

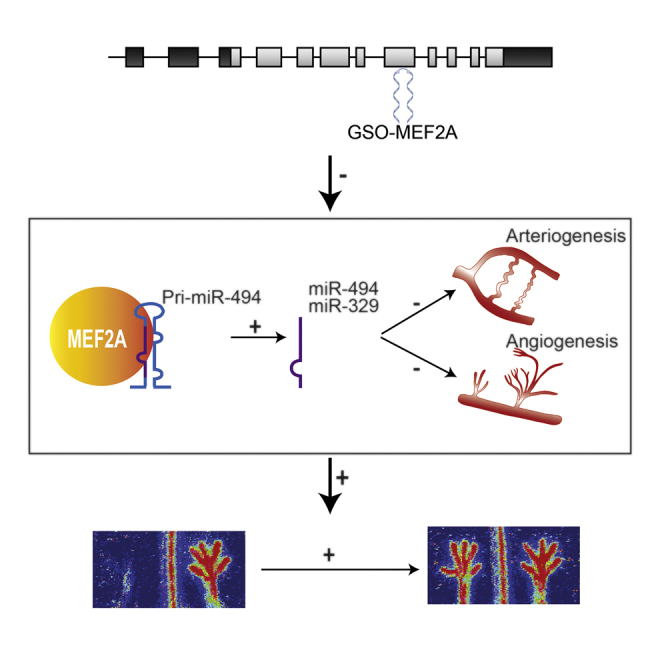
Abstract
Improving the efficacy of neovascularization is a promising strategy to restore perfusion of ischemic tissues in patients with peripheral arterial disease. The 14q32 microRNA cluster is highly involved in neovascularization. The Mef2a transcription factor has been shown to induce transcription of the microRNAs within this cluster. We inhibited expression of Mef2a using gene-silencing oligonucleotides (GSOs) in an in vivo hind limb ischemia model. Treatment with GSO-Mef2a clearly improved blood flow recovery within 3 days (44% recovery versus 25% recovery in control) and persisted until 14 days after ischemia induction (80% recovery versus 60% recovery in control). Animals treated with GSO-Mef2a showed increased arteriogenesis and angiogenesis in the relevant muscle tissues. Inhibition of Mef2a decreased expression of 14q32 microRNAs miR-329 (p = 0.026) and miR-494 (trend, p = 0.06), but not of other 14q32 microRNAs, nor of 14q32 microRNA precursors. Because Mef2a did not influence 14q32 microRNA transcription, we hypothesized it functions as an RNA-binding protein that influences processing of 14q32 microRNA miR-329 and miR-494. Mef2A immunoprecipitation followed by RNA isolation and rt/qPCR confirmed direct binding of MEF2A to pri-miR-494, supporting this hypothesis. Our study demonstrates a novel function for Mef2a in post-ischemic neovascularization via post-transcriptional regulation of 14q32 microRNAs miR-329 and miR-494.
Keywords: MEF2A, miR-329, miR-494, 14q32, neovascularization, arteriogenesis, angiogenesis, posttranscriptional regulation, RNA binding protein, ischemia
Graphical Abstract
Introduction
Patients with peripheral arterial disease (PAD) suffer from reduced blood supply toward the extremities caused by the build-up of obstructive atherosclerotic plaques in the arterial wall. Restoration of blood flow in these patients is imperative for the prevention of critical limb ischemia and limb loss. This can be accomplished by stimulating post-ischemic neovascularization, i.e., arteriogenesis and angiogenesis. Arteriogenesis, the outward remodeling of pre-existent collateral arterioles, is initiated upon increases in shear stress through the arterioles.1 Angiogenesis is driven by ischemia and leads to sprouting of new capillaries from existing blood vessels into the ischemic tissue.2 Both arteriogenesis and angiogenesis are multifactorial processes that involve, for example, the activation of endothelial cells (ECs), proliferation of vascular cell types (especially smooth muscle cells [SMCs]), and recruitment of immune cells.
MicroRNAs are short, non-coding RNA molecules that regulate the expression of their target genes at the post-transcriptional level.3 Each microRNA has multiple target genes, and microRNA binding to a target messenger RNA downregulates the expression of that gene. The ability of microRNAs to target numerous genes makes them attractive targets for the regulation of multifactorial physiological processes. In the past decade, microRNAs have emerged as key regulators in the development and progression of cardiovascular disease.4, 5, 6, 7, 8, 9 Recently, we have shown the involvement of multiple microRNAs from a single microRNA cluster located on human chromosome 14q32 in post-ischemic neovascularization.10 The 14q32 locus contains the largest known mammalian microRNA gene cluster and contains 54 microRNAs in humans (12F1 locus in mice containing 61 microRNAs).11 In our previous study, we showed that inhibition of microRNAs miR-329, miR-487b, miR-494, and miR-495 from the 14q32 microRNA cluster led to improved blood flow recovery after induction of hind limb ischemia in mice.10 Arteriogenesis and angiogenesis were both increased in the adductor and soleus muscles of these mice, respectively. Myocyte enhancer factor 2a (Mef2a) was confirmed as a target gene of miR-329, and Mef2a expression was upregulated in the ligated hind limb after miR-329 inhibition. Interestingly, MEF2A itself was reported to target miR-329 and the other 14q32 microRNAs.12 Several studies reported transcriptional regulation of the 14q32 microRNA cluster by the MEF2 family of transcription factors and, in particular, by MEF2A.12, 13, 14
The MEF2 family of transcription factors consists of four members, namely, MEF2A, MEF2B, MEF2C, and MEF2D. Although mainly studied for their role in muscle development and differentiation, MEF2 family members are also expressed in ECs and vascular SMCs.15, 16 MEF2A and MEF2C are the predominant isoforms expressed in vascular cells. In ECs, MEF2 proteins have been shown to control vascular integrity by promoting EC survival, whereas in SMCs, MEF2 proteins are expressed in cells with an activated phenotype and control differentiation of SMCs.17, 18 Moreover, inhibition of MEF2A induced phenotypic switching of vascular SMCs, leading to proliferation and migration of these cells.19
The aim of this study was to evaluate the effect of Mef2a inhibition on post-ischemic blood flow recovery. We hypothesized that Mef2a could act as a novel switch in neovascularization via regulation of 14q32 microRNAs. We show here that inhibition of Mef2a, via subsequent inhibition of 14q32 microRNAs miR-329 and miR-494, but not other 14q32 microRNAs, indeed improves post-ischemic blood flow recovery in vivo.
Results
Gene Silencing Oligonucleotide Mediated Inhibition of Mef2a Expression
We tested several gene silencing oligonucleotides (GSOs) in vitro for their capacity to inhibit Mef2a mRNA expression in murine primary SMCs (GSO-1, GSO-2, GSO-3, and GSO-4; Figure 1A). Expression of Mef2a was significantly inhibited by GSO-2 (35% expression of control, p = 0.0005) and GSO-3 (30% expression of control, p = 0.0094), but not by treatment with GSO-1 or GSO-4 (Figure 1B). For in vivo experiments, we selected GSO-3 for inhibition of Mef2a (GSO-Mef2a from here on), which gave the strongest downregulation of Mef2a expression.
Figure 1.

Inhibition of Mef2a by GSOs
(A) Four different GSOs were developed and tested for their capacity to inhibit Mef2a expression in vitro. GSOs were designed to target different exons on the murine Mef2a mRNA sequence. (B) Inhibition of Mef2a in primary murine vascular smooth muscle cells for 48 hr by GSOs (10 ng/μL). Mean expression levels, from at least three independent experiments, relative to HPRT and as a percentage of GSO-control are shown here (±SEM). **p < 0.01; ***p < 0.001.
In Vivo Inhibition of Mef2a Using GSOs
Immunohistochemical staining of murine adductor muscle tissue demonstrated that MEF2A is expressed in this tissue, particularly in the nuclei of cells (Figure 2A). Expression of Mef2a was measured in the adductor and gastrocnemius muscles of animals, 14 days after induction of ischemia and 4 days after the final GSO injection. Expression of Mef2a was downregulated in both adductor and gastrocnemius muscle tissue of GSO-Mef2a-treated mice compared to control-treated animals (Figures 2B and 2C, respectively). Although inhibition of Mef2a was less strong than in vitro cell cultures, inhibition was highly significant (72% expression of control, p = 0.00052 in the adductor, and 78% expression of control, p < 0.0001 in the gastrocnemius muscle). GSO-Mef2a treatment furthermore reduced MEF2A protein levels in the adductor muscle by 30% (Figure 2D).
Figure 2.

In Vivo Inhibition of Mef2a by GSO-Mef2a
(A) Immunohistochemical staining of MEF2A in the adductor muscle of GSO-Mef2a- and GSO-control-treated mice. (B) mRNA expression of Mef2a in the adductor muscle of C57BL/6 mice treated with GSO-Mef2a or GSO-control, 14 days after double ligation of the left femoral artery. Per group, adductor muscle tissue of ten animals was used. (C) Expression of Mef2a in the gastrocnemius muscle of C57BL/6 mice treated with GSO-Mef2a or GSO-control, 14 days after ischemia induction. Per group, gastrocnemius muscle tissue of ten animals was used. Mean expression levels relative to HPRT are shown here (±SEM). (D) MEF2A and actin protein expression and quantification in adductor muscle lysates of GSO-Mef2a- (M) and GSO-control (C)-treated mice (n = 2 animals per group). Mean expression as a percentage of control is shown here (±SEM). ***p < 0.001; ****p < 0.0001.
Improved Blood Flow Recovery upon Mef2a Inhibition
The effect of Mef2a inhibition on blood flow recovery was evaluated at different time points after ligation of the femoral artery (pre-operative, post-operative, day 3, day 7, day 10, and day 14 after ischemia induction; Figure 3). Within 3 days after hind limb ischemia, animals injected with GSOs against Mef2a already showed improved blood flow recovery (44% recovery in GSO-Mef2a animals versus 25% recovery in GSO-control animals; Figure 3). This increase in perfusion persisted over time. At time of sacrifice, GSO-Mef2a-treated animals showed 80% recovery in blood flow compared to 60% recovery of perfusion in GSO-control-treated animals.
Figure 3.

Blood Flow Recovery after In Vivo Mef2a Inhibition
(A) LDPI from paws of mice subjected to unilateral hind limb ischemia showing blood flow recovery in these animals over time. (B) Quantification of LDPI measurements over time in mice (11 per group) treated with GSO-Mef2a or GSO-control (1 mg per mouse injected i.p. at different time points as described in Materials and Methods). Data are calculated as the ratio of the left (ischemic) over the right (non-ischemic) paw and presented as mean ± SEM. p values are shown for each time point.
In Vivo Inhibition of Mef2a Increases Arteriogenesis
Alpha smooth muscle actin (α-SMA+) staining was used to visualize collateral vessels in the adductor muscle. Significant increases in arteriole diameters were observed between the left and right adductor muscles for GSO-Mef2a-treated mice compared to GSO-control-treated mice (increase in arteriole diameter in left over right adductor [LA/RA ratio] was 2.057 compared to the LA/RA ratio of 1.16 in controls, p = 0.0060; Figure 4A). Both total α-SMA+ area per section (LA GSO-Mef2a 846 ± 99 μm2 versus RA GSO-Mef2a 404 ± 37 μm2, p = 0.00043, and LA GSO-control 654 ± 68 μm2 versus RA GSO-control 480 ± 27 μm2, p = 0.05) as well as mean lumen area per α-SMA+ collateral (LA GSO-Mef2a 342 ± 25 μm2 versus RA GSO-Mef2a 170 ± 9 μm2, p = 0.0002, and LA GSO-control 237 ± 47 μm2 versus RA GSO-control 217 ± 18 μm2, p = 0.54) were increased in the left adductor muscles of GSO-Mef2a-treated animals compared to the right adductor muscles, which was not the case for GSO-control-treated animals (Figures 4B and 4C, respectively). Moreover, α-SMA+ area per collateral was increased in the left adductor of GSO-Mef2a-treated animals compared to controls (GSO-Mef2a 342 ± 25 μm2 versus GSO-control 237 ± 47 μm2, p = 0.03; Figure 4C and representative images in Figure 4D). The number of α-SMA+ vessels between the left and right paw were similar in the GSO-Mef2a group (LA/RA ratio 1.04), whereas GSO-control-treated animals had more α-SMA+ vessels in the left compared to the right paw (LA/RA ratio 1.27; Figure 4E). Arteriogenesis is the enlargement of pre-existing arterioles to form collateral arteries. Therefore, not the increase in number of collateral arteries, but rather the increase in arteriole diameters demonstrate increased arteriogenesis in GSO-Mef2a-treated mice. Furthermore, because an inflammatory environment facilitates extracellular matrix rearrangement and outward remodeling of collateral arterioles, we also looked at the number of inflammatory cells surrounding the collaterals. There was a trend toward an increase in the number of perivascular CD45+ leukocytes around the remodeling collaterals of GSO-Mef2a-treated animals compared to controls (64% increase compared to GSO-control animals, p = 0.09; Figure 4F).
Figure 4.

In Vivo Arteriogenesis after Mef2a Inhibition
Immunofluorescence staining of paraffin-embedded adductor muscle group of C57BL/6 mice treated with either GSO-control (n = 11 animals) or GSO-Mef2a (n = 10 animals), 14 days after ischemia induction, using anti-αSMA (turquoise) antibodies. (A) Quantification of the increase in diameter of α-SMA+ arterioles between the left and the right adductor muscles of mice. (B and C) Total α-SMA+ area per section (B) as well as mean lumen area per α-SMA+ collateral (C) are shown. (D) Representative images of α-SMA staining in unligated and ligated adductor muscle tissues of mice treated with GSO-Mef2a or GSO-control. (E) Number of total collateral arterioles in left (ligated) over right (unligated) adductor muscles of mice. (F) Quantification of the number of CD45+ cells around remodeling collateral arterioles in the adductor muscle of GSO-control- or GSO-Mef2a-treated mice, 14 days after induction of ischemia. From each muscle, eight representative images were used for quantification. Data are presented as mean ± SEM. *p < 0.05; *p < 0.001; ***p < 0.0005.
In Vivo Angiogenesis upon Mef2a Inhibition
Capillary formation was evaluated in the left (ischemic) and right (normoxic) soleus muscles of GSO-treated mice, 14 days after induction of ischemia. Muscle tissues were stained with anti-CD31 to visualize capillaries. Inhibition of Mef2a increased capillary formation in the soleus muscles of these mice (1.4-fold, p = 0.04) compared to the soleus muscles of GSO-control-treated mice (Figure 5). We also investigated the effect of Mef2a inhibition on ex vivo angiogenesis using aortic explants. GSO-mediated inhibition of Mef2a led to a mild increase in the number of sprouts from aortic explants compared to GSO-control (Figure S1).
Figure 5.

In Vivo Angiogenesis after Mef2a Inhibition
(A) Representative images of CD31 staining in the right (non-ischemic) and left (ischemic) soleus muscles of C57BL/6 mice treated with GSO-Mef2a (n = 10 animals) or GSO-control (n = 11 animals). (B) Quantification of the capillary density in the soleus muscle, defined as the increase in CD31+ area between the left and right soleus muscles, shown as a percentage relative to the increase in mice treated with GSO-control. From each soleus muscle, six representative photographs were used for quantification. Data are presented as mean ± SEM. *p < 0.05.
Mef2a Targets miR-329 and miR-494 of the 14q32 MicroRNA Cluster
We determined expression of 14q32 microRNAs miR-329, miR-487b, miR-494, miR-495, and miR-410 in the adductor muscles of GSO-Mef2a- and GSO-control-treated mice. Inhibition of Mef2a led to significant downregulation of 14q32 microRNA miR-329 (Figure 6A; p = 0.0026) and a borderline significant downregulation of miR-494 (Figure 6B; p = 0.06). However, expression of 14q32 microRNAs miR-487b, miR-410, and miR-495 was not inhibited in animals treated with GSO-Mef2a (Figures 6C–6E). MicroRNAs are transcribed as primary microRNAs (pri-miRs) before being processed into pre-microRNAs (pre-miRs) and subsequently into mature microRNAs. To determine whether Mef2a does influence transcription of multiple 14q32 microRNAs, we also measured expression of the pri-miR transcripts and the intermediate pre-miRs in adductor muscle tissue of GSO-treated animals. No differences were observed in pri-miR and pre-miR levels of any of the microRNAs between GSO-Mef2a- and GSO-control-treated animals, indicating that the regulation of mature miR-329 and miR-494 levels was indirect (Figure 7A).
Figure 6.

Regulation of 14q32 MicroRNA Expression In Vivo
(A–E) Expression of 14q32 microRNAs miR-329 (A), miR-494 (B), miR-487b (C), miR-410 (D), and miR-495 (E) in adductor muscle tissue of GSO-Mef2a- or GSO-control-treated animals at 14 days after hind limb ischemia. Per group, adductor muscle tissue of four mice was used. Mean expression levels are shown here relative to expression of let-7c (±SEM). *p < 0.05.
Figure 7.

Regulation of 14q32 Pri-miR and Pre-miR Expression In Vivo
(A) Expression of pri-miR and pre-miR transcripts of 14q32 microRNAs miR-329, miR-487b, miR-494, and miR-495 in adductor muscle tissue of GSO-Mef2a- or GSO-control-treated animals at 14 days after hind limb ischemia. Per group, adductor muscle tissue of 11 mice was used. Mean expression levels are shown here relative to expression of snRNA-U6 (±SEM). (B) RNA-binding protein immunoprecipitation with either MEF2A antibodies or negative control rabbit IgG followed by qRT-PCR on pri-miR transcripts of 14q32 microRNAs miR-494 and miR-487b. Data are normalized against 10% input.
Mef2a Binds to the Pri-miR Transcript of 14q32 MicroRNA miR-494, but Not to miR-487b
To investigate whether Mef2a could regulate post-transcriptional processing of 14q32 microRNAs, we performed RNA-binding protein immunoprecipitation (RIP) assays. RIP experiments on 3T3 cell lysates performed with anti-MEF2A antibodies demonstrated specific binding of MEF2A to the pri-miR-494 transcript, but not to the pri-miR-487b transcript (Figure 7B). Expression of pri-miR-329 was too low to confirm or exclude MEF2A binding in these cells.
Discussion
We demonstrate here that the Mef2a transcription factor plays a role in post-ischemic neovascularization. Inhibition of MEF2A improved blood flow recovery in animals after unilateral ligation of the femoral artery. Previous reports have demonstrated the role of Mef2a in other forms of vascular remodeling as well. One study showed that the expression of MEF2 members Mef2a, Mef2b, and Mef2d was increased in the neointima of rat carotid arteries after balloon injury.18 In another study, overexpression of a dominant-negative mutant form of Mef2a reduced neointima formation, inhibited macrophage infiltration, and decreased MCP1 expression upon wire injury in rats.20 However, to our knowledge, a role of Mef2a in post-ischemic neovascularization is a completely novel finding.
We hypothesized that inhibition of Mef2a expression would improve post-ischemic neovascularization via subsequent downregulation of 14q32 microRNAs because Mef2a was reported to control 14q32 microRNA expression and because we have previously shown that 14q32 microRNAs have anti-angiogenic and anti-arteriogenic functions.10, 12 Inhibition of Mef2a in our study revealed decreased expression of 14q32 microRNA miR-329 and a trend toward decreased expression of miR-494, but not of several other 14q32 microRNAs studied here. Indeed, animals treated with GSO-Mef2a showed ∼80% recovery of blood flow, 14 days after ischemia induction. However, this effect was not greater than inhibition of single 14q32 microRNAs miR-329 and miR-494 because inhibition of these microRNAs led to (nearly) complete restoration of blood flow within 7 and 10 days, respectively.
In addition to the mature miR levels, we also determined expression levels of pri-miR transcripts and pre-miR intermediates of several 14q32 microRNAs. Pri-miR levels directly indicate the level of transcription of the 14q32 microRNA genes, which were expected to be influenced by the transcription factor Mef2a. Pre-miRs are an intermediate form of the microRNA transcript, and changes in expression of the pri-miR to the pre-miR or from pre-miR to the mature microRNA indicate changes in post-transcriptional processing of the microRNA. Pri-miR and pre-miR levels for miR-329, miR-487b, miR-494, and miR-495 showed no difference in expression levels between the GSO-treated groups. These data indicate that there is no direct transcriptional regulation by MEF2a of the 14q32 microRNAs studied here in this model and that the effects on mature miR-329 and miR-494 levels were achieved through other mechanism than transcription. Using RIP experiments, we demonstrated specific binding of MEF2A to the pri-miR-494 transcript, but not to pri-miR-487b, a microRNA that was not regulated by Mef2a inhibition in our study. Because pri-miR-329 levels were too low in 3T3 cells, we could not confirm or exclude MEF2A binding. Nonetheless, we show here for the first time that MEF2A may not only function as a transcription factor, but potentially also as an RNA-binding protein. Future experiments will have to determine whether MEF2A can regulate post-transcriptional processing of other 14q32 microRNAs as well as non-14q32 microRNAs. For example, a recent study demonstrated that MEF2A, via miR-143, regulates proliferation, migration, and H2O2-induced senescence of vascular SMCs.21 This work by Zhao et al. indeed shows that MEF2A can also regulate expression of microRNAs outside the 14q32 gene cluster.
In the study by Snyder et al.,12 microarray analysis of injured muscle tissue from Mef2a knockout mice revealed downregulation of multiple microRNAs located on mouse chromosome 12F1 (14q32 in humans), indicating regulation of the 12F1/14q32 region by Mef2a, at least in response to injury. To evaluate the role of Mef2a in skeletal muscle regeneration, the authors induced muscle injury in mice.12 In our study, we made use of the hind limb ischemia model to investigate the role of Mef2a in post-ischemic neovascularization, in which no direct damage to the muscle itself was introduced. Furthermore, Snyder et al. determined the expression of 14q32 microRNAs in (injured) muscle tissue of Mef2a knockout mice, whereas we measured expression of 14q32 microRNAs in C57BL/6 muscle tissue, wherein Mef2a expression was reduced by ∼25% only using GSOs.12 These differences in the chosen models may explain the observed discrepancies in the regulation of 14q32 microRNAs by Mef2a.
Following the improved blood flow recovery in GSO-Mef2a-treated mice, we observed increased arteriogenesis in the adductor muscle of these mice. Phenotypic switching of SMCs, from a contractile to a proliferative state, is an important process in arteriogenesis and other forms of vascular remodeling.22 Our results would imply that inhibition of Mef2a stimulates proliferation of SMCs because GSO-Mef2a-treated animals showed increased αSMA+ collateral areas compared to GSO-control-treated mice. These findings are in line with the study by Zhao et al.,19 who reported increased proliferation and migration of human SMCs and downregulation of SMC markers upon MEF2A siRNA treatment. Firulli et al.18 reported that the high levels of MEF2A expression in the neointima of rats upon balloon injury correlated with an activated SMC phenotype rather than a mere differentiated phenotype. In addition, we observed increased angiogenesis in the ischemic soleus muscle of GSO-Mef2a-treated animals. Collectively, our data suggest that Mef2a plays a role in SMC differentiation and proliferation as well as EC proliferation.
In conclusion, our study demonstrates a novel function for MEF2A in post-ischemic neovascularization and provides a link among MEF2A, 14q32 microRNAs miR-329 and miR-494, and arteriogenesis and angiogenesis. Furthermore, we show for the first time that Mef2a is not only a DNA-binding protein that regulates transcription, but also an RNA-binding protein that may regulate post-transcriptional processing of microRNAs. MEF2A acts as a novel switch in vascular remodeling and neovascularization, and MEF2A inhibition increases blood flow recovery and tissue perfusion. However, because the effects of direct 14q32 microRNA inhibition on post-ischemic neovascularization are much larger, MEF2A inhibition is unlikely to be a promising therapeutic strategy to increase neovascularization and blood flow toward the extremities in patients with PAD.
Materials and Methods
Mef2a Inhibitors
GSOs were designed with reverse complementarity to the murine Mef2a target mRNA sequence and synthesized at Idera Pharmaceuticals.23 As negative control, a scrambled sequence was used, designed not to target any known murine mRNA. GSOs were made up of two single-stranded DNA strands, which were 2′O-methylated. The two DNA strands were linked together at their 5′ ends by a phosphothioate linker in order to prevent Toll-like-receptor-mediated immune activation. Sequences of Mef2a GSOs are given in Figure 1.
Hind Limb Ischemia Model
This study was performed in accordance with Dutch government guidelines and the Directive 2010/63/EU of the European Parliament. All experiments were approved by the committee on animal welfare of the Leiden University Medical Center (approval reference number 12029). C57BL/6 male mice, aged 8 to 12 weeks (Harlan), were housed in groups of four or five mice, with free access to water and regular chow. For Mef2a-inhibition experiments, mice were given intraperitoneal (i.p.) injections of 1 mg (∼40 mg/kg) GSO in PBS or PBS alone at 10, 7, and 4 days directly before surgery and at 3, 7, and 10 days after surgery. Mice were anesthetized by i.p. injection of midazolam (8 mg/kg, Roche Diagnostics), medetomidine (0.4 mg/kg, Orion), and fentanyl (0.08 mg/kg, Janssen Pharmaceuticals). Unilateral hind limb ischemia was induced by electrocoagulation of the left femoral artery proximal to the superficial epigastric artery and proximal to the bifurcation of the popliteal and saphenous artery (double ligation model).24 After surgery, anesthesia was antagonized with flumazenil (0.7 mg/kg, Fresenius Kabi), atipamezole (3.3 mg/kg, Orion), and buprenorphine (0.2 mg/kg, MSD Animal Health).
Laser Doppler Perfusion Measurements
Blood flow recovery to the ligated hind limb was measured over time using laser Doppler perfusion imaging (LDPI) (Moor Instruments) before and directly after surgery and at 3, 7, 10, and 14 days after surgery. For LDPI measurements, mice were anesthetized by i.p. injection of midazolam (8 mg/kg, Roche Diagnostics) and medetomidine (0.4 mg/kg, Orion). Before each measurement, mice were placed in a double-glazed pot and perfused with water at 37°C for 5 min. After LDPI, anesthesia was antagonized by subcutaneous injection of flumazenil (0.7 mg/kg, Fresenius Kabi) and atipamezole (3.3 mg/kg, Orion). LDPI measurements in the ligated paw were normalized to measurements of the unligated paw as the internal control. After the last LDPI measurement at day 14, analgesic fentanyl (0.08 mg/kg, Janssen Pharmaceuticals) was administered subcutaneously and mice were sacrificed via cervical dislocation. The adductor, gastrocnemius, and soleus muscles were harvested and either snap-frozen or fixed in 4% paraformaldehyde (PFA).
Cell Culture
Primary Murine SMCs
Primary murine SMCs were isolated from mouse aortae of C57BL/6 mice. Aortae were cut into small pieces and embedded on gelatin-coated six-well plates. Culture medium was added to the wells with aortic fragments (DMEM [Invitrogen, GIBCO], 10% heat inactivated fetal bovine serum [PAA], and 1% penicillin/streptomycin). Outgrowth of SMCs occurred within 1 week, after which cells were passed using Trypsin-EDTA (Sigma) and transferred to fresh six-well plates. SMCs were characterized by positive α-SMA staining as described previously.10 Cells were cultured at 37°C in a humidified 5% CO2 environment.
3T3 Cells
3T3 cells were cultured at 37°C in a humidified 5% CO2 environment. Culture medium consisted of DMEM GlutaMAX (GIBCO) supplemented with 10% heat inactivated fetal bovine serum (PAA) and 1% penicillin/streptomycin (PAA). Culture medium was refreshed every 2 to 3 days. Cells were passed using trypsin-EDTA (Sigma) at 90% confluency.
In Vitro GSO-Mediated Inhibition of Mef2a Expression
For GSO experiments, murine SMCs were plated at 50,000 cells per well of a 12-well plate. After 24 hr, GSOs against Mef2a were added to the culture media at a concentration of 10 ng/μL for 48 hr. After 48 hr, cells were washed with PBS and TRIzol (Invitrogen) was added to the cells for RNA isolation.
qRT-PCR
mRNA qRT-PCR
Adductor and gastrocnemius muscles from 14 days after surgery were homogenized by grounding with a pestle and mortar in liquid nitrogen. Total RNA was isolated using a standard TRIzol-chloroform extraction protocol. RNA concentration and purity were determined by nanodrop (Nanodrop Technologies). RNA was reverse transcribed using high-capacity RNA to cDNA RT kits (Life Technologies). Relative quantitative mRNA PCR was performed on reverse-transcribed cDNA using Mef2a Taqman gene expression assays. For quantification of pri-miR and pre-miR levels of microRNAs, SYBR green dye (QIAGEN) was used, and primers were designed using Primer3. Sequences of primers are listed in Table S1. qPCRs were run on a 7900HT Fast Real-Time PCR system (Applied Biosystems), and amplification efficiencies were checked by standard curves. Data were normalized using a stably expressed endogenous control (HPRT for Mef2a quantification and snRNA-U6 for pri-miR and pre-miR quantification).
MicroRNA qRT-PCR
microRNA quantification was performed using Taqman microRNA assays (Applied Biosystems) according to the manufacturer’s protocol. Relative qPCR was performed on the Viia7 system (Applied Biosystems), and amplification efficiencies were checked by standard curves. Data were normalized using a stably expressed endogenous control (mmu-let-7c), as previously described.25
Immunohistochemistry
CD31
6-μm-thick fresh-frozen cross-sections of soleus muscle were fixed in ice-cold acetone and stained with anti-CD31 (BD PharMingen). Sections were counterstained with hematoxylin. Quantification of the CD31-positive area was performed on sections photographed randomly (six representative images per muscle per mouse, ten animals per group) using image analysis (ImageJ 1.48v, NHI), as described previously.26
α-SMA, CD31, and CD45 Triple Staining
5-μm-thick paraffin-embedded cross-sections of adductor muscle were re-hydrated, and antigen retrieval was performed using citrate buffer. SMCs were stained using primary antibodies against α-SMA (DAKO), ECs were stained with CD31 antibodies (BD PharMingen), and leukocytes were stained using CD45 antibodies (Abcam). Alexa Fluor 647, Alexa Fluor 488, and Alexa Fluor 594 antibodies (Life Technologies and Invitrogen [Alexa Fluor 594]) were used to visualize SMCs, ECs, and leukocytes, respectively. Finally, sections were mounted in Fluoroshield with DAPI (Sigma-Aldrich). The Pannoramic MIDI digital slide scanner (3DHistech) was used to create high-resolution images of the adductor muscles. Snapshots were taken using the Pannoramic viewer software (3DHistech) with 20× magnification. The amount and size of α-SMA positive collaterals was measured using ImageJ (ImageJ 1.48v, NHI).
MEF2A
5-μm-thick paraffin-embedded cross-sections of adductor muscle were re-hydrated, and endogenous peroxidase activity was blocked. Antigen retrieval was performed with Tris-EDTA (pH 9.0) at 100°C for 10 min. Adductor muscles were stained with anti-MEF2A antibodies (Abcam, ab86755) to visualize MEF2A expression and counterstained with hematoxylin.
Western Blot Analysis
Immunoblotting was performed to quantify MEF2A expression in vivo. Homogenized adductor muscle tissue of GSO-control- (n = 2) and GSO-Mef2a-treated animals (n = 2) were lysed on ice for 30 min with lysis buffer. Lysates were briefly centrifuged to remove debris. Equal amounts of protein (20 μg per sample) were loaded onto a PAGE minigel (Mini-PROTEAN TGX precast gel, Biorad) and run at 70 V for 10 min and subsequently at 120 V for 90 min. Fractionated tissue extracts were transferred to a nitrocellulose membrane using a transfer apparatus according to the manufacturer’s protocol (Bio-Rad). Incubation with primary rabbit anti-MEF2A antibody (Abcam, ab86755 1:100) was followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody goat anti-rabbit immunoglobulin G (IgG) (CST7074, Cell Signaling Technology). Signals were visualized using the SuperSignal ELISA Pico chemiluminescent substrate (ThermoScientific). Blots were stripped for 15 min using Restore Stripping Buffer (ThermoScientific) and incubated with anti-actin (C-11) (SC1615, Santa-Cruz) for normalization. The proportional expression of MEF2A per lane was normalized against the proportional expression of actin per lane (Biorad Image Lab Software 5.2.1).
Aortic Ring Assay
Mouse aortic ring assays were performed as described previously.27 In brief, the thoracic aorta was removed from 8- to 10-week-old mice and transferred to a 15-mL tube containing Opti-MEM (GIBCO).
Surrounding fat and branching vessels were carefully removed, and aortas were flushed with Opti-MEM (GIBCO). Collagen Type I (Millipore) was diluted to a final concentration of 1 mg/mL with DMEM (GIBCO), and pH was adjusted with 5 N NaOH. 96-well plates were coated with 75 μL of collagen matrix. After serum starvation overnight in Opti-MEM, aortic rings (0.5 to 1 mm) were transferred to the 96-well plate, and after 1 hr, 150 μL of Opti-MEM supplemented with 2.5% FBS (PAA), penicillin-streptomycin (PAA), 30 ng/mL vascular endothelial growth factor (VEGF) (Millipore), and GSOs (15 ng/μL) was added to each well. Microvessel outgrowth was quantified after 5 days by live phase-contrast microscopy (Axiovert 40C, Carl Zeiss). Starting from a specific point on the ring, each microvessel emerging from the ring was counted as a sprout and individual branches arising from each microvessel were counted as a separate sprout working around the ring clockwise.
RIP
RIP was performed using the EZMagna RIP kit (Millipore), according to the manufacturer’s instructions. 3T3 cells were grown to 90% confluency and lysed in complete RIP lysis buffer. Cell lysates were incubated with RIP buffer containing magnetic beads conjugated with antibodies against MEF2A (Abcam ab86755) and rabbit control IgG (Millipore PP64B). Before immunoprecipitation, 10% of cell lysate was taken and served as 10% input control. Next, samples were treated with proteinase K to digest protein, and RNA was isolated using a standard TRIzol-chloroform extraction protocol.
Statistical Analysis
Results are expressed as mean ± SEM. In vitro experiments were performed in triplicate and represent at least three independent experiments. Differences between groups were tested using Student’s t tests. p values of < 0.05 were considered statistically significant.
Author Contributions
Conceptualization, A.Y.N.; Methodology, M.R.d.V.; Formal Analysis, S.M.J.W.; Investigation, S.M.J.W., M.R.d.V., and E.A.B.P.; Resources, S.A.; Writing – Original Draft, S.M.J.W.; Writing – Review and Editing, P.H.A.Q. and A.Y.N.; Visualization, S.M.J.W.; Supervision, P.H.A.Q. and A.Y.N.; Project Administration, P.H.A.Q. and A.Y.N.; Funding Acquisition, P.H.A.Q. and A.Y.N.
Conflicts of Interest
S.A. is employed by Idera Pharmaceuticals.
Acknowledgments
This work was supported by the Netherlands Institute for Regenerative Medicine (NIRM) (FES0908), the Netherlands Organization for Scientific Research (NWO) (Veni-916.12.041), and the Dutch Heart Foundation (Dr. E. Dekker Senior Postdoc, 2014T102). We thank W. Razawy, W. Wong, T. Bezhaeva, and C.M. Janssen for their technical support. We thank R.A. Boon for sharing the protocol on RNA-binding protein immunoprecipitation.
Footnotes
Supplemental Information includes one figure and one table and can be found with this article online at http://dx.doi.org/10.1016/j.omtn.2017.03.003.
Supplemental Information
References
- 1.van Oostrom M.C., van Oostrom O., Quax P.H., Verhaar M.C., Hoefer I.E. Insights into mechanisms behind arteriogenesis: what does the future hold? J. Leukoc. Biol. 2008;84:1379–1391. doi: 10.1189/jlb.0508281. [DOI] [PubMed] [Google Scholar]
- 2.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 3.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 4.van Rooij E., Olson E.N. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat. Rev. Drug Discov. 2012;11:860–872. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ono K., Kuwabara Y., Han J. MicroRNAs and cardiovascular diseases. FEBS J. 2011;278:1619–1633. doi: 10.1111/j.1742-4658.2011.08090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thum T., Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ. Res. 2015;116:751–762. doi: 10.1161/CIRCRESAHA.116.303549. [DOI] [PubMed] [Google Scholar]
- 7.Boon R.A., Dimmeler S. MicroRNAs in myocardial infarction. Nat. Rev. Cardiol. 2015;12:135–142. doi: 10.1038/nrcardio.2014.207. [DOI] [PubMed] [Google Scholar]
- 8.Feinberg M.W., Moore K.J. MicroRNA regulation of atherosclerosis. Circ. Res. 2016;118:703–720. doi: 10.1161/CIRCRESAHA.115.306300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welten S.M., Goossens E.A., Quax P.H., Nossent A.Y. The multifactorial nature of microRNAs in vascular remodelling. Cardiovasc. Res. 2016;110:6–22. doi: 10.1093/cvr/cvw039. [DOI] [PubMed] [Google Scholar]
- 10.Welten S.M., Bastiaansen A.J., de Jong R.C., de Vries M.R., Peters E.A., Boonstra M.C., Sheikh S.P., La Monica N., Kandimalla E.R., Quax P.H. Inhibition of 14q32 microRNAs miR-329, miR-487b, miR-494, and miR-495 increases neovascularization and blood flow recovery after ischemia. Circ. Res. 2014;115:696–708. doi: 10.1161/CIRCRESAHA.114.304747. [DOI] [PubMed] [Google Scholar]
- 11.Seitz H., Royo H., Bortolin M.L., Lin S.P., Ferguson-Smith A.C., Cavaillé J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14:1741–1748. doi: 10.1101/gr.2743304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snyder C.M., Rice A.L., Estrella N.L., Held A., Kandarian S.C., Naya F.J. MEF2A regulates the Gtl2-Dio3 microRNA mega-cluster to modulate WNT signaling in skeletal muscle regeneration. Development. 2013;140:31–42. doi: 10.1242/dev.081851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark A.L., Naya F.J. MicroRNAs in the myocyte enhancer factor 2 (MEF2)-regulated Gtl2-Dio3 noncoding RNA locus promote cardiomyocyte proliferation by targeting the transcriptional coactivator cited2. J. Biol. Chem. 2015;290:23162–23172. doi: 10.1074/jbc.M115.672659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiore R., Khudayberdiev S., Christensen M., Siegel G., Flavell S.W., Kim T.K., Greenberg M.E., Schratt G. Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009;28:697–710. doi: 10.1038/emboj.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edmondson D.G., Lyons G.E., Martin J.F., Olson E.N. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994;120:1251–1263. doi: 10.1242/dev.120.5.1251. [DOI] [PubMed] [Google Scholar]
- 16.Potthoff M.J., Olson E.N. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi M., Kim S.W., Imanaka-Yoshida K., Yoshida T., Abel E.D., Eliceiri B., Yang Y., Ulevitch R.J., Lee J.D. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Firulli A.B., Miano J.M., Bi W., Johnson A.D., Casscells W., Olson E.N., Schwarz J.J. Myocyte enhancer binding factor-2 expression and activity in vascular smooth muscle cells. Association with the activated phenotype. Circ. Res. 1996;78:196–204. doi: 10.1161/01.res.78.2.196. [DOI] [PubMed] [Google Scholar]
- 19.Zhao W., Zhao S.P., Peng D.Q. The effects of myocyte enhancer factor 2A gene on the proliferation, migration and phenotype of vascular smooth muscle cells. Cell Biochem. Funct. 2012;30:108–113. doi: 10.1002/cbf.1823. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki E., Satonaka H., Nishimatsu H., Oba S., Takeda R., Omata M., Fujita T., Nagai R., Hirata Y. Myocyte enhancer factor 2 mediates vascular inflammation via the p38-dependent pathway. Circ. Res. 2004;95:42–49. doi: 10.1161/01.RES.0000134631.75684.4A. [DOI] [PubMed] [Google Scholar]
- 21.Zhao W., Zheng X.L., Peng D.Q., Zhao S.P. Myocyte enhancer factor 2A regulates hydrogen peroxide-induced senescence of vascular smooth muscle cells via microRNA-143. J. Cell. Physiol. 2015;230:2202–2211. doi: 10.1002/jcp.24948. [DOI] [PubMed] [Google Scholar]
- 22.Schirmer S.H., van Nooijen F.C., Piek J.J., van Royen N. Stimulation of collateral artery growth: travelling further down the road to clinical application. Heart. 2009;95:191–197. doi: 10.1136/hrt.2007.136119. [DOI] [PubMed] [Google Scholar]
- 23.Bhagat L., Putta M.R., Wang D., Yu D., Lan T., Jiang W., Sun Z., Wang H., Tang J.X., La Monica N. Novel oligonucleotides containing two 3′-ends complementary to target mRNA show optimal gene-silencing activity. J. Med. Chem. 2011;54:3027–3036. doi: 10.1021/jm200113t. [DOI] [PubMed] [Google Scholar]
- 24.Hellingman A.A., Bastiaansen A.J., de Vries M.R., Seghers L., Lijkwan M.A., Löwik C.W., Hamming J.F., Quax P.H. Variations in surgical procedures for hind limb ischaemia mouse models result in differences in collateral formation. Eur. J. Vasc. Endovasc. Surg. 2010;40:796–803. doi: 10.1016/j.ejvs.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 25.Nossent A.Y., Eskildsen T.V., Andersen L.B., Bie P., Brønnum H., Schneider M., Andersen D.C., Welten S.M., Jeppesen P.L., Hamming J.F. The 14q32 microRNA-487b targets the antiapoptotic insulin receptor substrate 1 in hypertension-induced remodeling of the aorta. Ann. Surg. 2013;258:743–751. doi: 10.1097/SLA.0b013e3182a6aac0. discussion 752–753. [DOI] [PubMed] [Google Scholar]
- 26.Bastiaansen A.J., Karper J.C., Wezel A., de Boer H.C., Welten S.M., de Jong R.C., Peters E.A., de Vries M.R., van Oeveren-Rietdijk A.M., van Zonneveld A.J. TLR4 accessory molecule RP105 (CD180) regulates monocyte-driven arteriogenesis in a murine hind limb ischemia model. PLoS ONE. 2014;9:e99882. doi: 10.1371/journal.pone.0099882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker M., Robinson S.D., Lechertier T., Barber P.R., Tavora B., D’Amico G., Jones D.T., Vojnovic B., Hodivala-Dilke K. Use of the mouse aortic ring assay to study angiogenesis. Nat. Protoc. 2011;7:89–104. doi: 10.1038/nprot.2011.435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.