


Abstract
Mutation ‘hotspot’ regions in the genome are susceptible to genetic instability, implicating them in diseases. These hotspots are not random and often co-localize with DNA sequences potentially capable of adopting alternative DNA structures (non-B DNA, e.g. H-DNA and G4-DNA), which have been identified as endogenous sources of genomic instability. There are regions that contain overlapping sequences that may form more than one non-B DNA structure. The extent to which one structure impacts the formation/stability of another, within the sequence, is not fully understood. To address this issue, we investigated the folding preferences of oligonucleotides from a chromosomal breakpoint hotspot in the human c-MYC oncogene containing both potential G4-forming and H-DNA-forming elements. We characterized the structures formed in the presence of G4-DNA-stabilizing K+ ions or H-DNA-stabilizing Mg2+ ions using multiple techniques. We found that under conditions favorable for H-DNA formation, a stable intramolecular triplex DNA structure predominated; whereas, under K+-rich, G4-DNA-forming conditions, a plurality of unfolded and folded species were present. Thus, within a limited region containing sequences with the potential to adopt multiple structures, only one structure predominates under a given condition. The predominance of H-DNA implicates this structure in the instability associated with the human c-MYC oncogene.
INTRODUCTION
Genetic instability underlies the etiology of many human disorders, including cancer. Genetic instabilities initiated by DNA double-strand breaks (DSBs) and subsequent chromosomal alterations often occur at common ‘hotspots’ in the genome that co-localize with DNA sequences capable of adopting alternative DNA structures (i.e. non-B DNA, such as H-DNA and G4-DNA) (1–3). The mutagenic potential of non-B DNA-forming sequences has been demonstrated in various model systems, implicating these structure-forming sequences as an endogenous source of genetic instability (4–8). Recently, we mapped H-DNA-forming sequences (HFS) within ±100 bp (‘bins’) of >19,000 translocation breakpoints (from sequenced human cancer genomes), and found that HFS were substantially enriched at translocations breakpoints compared to random-pick control sequences (1). Further, the number of HFS in each bin surrounding the translocation breakpoints was consistently higher than control, further implicating H-DNA in cancer development. Specifically, in cancers such as Burkitt's lymphoma, L3 type acute lymphoblastic leukemias, and plasmacytoma, H-DNA-forming sequences co-localize with common translocation breakpoints in the human c-MYC gene (9–11); whereas, G4-DNA-forming sequences map to the BCL-2 gene major breakpoint region of a translocation common in follicular lymphomas (12,13), and also within the nuclease hypersensitive locus near the H-DNA-forming sequence in the c-MYC gene (14,15). Although the mutations in these cancers are known (i.e. specific translocation), the initiating mutagenic mechanisms are still unclear.
A polypurine-polypyrimidine mirror-repeat can adopt an intramolecular triplex structure via Hoogsteen bonding (16,17), when one half of the mirror symmetry (Figure 1A) binds to the major groove of a purine-rich tract of the underlying duplex, leaving a single-stranded region. If the purine-rich tract serves as the third strand, a divalent (e.g. Mg2+) cation-dependent R*RY type (R: purine, Y: pyrimidine, *Hoogsteen bonds) triplex may form (e.g. Figure 1B). Alternatively, an acidic pH-dependent Y*RY type of triplex may form if the pyrimidine-rich strand serves as the third strand. Two isomers may further exist for each type of triplex, depending on which end of the DNA (5΄- or 3΄) the Hoogsteen hydrogen bonding occurs, giving rise to four possible triplex isomers (18,19). G4-DNA or G-quadruplex DNA forms at tandem repeats of guanine (G) sequences (Figure 1A) (20). Either intra- or intermolecularly, four G bases associate in a square-planar cyclic array, stabilized by Hoogsteen bonds to form a G-tetrad that can stack, and thereby form G4-DNA structures stabilized by monovalent cations, such as K+ or Na+. There are a number of possible topologies for intramolecular G4-DNA structures (14,21) and they are difficult to predict, but one common motif for K+-stabilized G4-DNA is a parallel-stranded topology (Figure 1C).
Figure 1.

Schematic of potential non-B DNA structures formed at the c-MYC sequence used in this study. (A) Sequence from a translocation breakpoint hotspot in the human c-MYC gene with mirror repeat symmetry (red arrows) that can form (B) H-DNA, and guanine repeats (black bars) that can form (C) G-quadruplex or G4-DNA.
One of the challenges in studying non-B DNA in vivo is that there are regions in the genome that contain multiple, overlapping sequences with the potential to form more than one non-B DNA structure (22). One example is found in the oncogenic human c-MYC promoter region, where clusters of non-B DNA-forming sequences (e.g. H-DNA, Z-DNA, G4-DNA) surround the promoter P0, which is highly transcribed in some cancer cells (23,24). And therefore, the extent to which one (or more) non-B DNA structure impacts the formation and/or stabilization of another non-B DNA structure (within the sequence) is not fully understood. Given that non-B DNA formation relaxes negative supercoiling that is required for the subsequent formation of additional non-B DNA structures, we speculate that in a region of DNA containing more than one potential non-B DNA-forming sequence, one tract may form a specific non-B DNA structure at a given time and condition, thus, subsequently inhibiting the formation of other mutagenic non-B DNA structures.
Here, we focused on a short sequence derived from a chromosomal breakpoint hotspot in the human c-MYC gene (Figure 1A), ∼400 base pairs (bp) downstream of the P0 promoter (25–27). This 23-bp duplex sequence contains both a mirror symmetry and a tandem repeat of Gs, which could potentially form H-DNA or G4-DNA, respectively (25). This sequence was previously found to stimulate genetic instability by its propensity to adopt an H-DNA structure (6). In order to address the issue of competing non-B DNA structures formed by this sequence, we have investigated the folding preferences of oligonucleotide constructs (Table 1) containing the G-rich polypurine strand and one copy of the H-DNA-forming mirror repeat. By employing multiple spectroscopic, nuclease and chemical probing, and polyacrylamide gel electrophoresis (PAGE)-based techniques, we have characterized the preferred structures formed by these constructs in the presence of either G-quadruplex-stabilizing K+ ions, and/or H-DNA-stabilizing Mg2+ ions. We demonstrate that under conditions favorable for H-DNA formation, a stable G-rich, R*RY-type intramolecular triplex DNA structure is formed; whereas, under G-quadruplex-forming conditions, a plurality of unfolded and folded species is present.
Table 1. Oligonucleotides used in this study.
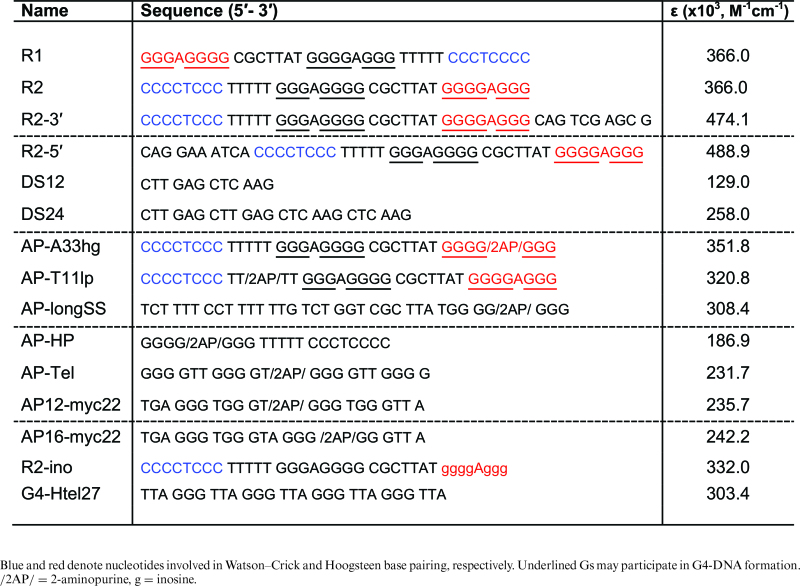
|
MATERIALS AND METHODS
Materials
Oligo-deoxyribonucleotides (oligonucleotides) used for the gel mobility assays, circular dichroism (CD), and chemical modification analyses were PAGE- or HPLC-purified and obtained in lyophilized form from either Midland Certified Reagent Company, Inc. (Midland, TX, USA) or Integrated DNA Technologies (IDT, Coralville, IA, USA). The RP (Reverse Phase)-HPLC-purified oligonucleotides containing an internal 2-aminopurine (2AP) modification were synthesized by Midland Certified Reagent Company, Inc. Oligonucleotides were kept at –20°C until resuspended. The pH of solutions was adjusted using a Pinnacle Series M540P pH meter.
Methods
Experimental design
We conducted the experiments in the presence of either H-DNA/triplex-forming (20 mM sodium cacodylate pH 7.0, 100 mM NaCl, 0.1 mM EDTA, 10 mM MgCl2) or G4-forming (20 mM sodium cacodylate pH 7.0, 0.1 mM EDTA, 110 mM KCl) buffers to assay for H-DNA (intramolecular triplex) and G-quadruplex-formation, respectively. Sodium cacodylate was used as the buffer because its pH varies minimally with temperature, particularly during thermal melting studies. However, to assay under physiological conditions, we used Cellgro's Dulbecco's Phosphate Buffered Saline (DPBS, Cat. # 20-030-CV) containing 81 mM Na2HPO4, 15 mM KH2PO4, 27 mM KCl, 9 mM CaCl2, 5 mM MgCl2·6H2O, 1.4 M NaCl. Oligonucleotide solutions were annealed in the presence of an appropriate buffer by heating to 95°C and then slow cooling (3–4 h, on a heat block) to room temperature. The annealed DNA solutions were kept at 4°C prior to their use in experiments. We determined that full-length oligonucleotides migrated as single bands by using denaturing PAGE [8 M urea, 89 mM Tris-borate, 2 mM EDTA (TBE)] on 20% gels. The oligonucleotides were 5΄-end labeled with [γ-32P]-ATP (Perkin-Elmer, Boston, MA) using T4 polynucleotide kinase (New England Biolabs, Ipswich, MA, USA) for 60 min at 37°C, then heat inactivated at 65°C for 20 min. The unincorporated nucleotides were removed by size-exclusion chromatography via MicroSpin G-25 columns (GE, Buckinghamshire, UK). Concentrations of oligonucleotide stock solutions prepared in nuclease-free water were calculated using molar extinction coefficient (ε, Table 1) for each oligonucleotide from absorbances measured at 260 nm using a NanoDrop 2000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
Gel mobility assay using native PAGE
To determine the buffer-specific mobility of the oligonucleotides and assess intramolecular structure formation, oligonucleotides were subjected to native PAGE on 20% native minigels (19:1 acrylamide/bisacrylamide) using the corresponding buffers. DNA at a concentration of 15 μM was annealed, then ∼100 pg was mixed with Bio-Rad Nucleic Acid Loading Buffer (15 μl loading volume) and electrophoresed on a 20% native polyacrylamide gel at room temperature at 40–50 V using 1× Tris-Borate (89 mM) supplemented with the appropriate cation (10 mM Mg2+ or 50 mM K+, depending on the tested condition). DNA mobility was visualized on a Chemidoc XRS+ Gel Imaging System (Bio-Rad, Hercules, CA, USA) using Image Quant TL v7 software after SYBR Gold (emission filter: 520–550 nm) staining for 30–45 min. Native gels also included 10- and 20-base pair markers, T30/T40 single-strand polydT markers and DS12/DS24 double-strand markers.
Circular dichroism (CD) spectroscopy
To probe for secondary structure formation, CD spectra of annealed DNA (10 μM, 250 μl) in a 0.1 cm quartz cuvette were recorded at 25°C using a Jasco J-815 (Jasco Inc., Easton, MD) CD spectrometer equipped with a Peltier temperature controller. The 200–350 nm range was scanned three times at a rate of 100 nm/min with 2 s response time. Buffer-corrected CD data, collected as mdeg as a function of wavelength, were processed using the Jasco Spectra Manager software and exported to Graph Pad Prism (La Jolla, CA). For the spectral titration experiments to increase K+ concentration in solution, aliquots of K+ stock solution were added to the DNA solution previously annealed in triplex-forming buffer. The CD spectrum was monitored after each K+ addition. The final solution, with a final concentration of 160 mM K together with the initial 10 mM Mg2+ and 100 mM Na+, was boiled and annealed, and then subjected to CD.
Chemical modification (using oligonucleotides) and nuclease sensitivity assay (using plasmid DNA) experiments
To map DNA structure formation at the nucleotide level, we performed chemical cleavage/protection assays using dimethyl sulfate (DMS) and potassium permanganate (KMnO4) to probe for guanine (G) and thymine (T) reactivity, respectively. Oligonucleotides were 5΄-radiolabeled as described above, prior to annealing in the appropriate buffer. Ten picomoles of the radiolabeled and annealed DNA were resuspendend in buffer to attain a final reaction volume of 25 and 40 μl for DMS (1.5 μg tRNA also added) and KMnO4 treatments, respectively. For reactions involving non-structured/denatured DNA, the solution was boiled at 100°C for 1 min and then quick-chilled on ice. The samples were allowed to equilibrate at 24°C for 15 min prior to chemical treatment. Then, 5 μl of a freshly prepared 2.5% aqueous solution of DMS was added and the reaction allowed to proceed for 10 min. In the other samples, 5 μl of freshly prepared 0.56 mM KMnO4 was introduced for 15 min. The reactions were stopped by adding a solution composed of 3 μl β-mercaptoethanol and 20 μg of tRNA, followed by ethanol precipitation and piperidine treatment (100 μl, 1 M piperidine, 90°C, 30 min) as previously described (28). The cleavage products were resuspended in formamide loading buffer, heated at 100°C for 1 min, then quick chilled prior to loading onto a 20% denaturing polyacrylamide gel. The level of protection was calculated by first normalizing the signals against the same band from the denatured sample and then, against a similar nucleotide that is known not to participate in secondary structure formation, i.e. T8 from the overhang sequence and G33 from the long loop (CGCTTAT), from the same treatment to serve as the reference signal ( = 1). The difference was calculated for the bands in question, and multiplied by 100 to quantify the percentage of protection at a particular base. The error bars were calculated from the sample standard deviation divided by the square root of the total number of trials.
To probe structure-formation in the context of plasmid DNA under the different buffer conditions, we incorporated non-structure-forming (B-DNA: TCGAGTTGGGAGGGGCGCTTATGGAGGGGGGCGCCTTC) and the R2-5΄ sequences (MycR25: AATTCCAGGAAATCACCCCTCCCTTTTTGGGAGGGGCGCTTATGGGGAGGGC) into a shuttle vector backbone (psp189 plasmid) as described (6). We prepared supercoiled monomer plasmids by excising the gel fragment containing the monomer, followed by plasmid isolation, transformation into DH5α Escherichia coli cells and subsequent plasmid re-isolation. Plasmids were sequenced to check for sequence fidelity and tested for supercoiling levels prior to use. Supercoiled monomer plasmids (1 μg) were incubated under either triplex- or G4-DNA-forming buffers at 37°C, for 12–18 h. They were then treated with S1 nuclease (37.5 U) for 30 min on ice using a buffer containing 30 mM sodium acetate, pH 4.6, 1 mM ZnCl2, 5% (v/v) glycerol, 50 mM NaCl, and 4 mM MgCl2. The plasmids were extracted with phenol-chloroform and then ethanol precipitated. The resuspended DNA was then digested with restriction enzyme Stu1 (10 U, 1 h, 37°C), and the resulting fragments separated on a 1.4% agarose gel. To probe for smaller fragments, samples were digested with BsrBI after S1 treatment. Fragments were dephosphorylated using Antarctic phosphatase (5 U, 37°C, 30 min; inactivated at 80°C, 2 min), radiolabeled as described above using T4PNK, and separated on a 20% polyacrylamide gel.
2-Aminopurine (2AP)-based fluorescence experiments
To further probe for the local DNA conformation in solution, fluorescence emission studies were conducted using 500 nM (purged with Ar gas for 2 min) of the annealed 2AP-containing oligonucleotides using either a low-salt, triplex-forming (10 mM sodium cacodylate pH 7.0, 10 mM NaCl, 0.1 mM EDTA, 5 mM MgCl2) or a low-salt, G4-forming (10 mM sodium cacodylate pH 7.0, 0.1 mM EDTA, 15 mM KCl) buffer. The fluorescence emission (excitation at 305 nm) at 25°C was monitored from 325 to 500 nm using a Varian Cary Eclipse Fluorescence spectrophotometer (Walnut Creek, CA) equipped with a Peltier temperature control system with the emission and excitation slits set at 5 and 10 nm, respectively. The temperature-dependent fluorescence (excitation at 305 nm, emission at 370 nm) was monitored every 5° from 10 to 90°C, with a heating rate of 1°C/min. A quartz 4 mm microsquare fluorimeter cell (Starna Cells) was used in all of the fluorescence experiments. The error bars were calculated from the sample standard deviation divided by the square root of the total number of trials.
UV-thermal denaturation experiments
To determine thermal melting temperatures of the buffer-dependent structures, DNA solutions were annealed in the presence of either triplex- or G4-DNA-forming buffers. The thermal denaturation of 1.7 μM annealed DNA in a 1 cm quartz cuvette was monitored from 20 to 100°C at 260 nm, and at 295 nm, using a Cary 100 UV-Vis equipped with a Peltier temperature controller using a ramp rate of 0.4°C/min. The starting point of the melting curves was normalized to Abs = 1 and the thermal melting temperature (Tm) calculated as the maximum value of the first derivative of the absorbance versus temperature graph using Graph Pad Prism (La Jolla, CA) program.
RESULTS
In this study, we characterized the DNA secondary structure formation of oligonucleotides encompassing a 23-bp translocation breakpoint hotspot in the human c-MYC oncogene containing both G4-forming (i.e. tandem guanine repeats) and H-DNA-forming (i.e. presence of mirror symmetry) sequences. These oligonucleotides were constructed to allow intramolecular triplex formation with the purine (R)-rich repeat as the ‘third strand’, involved in reverse Hoogsteen H-bonding (R*RY type of triplex). Unless otherwise noted, the sequences are denoted as ‘R’ (R1, R2, etc., Table 1) where R1 is expected to have the third strand on the 5΄-side, and R2 is predicted to have the third strand on the 3΄-side. R2-3΄ and R2-5΄ possess overhang sequences in the 3΄- and 5΄-sides, respectively (Supplementary Figure SI-1). Gel mobility assays using native PAGE, CD, chemical footprinting and 2AP fluorescence analyses indicated that intramolecular DNA secondary structures were formed. Under H-DNA-forming conditions, the intramolecular triplex DNA structure predominated, while under G4-DNA-forming conditions, a combination of folded and unfolded species was present.
Native PAGE and CD spectroscopy indicate that a stable, K+-resistant, intramolecular triplex forms under H-DNA-forming conditions, and intra- and intermolecular parallel species occur under G4-DNA-forming conditions
To assess the formation of intramolecular secondary DNA structures, gel mobility assays using native PAGE were performed on the DNA samples annealed under H-DNA- or G4-DNA-forming conditions. Here, the intermolecular structures or more relaxed DNA fragments will migrate slower than the more compact, folded intramolecular DNA structures. The results revealed that under H-DNA-forming conditions, the annealed 36-mer samples (R1, R2, Table 1, Supplementary Figure SI-1) migrated at a mobility comparable to that of an 18 base-pair (bp) oligonucleotide and well below the T30 single-strand control. Whereas, the 46-mer samples (R2-3΄, R2-5΄, Table 1, Supplementary Figure SI-1) migrated comparable to a 25-bp oligonucleotide and below the T40 single-strand control (Figure 2A). The faster mobility of the annealed samples through the native gel, which is similar to their half-length if they had been double-stranded, indicated that they formed compact, intramolecular structures (29,30). Intramolecular structure formation was further supported by the consistent mobilities of the annealed DNA samples up to 100 μM oligonucleotide concentration (Supplementary Figure SI-2). Under G4-DNA-forming conditions, there appeared to be a combination of species composed of compact, high mobility species and slower migrating, potentially intermolecular species (Figure 2C).
Figure 2.

Characterization of non-B DNA structure formation by native PAGE and CD spectroscopy. Native gel electrophoresis results showing folded mobilities under (A) H-DNA/triplex-forming and (C) G4-DNA-forming conditions. R1, R2 = 36-mer; R2-3΄, R2-5΄ = 46-mer; M10 = 10-bp marker; T30/T40 = single-strand polydT markers; DS12/DS24 = double-strand markers. Circular dichroism (CD) spectroscopy results showing (B) a negative peak in the 210–220 nm region (oval) indicates triplex formation under triplex-forming conditions. (D) A negative peak at 240 nm and positive peak at 260 nm suggest parallel G4-DNA formation under G4-DNA-forming conditions.
CD is based on a molecule's absorption of circularly polarized light and generates characteristic signatures for specific nucleic acid secondary structures and thus, is a useful tool to determine the conformation/secondary structure of DNA (31,32). To probe for the types of structures identified via native gel electrophoresis, CD spectroscopy was performed on the annealed samples under either H-DNA-forming or G4-forming conditions. Under H-DNA-forming conditions, R1, R2, R2-3΄ and R2-5΄, all exhibited a strong positive peak at ∼260 nm, a negative peak at ∼240 and a negative peak at ∼210 nm (Figure 2B). The negative peak at 210–220 nm (oval, Figure 2B) is considered a characteristic predictor of triplex formation (30,33–35), specifically for the binding of the third or Hoogsteen-paired strand (36). Thus, based on the CD results, the structure formed by the intramolecular structure under H-DNA-forming conditions is consistent with intramolecular triplex/H-DNA.
On the other hand, under G4-forming conditions, R1, R2, R2-3΄ and R2-5΄ all showed negative peaks at ∼240 nm, R1 and R2 had a positive peak at ∼260 nm, and R2-3΄ and R2-5΄, had a positive peak at ∼270 nm (Figure 2D). The presence of a strong positive peak at 260–265 nm with a weak negative signal at 240 nm is indicative of parallel G-quadruplex structures, with all the strands having the same 5΄-3΄ orientation and with an anti- glycosidic bond conformation (37–39). Thus, under G4-DNA-forming conditions, the tested inter- or intramolecular DNA may be forming G4-DNA structures with a parallel topology. It should be noted that neither H-DNA nor parallel-G4 features were found in the single-strand (T40) and double-strand (DS24) control sequences, both of which exhibited conservative CD spectra with positive peaks at ∼278 nm and negative peaks at ∼255 nm. DS24 exhibited a positive peak at a slightly shorter wavelength than T40, which is indicative of double-stranded DNA rather than single-stranded DNA (40).
K+ is known to stabilize G4-DNA, and can destabilize triplex structures (41). To determine the stability of the H-DNA structure and to determine whether an equilibrium between H-DNA and G4-DNA occurred under H-DNA-forming conditions, we titrated aliquots of K+ into a pre-annealed solution of R1 or R2 under H-DNA-forming conditions, and monitored shifts in the H-DNA CD signature (negative peak at ∼210 nm) after each addition. We found that the addition of K+, even at a final concentration of 160 mM, resulted in little to no change in the H-DNA CD signature for R1 (Figure 3A) and R2 (Figure 3D). This is in contrast to what we observed when we titrated K+ into known G4-forming sequences (Htel 27 and cMyc22, similarly annealed under H-DNA-forming conditions) where we detected drastic changes in their CD signatures accompanying G4-formation (Supplementary Figure SI-3). However, after the final solution (containing Na+, Mg2+ and K+ ions) was subsequently heated to 95°C and slowly re-annealed, the triplex signature at 210 nm was markedly diminished for both R1 (Figure 3B) and R2 (Figure 3E). Their CD signatures approximate that of the CD spectra when the motifs were heated and annealed in the presence of 110 mM K+ alone (R1, Figure 3C; R2, Figure 3F), with weak/absent negative signals in the 210 nm region. These results indicate that the structure formed under H-DNA-forming conditions was predominantly intramolecular triplex and that it was stable even in the presence of added K+. However, upon heating, the sequences were exposed to both Mg2+ and K+, in which case, K+ appeared to inhibit the formation of H-DNA.
Figure 3.

The structure formed under H-DNA/triplex-forming conditions is stable in the presence of K+, except when heated. Using CD spectroscopy, the DNA structure annealed under triplex-forming conditions (100 mM Na+, 10 mM Mg2+) remained unperturbed with increasing concentrations of K+ for both the (A) R1 and (D) R2 motifs. When DNA was annealed in the presence of 160 mM K+, competing with the triplex-forming conditions (100 mM Na+ + 10 mM Mg2+), the triplex signature at 210 nm was diminished, (B) R1 and (E) R2. CD spectra of (C) R1 and (F) R2 when annealed under triplex-forming conditions (black) or 110 mM K+ (red) alone. The CD spectra obtained in the presence of competing conditions (B and E) approaches that of the signature under 110 mM K+ alone (C and F, red).
To further confirm that the CD signatures obtained at 25°C were structure related, we also obtained CD data for R2 (under both H-DNA-forming and G4-forming conditions) at 90°C. The CD spectra for R2 under both conditions at 90°C were similar (Supplementary Figure SI-4), suggesting that the DNA was in a random coil/denatured state. Thus, the different CD spectra observed for R2 at 25°C under H-DNA-forming or G4-forming conditions signify the occurrence of two distinct structural topologies under these conditions (Supplementary Figure SI-4).
Importantly, under physiologically relevant buffer conditions, the H-DNA structure was also favored. We investigated the gel mobility and CD spectra of the various motifs R1, R1, R2-5΄ and R2-3΄ in Dulbecco's PBS (see Methods). All of these constructs ran as high mobility species by gel electrophoresis and possessed the CD signature characteristic of that of an intramolecular triplex (Supplementary Figure SI-5). The results suggest that intramolecular triplex/H-DNA also formed in this ‘physiological’ buffer composition.
2-Aminopurine (2AP) substitution corroborates Hoogsteen bonding under H-DNA-forming conditions, and suggests loop formation under G4-DNA-forming conditions
When incorporated into DNA, 2-aminopurine (2AP) induces little or no structural perturbation (42). It has been incorporated into loops of G4-DNA (43,44) and into duplex and triplex DNA (45) without destabilizing the structures. Thus, it is useful as a base modification in conformational studies of nucleic acid secondary structures. Also, 2AP fluorescence increases with solvent exposure and decreases with base stacking and H-bonding, (42,45) allowing one to probe local conformation of a DNA structure in solution by strategically substituting a single 2AP within the sequences. To further confirm the presence of DNA secondary structures consistent with the other techniques that we have employed in this study, in particular the binding of the Hoogsteen paired strand (third strand) to the duplex to form triplex DNA under H-DNA-forming conditions, and possible G4-DNA formation under G4-forming conditions, we substituted the last adenine of the R2 sequence to obtain the 2AP-containing sequence, AP-A33hg (Table 1).
Guanines are known to quench 2AP fluorescence (46–48). To confirm that the 2AP substitution, which was in between two guanines, was not quenched, we performed preliminary studies involving a single-strand and a duplex/hairpin containing the 2AP flanked by guanines. We found that there was a substantial signal difference (data not shown) between the single-strand and the duplex DNA that allowed us to proceed with the experiments. We used other R2-derived sequences as controls, such as a sequence with 2AP substituted in the loop (AP-T11lp), a 2AP-substituted single-strand (AP-longSS), and a duplex/hairpin (AP-HP). We also employed G4-forming control sequences, AP-Tel (mixed parallel/anti-parallel), AP12-myc22 and AP16-myc22 (parallel), which contained a 2AP-substitution in one of the loop nucleobases (Table 1). Lastly, we performed the 2AP experiments under reduced salt concentrations (see Methods) because our initial experiments also showed that the 2AP single-strand control possessing 3–4 runs of guanines underwent intermolecular interactions using our standard concentration of monovalent cation (∼100 mM). Reducing the concentration to 10 mM Na+ (with 10 mM sodium cacodylate, 0.1 mM EDTA) prevented such intermolecular interactions for the single-strand control. Consequently, we also reduced the levels of Mg2+ and K+ under the low-salt, H-DNA-forming and G4-DNA-forming conditions, respectively, to allow comparison.
Under low-salt, H-DNA-forming conditions, the 325–500 nm fluorescence emission of AP-A33hg was quenched similarly to AP-HP (Figure 4A). This suggested that the 2AP in AP-A33hg was within a structured environment and the 2AP was not exposed to the solution. Also, its steady-state emission intensity at 370 nm, upon excitation at 305 nm, increased with temperature, and again similar to that of AP-HP (Figure 4A and B), although AP-HP had a sharper increase in fluorescence intensity at 65°C, whereas AP-A33hg showed a steady rise starting at 35°C. This temperature-dependent trend indicated that the 2AP in AP-A33hg, like that of the 2AP within an AP-HP duplex/hairpin, was initially within a rigid secondary structure and the subsequent structure denaturation when the solution was heated, resulting in solvent exposure of the 2AP and thus, the increase in fluorescence. We interpreted this to mean that the guanine tract with the 2AP substitution in AP-A33hg is involved in Hoogsteen base pairing (serving as the third strand) to the duplex consistent with intramolecular triplex formation under triplex/H-DNA-forming conditions. This Hoogsteen base pair interaction also subsequently strengthened the base-stacking within this third strand which protected the 2AP from being exposed to the solution, and hence its quenched fluorescence. On the other hand, AP-T11lp and AP-longSS were both fluorescent at ambient temperature but demonstrated a decrease in fluorescence intensity at higher temperatures. This decrease in fluorescence intensity can be ascribed to increased collisions of the accessible 2AP (in the loop for AP-T11lp and as a single-strand in AP-longSS) with water and other molecules in solution, due to increased kinetic energy upon heating, thus, quenching its fluorescence.
Figure 4.

2AP substitution corroborates Hoogsteen bonding under H-DNA/triplex-forming conditions, and suggests loop formation under G4-DNA-forming conditions. Under H-DNA-forming conditions, the fluorescence emission of AP-A33hg is (A) quenched and follows a (B) temperature-dependent increase in fluorescence, comparable to that of a hairpin-forming structure, AP-HP. AP-longSS and AP-T11lp are the single-strand and loop controls, respectively. Under G4-DNA-forming conditions, (C) AP-A33hg is fluorescent and follows a (D) temperature-dependent decrease in fluorescence, suggestive of a loop conformation.
Under low-salt, G4-forming conditions, AP-A33hg was highly fluorescent at room temperature, and upon heating, a sharp sigmoidal decrease in fluorescence (245 fluorescence units within 30°C) centered at 45°C was observed (Figure 4C and D). Conversely, AP-HP and AP-longSS displayed similar trends as observed under H-DNA-forming conditions, showing a temperature-dependent decrease in fluorescence. When we compared the temperature-dependent fluorescence of AP-A33hg with known G4-forming, 2AP-loop-substituted sequences (as 1-nt loop in AP16-myc22, as part of 2-nt loop in AP12-myc22, as part of 2–4 nt loop in AP-Tel) under G4-DNA-forming conditions (Supplementary Figure SI-6A and B), AP-A33hg displayed the same trend as that of AP16-myc22; however, the fluorescence intensity of AP16-myc22 at room temperature was approximately 4-times more intense than AP-A33hg. Also the sharp, sigmoidal melting transition for AP16-myc22 occurred at a higher temperature (∼75°C) (Supplementary Figure SI-6A and B). In contrast, the fluorescence behavior of AP-Tel was quite different; it showed low fluorescence at all temperatures. From these fluorescence-based substitution studies of known G-quadruplex-forming sequences, we conclude that 2AP substitution in short, cross-over loops, such as in the case of AP16-myc22, affords highly fluorescent species, while substitution in longer, lateral loops led to quenched fluorescence possibly due to base stacking within the loop. Thus, the highly fluorescent species observed in AP-A33hg under G4-DNA-forming conditions is likely a G4-DNA structure in which the 2AP resides in a short, cross-over loop. However, there are other species present under G4-DNA-forming conditions, as indicated by PAGE results for R2 (Figure 2C), and these species may be only weakly fluorescent, accounting for the lower fluorescence intensity for AP-A33hg compared to AP16-myc22 and AP12-myc22. These weakly fluorescent species may be G4-DNA structures in which the 2AP resides in a lateral loop or non-G4-DNA species in which the 2AP is quenched by stacking interactions with adjacent residues.
As a control for the conditions, we also monitored the steady-state fluorescence of the 2AP motifs under a cation-free, Tris–HCl buffer, and we detected nearly identical levels of fluorescence for the same concentrations of AP-A33hg, AP-Tel and AP-LongSS (data not shown). This confirms that the different levels of fluorescence seen under triplex/H-DNA- or G4-DNA-forming conditions were due to structure formation.
Oligonucleotide chemical modification and plasmid nuclease sensitivity experiments further support triplex formation
Chemical modification experiments map the nucleobases according to their reactivities; hence, DNA secondary structures can be probed using modifying agents (e.g. DMS for guanines and KMnO4 for thymines). DMS incorporates a methyl group on an exposed N7 of G. However, in an H-DNA or G4-DNA structure, if a G is involved in a Hoogsteen bonding (Figure 1B and C), then the N7 is protected and cannot be modified (49). KMnO4, on the other hand, reacts with exposed C5-6 position (double bond) of thymines (Ts) if they are not within a duplex or base-stacked, i.e. in a loop (50,51). Subsequent treatment with a base cleaves the backbone of modified Gs or Ts, the bands of which can be resolved by denaturing PAGE (52). Thus, the absence of or low-intensity G- or T- band (‘protection’) in the gel indicates involvement in Hoogsteen bonding or duplex-formation.
Under triplex/H-DNA-forming conditions, we found that bases G24 to G30 of R2-5΄ were protected from DMS modification, indicating that they were involved in Hoogsteen bonding. T15, which was also protected from KMnO4 treatment, signified that it is within a duplex structure (Figure 5A, Supplementary Figure SI-7). These combined results of the G24-30 and T15 protection, confirmed that R2-5΄ formed an intramolecular triplex structure under H-DNA-forming conditions, consistent with the 2AP fluorescence results. The strong level of protection also indicated that the H-DNA structure formed was highly stable and was adopted by the majority of the population of molecules.
Figure 5.

Chemical probing and nuclease sensitivity assay of non-B DNA structures. Chemical modification results for R2-5΄ under (A) H-DNA/triplex-forming (‘Triplex conditions’) or (B) G4-forming conditions (‘G4-DNA conditions’). The ‘protection’ of T15, together with that of G24-31 (panel A), indicates that a triplex is formed under triplex-forming conditions. Under G4-DNA-forming conditions (panel B), no distinct level of protection for the G's that may be involved in G4-DNA were seen, possibly due to competing structures that precluded distinct protection patterns. (C) Nuclease sensitivity assay using plasmids containing inserts of non-structure-forming (B-DNA) and R2-5΄ sequences (MycR25) showing similar fragment patterns for MycR25 under triplex- or G4-DNA-forming conditions. Both conditions yielded a 1.4 kb band (MycR25, lane 4, with arrow) not present in the B-DNA plasmid indicative of H-DNA structure formation. Lane 1: DNA only, Lane 2: DNA + StuI, Lane 3: DNA + S1, Lane 4: DNA + S1 + StuI.
On the contrary, under G4-DNA-forming conditions, we did not detect a distinct level of protection for the G's that may be involved in G4-DNA formation (Figure 5B, Supplementary Figure SI-7). The protection levels of G29-30 and G39-40 were only slightly comparable to that of T15, which is not expected to participate in G4-DNA formation. It is possible that there are competing parallel G4 structures formed under these conditions that preclude strong and constant protection patterns. Alternatively, as the gel mobility assay results are consistent with the presence of multiple species, it is likely that G4-DNA species may form to some extent, giving rise to the observed weak protection patterns, but a single stable G4-DNA structure is not the predominant species under these conditions.
Translating the probe for structure-formation in the context of plasmid DNA, we incorporated non-structure-forming (B-DNA), and the R2-5΄ sequences (MycR25) into a shuttle vector backbone (psp189 plasmid). The plasmids were incubated with either the triplex- or G4-DNA-forming buffers prior to digestion with S1 nuclease followed by StuI enzyme digestion. We expected that in the presence of a secondary structure such as H- or G4-DNA, there are single-stranded regions, which are substrates for S1 nuclease. Subsequent treatment with the restriction enzyme StuI will yield a fragment with known size. Independent of the buffer used for pre-incubation, the treatments yielded for MycR25 a ∼1.4 kb band (Figure 5C, marked with arrows) not present in the plasmid lacking the MycR25 insert (Figure 5C, B-DNA, lane 4). We can conclude that the insert in MycR25 formed the same structure yielding a single-stranded region that is sensitive to S1 nuclease digestion, regardless of the buffer condition. This structure is consistent with H-DNA, as we did not detect additional fragments expected for a G4 structure after probing for smaller fragments (∼20–30 bp) on a 20% polyacrylamide gel resulting from a S1-BsrBI treatment. In a related investigation, sequencing of this 1.4 kb band confirmed the presence of the triplex-forming sequence and showed that S1 nuclease cut in close proximity to that sequence (Zewail-Foote et al., unpublished results).
Thermal melting experiments reveal a Mg2+-stabilized structure under H-DNA-forming conditions, and an absence of hypochromicity at A295 under G4-DNA-forming conditions
To further characterize the DNA structures formed by the oligonucleotides comprised of the c-MYC sequence, we monitored the absorbance of the R2 sequence at 260 nm (triplex/H-DNA condition) and 295 nm (G4-DNA condition), as a function of temperature. This thermal melting data also allowed us to calculate the thermal melting temperature (TM), which is indicative of the stability of the structure formed in solution. Under H-DNA-forming conditions (with 10 mM Mg2+), if a triplex/H-DNA was formed, then we would expect the absorbance at 260 to increase (hyperchromic) as the H-DNA structure dissociates/melts. In addition, some triplexes (H-DNA structures) also exhibit biphasic melting with separate TM's for the transitions (triplex-to-duplex transition followed by the duplex-to-random coil transition) (53). We observed that the c-MYC-derived H-DNA structure formed here was very stable and Mg2+-dependent, melting at ∼90°C, which was destabilized down to 78°C in the absence of the divalent ion (Figure 6A). Native gel electrophoresis and CD experiments revealed that in the absence of Mg2+, R2 migrates with a similar mobility and possesses a CD signature (Supplementary Figure SI-8A-C) similar to that of R2 in the presence of Mg2+, implying that triplex formation occurs even in the absence of Mg2+. The results of the thermal melting experiments demonstrated the structure-stabilizing effect of a divalent cation. We then substituted the guanines in the third strand with inosine (a guanine derivative that lacks the exocyclic 2-amino group), R2-ino, to promote hairpin formation by destabilizing the formation of an intramolecular triplex (54,55). This was indicated by the slower mobility of R2-ino compared to that of R2 in the presence (and absence) of Mg2+ (Supplementary Figure SI-8A and B). The TM for R2-ino in the presence of Mg2+ was ∼80°C, similar to the TM of the R2 in the absence of Mg2+ (Figure 6A). This similarity of TM suggests that H-DNA formation is stabilized by both Mg2+ and H-bonding donors (exocyclic 2-amino groups present in guanines). We also observed that the H-DNA structure melted in a single-step, monophasic manner such that the triplex-to-random coil was the transition solely observed at TM ∼90°C, which is similar to previously reported antiparallel, purine-motif, R*RY triplex structures (30,56–59).
Figure 6.

Thermal melting analyses results. Thermal melting analyses monitoring R2 absorbances at (A) 260 nm and (B) 295 nm to assess stabilities under H-DNA-forming and G4-DNA-forming conditions, respectively. Results show a Mg2+- and exocyclic amino-stabilized structure under H-DNA-forming conditions whereas, under G4-DNA-forming conditions, no hypochromicity was detected at A295 suggesting that G4-DNA is not the predominant structure under these conditions.
Under G4-DNA-forming conditions, if intra- or intermolecular G4-DNA structures were formed, then we would expect their dissociation to be accompanied by a decrease in absorbance (hypochromic) at 295 nm, an opposite trend to the melting of a duplex or triplex structure (60). In certain cases, a mixture of stable conformers may afford biphasic melting profiles (61). However, we found that the R2's melting absorbance at 295 nm was hyperchromic with a steady upward trend (Figure 6B). Its trend was unlike that of a known G4-DNA forming sequence (G4-Htel27), which exhibited the G4-DNA characteristic hypochromic melting at 295 nm (60) (Figure 6B), indicating that stable G4-DNA structures are not predominant in solution under these conditions. On the other hand, the TM at 260 nm was determined to be ∼75°C, perhaps consistent with a hairpin structure or other less stable structures.
DISCUSSION
To address the question as to which non-B DNA structure forms in a region containing multiple competing potential non-B DNA-forming sequence elements, we focused on a 23-bp translocation breakpoint hotspot in the human c-MYC oncogene. This region consists of two overlapping sequence motifs associated with the formation of non-B DNA structures: a purine-rich mirror repeat associated with triplex or H-DNA formation, and a G-rich motif associated with G-quadruplex DNA formation. In the context of genomic DNA, superhelical stress can promote duplex unwinding, possibly allowing a competition between complete duplex separation and partial duplex separation. Complete duplex separation can lead to G-quadruplex formation on the G-rich strand, with possible i-motif formation on the complementary C-rich strand (14). Whereas, partial duplex separation can lead to H-DNA formation resulting from the binding of the single-stranded half of the mirror repeat to the remaining duplex portion of the other half of the mirror repeat. In order to better understand the factors that might affect this competition, model oligonucleotides were constructed that encompassed the non-B DNA-forming sequence of this region to detect potential hairpin, H-DNA or G-quadruplex structures. And because the optimal in vitro conditions for H-DNA formation and G4-DNA formation are significantly different, we performed analyses of structure-formation under their respective optimal conditions. These studies indeed demonstrated the propensity of different structures to predominate under these different conditions and allowed the characterization of these different species. We observed that once a structure was formed (i.e. H-DNA structure), it was stable against a competing structure (i.e. G4-DNA). More importantly, this H-DNA structure was found to form in vitro under physiologically relevant buffer conditions.
Our results indicated that under conditions known to stabilize H-DNA/triplex-DNA structures, where Na+ ions neutralize the phosphate backbone, whereas Mg2+ ions stabilize the binding of the third strand (30), the oligonucleotides formed an intramolecular triplex/H-DNA structure. The intramolecular triplex is presumed to be composed of a duplex hairpin connected via the T5 loop, with the G-rich, third strand folding back in an antiparallel fashion to the underlying duplex via reverse Hoogsteen base pairing, facilitated by a -CGCTTAT- loop (Supplementary Figure SI-1). The oligonucleotides folded intramolecularly (Figure 2A) as evidenced by their mobility in a native gel, and this mobility was independent of DNA concentration. These intramolecular structures also possessed CD signatures that characterize triplex formation (Figure 2B). These observations were also seen when we tested for a more physiological buffer in vitro, e.g. Dulbecco's PBS. The third strand of the R2 sequence also appeared to bind to the duplex, because a strategically substituted 2AP in the third strand had a quenched steady-state fluorescence (Figure 4A), which then increased as a function of temperature (Figure 4B). This quenched fluorescence is consistent with 2AP having been protected from solution and thus, being within a rigid, structured environment (i.e. secondary structure such as H-DNA) which then denatured/melted as the temperature increased, exposing the 2AP to the solution with a concomitant increase in fluorescence. This fluorescence increase of the 2AP in the third strand (Figure 4B) did not show as much cooperativity as that of the 2AP within a control hairpin duplex, perhaps due to the greater dissociative propensity of the reverse Hoogsteen base pairs compared to the Watson-Crick base pairs. In a study assessing the dynamics of reverse Hoogsteen base pairs in a similar antiparallel triplex compared to Watson-Crick base pairs, it was found that the dynamics of the triplex reverse Hoogsteen base pair was markedly faster, with shorter lifetimes (i.e. 15 ms for a G*G reverse Hoogsteen base pair versus 76 ms for a GC Watson–Crick base pair) and larger apparent dissociation constant (an order of magnitude larger for the G*G reverse Hoogsteen base pair) (62) than a GC Watson-Crick base pair. With such greater opening dynamics for the reverse Hoogsteen base pairs, it follows that the 2AP in the third strand of the H-DNA structure would tend to dissociate more readily compared with the 2AP within a Watson-Crick hairpin duplex. Moreover, it is interesting to note that we detected such structure-dependent fluorescent trends even at lower salt concentrations in which we performed the 2AP studies, further indicating that this H-DNA structure formation does not require a high concentration of divalent cations.
The 2AP-related results also eliminated the possibility that only a hairpin, composed of the Watson-Crick duplex alone (with a dangling, non-binding third strand) was present in solution. If this were the case, then the 2AP (in AP-A33hg) would have had a similar fluorescence trend with that of the single-strand AP-longSS (2AP of which has the same flanking sequences as that of the third strand of the triplex), which was not seen in our experiments. Furthermore, this was confirmed when we measured the TM of such a hairpin duplex with an inosine-substituted, destabilized third strand (R2-ino) (Figure 6A). Its TM in the presence of Mg2+ (∼80°C) was very similar to the TM of R2 in the absence of Mg2+(∼78°C). The TM's in both cases were markedly low and destabilized compared to the Hoogsteen bonded, Mg2+-dependent structure (i.e. an H-DNA intramolecular triplex structure) with a TM ∼90°C. These findings reiterate that stable triplex formation by binding of the third strand to the hairpin stem occurred in the presence of Hoogsteen bonding capability (via G's exocyclic amino group) and of Mg2+ ions, under triplex/H-DNA-forming conditions. Consistent with these results, the ‘protection’ patterns from DMS and KMnO4 chemical modification assays of the R2-5΄ sequence also indicated the formation of an H-DNA structure. The T15 protection, together with protection of the G24–G30 region, confirmed H-DNA formation and eliminated the possibility of a G4-DNA structure under H-DNA-forming conditions (Figure 5A). If a G4-DNA structure had formed, then only the guanines would be protected and the T15 would not be involved in the structure and hence, unprotected, which was not the case in our experiments. To further eliminate the possibility of G4-DNA formation, we also monitored the temperature-dependent absorbance at 295 nm under H-DNA-forming conditions (Figure 6B). Here, we did not observe any hypochromic trend upon denaturation of the structure, which indicates that a G4-DNA structure was not present in these solution studies.
On the other hand, under G4-DNA-forming conditions, a single, well-defined structure was not observed. Gel mobility assays showed the presence of both high- and low-mobility species in solution (Figure 2C). CD spectroscopy results revealed a signature characteristic of parallel G4-DNA (Figure 2D). This parallel CD signature is not consistent with the formation of bimolecular G4-DNA structures that could have resulted from the interaction between two 5΄-GGGGAGGG-3΄ dangling tails (if the first and second tract of guanines had been involved in a duplex hairpin with a T5 loop). The bimolecular G4-DNA, if formed, would presumably adopt an antiparallel orientation, which we did not observe via CD spectroscopy measurements. Thus, it appears that a mixture of species including either intra- or intermolecular parallel-stranded G4-DNA structures were formed under these conditions, rather than a single stable G4-DNA structure. Fluorescence studies of AP-A33hg, bearing a 2AP moiety in the 3΄-terminal G-track revealed a cooperative denaturation implying G4-DNA structure formation (Figure 4D), with the 2AP as part of a short crossover loop. However, the results from chemical modification and thermal melting studies were inconsistent with exclusive G4-DNA formation. We did not observe marked protection from DMS for the guanine-repeats (Figure 5B). Further, we did not observe a cooperative and hypochromic trend, characteristic of G4-DNA denaturation (Figure 6B) in the temperature-dependent absorbance monitored at 295 nm, although a hyperchromic transition at 260 nm was observed (Figure 6A). Taken together, our results indicate that any G4-DNA structures that may have formed under G4-DNA-forming conditions were accompanied by other, less well-defined structures including denatured, or partially denatured structures.
In the context of competing structures, the intramolecular structure formed under H-DNA-forming conditions was very stable once formed, such that the presence of up to 160 mM K+, which can promote formation of G4-DNA as a competing structure, did not perturb the H-DNA structure as monitored by CD spectroscopy (Figure 3A and D). However, we also found that after the H-DNA structure was denatured (after heating), and upon simultaneous exposure to both K+ and Mg2+, that K+ (at higher concentrations) was able to inhibit the formation of H-DNA (Figure 3B and E), similar to a previous report on the stability of an antiparallel triplex (56).
While most of these studies were conducted in vitro, they are a part of an ensemble of approaches used in our laboratory (e.g. plasmid and in vivo systems) to study non-B DNA structure formation. Thus, we also incorporated non-structure-forming (B-DNA), and the R2-5΄ sequences (MycR25) into a shuttle vector backbone and found that the fragment patterns were similar regardless of the buffer (H-DNA versus G4-DNA-forming) in which the plasmids were pre-incubated, and without multiple fragments expected from cleaved G4-DNA. These results support the conclusion that H-DNA was the predominant structure formed even in the presence of the G4-DNA-forming elements consistent with the other data presented herein. Because acidic conditions were required in the S1 nuclease assay, we could not rule out the possibility that the H-DNA may have isomerized to its low pH-dependent, pyrimidine (Y*RY type) triplex isomer. We have performed nuclease sensitivity studies using plasmids with varying propensities to form H-DNA structures, including the wild-type c-MYC chromosomal breakpoint hotspot sequence (6). We found that an H-DNA structure formed in the plasmid carrying the wild-type c-MYC chromosomal breakpoint hotspot sequence in the solution after cleaving with P1 nuclease (at physiological pH, pH 7.1), also consistent with the data herein.
Our current approach is mechanistically informative on structure formation, and to an extent is predictive in cells. The new details we obtained regarding structure formation, using a wide range of techniques (PAGE, thermal melting, CD and fluorescence spectroscopy, and chemical and enzymatic footprinting) were possible because we focused on the short, but relevant, hotspot region from the human c-MYC DNA sequence. The conditions with which we carried out these studies were also chosen to be compatible with these techniques while tending towards physiologically relevant conditions. Thus, intracellular concentrations of K+ (approximately 0.15 M) (63–65) are similar to those we employed for studying the formation of G4-DNA and in testing stability of the pre-formed H-DNA structures. Intracellular concentrations of ‘free’ Mg2+ of up to 1 mM (estimated bound concentration of ∼8 mM) (66) are lower than the 5–10 mM Mg2+ employed in these studies. In vitro, the lower, physiological Mg2+ concentration can only partially stabilize G*GC triads (67,68); however, there are a number of other factors that can act to stabilize intramolecular triplex/H-DNA formation in the presence of limited ‘free’ Mg2+ in cells. These include possible transient increases in Mg2+ levels due to the release of bound Mg2+, e.g. from a number of physiological chelators (69), for example, a major Mg2+ binder, ATP (adenosine triphosphate) with an intracellular concentration of 1–10 mM (65,70) releases the divalent ion after ATP-signalling events (69). In cells, there are also other biologically-relevant divalent ions such as Zn2+, Mn2+ and Co2+, which can be even more potent than Mg2+ in supporting triplex formation (68,71–73). In addition, H-DNA formation may be facilitated by the presence of triplex-specific binding proteins that may promote DNA bending accompanying triplex formation (74,75). These conditions/factors are simultaneously available in cells, together with the assured presence of negative supercoiling and other cationic macromolecules such as polyamines, which are also known to stabilize H-DNA structures (76). Moreover, there have been reports of the formation of triplexes in the absence of divalent cations (30,58,77), where Mg2+ stabilized the triplex structure through inhibition of the dissociation of the Hoogsteen strand (77). Additionally, the formation and dynamics of non-B DNA structures may also be affected by factors such as DNA helicases that can unwind these structures (78,79). The studies presented here provide the basis for evaluating these additional factors and establish the predominant non-B DNA structure of interest.
These characterizations indicated that in the presence of multiple, overlapping non-B DNA-forming sequences tested herein, only the intramolecular triplex predominated under H-DNA-forming conditions. The predominance of H-DNA formation implicates this structure in the genome instability associated with the human c-MYC oncogene region. Our current results are consistent with our previous report demonstrating that upon modifications of the wild-type c-MYC sequence (symmetrical permutation or nucleotide substitution to promote exclusive H-DNA or G4-DNA formation), only the sequence forming H-DNA impeded transcription similar to that of the wild-type sequence (25). This follows that the H-DNA, and not the G4-DNA, modulates transcription in the chromosomal breakpoint hotspot sequence of the human c-MYC oncogene. Having demonstrated the same structure formation within the context of an oligonucleotide, our current results now provide a well-characterized oligonucleotide intramolecular triplex model with a cancer-relevant sequence: that of a mutational hotspot in the c-MYC oncogene implicated in haematological diseases such as Burkitt's lymphoma and leukemia (9–11,80,81). These models will aid in addressing challenges regarding the possible effect/s of alternative DNA structures on genetic instability in genetic diseases. In particular, they can be used as substrates in screening small-molecule libraries for potential ligands that may be used, either to visualize the occurrence of H-DNA in vivo, or to modulate the kinetics of H-DNA structure formation, leading to a better understanding of the biological roles of H-DNA and its propensity for targeting which may be helpful for therapies against genetic diseases. Further, the models can also be used as a proof-of-principle in elucidating the various proteins involved in mechanistic processing of H-DNA via protein–DNA interaction experiments. For example, in a recent related investigation (Zhao et al., unpublished data, 2017), we were able to demonstrate for the first time that R2-5΄, as an intramolecular triplex/H-DNA, is a novel substrate of DNA structure-specific repair-related nucleases such as ERCC1-XPF and XPG, and DNA replication-related flap endonuclease 1 (FEN1), indicating that non-B DNA structures are indeed processed by DNA repair mechanisms within cells, and thus, merits further investigation. Thus, these results will provide insight into the basic processing of non-B DNA and will assist in elucidating the mechanisms of genetic instability induced at endogenous mutation hotspots.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Tyler Lieberthal of Imperial College London for the help with the illustrations; Dominic McBrayer, Olivia Drummond, Sarah Coe and Albert Lee for technical assistance; Wendi David and the members of the Vasquez laboratory for their valuable discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health/National Cancer Institute (NIH/NCI) [CA093729 to K.M.V.]; Cancer Prevention Research Institute of Texas (CPRIT) fellowship [ID RP140108 to I.M.A.d.M.]. M.Z.-F. acknowledges grant support for undergraduate student research from the Jean Dreyfus Boissevain Lectureship for Undergraduate Institutions, the Robert A. Welch Foundation [AF-0005], and the Howard Hughes Medical Institute through the Undergraduate Science Education Program [52007558]. Funding for open access charge: NIH/NCI [CA093729 to K.M.V.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bacolla A., Tainer J.A., Vasquez K.M., Cooper D.N.. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 2016; 44:5673–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Javadekar S.M., Raghavan S.C.. Snaps and mends: DNA breaks and chromosomal translocations. FEBS J. 2015; 282:2627–2645. [DOI] [PubMed] [Google Scholar]
- 3. Zhao J., Bacolla A., Wang G., Vasquez K.M.. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010; 67:43–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang G., Carbajal S., Vijg J., DiGiovanni J., Vasquez K.M.. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008; 100:1815–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang G., Christensen L.A., Vasquez K.M.. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:2677–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang G., Vasquez K.M.. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:13448–13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nag D.K., Kurst A.. A 140-bp-long palindromic sequence induces double-strand breaks during meiosis in the yeast Saccharomyces cerevisiae. Genetics. 1997; 146:835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Glickman B.W., Ripley L.S.. Structural intermediates of deletion mutagenesis: a role for palindromic DNA. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilda M., Busch K., Klose I., Keller T., Woessmann W., Kreuder J., Harbott J., Borkhardt A.. Level of MYC overexpression in pediatric Burkitt's lymphoma is strongly dependent on genomic breakpoint location within the MYC locus. Genes Chromosomes Cancer. 2004; 41:178–182. [DOI] [PubMed] [Google Scholar]
- 10. Akasaka T., Akasaka H., Ueda C., Yonetani N., Maesako Y., Shimizu A., Yamabe H., Fukuhara S., Uchiyama T., Ohno H.. Molecular and clinical features of non-Burkitt's, diffuse large-cell lymphoma of B-cell type associated with the c-MYC/immunoglobulin heavy-chain fusion gene. J. Clin. Oncol. 2000; 18:510–518. [DOI] [PubMed] [Google Scholar]
- 11. Saglio G., Grazia Borrello M., Guerrasio A., Sozzi G., Serra A., di Celle P.F., Foa R., Ferrarini M., Roncella S., Borgna Pignatti C. et al. . Preferential clustering of chromosomal breakpoints in Burkitt's lymphomas and L3 type acute lymphoblastic leukemias with a t(8;14) translocation. Genes Chromosomes Cancer. 1993; 8:1–7. [DOI] [PubMed] [Google Scholar]
- 12. Nambiar M., Goldsmith G., Moorthy B.T., Lieber M.R., Joshi M.V., Choudhary B., Hosur R.V., Raghavan S.C.. Formation of a G-quadruplex at the BCL2 major breakpoint region of the t(14;18) translocation in follicular lymphoma. Nucleic Acids Res. 2011; 39:936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nambiar M., Raghavan S.C.. How does DNA break during chromosomal translocations. Nucleic Acids Res. 2011; 39:5813–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qin Y., Hurley L.H.. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie. 2008; 90:1149–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ambrus A., Chen D., Dai J., Jones R.A., Yang D.. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry. 2005; 44:2048–2058. [DOI] [PubMed] [Google Scholar]
- 16. Mirkin S.M., Frank-Kamenetskii M.D.. H-DNA and related structures. Annu. Rev. Biophys. Biomol. Struct. 1994; 23:541–576. [DOI] [PubMed] [Google Scholar]
- 17. Jain A., Wang G., Vasquez K.M.. DNA triple helices: biological consequences and therapeutic potential. Biochimie. 2008; 90:1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kang S.M., Wohlrab F., Wells R.D.. Metal ions cause the isomerization of certain intramolecular triplexes. J. Biol. Chem. 1992; 267:1259–1264. [PubMed] [Google Scholar]
- 19. Hoyne P.R., Gacy A.M., McMurray C.T., Maher L.J. 3rd. Stabilities of intrastrand pyrimidine motif DNA and RNA triple helices. Nucleic Acids Res. 2000; 28:770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S.. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006; 34:5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fogolari F., Haridas H., Corazza A., Viglino P., Cora D., Caselle M., Esposito G., Xodo L.E.. Molecular models for intrastrand DNA G-quadruplexes. BMC Struct. Biol. 2009; 9:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang G., Vasquez K.M.. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair (Amst.). 2014; 19:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eick D., Polack A., Kofler E., Lenoir G.M., Rickinson A.B., Bornkamm G.W.. Expression of P0- and P3-RNA from the normal and translocated c-myc allele in Burkitt's lymphoma cells. Oncogene. 1990; 5:1397–1402. [PubMed] [Google Scholar]
- 24. Hoover R.G., Kaushal V., Lary C., Travis P., Sneed T.. c-myc transcription is initiated from P0 in 70% of patients with multiple myeloma. Curr. Top. Microbiol. Immunol. 1995; 194:257–264. [DOI] [PubMed] [Google Scholar]
- 25. Belotserkovskii B.P., De Silva E., Tornaletti S., Wang G., Vasquez K.M., Hanawalt P.C.. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J. Biol. Chem. 2007; 282:32433–32441. [DOI] [PubMed] [Google Scholar]
- 26. Wang G., Vasquez K.M.. Non-B DNA structure-induced genetic instability. Mutat. Res. 2006; 598:103–119. [DOI] [PubMed] [Google Scholar]
- 27. Marcu K.B., Bossone S.A., Patel A.J.. myc Function and regulation. Annu. Rev. Biochem. 1992; 61:809–860. [DOI] [PubMed] [Google Scholar]
- 28. Maxam A.M., Gilbert W.. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980; 65:499–560. [DOI] [PubMed] [Google Scholar]
- 29. Pedroso I.M., Duarte L.F., Yanez G., Burkewitz K., Fletcher T.M.. Sequence specificity of inter- and intramolecular G-quadruplex formation by human telomeric DNA. Biopolymers. 2007; 87:74–84. [DOI] [PubMed] [Google Scholar]
- 30. Kaushik S., Kaushik M., Svinarchuk F., Malvy C., Fermandjian S., Kukreti S.. Presence of divalent cation is not mandatory for the formation of intramolecular purine-motif triplex containing human c-jun protooncogene target. Biochemistry. 2011; 50:4132–4142. [DOI] [PubMed] [Google Scholar]
- 31. Kypr J., Kejnovska I., Renciuk D., Vorlickova M.. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009; 37:1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baase W.A., Johnson W.C. Jr. Circular dichroism and DNA secondary structure. Nucleic Acids Res. 1979; 6:797–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Avino A., Frieden M., Morales J.C., Garcia de la Torre B., Guimil Garcia R., Azorin F., Gelpi J.L., Orozco M., Gonzalez C., Eritja R.. Properties of triple helices formed by parallel-stranded hairpins containing 8-aminopurines. Nucleic Acids Res. 2002; 30:2609–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glick G.D. Design, synthesis, and analysis of conformationally constrained nucleic acids. Biopolymers. 1998; 48:83–96. [DOI] [PubMed] [Google Scholar]
- 35. Xodo L.E., Manzini G., Quadrifoglio F.. Spectroscopic and calorimetric investigation on the DNA triplex formed by d(CTCTTCTTTCTTTTCTTTCTTCTC) and d(GAGAAGAAAGA) at acidic pH. Nucleic Acids Res. 1990; 18:3557–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sugimoto N., Wu P., Hara H., Kawamoto Y.. pH and cation effects on the properties of parallel pyrimidine motif DNA triplexes. Biochemistry. 2001; 40:9396–9405. [DOI] [PubMed] [Google Scholar]
- 37. Karsisiotis A.I., Hessari N.M.A., Novellino E., Spada G.P., Randazzo A., Webba da Silva M.. Topological characterization of nucleic acid G-quadruplexes by UV absorption and circular dichroism. Angew. Chem. 2011; 123:10833–10836. [DOI] [PubMed] [Google Scholar]
- 38. Dapic V., Abdomerovic V., Marrington R., Peberdy J., Rodger A., Trent J.O., Bates P.J.. Biophysical and biological properties of quadruplex oligodeoxyribonucleotides. Nucleic Acids Res. 2003; 31:2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hardin C.C., Henderson E., Watson T., Prosser J.K.. Monovalent cation induced structural transitions in telomeric DNAs: G-DNA folding intermediates. Biochemistry. 1991; 30:4460–4472. [DOI] [PubMed] [Google Scholar]
- 40. Rajendran A., Nair B.U.. Unprecedented dual binding behaviour of acridine group of dye: a combined experimental and theoretical investigation for the development of anticancer chemotherapeutic agents. Biochim. Biophys. Acta. 2006; 1760:1794–1801. [DOI] [PubMed] [Google Scholar]
- 41. Cheng A.J., Van Dyke M.W.. Monovalent cation effects on intermolecular purine-purine-pyrimidine triple-helix formation. Nucleic Acids Res. 1993; 21:5630–5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gray R.D., Petraccone L., Buscaglia R., Chaires J.B.. 2-aminopurine as a probe for quadruplex loop structures. Methods Mol. Biol. 2010; 608:121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kimura T., Kawai K., Fujitsuka M., Majima T.. Monitoring G-quadruplex structures and G-quadruplex-ligand complex using 2-aminopurine modified oligonucleotides. Tetrahedron. 2007; 63:3585–3590. [Google Scholar]
- 44. Johnson J., Okyere R., Joseph A., Musier-Forsyth K., Kankia B.. Quadruplex formation as a molecular switch to turn on intrinsically fluorescent nucleotide analogs. Nucleic Acids Res. 2013; 41:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shchyolkina A.K., Kaluzhny D.N., Borisova O.F., Hawkins M.E., Jernigan R.L., Jovin T.M., Arndt-Jovin D.J., Zhurkin V.B.. Formation of an intramolecular triple-stranded DNA structure monitored by fluorescence of 2-aminopurine or 6-methylisoxanthopterin. Nucleic Acids Res. 2004; 32:432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bandwar R.P., Patel S.S.. Peculiar 2-aminopurine fluorescence monitors the dynamics of open complex formation by bacteriophage T7 RNA polymerase. J. Biol. Chem. 2001; 276:14075–14082. [DOI] [PubMed] [Google Scholar]
- 47. Wan C., Fiebig T., Schiemann O., Barton J.K., Zewail A.H.. Femtosecond direct observation of charge transfer between bases in DNA. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:14052–14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kelley S.O., Barton J.K.. Electron transfer between bases in double helical DNA. Science. 1999; 283:375–381. [DOI] [PubMed] [Google Scholar]
- 49. Rokita S.E. Glick G. Chemical reagents for investigating the major groove of DNA. Curr. Protoc. Nucleic Acid Chem. 2001; NY: Wiley; 6.6.1–6.6.16. [DOI] [PubMed] [Google Scholar]
- 50. Kahl B.F., Paule M.R.. The use of diethyl pyrocarbonate and potassium permanganate as probes for strand separation and structural distortions in DNA. Methods Mol. Biol. 2009; 543:73–85. [DOI] [PubMed] [Google Scholar]
- 51. Haner R., Dervan P.B.. Single-strand DNA triple-helix formation. Biochemistry. 1990; 29:9761–9765. [DOI] [PubMed] [Google Scholar]
- 52. Maxam A.M., Gilbert W.. A new method for sequencing DNA. Proc. Natl. Acad. Sci. U.S.A. 1977; 74:560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gondeau C., Maurizot J.C., Durand M.. Circular dichroism and UV melting studies on formation of an intramolecular triplex containing parallel T*A:T and G*G:C triplets: netropsin complexation with the triplex. Nucleic Acids Res. 1998; 26:4996–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mills M., Volker J., Klump H.H.. Triple helical structures involving inosine: there is a penalty for promiscuity. Biochemistry. 1996; 35:13338–13344. [DOI] [PubMed] [Google Scholar]
- 55. Cubero E., Guimil-Garcia R., Luque F.J., Eritja R., Orozco M.. The effect of amino groups on the stability of DNA duplexes and triplexes based on purines derived from inosine. Nucleic Acids Res. 2001; 29:2522–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gondeau C., Maurizot J.C., Durand M.. Spectroscopic investigation of an intramolecular DNA triplex containing both G.G:C and T.A:T triads and its complex with netropsin. J. Biomol. Struct. Dyn. 1998; 15:1133–1145. [DOI] [PubMed] [Google Scholar]
- 57. Scaria P.V., Shafer R.H.. Calorimetric analysis of triple helices targeted to the d(G3A4G3).d(C3T4C3) duplex. Biochemistry. 1996; 35:10985–10994. [DOI] [PubMed] [Google Scholar]
- 58. Chen F.M. Intramolecular triplex formation of the purine.purine.pyrimidine type. Biochemistry. 1991; 30:4472–4479. [DOI] [PubMed] [Google Scholar]
- 59. Pilch D.S., Levenson C., Shafer R.H.. Structure, stability, and thermodynamics of a short intermolecular purine-purine-pyrimidine triple helix. Biochemistry. 1991; 30:6081–6088. [DOI] [PubMed] [Google Scholar]
- 60. Mergny J.L., Phan A.T., Lacroix L.. Following G-quartet formation by UV-spectroscopy. FEBS Lett. 1998; 435:74–78. [DOI] [PubMed] [Google Scholar]
- 61. Rachwal P.A., Fox K.R.. Quadruplex melting. Methods. 2007; 43:291–301. [DOI] [PubMed] [Google Scholar]
- 62. Warmlander S., Sandstrom K., Leijon M., Graslund A.. Base-pair dynamics in an antiparallel DNA triplex measured by catalyzed imino proton exchange monitored via 1H NMR spectroscopy. Biochemistry. 2003; 42:12589–12595. [DOI] [PubMed] [Google Scholar]
- 63. Moore R.D., Morrill G.A.. A possible mechanism for concentrating sodium and potassium in the cell nucleus. Biophys. J. 1976; 16:527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Paine P.L., Pearson T.W., Tluczek L.J., Horowitz S.B.. Nuclear sodium and potassium. Nature. 1981; 291:258–259. [DOI] [PubMed] [Google Scholar]
- 65. Ammann H., Noel J., Tejedor A., Boulanger Y., Gougoux A., Vinay P.. Could cytoplasmic concentration gradients for sodium and ATP exist in intact renal cells. Can. J. Physiol. Pharmacol. 1995; 73:421–435. [DOI] [PubMed] [Google Scholar]
- 66. Traut T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994; 140:1–22. [DOI] [PubMed] [Google Scholar]
- 67. Singleton S.F., Dervan P.B.. Equilibrium association constants for oligonucleotide-directed triple helix formation at single DNA sites: linkage to cation valence and concentration. Biochemistry. 1993; 32:13171–13179. [DOI] [PubMed] [Google Scholar]
- 68. Lyamichev V.I., Voloshin O.N., Frank-Kamenetskii M.D., Soyfer V.N.. Photofootprinting of DNA triplexes. Nucleic Acids Res. 1991; 19:1633–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grubbs R.D. Intracellular magnesium and magnesium buffering. Biometals. 2002; 15:251–259. [DOI] [PubMed] [Google Scholar]
- 70. Beis I., Newsholme E.A.. The contents of adenine nucleotides, phosphagens and some glycolytic intermediates in resting muscles from vertebrates and invertebrates. Biochem J. 1975; 152:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Blume S.W., Lebowitz J., Zacharias W., Guarcello V., Mayfield C.A., Ebbinghaus S.W., Bates P., Jones D.E. Jr, Trent J., Vigneswaran N. et al. . The integral divalent cation within the intermolecular purine*purine. pyrimidine structure: a variable determinant of the potential for and characteristics of the triple helical association. Nucleic Acids Res. 1999; 27:695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wan C., Cui M., Song F., Liu Z., Liu S.. Evaluation of effects of bivalent cations on the formation of purine-rich triple-helix DNA by ESI-FT-MS. J. Am. Soc. Mass Spectrom. 2009; 20:1281–1286. [DOI] [PubMed] [Google Scholar]
- 73. Bernues J., Beltran R., Casasnovas J.M., Azorin F.. Structural polymorphism of homopurine–homopyrimidine sequences: the secondary DNA structure adopted by a d(GA.CT)22 sequence in the presence of zinc ions. EMBO J. 1989; 8:2087–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zain R., Sun J.S.. Do natural DNA triple-helical structures occur and function in vivo?. Cell. Mol. Life Sci. 2003; 60:862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Buske F.A., Mattick J.S., Bailey T.L.. Potential in vivo roles of nucleic acid triple-helices. RNA Biol. 2011; 8:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Soyfer V.N., Potaman V.N.. Triple-Helical Nucleic Acids. 1995; NY: Springer-Verlag. [Google Scholar]
- 77. Vasquez K.M., Wensel T.G., Hogan M.E., Wilson J.H.. High-affinity triple helix formation by synthetic oligonucleotides at a site within a selectable mammalian gene. Biochemistry. 1995; 34:7243–7251. [DOI] [PubMed] [Google Scholar]
- 78. Jain A., Bacolla A., Chakraborty P., Grosse F., Vasquez K.M.. Human DHX9 helicase unwinds triple-helical DNA structures. Biochemistry. 2010; 49:6992–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jain A., Bacolla A., del Mundo I.M.A., Zhao J., Wang G., Vasquez K.M.. DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res. 2013; 41:10345–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Firulli A.B., Maibenco D.C., Kinniburgh A.J.. The identification of a tandem H-DNA structure in the c-myc nuclease sensitive promoter element. Biochem. Biophys. Res. Commun. 1992; 185:264–270. [DOI] [PubMed] [Google Scholar]
- 81. Kinniburgh A.J. A cis-acting transcription element of the c-myc gene can assume an H-DNA conformation. Nucleic Acids Res. 1989; 17:7771–7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.