Rev. 2014;(10):CD008825. doi: 10.1002/14651858.CD008825.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parmar H, Patkar D, Shah J, Maheshwari M. Diastematomyelia with terminal lipomyelocystocele arising from one hemicord: case report. Clin Imaging. 2003;27:41–43. doi: 10.1016/s0899-7071(02)00522-3. [DOI] [PubMed] [Google Scholar]
- 23.Naidich TP, Blaser SI, Delman BN, McLone DG, Dias MS, Zimmerman RA, Raybaud CA, Birschansky SB, Altman NR, Braffman BH. Embryology of the spine and spinal cord. ASNR. 2002:3–13. [Google Scholar]
- 24.Naidich TP, McLone DG, Mutluer S. A new understanding of dorsal dysraphism with lipoma (lipomyeloschisis): radiologic evaluation and surgical correction. AJR Am J Roentgenol. 1983;140:1065–1078. doi: 10.2214/ajr.140.6.1065. [DOI] [PubMed] [Google Scholar]
- 25.Lee KS, Gower DJ, McWhorter JM, Albertson DA. The role of MR imaging in the diagnosis and treatment of anterior sacral meningocele. Report of two cases. J Neurosurg. 1988;69:628–631. doi: 10.3171/jns.1988.69.4.0628. [DOI] [PubMed] [Google Scholar]
- 26.Byrd SE, Harvey C, Darling CF. MR of terminal myelocystoceles. Eur J Radiol. 1995;20:215–220. doi: 10.1016/0720-048x(95)00659-e. [DOI] [PubMed] [Google Scholar]
- 27.Peacock WJ, Murovic JA. Magnetic resonance imaging in myelocystoceles. Report of two cases. J Neurosurg. 1989;70:804–807. doi: 10.3171/jns.1989.70.5.0804. [DOI] [PubMed] [Google Scholar]
- 28.McLone DG, Naidich TP. Terminal myelocystocele. Neurosurgery. 1985;16:36–43. [PubMed] [Google Scholar]
- 29.Tortori-Donati P, Fondelli MP, Rossi A, Raybaud CA, Cama A, Capra V. Segmental spinal dysgenesis: neuroradiologic findings with clinical and embryologic correlation. AJNR Am J Neuroradiol. 1999;20:445–456. [PMC free article] [PubMed] [Google Scholar]
- 30.Uchino A, Mori T, Ohno M. Thickened fatty filum terminale: MR imaging. Neuroradiology. 1991;33:331–333. doi: 10.1007/BF00587817. [DOI] [PubMed] [Google Scholar]
- 31.Brown E, Matthes JC, Bazan C, Jinkins JR. Prevalence of incidental intraspinal lipoma of the lumbosacral spine as determined by MRI. Spine (Phila Pa 1976) 1994;19:833–836. doi: 10.1097/00007632-199404000-00018. [DOI] [PubMed] [Google Scholar]
- 32.El-Hefnawy AS, Wadie BS. Effect of detethering on bladder function in children with myelomeningocele: Urodynamic evaluation. J Pediatr Neurosci. 2009;4:70–72. doi: 10.4103/1817-1745.57324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanev PM, Park TS. Dermoids and dermal sinus tracts of the spine. Neurosurg Clin N Am. 1995;6:359–366. [PubMed] [Google Scholar]
- 34.Thompson DN. Spinal inclusion cysts. Childs Nerv Syst. 2013;29:1647–1655. doi: 10.1007/s00381-013-2147-z. [DOI] [PubMed] [Google Scholar]
- 35.Coleman LT, Zimmerman RA, Rorke LB. Ventriculus terminalis of the conus medullaris: MR findings in children. AJNR Am J Neuroradiol. 1995;16:1421–1426. [PMC free article] [PubMed] [Google Scholar]
- 36.Dias MS, Walker ML. The embryogenesis of complex dysraphic malformations: a disorder of gastrulation? Pediatr Neurosurg. 1992;18:229–253. doi: 10.1159/000120670. [DOI] [PubMed] [Google Scholar]
- 37.Harris CP, Dias MS, Brockmeyer DL, Townsend JJ, Willis BK, Apfelbaum RI. Neurenteric cysts of the posterior fossa: recognition, management, and embryogenesis. Neurosurgery. 1991;29:893–897; discussion 897-898. doi: 10.1097/00006123-199112000-00015. [DOI] [PubMed] [Google Scholar]
- 38.Simon JA, Olan WJ, Santi M. Intracranial neurenteric cysts: a differential diagnosis and review. Radiographics. 1997;17:1587–1593. doi: 10.1148/radiographics.17.6.9397466. [DOI] [PubMed] [Google Scholar]
- 39.Rufener S, Ibrahim M, Parmar HA. Imaging of congenital spine and spinal cord malformations. Neuroimaging Clin N Am. 2011;21:659–676, viii. doi: 10.1016/j.nic.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 40.Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: Part I: A unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451–480. doi: 10.1227/00006123-199209000-00010. [DOI] [PubMed] [Google Scholar]
- 41.Barkovich AJ, Edwards Ms, Cogen PH. MR evaluation of spinal dermal sinus tracts in children. AJNR Am J Neuroradiol. 1991;12:123–129. [PMC free article] [PubMed] [Google Scholar]
- 42.Estin D, Cohen AR. Caudal agenesis and associated caudal spinal cord malformations. Neurosurg Clin N Am. 1995;6:377–391. [PubMed] [Google Scholar]
- 43.Rufener SL, Ibrahim M, Raybaud CA, Parmar HA. Congenital spine and spinal cord malformations--pictorial review. AJR Am J Roentgenol. 2010;194:S26–S37. doi: 10.2214/AJR.07.7141. [DOI] [PubMed] [Google Scholar]
- 44.Zana E, Chalard F, Mazda K, Sebag G. An atypical case of segmental spinal dysgenesis. Pediatr Radiol. 2005;35:914–917. doi: 10.1007/s00247-005-1483-x. [DOI] [PubMed] [Google Scholar]
- 45.Danzer E, Hubbard AM, Hedrick HL, Johnson MP, Wilson RD, Howell LJ, Flake AW, Adzick NS. Diagnosis and characterization of fetal sacrococcygeal teratoma with prenatal MRI. AJR Am J Roentgenol. 2006;187:W350–W356. doi: 10.2214/AJR.05.0152. [DOI] [PubMed] [Google Scholar]
- 46.Kocaoglu M, Frush DP. Pediatric presacral masses. Radiographics. 2006;26:833–857. doi: 10.1148/rg.263055102. [DOI] [PubMed] [Google Scholar]
- 47.Venkataramana NK. Spinal dysraphism. J Pediatr Neurosci. 2011;6:S31–S40. doi: 10.4103/1817-1745.85707. [DOI] [PMC free article] [PubMed] [Google Scholar]