

Abstract
Monocarboxylate transporter 8 (MCT8) is a thyroid hormone (TH)-specific transporter. Mutations in the MCT8 gene are associated with Allan-Herndon-Dudley Syndrome (AHDS), consisting of severe psychomotor retardation and disturbed TH parameters. To study the functional consequences of different MCT8 mutations in detail, we combined functional analysis in different cell types with live-cell imaging of the cellular distribution of seven mutations that we identified in patients with AHDS. We used two cell models to study the mutations in vitro: 1) transiently transfected COS1 and JEG3 cells, and 2) stably transfected Flp-in 293 cells expressing a MCT8-cyan fluorescent protein construct. All seven mutants were expressed at the protein level and showed a defect in T3 and T4 transport in uptake and metabolism studies. Three mutants (G282C, P537L, and G558D) had residual uptake activity in Flp-in 293 and COS1 cells, but not in JEG3 cells. Four mutants (G221R, P321L, D453V, P537L) were expressed at the plasma membrane. The mobility in the plasma membrane of P537L was similar to WT, but the mobility of P321L was altered. The other mutants studied (insV236, G282C, G558D) were predominantly localized in the endoplasmic reticulum. In essence, loss of function by MCT8 mutations can be divided in two groups: mutations that result in partial or complete loss of transport activity (G221R, P321L, D453V, P537L) and mutations that mainly disturb protein expression and trafficking (insV236, G282C, G558D). The cell type-dependent results suggest that MCT8 mutations in AHDS patients may have tissue-specific effects on TH transport probably caused by tissue-specific expression of yet unknown MCT8-interacting proteins.
In 2003, monocarboxylate transporter 8 (MCT8) was identified as a specific thyroid hormone (TH) transporter (1). It is expressed in tissues throughout the body, including the brain (2), which is critically dependent on TH during the different stages of its development (3). Shortly after the identification of MCT8, mutations in the MCT8 gene were linked to Allan-Herndon-Dudley Syndrome (AHDS) (4–6), in which severe psychomotor retardation is combined with high serum T3, low T4, and slightly increased TSH levels. Even though all patients have high serum T3 levels, psychomotor development varies between the mutations identified, ranging from a total absence of motor skills to the ability to walk independently (2, 6–9).
Multiple studies have been published regarding the pathogenesis of AHDS caused by MCT8 mutations using Mct8 knockout (KO) mice and in vitro cell systems. Although Mct8 KO mice have similarly disturbed TH serum levels as AHDS patients, they do not have an obvious neurologic phenotype (10–12). This may be explained by a lack of compensatory transport via alternative transporters in humans (12). Although Mct8 KO mice are useful for studying the disturbed TH serum levels in patients with AHDS, the cellular pathogenic mechanism cannot be explored. This has been studied using different in vitro cell models. These studies have shown a genotype-phenotype relationship for some mutations in MCT8, as well as a relationship between plasma membrane (PM) localization and transport functionality (9, 13).
However, although most mutations identified in MCT8 patients have been functionally tested in vitro by uptake assays, it is not clear what the cellular distribution of wild-type (WT) MCT8 and mutants is. Furthermore, it is unclear whether different mutations may have different effects on transport characteristics in different tissues. Therefore, the objective of our study was to clarify the cellular pathogenic mechanisms of mutations in MCT8 by studying the effects of seven mutations identified in AHDS patients on the subcellular localization and transport activity of MCT8 in three different cell types.
Our results suggest that pathogenic mechanisms differ between different MCT8 mutations and different cell types and improve our understanding of the pathogenic mechanism of MCT8 mutations.
Materials and Methods
Materials
Oligonucleotides were obtained from Integrated DNA Technologies (Leuven, Belgium); pECFP-N1 vector from Clontech (Breda, The Netherlands); SYBR Green Eurogentec (Maastricht, The Netherlands); pcDNA5/FRT vector, pOG44 vector, Flp-in 293 cells (human embryonic kidney cells), culture medium, fetal bovine serum, and antibiotics from Invitrogen (Bleiswijk, The Netherlands); QuikChange XL-II Site-Directed Mutagenesis kits from Stratagene (Amstelveen, The Netherlands); transfection reagent X-tremeGENE 9 from Roche (Almere, The Netherlands); culture dishes from Corning (Schiphol, The Netherlands); sodium dodecyl sulfate (SDS) gels from Thermo Fisher (Breda, The Netherlands); nitrocellulose membrane from GE Healthcare (Zeist, The Netherlands); iodothyronines, BSA, d-glucose, and Na2SeO3 from Sigma Aldrich (Zwijndrecht, The Netherlands); Na125I from IDB Holland BV (Baarle-Nassau, The Netherlands); and JEG3 cells from the European Collection of Cell Cultures (Salisbury, United Kingdom). [125I]Iodothyronines were prepared in our laboratory as described previously (14).
Plasmids
The cloning of pcDNA3-hMCT8 and pCIneo-hD3 has been described previously (15). The hMCT8-cyan fluorescent protein (CFP) construct was constructed similar to hMCT8-yellow fluorescent protein described previously (16). The MCT8-CFP chimeric insert was cloned into the pcDNA5/FRT vector using HindIII and NotI restriction sites. The seven mutations identified in AHDS patients (Table 1), were introduced into WT MCT8-pcDNA3 and in WT MCT8-CFP-pcDNA5/FRT by site-directed mutagenesis following the QuikChange protocol. All mutations were confirmed by sequencing.
Table 1.
Primers Used for Introduction of the MCT8 Mutations
| Mutation | Location | Reference | Primers | Tm (°C) | |
|---|---|---|---|---|---|
| G221R | TMD 2 | 29 | Forward | 5′-CAAGCAGCATGGGTCAGAGCCCTCGCGATG-3′ | 81.8 |
| Reverse | 5′-CATCGCGAGGGCTCTGACCCATGCTGCTTG-3′ | ||||
| InsV236 | TMD 2 | 30 | Forward | 5′-CTTCTGTTCTCCCATTGTGGTGAGTATATTCACTGACCG-3′ | 80.8 |
| Reverse | 5′-CGGTCAGTGAATATACTCACCACAATGGGAGAACAGAAG-3′ | ||||
| G282C | TMD 4 | 31 | Forward | 5′-GATTCTCTTTGGTTGTTGCTGTTCCTTCGCCTTTC-3′ | 78 |
| Reverse | 5′-GAAAGGCGAAGGAACAGCAACAACCAAAGAGAATC-3′ | ||||
| P321L | TMD 5 | 29 | Forward | 5′-CATTTTCTCCATGTCCTTCCTCTTCCTCATCAGAATGCTG-3′ | 80.1 |
| Reverse | 5′-CAGCATTCTGATGAGGAAGAGGAAGGACATGGAGAAAATG-3′ | ||||
| D453V | TMD8/ICL 4 | 33 | Forward | 5′-CAGGCCACATCAGTGTCTCCATCCCTGGAC-3′ | 80.6 |
| Reverse | 5′-GTCCAGGGATGGAGACACTGATGTGGCCTG-3′ | ||||
| P537L | TMD 11 | 32 | Forward | 5′-CCAATGATTGCTGGGCTCCCCATTGCAGGCCTAC-3′ | 82.8 |
| Reverse | 5′-GTAGGCCTGCAATGGGGAGCCCAGCAATCATTGG-3′ | ||||
| G558D | TMD 12 | 7 | Forward | 5′-GCCTTCTACTTTGCCGATGTGCCCCCCATCATCG-3′ | 82.8 |
| Reverse | 5′-CGATGATGGGGGGCACATCGGCAAAGTAGAAGGC-3′ | ||||
Tm, melting temperature; TMD, transmembrane domain; ICL, intracellular loop.
Cell culture
Transient transfection assays
JEG3 and COS1 cells were cultured in culturing medium (DMEM/F12+glutamax containing 9% fetal bovine serum, 1% penicillin/streptomycin, and 100 nM Na2SeO3). Cells were plated in six-well plates for the uptake experiments and in 24-well plates for metabolism studies. In both assays, cells were maintained in the DMEM described, but without penicillin/streptomycin.
For uptake studies, cells were transfected in duplicate with 250 ng empty pcDNA3 as a negative control, WT pcDNA3-MCT8 as a positive control, or mutant pcDNA3-MCT8 together with 250 ng pSG5-CRYM. As described previously (17), μ-crystallin (CRYM) is a cytoplasmic TH-binding protein that strongly reduces TH efflux. All transfections were carried out using X-tremeGENE 9 following the manufacturer's protocol.
For metabolism studies, cells were cotransfected with 100 ng pCIneo-D3 and 100 ng pcDNA3 or WT or mutant pcDNA3-MCT8.
All experiments using COS1 and JEG3 cells were carried out after 48 hours transfection time.
Generation of MCT8-CFP stably expressing Flp-in 293 cells
We transfected Flp-in 293 cells with the Flp-recombinase-containing vector pOG44 and pcDNA5/FRT-MCT8-CFP in a 9:1 ratio using X-tremeGENE9 following the Invitrogen Flp-in 293 protocol. Selection of MCT8-CFP-expressing cells was carried out by addition of 100 μg/mL hygromicin B. Subsequently we chose six colonies per construct. MCT8-CFP expression levels of the colonies were determined by SYBR Green quantitative PCR. These MCT8 expression levels were used to select colonies with equal expression levels of the different constructs. The cells were maintained under 100 μg/mL hygromycin B.
TH uptake experiments
TH uptake experiments were carried out in 48 hour-transfected COS1 and JEG3 cells and confluent, stably transfected Flp-in 293 cells plated in six-well plates. First, cells were washed with the assay buffer (d-PBS + Ca2+/Mg2+ + 0.1% BSA + 0.1% d-glucose). Subsequently, cells were incubated for 10 minutes at 37°C with 1 nM (105 cpm) [125I]T3 or [125I]T4 in 1.5 mL assay buffer. After incubation, cells were washed with the assay buffer and lysed with 0.1 M NaOH. The radioactive cell lysates were counted and protein levels were determined using the Bradford protein assay.
TH metabolism
The cells were washed with assay buffer and subsequently incubated for 4 hours at 37°C with 1 nM (5 × 105 cpm) [125I]T3 or [125I]T4 in 400 μL assay buffer. After incubation, 100 μL medium was added to 100 μL ice-cold ethanol and incubated for 30 minutes on ice. After centrifugation, 100 μL supernatant was mixed with 100 μL 0.02 M ammonium acetate (pH 4.0). This mixture was analyzed by HPLC as described previously (15).
Western blotting
Transiently transfected (48 h) COS1 and JEG3 cells and stably transfected Flp-in 293 cells in six-well plates were washed with d-PBS, harvested in 200 μL 100 mM phosphate buffer (pH 7.2) containing 2 mM EDTA, and sonicated on ice. Cell lysate proteins (20–50 μg) were separated on 8% or 10% precise Tris-HEPES SDS-GEL (Fisher Scientific, Landsmeer, The Netherlands), and transferred to a nitrocellulose membrane (GE Healthcare, Diegem, Belgium). The membrane was blocked for 2 hours at 37°C in d-PBS/0.1% Tween/5% milk, washed four times for 10 minutes in d-PBS/0.1% Tween, and incubated overnight at 4°C in d-PBS/0.1% Tween/1% BSA with 1:10 000 N-terminal rabbit anti-SLC16A2 antibody (Sigma) and 1:20 000 mouse anti-hGAPDH antibody MAB374 (Chemicon International, Amsterdam, The Netherlands). The next day, the membranes were rinsed four for 10 minutes with d-PBS/0.1%Tween, and incubated for 1 hour at room temperature in d-PBS/0.1% Tween/1% BSA with 1:20 000 IRDye 800 CW goat antirabbit IgG and 1:20,000 IRDye 680 LT antimouse IgG (LI-COR, Westburg, Leusden, The Netherlands). Finally, the blots were rinsed four times for 10 minutes with d-PBS/0.1% Tween and additionally for 10 minutes in d-PBS alone, before scanning by the Odyssey (LI-COR). Analyses were carried out using the Odyssey 3.0 software.
Live-cell confocal microscopy
To study the cellular distribution of MCT8-CFP proteins, cells were plated on poly-d-lysine (Sigma)-coated 24-mm cover slips in six-well plates. COS1 and JEG3 cells were transfected as described above. Intact cells were stained with Hoechst 33342 (Invitrogen) for nuclear staining and deep red CellMASK (Invitrogen) for PM staining. To determine the intracellular localization, cells were incubated for 24 hours with CellLight ER-RFP *BacMAm 2.0 (Invitrogen). After staining, intact cells were imaged at 37°C using a Zeiss LSM510meta confocal laser scanning microscope and a 40 × 1.3 Plan-Neofluar oil immersion objective. The 405-nm laser line was used together with a BP 420–480 emission filter for Hoechst, the 458-nm laser line was used together with the LP 475 emission filter for CFP, the 543 laser line was used together with the BP 560–615 emission filter for CellLight ER-RFP, and the 633 laser line was used together with the LP 650 emission for CellMASK. Images were deconvolved and corrected for chromatic shifts using Huygens professional software V4.1 (Scientific Volume Imaging, Hilversum, The Netherlands).
Fluorescence recovery after photobleaching (FRAP) experiments were carried out to study the mobility of WT MCT8 and mutants in the PM using the FRAP wizard of a Leica SP5 confocal microscope with LAS AF software. A small PM region of interest (ROI) was photo-bleached at maximal power for five iterations using the FRAP booster (excitation, 458 nm; and emission spectrum, 470–550 nm). All experiments were carried out at 37°C in culturing medium, using a 63 × 1.4 Plan-Apochromat objective. As a control for reversible photobleaching, FRAP experiments were carried out on fixed cells. The cells were covered with 4% paraformaldehyde in d-PBS without magnesium and calcium for 10 minutes at 37°C, rinsed three times for 5 minutes with d-PBS + Mg2+/Ca2+, and cover slips were mounted on glass slides using Vectashield H-1200 (Brunschwig, Amsterdam, The Netherlands).
FRAP data analysis was done using LAS AF software. An ROI was drawn over the bleached area of the PM, and the average intensity over time was plotted. The traces were normalized to the fluorescence intensity before bleaching. A background correction was done by subtracting the average fluorescence outside the cell from the recovery curves. To optimize visualization of the images in Figure 7, the contrast was enhanced and a Gaussian blur was applied (18).
Figure 7.

Live Cell Qualitative FRAP Recovery. A, Images of stably transfected WT-CFP (upper panel), mutant P321L (middle panel), and mutant P537L (lower panel). Images were captured before bleaching (Prebleach), immediately after bleaching (Postbleach), and at increasing points in time to 300 seconds after bleaching. The scale bar corresponds to 10 μm. B, Qualitatively normalized recovery curves of the measured fluorescence of the CFP constructs in panel A that moved into the bleached ROI. Each recovery curve represents mean ± SEM of cells (n = 6) produced from the raw data. A Gaussian blur was applied to the images, and contrast was enhanced to optimize visualization.
Statistical analysis
Results are presented as means and SEM of at least three experiments, each performed in duplicate. Statistical analysis was carried out in GraphPad Prism version 5.01 for Windows (GraphPad Software, San Diego, California). Statistical significance of the differences in TH transport between cells transfected with MCT8 mutants or empty vector was tested using one-way ANOVA followed by Bonferroni post test with a cut-off level of P < .05.
Results
Uptake of T3 and T4 by MCT8 in COS1, JEG3 and Flp-in 293 cells
Figure 1 shows the 10-minute uptake of [125I]T3 by COS1 (panel A) and JEG3 (panel B) cells that had been cotransfected for 48 hours with pSG5-CRYM and WT or mutant pcDNA3-hMCT8. Transfection with WT MCT8 increased [125I]T3 uptake 5.4-fold in COS1 cells, and 4.4-fold in JEG3 cells compared with cells transfected with empty vector. In COS1 cells, [125I]T3 uptake was significantly but modestly increased by mutants G221R (1.2-fold), insV236 (1.2-fold) and P321L (1.3-fold). Slightly higher [125I]T3 uptake was facilitated by mutants G282C (1.7-fold) and P537L (1.5-fold). The highest T3 uptake was mediated by mutant G558D (2.3-fold). COS1 cells transfected with D453V did not show increased uptake. [125I]T3 uptake by JEG3 cells transfected with the different MCT8 mutants did not significantly differ from background. Similar results were obtained in 125I-T4 uptake experiments with transfected COS1 and JEG3 cells (Figure 1, C and D).
Figure 1.

Uptake of [125I]T3 (A and B) and [125I]T4 (C and D) after 10-minute Incubation in COS1 (A and C) or JEG3 (B and D) Cells Cotransfected with pSG5-CRYM and WT or Mutant pcDNA3-hMCT8. Uptake is shown as percentage of added [125I]T3 or [125I]T4 corrected for protein concentrations. Results are presented as mean ± SEM (n = 3). Significance represents empty vector versus mutant hMCT8. *P < .05; **P < .01; ***P < .001.
To study the effects of the mutations on the cellular localization of MCT8, we generated MCT8-CFP constructs that were tested for [125I]T3 and [125I]T4 transport in COS1 cells (Figure 2, A and D) and JEG3 (Figure 2, B and E) transiently cotransfected with CRYM. The results were compared with the [125I]T3 and [125I]T4 uptake described above for MCT8 constructs without CFP tag in COS1 cells (Figure 1). In general, the MCT8-CFP constructs were somewhat less active in transporting [125I]T3 and [125I]T4 than the constructs without CFP tag. Although the fold uptake was lower, the pattern of the activity of WT-MCT8 and mutants was not affected by the CFP tag.
Figure 2.

Cellular Uptake of [125I]T3 (A–C) and [125I]T4 (D–F) after 10-Minute Incubation in Transiently Cotransfected COS1 Cells (A and D) and JEG3 Cells (B and E) with pSG5-CRYM and WT or Mutant pcDNA5/FRT-hMCT8-CFP, and Cellular Uptake in Stably Transfected Flp-in 293 Cells (C and F). The uptake is shown as percentage of added T3 or T4 and corrected for protein. Results are presented as mean ± SEM (n = 3). Significance represents empty vector vs mutant hMCT8. *P < .05; **P < .01; ***P < .001.
To study the function and cellular distribution of MCT8 mutants in cells with uniform, moderate expression levels, stably transfected MCT8-CFP Flp-in 293 cell lines were generated. Because the MCT8 expression in these cells is much lower than the large overexpression in transiently transfected JEG3 and COS1 cells, stable transfected Flp-in 293 cells appear to represent a better model for the study of the pathogenic mechanisms of MCT8 mutations. These cell lines were also functionally tested for uptake of T3 (Figure 2C) and T4 (Figure 2F) during 10-minute incubations using cells stably expressing CFP alone as control. Stable WT MCT8 expressing Flp-in 293 cells showed a 2.0-fold higher T3 uptake than control cells. This smaller MCT8-induced fold increase in T3 uptake compared with transiently transfected COS1 and JEG3 cells is largely explained by the 2–4 fold higher endogenous T3 uptake of nontransfected Flp-in 293 cells. T3 uptake was increased significantly but very modestly by the mutants insV236-CFP (1.1-fold), G282C-CFP (1.3-fold), and P321L-CFP (1.3-fold). A slightly greater increase in T3 uptake was mediated by P537L-CFP (1.6-fold) and by G558D-CFP (1.4-fold).
T4 uptake experiments in these Flp-in cell lines also showed residual uptake by the same constructs, but the fold increase was higher for T4 than for T3 uptake. T4 uptake was increased 2.9-fold by WT MCT8-CFP, 1.3-fold by G282C-CFP, 1.5-fold by P321L-CFP, 1.8-fold by P537L-CFP, and G558D-CFP by 1.6-fold.
Attempts to produce stably MCT8-CFP expressing Flp-in 293 cell lines were repeatedly unsuccessful for the G221R and D453V mutants.
T3 and T4 metabolism in MCT8 plus D3-transfected COS1 and JEG3 cells
Because the uptake assay reflects the initial uptake of TH across the PM by MCT8, metabolism assays reflect the steady state of the transporter. Therefore, the metabolism of [125I]T3 and [125I]T4 was studied in cells cotransfected with MCT8 and D3, because deiodination of TH takes place intracellularly, to determine effective TH transport across the PM by mutant MCT8 in the steady state. Figure 3 presents the metabolism of [125I]T3 during 4-hour incubation with transfected COS1 (panel A) and JEG3 (panel B) cells cotransfected with D3 and WT or mutant MCT8. Cotransfection with WT MCT8 increased [125I]T3 metabolism 2.5-fold over COS1 cells transfected with D3 alone. No [125I]T3 metabolism was detected in cells cotransfected with the G221R or P321L. [125I]T3 metabolism was increased modestly by insV236 (1.5-fold) and D453V (1.4-fold), and slightly more by G282C (1.8-fold), P537L (1.8-fold), and G558D (2.1-fold). In JEG3 cells, cotransfection with WT MCT8 resulted in a 2.5-fold greater [125I]T3 metabolism during 4 hours than in cells transfected with D3 alone. T3 metabolism in JEG3 cells cotransfected with the different MCT8 mutants did not significantly differ from that in cells transfected with D3 alone, except for mutant D453V (1.3-fold increase). Similar results were obtained after 4 hours incubation with [125I]T4 in COS1 (Figure 3C) and in JEG3 (Figure 3D) cells.
Figure 3.

Metabolism of [125I]T3 (A and B) and [125I]T4 (C and D) after 4-Hour Incubation in COS1 (A and C) or JEG3 (B and D) Cells Cotransfected with pCIneo-hD3 and WT or Mutant pcNDA3-hMCT8. Metabolism is shown as percentage T1, T1S, T2, and rT3 in the incubation medium. Results are presented as mean ± SEM (n = 3). Significance represents hD3 without a transporter vs hD3 cotransfected with mutant hMCT8. *P < .05; **P < .01; ***P < .001
Western blotting
Figure 4 shows the immunoblotting of transfected cell lysates from COS1 (panels A and B), JEG3 (panels C and D), and Flp-in 293 (panel E) cells. The housekeeping protein GAPDH was used as a control for loading. Transfection with WT hMCT8 resulted in two bands: a monomer band of 60 000 and a weaker homodimer band of 120 000. Cells transfected with WT hMCT8-CFP also expressed a monomer and a weaker homodimer band. The WT and mutant MCT8-CFP proteins showed considerably weaker bands than the MCT8 proteins without CFP tag. The lanes loaded with lysates of cells transfected with empty vectors showed no MCT8-specific bands in all three cell lines.
Figure 4.
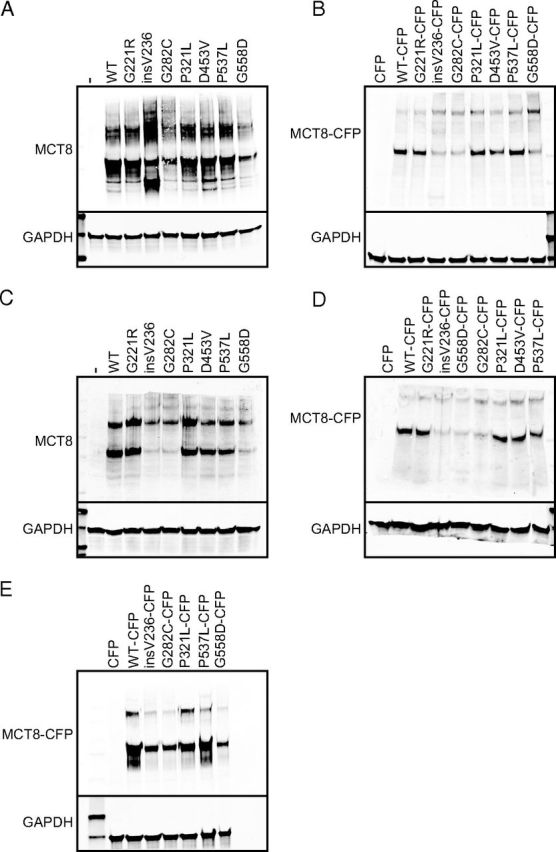
Western Blots of Whole-Cell Lysates of Transiently Transfected COS1 (A and B) and JEG3 (C and D) Cells and Stably Transfected Flp-in 293 Cells (E). The cells were transfected with WT or mutant hMCT8 without and with CFP tag. The MCT8 constructs with and without CFP tag show a specific monomer band and a homodimer band. The blots show a specific MCT8 monomer band of 60 000 (ie, without CFP) and approximately 87 000 (ie, with CFP) and a homodimer band of 120 000 (ie, without CFP) and 174 000 (ie, with CFP). hGAPDH was used as a housekeeping protein. A and B, Lanes in 10% SDS-gels (A) and in 8% gels (B) were loaded with 25 μg protein of whole-cell lysates of transiently transfected COS1 cells. C and D, Lanes in 10% (C) and 8% (D) SDS-gel were loaded with 25 μg protein of transfected JEG3 cells with WT or mutant MCT8 with or without CFP tag. E, 50 μg protein of stably transfected Flp-in 293 was loaded onto 10% SDS-gel per lane. Immunoblots were carried out at least two times per cell line.
Expression levels of the mutants G221R, P321L, D453V, and P537L were almost identical to those for WT in COS1 cells, but mutants insV236, G282C, and G558D were expressed at slightly lower protein levels (Figure 4A). Similar relative findings were found for proteins with CFP tag (Figure 4B). In addition to the monomer and dimer bands, insV236 without CFP also shows two smaller MCT8-specific bands probably representing proteolytic fragments. In JEG3 cells (Figure 4, C and D), the G221R and P321L proteins were expressed at the same level as WT MCT8. Unlike the findings obtained in COS1 cells, the D453V and P537L were less expressed than WT at both monomer and homodimer level. Mutants insV236, G282C, and G558D showed faint bands, especially at the monomer level. A similar protein expression was seen for the proteins with CFP tag in JEG3 cells (Figure 4D). The stably transfected Flp-in 293 cells (Figure 4E) had equally intense bands for WT MCT8, and the P321L and P537L mutants, and faint bands for insV236, G282C, and G558D, also indicating a lower expression of the last three mutants at the protein level in Flp-in 293 cells. Thus, all MCT8 mutants without and with CFP tag are expressed at the protein level, although they show considerable variability between different cell lines.
Live-cell confocal microscopy
To explain the loss of function of the MCT8 mutants, we studied the cellular distribution of CFP-tagged WT and mutant MCT8 in intact cells by confocal microscopy. Figure 5 shows the cellular distribution of the stably expressed MCT8-CFP proteins in Flp-in293 cells (panel A) and the transiently expressed fusion proteins in JEG3 cells (panel B). A clear colocalization of WT MCT8-CFP with the PM marker was seen, indicating the expression of CFP-tagged WT MCT8 at the PM and confirming the robustness of our model. The CFP-tagged G221R, P321L, D453V, and P537L mutants showed partial colocalization with the PM marker, suggesting substantial expression of these mutants at the PM. In contrast, the mutant insV236, G282C, and G558D fusion proteins showed minimal if any colocalization with the PM marker, indicating a defect in the trafficking of these mutants to the PM.
Figure 5.
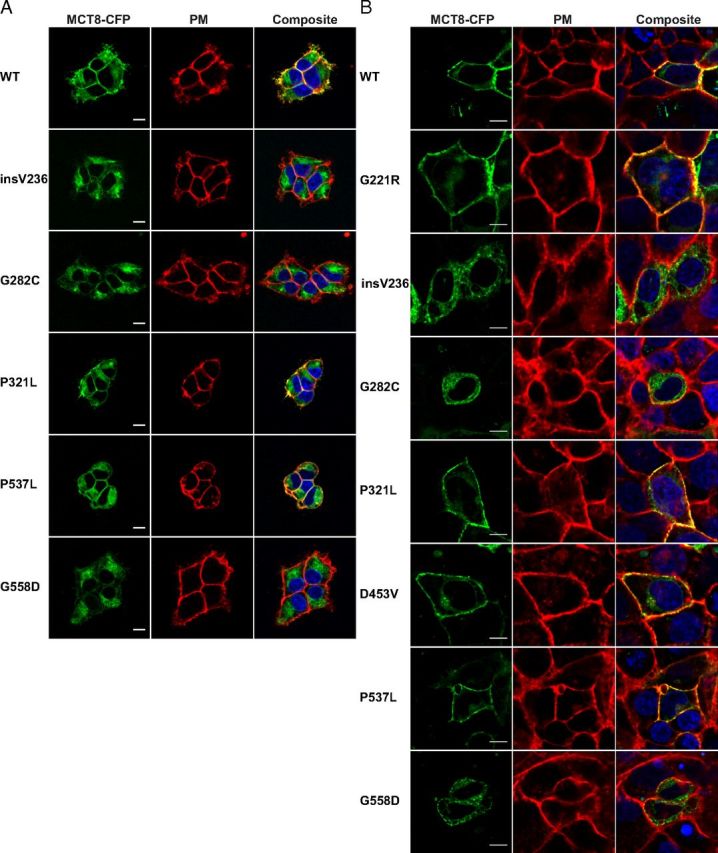
Live Cell Confocal Imaging of Stably Transfected Flp-in 293 Cells (A) and Transiently Transfected JEG3 Cells (B). The cellular distribution of MCT8-CFP proteins is shown in green, nuclear staining with Hoechst in blue, and PM staining with CellMASK in red. The yellow signal in the composite image reflects colocalization of the PM marker and MCT8-CFP, indicating that MCT8-CFP is expressed at the PM. Partial colocalization was seen for WT MCT8, G221R, P321L, D453V, and P537L. Minimal if any colocalization was observed for insV236, G282C, and G558D. Images were deconvolved and corrected for chromatic shift using Huygens. Contrast was enhanced to optimize visualization. Scale bar, 10 μm.
Next we studied whether the mutant proteins, which do not substantially reach the PM, were retained in the endoplasmic reticulum (ER). Indeed, CFP-tagged WT MCT8 and mutants colocalized with the ER marker but not strictly (Figure 6).
Figure 6.
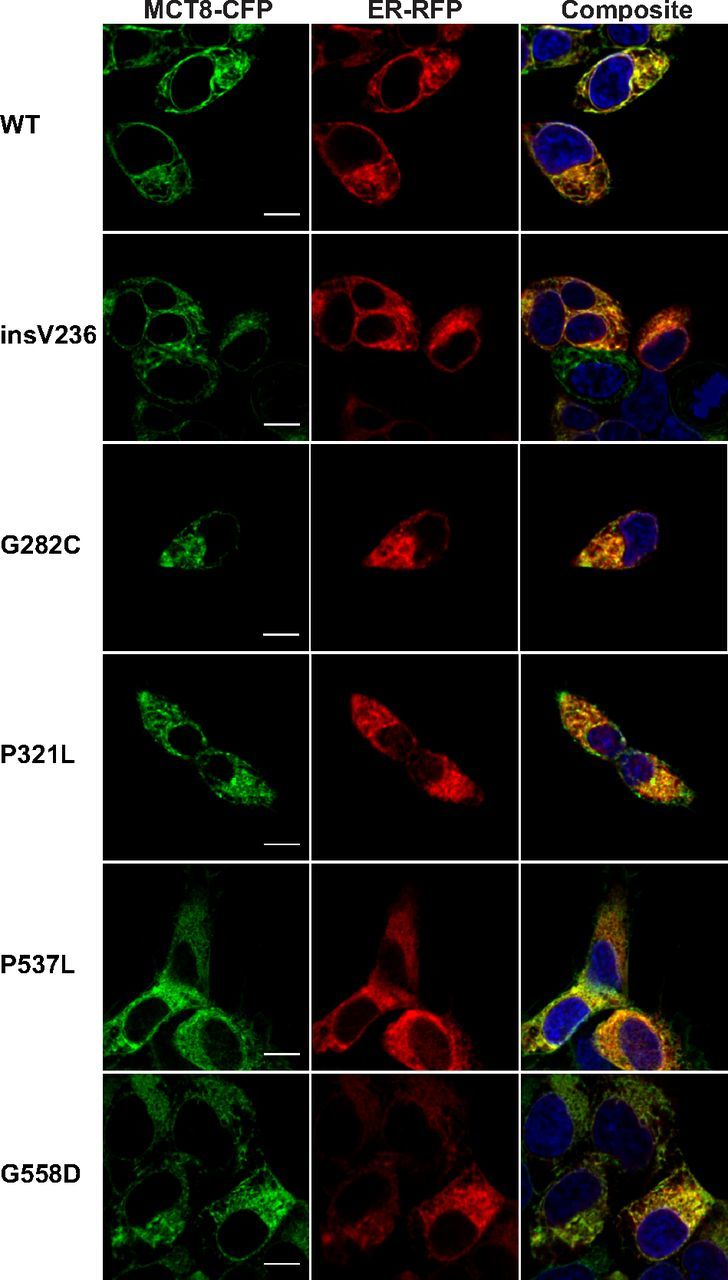
Live Cell Confocal Images of MCT8-CFP Stably Transfected Flp-in 293 Cells. The cellular distribution of MCT8-CFP proteins is shown in green, nuclear staining with Hoechst in blue, and ER staining with CellLight ER-RFP in red. The yellow signal in the composite image reflects colocalization of MCT8-CFP and the ER marker. CFP-tagged WT MCT8 and mutants colocalized with the ER marker but not strictly. Images were deconvolved and corrected for chromatic shift using Huygens. Contrast was enhanced to optimize visualization. Scale bar, 10 μm.
FRAP
Photobleaching experiments of the MCT8-CFP chimeras in the stable cell lines were carried out to investigate the mobility of WT MCT8-CFP and the two mutants that are present at the PM. A small ROI of the PM was bleached and the fluorescent recovery in this region was measured during 5 minutes. Figure 7A shows representative sequential images of diffusion of unbleached WT MCT8-CFP (upper panel), P321L-CFP (middle panel), and P537L-CFP (lower panel) into the bleached region. Ten seconds after photobleaching, the PM is visible as a thin fluorescent line in all three constructs. To exclude reversible photobleaching, FRAP experiments were carried out in fixed cells, which showed no recovery in the ROI (data not shown).
The degree of recovery after photobleaching reflects the mobility of the protein. A decrease in recovery indicates that a larger fraction of the protein is immobilized. The normalized recovery curves (Figure 7B) show initially a similar recovery pattern. However, at 20 seconds after bleaching the curve for P321L-CFP deflects, with a final recovery of approximately 60%, whereas the recovery of WT MCT8-CFP and P537L-CFP amounts to about 80%. These findings indicate that the P321L mutation decreases the mobility of CFP-tagged MCT8.
Discussion
This in vitro study demonstrates that pathogenic mechanisms differ between the seven MCT8 mutations (Table 2). Three of the mutants tested were localized mainly in the ER (insV236, G282C, G558D); four others reached the PM but showed partial (P537L) or complete (G221R, P321L, D453V) loss of transport function. We additionally showed that protein mobility of the P321L mutant, which did reach the PM, was altered. Interestingly, the consequences of different mutations were dependent on the cellular context (ie, cell type).
Table 2.
In-Vitro Functional Characteristics of MCT8 Mutations
| Mutation | Functional analysis |
Subcellular localization |
Main cellular pathogenic mechanism | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein expression |
Uptake |
Metabolism |
PM |
ER |
||||||||
| COS1 | JEG3 | Flp-in 293 | COS1 | JEG3 | Flp-in 293 | COS1 | JEG3 | Flp-in 293 | JEG3 | Flp-in 293 | ||
| G221R | n | n | n.d. | − | − | n.d. | − | − | n.d. | + | n.d. | Loss of transport activity |
| insV236 | low | low | low | − | − | − | + | − | − | − | + | Protein degradation and trafficking defect plus partial loss of transport activity |
| G282C | low | low | low | + | − | − | + | − | − | − | + | Decreased protein synthesis and/or increased protein degradation |
| P321L | n | n | n | − | − | + | − | − | + | + | + | Loss of transport activity |
| D453V | n | n | n.d. | − | − | n.d. | + | − | n.d. | + | n.d. | Loss of transport activity |
| P537L | n | n | n | + | − | + | + | − | + | + | + | Loss of transport activity |
| G558D | low | low | low | + | − | + | + | − | − | − | + | Decreased protein synthesis and/or increased protein degradation |
Functional analysis: n, normal, similar to WT; low, lower expression than WT; −, no significant difference compared with empty vector for T3 uptake; +, significant T3 uptake compared with empty vector; n.d., not done; −, no significant difference compared to D3 alone for T3 metabolism; +, significant T3 metabolism compared to D3 alone.
Subcellular localization: −, minimal if any colocalization; +, colocalization.
As suggested previously by our group (9), loss-of-function mutations of MCT8 could involve several mechanisms: impaired synthesis, disturbed protein folding, defective trafficking, rapid protein degradation, or loss of TH binding and transport. Impaired synthesis is not a likely mechanism in the MCT8 mutants tested in this study, because all seven mutant proteins were expressed in all three cell lines, although at varying levels. Although protein expression was lower for the insV236, G282C, and G558D mutants in all three cell lines, mRNA levels were not different between WT MCT8 and any of the mutants (data not shown). This suggests that the difference in protein levels cannot be explained by an impaired transcription and is consistent with previous studies showing similar mRNA levels but different protein levels between WT MCT8 and certain mutants (9). The lower protein levels suggest that the G282C, insV236, and G558D mutant proteins are degraded more rapidly than WT MCT8, thus contributing to their loss of function.
Of course, trafficking to the PM is crucial for MCT8 function. The uptake and metabolism data suggest that in COS1 cells the insV236, G282C, D453V, P537L, and G558D mutants are all expressed to some extent at the PM. P537L is the only one of the three active mutants that is fully expressed at the protein level and localized at the PM in Flp-in 293 cells. The main defect in the two other mutants showing partial activity in Flp-in cells (G282C, G558D) is related to decreased protein synthesis and/or increased protein degradation, perhaps associated with impaired protein folding and/or trafficking. Given their low protein expression and undetectable PM expression, the small amounts of these transporters reaching the PM must be quite active. This suggests that the activity of MCT8 to bind and transport TH is unchanged by the G282C and G558D mutations.
The P537L mutant is a less active transporter than the G282C and G558D mutants, is fully expressed at the protein level, and reaches the PM in significant quantities. The rate of T3 and T4 uptake in relation to the protein expression level and PM localization is therefore relatively low. This implies that the mechanism for the loss of function of the P537L mutant is an intrinsic defect in TH binding and/or transport. This mechanism applies to an even greater extent to the G221R, P321L, and D453V mutants, which were substantially expressed at the protein level and correctly routed to the PM but lacked any TH transport activity. Interestingly, this difference in impact between P537L and the others is supported by the protein mobility studies, which showed that the mobility of the inactive P321L in the PM was disturbed whereas the mobility of the active P537L mutant was similar to WT.
The loss-of-function caused by the insV236 mutation possibly involves a combined pathogenic mechanism of 1) protein folding and/or trafficking defect, and 2) partial loss of TH binding and/or transport. Increased protein degradation is suggested by the low mutant protein level in JEG3 and Flp-in 293 cells and proteolytic bands in COS1. A partial decrease in transport function is suggested by the lack of activity in uptake experiments combined with modest activity in metabolism studies of both T3 and T4 substrates in COS1 cells. It should be pointed out that the uptake studies mainly address the inward movement of substrates, whereas the metabolism data may rather reflect the intracellular TH concentration. It is not excluded that mutations have differential effects on TH uptake and efflux activities of MCT8.
Previous studies of the differences in function of MCT8 mutants showed a relation between transport activity and PM localization (8, 9, 13, 19). In general, our studies of the subcellular localization and functional properties of MCT8 mutants are in agreement with pathogenic mechanisms of other MCT8 mutations studied previously (8, 9, 13, 19). Our findings of the insV236 mutant in JEG3 cells show a similar loss of function as was found by Kinne et al. (13) in JEG1 cells.
The same group found in their cell lines also a cell type-dependent TH transport by MCT8 mutants that was dependent on surface translocation as determined in biotinylation experiments (13). An important difference between the cell lines we used is that JEG3 cells lack endogenous MCT8 expression, whereas COS1 and Flp-in 293 cells do express endogenous MCT8 (15). We therefore hypothesized that exogenous MCT8 mutants without intrinsic transport activity may heterodimerize with endogenous WT MCT8 and thus chaperone functional MCT8 to the PM. This hypothesis was tested in JEG3 cells by cotransfecting inactive MCT8 mutants with small amounts of WT MCT8. This did not result in an increased functional expression of MCT8 (data not shown).
An alternative explanation may lie in different expression profiles of interacting proteins in these cell lines. The JEG3 cells may lack some important MCT8 interacting proteins. The maturation and cellular surface localization of MCT1, MCT3, MCT4 (20, 21), and MCT12 (22) are dependent on the accessory protein basigin (CD147) whereas functional expression of MCT2 depends on embigin (23). Although MCT8 function is not dependent on basigin or embigin (24), it may well depend on other interacting proteins. The first modulating protein interaction has been shown recently between the proto-oncogene pituitary tumor-transforming gene-binding factor and MCT8 in the thyroid gland (25). Our findings of the larger immobile PM fraction of the P321L mutant may suggest a more rigid binding to interacting proteins than WT MCT8.
Another reason for the difference in functional studies between COS1 and JEG3 cells may be a difference in molecular chaperones within the ER. Studies of the cystic fibrosis transmembrane conductance regulator protein, which is mutated in cystic fibrosis, have shown that different molecular chaperones and cochaperones within the ER are involved in the folding and cellular quality control of cystic fibrosis transmembrane conductance regulator (26). Although JEG3 cells are capable of expressing functional WT MCT8, they may lack certain chaperone proteins that allow functional expression of even mutated MCT8 in COS1 cells. We studied the subcellular localization of WT and mutant MCT8 in JEG3 and COS1 cells using fusion proteins and by immunocytochemistry, but unfortunately PM localization of mutant and WT MCT8 was too low in either assay to detect a fluorescent signal at the PM in COS1 cells to carry out colocalization studies (data not shown). However, the similar PM localization of the mutants in combination with the difference in TH transport of the mutants in JEG3 and Flp-in 293 cells suggest that the folding of the transporters might be disturbed in JEG3 cells. Up to now we can only speculate on the explanation of cell type-dependent uptake.
Our studies and those of Schweizer's group of the cell type-dependent uptake of TH by MCT8 mutants suggest that choriocarcinoma JEG cell lines do not provide a representative model for all MCT8-expressing tissues. Studies of additional cell types may increase our insight into the pathogenic mechanism of MCT8 mutations. Assuming that interacting proteins are necessary for the proper expression and function of MCT8, the activity of MCT8 mutants in different tissues may depend on the expression of interacting proteins, such as scaffolding, ancillary, and chaperone proteins. Our findings suggest that this may well be the case for the G282C, P537L, and G558D mutants.
Given the cell-specific residual uptake by the mutations studied, one could argue that the Mct8 KO mouse is not a suitable model to study the pathogenic mechanism of mutations that do not result in an absent MCT8 expression. However, we have studied different patients with large deletions including the first exon of MCT8 that should result in the complete absence of MCT8 expression (4, 8, 19). The clinical phenotype of these patients is as severe as that of patients with other inactivating mutations (9). It may also be hypothesized that MCT8 mutations affect the expression of MCT8-related proteins at the cellular level. Some MCTs are known to form homodimers (17, 24, 27, 28) and even larger complexes with ancillary proteins (20–23). Therefore, it is possible that MCT8 dimerizes with other MCT family members, although evidence for the formation of such heterodimers has not been reported. If such heterodimers exist, it is possible that deletions or mutations in MCT8 have different effects on the functional expression of other MCT8 partners. Again, the association of large deletions and of various missense mutations in MCT8 with a similar, severe clinical phenotype argues against an important involvement of other MCTs.
Our model has some limitations because we use in vitro overexpression systems with generic promoters in genetically modified cell lines that may not reflect the in vivo MCT8 expression. The overexpression may induce artifacts in protein folding and trafficking. These limitations apply especially to the transient transfection studies using JEG3 and COS1 cells and less to the stable expression studies in Flp-in 293 cells. Nevertheless, these limitations should be considered in the translation to the in vivo situation. The MCT8 mutations studied here have all been identified in severely affected AHDS patients; none of them was reported to have any advanced psychomotor development, such as acquisition of speech or ability to walk (7, 29–32). Although all MCT8 mutations resulted in a severe phenotype, the cellular pathogenic mechanisms were different, ie, a defect in intrinsic transport activity for some mutations and impaired protein expression and trafficking for other mutations. Thus, an impaired TH transport by MCT8 mutants results in a severe clinical phenotype irrespective of the underlying pathogenic mechanism, suggesting that TH transport is the critical function of MCT8 with respect to brain development.
In conclusion, all seven MCT8 mutations studied impaired T3 and T4 uptake and metabolism in COS1 and Flp-in 293 cells, and especially in JEG3 cells. The MCT8 mutations could be divided in two major groups: mutations directly interfering the transport activity (ie, G221R, P321L, D453V, and P537L), and mutations predominantly disturbing protein expression and trafficking (ie, insV236, G282C, and G558D). This is the first study that provides insights in the mobility of WT and mutant MCT8 protein in cells by showing that although mutant proteins reach the PM, the protein mobility can be altered. The cell-type dependent residual uptake by G282C, P537L, and G558D supports the idea that in vivo TH transport by MCT8 mutants may be tissue specific depending on the expression of yet unknown MCT8-interacting proteins (13). This highlights the importance of understanding the pathogenic mechanism of MCT8 mutations in different cell types, which may help to identify proteins necessary for the folding of the protein and the trafficking to the PM. The identification of MCT8-interacting proteins might help us to define decisive therapeutic approaches for MCT8 patients.
Acknowledgments
This work was supported by the Smile Foundation with support from the Sherman Family.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AHDS
- Allan-Herndon-Dudley Syndrome
- CFP
- cyan fluorescent protein
- CRYM
- μ-crystallin
- ER
- endoplasmic reticulum
- FRAP
- fluorescence recovery after photobleaching
- KO
- knockout
- MCT8
- monocarboxylate transporter 8
- PM
- plasma membrane
- ROI
- region of interest
- SDS
- sodium dodecyl sulfate
- TH
- thyroid hormone
- WT
- wild type.
References
- 1. Friesema EC , Ganguly S , Abdalla A , Manning Fox JE , Halestrap AP , Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278(41):40128–40135. [DOI] [PubMed] [Google Scholar]
- 2. Friesema EC , Jansen J , Heuer H , Trajkovic M , Bauer K , Visser TJ. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab. 2006;2(9):512–523. [DOI] [PubMed] [Google Scholar]
- 3. Santisteban P , Bernal J. Thyroid development and effect on the nervous system. Rev Endocr Metab Disord. 2005;6(3):217–228. [DOI] [PubMed] [Google Scholar]
- 4. Friesema EC , Grueters A , Biebermann H , et al. . Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364(9443):1435–1437. [DOI] [PubMed] [Google Scholar]
- 5. Dumitrescu AM , Liao XH , Best TB , Brockmann K , Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74(1):168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwartz CE , May MM , Carpenter NJ , et al. . Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. 2005;77(1):41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frints SG , Lenzner S , Bauters M , et al. . MCT8 mutation analysis and identification of the first female with Allan-Herndon-Dudley syndrome due to loss of MCT8 expression. Eur J Hum Genet. 2008;16(9):1029–1037. [DOI] [PubMed] [Google Scholar]
- 8. Visser WE , Jansen J , Friesema EC , et al. . Novel pathogenic mechanism suggested by ex vivo analysis of MCT8 (SLC16A2) mutations. Hum Mutat. 2009;30(1):29–38. [DOI] [PubMed] [Google Scholar]
- 9. Jansen J , Friesema EC , Kester MH , Schwartz CE , Visser TJ. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology. 2008;149(5):2184–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trajkovic M , Visser TJ , Mittag J , et al. . Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007;117(3):627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dumitrescu AM , Liao XH , Weiss RE , Millen K , Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology. 2006;147(9):4036–4043. [DOI] [PubMed] [Google Scholar]
- 12. Wirth EK , Roth S , Blechschmidt C , et al. . Neuronal 3′,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci. 2009;29(30):9439–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kinne A , Roth S , Biebermann H , Kohrle J , Gruters A , Schweizer U. Surface translocation and tri-iodothyronine uptake of mutant MCT8 proteins are cell type-dependent. J Mol Endocrinol. 2009;43(6):263–271. [DOI] [PubMed] [Google Scholar]
- 14. Mol JA , Visser TJ. Synthesis and some properties of sulfate esters and sulfamates of iodothyronines. Endocrinology. 1985;117(1):1–7. [DOI] [PubMed] [Google Scholar]
- 15. Friesema EC , Kuiper GG , Jansen J , Visser TJ , Kester MH. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol. 2006;20(11):2761–2772. [DOI] [PubMed] [Google Scholar]
- 16. Visser WE , van Mullem AA , Jansen J , Visser TJ. The thyroid hormone transporters MCT8 and MCT10 transport the affinity-label N-bromoacetyl-[(125)I]T3 but are not modified by it. Mol Cell Endocrinol. 2011;337(1–2):96–100. [DOI] [PubMed] [Google Scholar]
- 17. Friesema EC , Jansen J , Jachtenberg JW , Visser WE , Kester MH , Visser TJ. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol. 2008;22(6):1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schindelin J , Arganda-Carreras I , Frise E , et al. . Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jansen J , Friesema EC , Kester MH , et al. . Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab. 2007;92(6):2378–2381. [DOI] [PubMed] [Google Scholar]
- 20. Kirk P , Wilson MC , Heddle C , et al. . CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000;19(15):3896–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Philp NJ , Ochrietor JD , Rudoy C , Muramatsu T , Linser PJ. Loss of MCT1, MCT3, and MCT4 expression in the retinal pigment epithelium and neural retina of the 5A11/basigin-null mouse. Invest Ophthalmol Vis Sci. 2003;44(3):1305–1311. [DOI] [PubMed] [Google Scholar]
- 22. Castorino JJ , Gallagher-Colombo SM , Levin AV , et al. . Juvenile cataract-associated mutation of solute carrier SLC16A12 impairs trafficking of the protein to the plasma membrane. Invest Ophthalmol Vis Sci. 2011;52(9):6774–6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilson MC , Meredith D , Fox JE , Manoharan C , Davies AJ , Halestrap AP. Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate transporter isoforms 1 and 4: the ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J Biol Chem. 2005;280(29):27213–27221. [DOI] [PubMed] [Google Scholar]
- 24. Visser WE , Philp NJ , van Dijk TB , et al. . Evidence for a homodimeric structure of human monocarboxylate transporter 8. Endocrinology. 2009;150(11):5163–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith VE , Read ML , Turnell AS , et al. . PTTG-binding factor (PBF) is a novel regulator of the thyroid hormone transporter MCT8. Endocrinology. 2012;153(7):3526–3536. [DOI] [PubMed] [Google Scholar]
- 26. Lukacs GL , Verkman AS. CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med. 2012;18(2):81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilson MC , Meredith D , Halestrap AP. Fluorescence resonance energy transfer studies on the interaction between the lactate transporter MCT1 and CD147 provide information on the topology and stoichiometry of the complex in situ. J Biol Chem. 2002;277(5):3666–3672. [DOI] [PubMed] [Google Scholar]
- 28. Alkemade A , Friesema EC , Kalsbeek A , Swaab DF , Visser TJ , Fliers E. Expression of thyroid hormone transporters in the human hypothalamus. J Clin Endocrinol Metab. 2011;96(6):E967–971. [DOI] [PubMed] [Google Scholar]
- 29. Vaurs-Barriere C , Deville M , Sarret C , et al. . Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol. 2009;65(1):114–118. [DOI] [PubMed] [Google Scholar]
- 30. Kakinuma H , Itoh M , Takahashi H. A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid. J Pediatr. 2005;147(4):552–554. [DOI] [PubMed] [Google Scholar]
- 31. Wood T , Hobson D , Browning B , et al. . The utilization of T3/T4 screening of males with MR of unknown etiology to identify patients with Allan-Herndon-Dudley syndrome. Barcelona, Spain: European Human Genetics Conference meeting; 2008, Abstract C07.6. [Google Scholar]
- 32. Papadimitriou A , Dumitrescu AM , Papavasiliou A , Fretzayas A , Nicolaidou P , Refetoff S. A novel monocarboxylate transporter 8 gene mutation as a cause of severe neonatal hypotonia and developmental delay. Pediatrics. 2008;121(1):e199–202. [DOI] [PubMed] [Google Scholar]
- 33. Friesema EC , Visser WE , Visser TJ. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol. 2010;322(1-2):107–113. [DOI] [PubMed] [Google Scholar]