

Abstract
White phosphorus is well known as a potent hepatotoxin and a severe local and systemic toxin causing damage to gastrointestinal, hepatic, cardiovascular, and renal systems. It is used in the manufacture of matches, fireworks, rodenticide, and fertilizers. Death results due to acute liver failure. Management of yellow phosphorus (YP) poisoning is supportive with no antidote available. Here, we present a case of acute YP poisoning in a 25-year-old female presenting with fulminant hepatic failure and duodenal perforation.
Keywords: Acute poisoning, antidote, duodenal perforation, hepatotoxin, intoxication, supportive treatment, yellow phosphorus
INTRODUCTION
Three forms of phosphorus are available – white, red, and black. “Yellow phosphorus” (YP) is formed by a small amount of red phosphorus resulting discoloration of white phosphorus.[1] YP is a general protoplasmic toxin[2] and is used in the manufacture of fireworks, rodenticide, and fertilizers. Rodenticides are available as powders or pastes containing 2%–5% of YP. The estimated dose of YP that is lethal to the liver is 1 mg/kg, and the ingestion of that amount results in death due to acute liver failure and cardiovascular collapse.[3] Although YP poisoning may result from industrial accidents in developed countries, it also occurs in adults who are attempting suicide and accidentally by oral intake in children, particularly on festival days, in developing countries. The only definitive treatment for acute liver failure due to the ingestion of YP is liver transplantation because no antidote or medical treatment is available to reverse the toxic effects on the liver.[4] Here, we present a case of acute YP poisoning in a 25-year-old female presenting with fulminant hepatic failure (FHF) and duodenal perforation.
CASE REPORT
A 25-year-old young female presented with ingestion of 5–6 pinches (around 2 g) rodenticide poison containing 3% phosphorous in the form of a paste. Multiple episodes of vomiting, abdominal pain, and hematemesis were the complaints on presentation. On evaluation, the patient had jaundice on the second day of admission. There was no evidence of hepatic encephalopathy. Serology for hepatitis B surface antigen, hepatitis C virus, dengue, Leptospira, and hepatitis was negative. Chest X-ray revealed mild right pleural effusion [Figure 1]. Abdominal sonography done on day 2 showed mild hepatomegaly and diffuse gallbladder (GB) wall edema [Figure 2]. Unenhanced computed tomography (CT) abdomen showed multiple patchy areas of homogeneous hyperattenuation (30–40 HU) in segment IVb, periportal region, and in the GB fossa [Figures 3 and 4]. GB wall appeared edematous [Figure 5]. Postcontrast, these regions showed enhancement (90–95 HU) suggesting patchy parenchymal hemorrhages of the liver. There was breach in the continuity of wall of second part of duodenum seen along the medial aspect with a contained pocket of air (pneumoperitoneum) suggesting contained duodenal perforation [Figure 6], with adjacent duodenal wall thickening [Figure 7] which could represent the site of viscus perforation and associated dirty fat density and local fluid between the duodenum and the pancreatic head [Figure 8]. Her total leukocyte count was 1.800 (109/L) on the day of admission, which improved to 4.400 (109/L) on the 7th day. She developed a spike of fever on day 5 of hospitalization. She had a temperature of 101.5°F, tachycardia (pulse of 150 counts/min), a respiratory rate of 30 counts/min, and a blood pressure of 100/70 mmHg. Her platelet counts were 1.53 (109/L) on the day of admission, which decreased daily from 1.28 (109/L) on the second day to 0.80 (109/L) on day 7 of admission. Platelet counts reached normal levels on day 11. Her renal function tests and electrolytes were normal (sodium 137 mmol/L, potassium 3.5 mmol/L, and chloride 100 mmol/L). Urine examination showed the presence of 48–50 red blood cells and 8–10 pus cells per high power field. Horseradish peroxidase 2 was negative. There was no hypoglycemia (blood glucose 108 mg/dl). Arterial blood gases revealed pH – 7.11, PCO2-20.6, PO2-75.4, and HCO3-6.3 suggesting metabolic acidosis. No cardiac dysrhythmias or electrocardiography changes were demonstrated. Intake and output were strictly monitored, and blood glucose was measured 6 hourly. Metabolic acidosis was corrected by sodium bicarbonate infusion. Central line insertion and endotracheal intubation were done, and the patient was treated with ventilatory support. Prothrombin time (PT) started deranging from the second day of admission with a crest of 38 s and international normalized ratio (INR) 2.98 noted on day 5. Total bilirubin levels mounted up to 10.5 mg/dl on day 2 of admission with liver enzymes serum glutamic pyruvic transaminase (SGPT) and serum glutamic oxaloacetic transaminase (SGOT) peaking to 1722 U/L and 2954 U/L, respectively. She was treated with intravenous (IV) Vitamin K, IV fluids, IV antibiotics, and fresh frozen plasma. In view of impending FHF, he was given N-acetyl cysteine, a loading dose of 150 mg/kg IV over 1 h, followed by 50 mg/kg IV over 4 h and 100 mg/kg IV over 16 h. Patient's PT/INR became stabilized. After bleeding parameters were stabilized, she was discharged from the hospital on day 15. On follow-up after 2 weeks, INR remained 1.11 with SGPT 29 (U/L), SGOT 27 (U/L), and total bilirubin 1.8 mg/dl. Laparoscopic surgical management of the perforated duodenal ulcer was done with Graham omental patch and a thorough abdominal lavage, and patient was started on proton-pump inhibitors.
Figure 1.
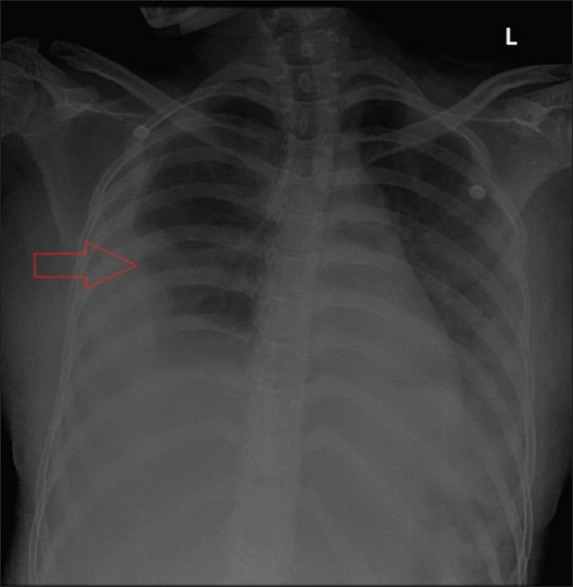
Chest X-ray in anteroposterior view showing mild right pleural effusion (arrow)
Figure 2.
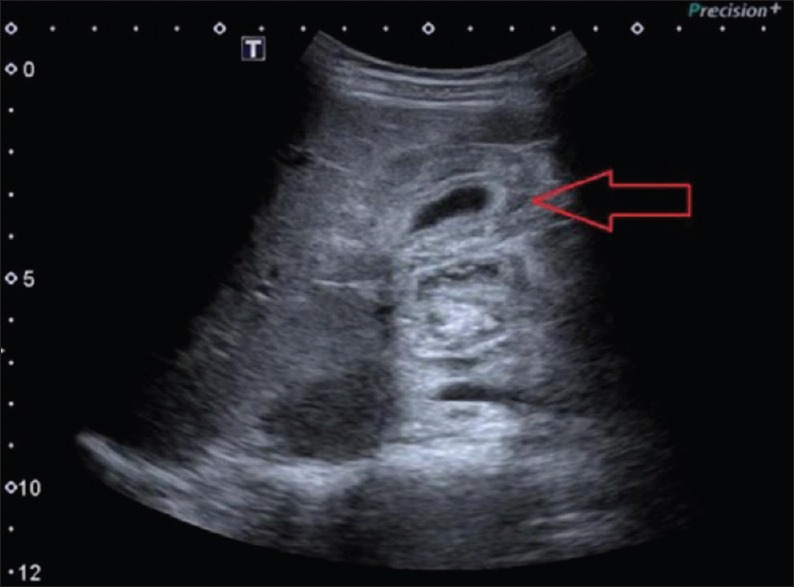
Abdominal ultrasonography image showing diffuse gallbladder wall edema (arrow)
Figure 3.
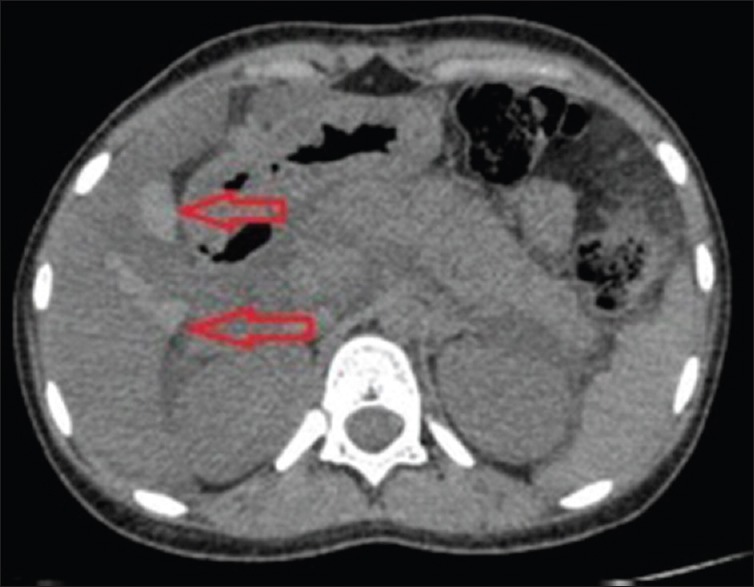
Axial unenhanced computerized tomography abdomen image showing multiple patchy areas of homogeneous hyperattenuation in segment IV, gallbladder fossa, and periportal region of the liver (arrows) suggesting parenchymal hemorrhages
Figure 4.
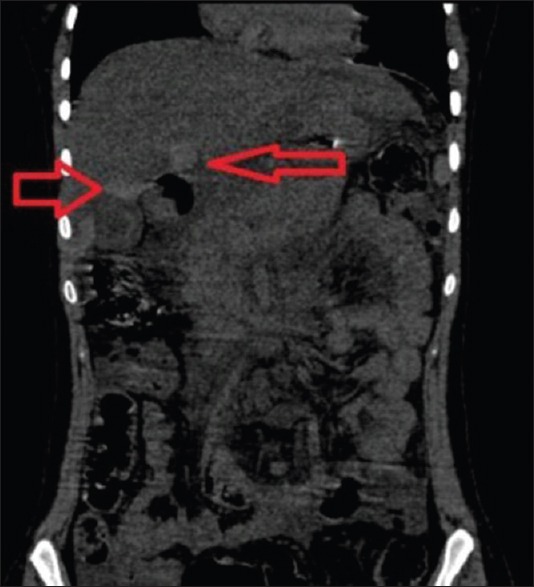
Coronal unenhanced computerized tomography abdomen image showing multiple patchy areas of homogeneous hyperattenuation in segment IV, gallbladder fossa, and periportal region of the liver (arrows) suggesting parenchymal hemorrhages
Figure 5.
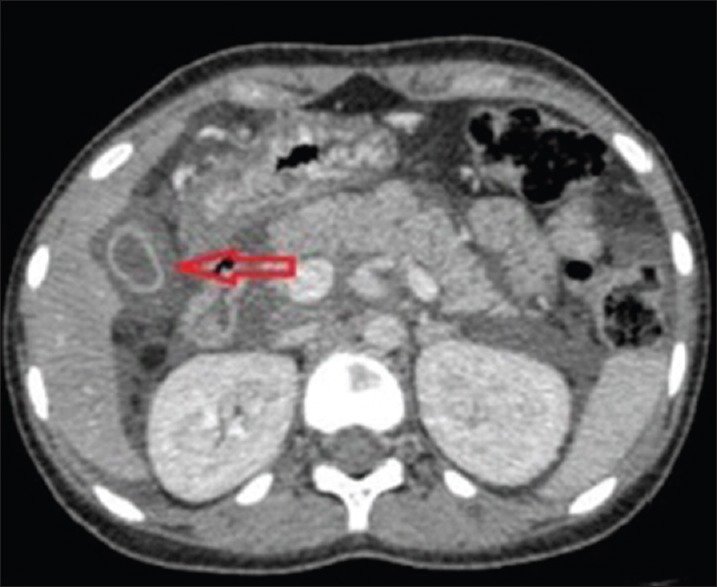
Axial contrast-enhanced computerized tomography abdomen image showing edematous gallbladder wall (arrow). Note free fluid in the abdomen
Figure 6.
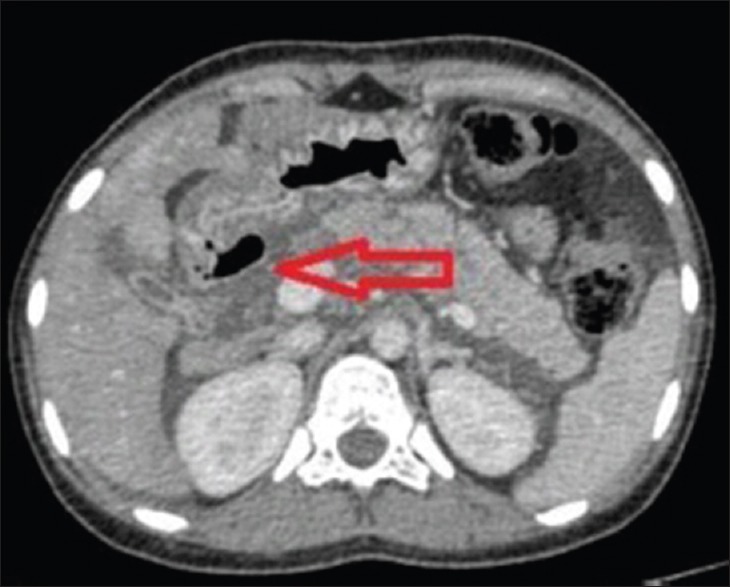
Axial contrast-enhanced computerized tomography abdomen image showing breach in the continuity of wall of second part of duodenum seen along the medial aspect with a contained pocket of air (arrow) suggesting pneumoperitoneum
Figure 7.
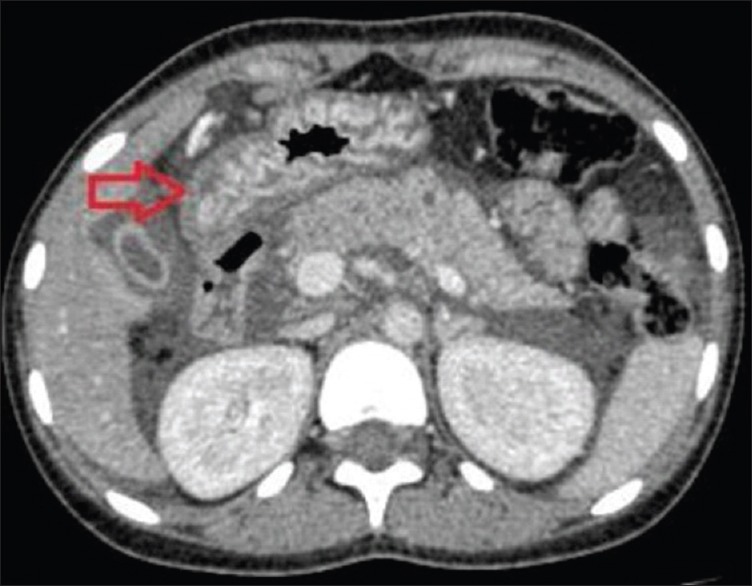
Axial contrast-enhanced computerized tomography abdomen image showing free gas within the peritoneal cavity adjacent to the first part of the duodenum with wall thickening (arrow) which could represent the site of viscus perforation in the region of the junction of the first and second part of the duodenum
Figure 8.
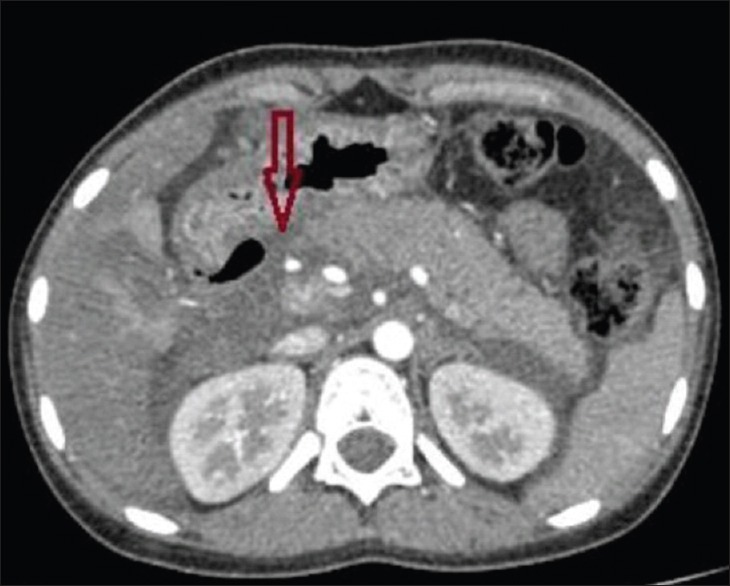
Axial contrast-enhanced computerized tomography abdomen image showing air foci in contact with the second part of the duodenum with abrupt wall thickening associated with adjacent dirty fat density and local fluid between the duodenum and the pancreatic head (arrow)
DISCUSSION
White phosphorus is an inorganic substance used in the production of fireworks, firecrackers, ammunition, rodenticide, and fertilizers. It causes direct hepatotoxicity typically resulting in periportal injury. There are several pastes and powders available in India that contain phosphorous. The usual fatal dose is approximately 1 mg/kg of body weight. Fulminant poisoning results from ingestion of more than 1 to 2 g.[5]
In our patient, 2 h following the ingestion of YP, she complained of nausea and repeated vomiting, for which she admitted to our hospital. Multiple episodes of vomiting, abdominal pain, and hematemesis were the complaints on presentation. She was provided symptomatic treatment in the form of stomach wash (gastric lavage) and antiemetics. On examination, her general condition was not satisfactory and she was stuporous. She had a temperature of 101.5°F, tachycardia (pulse of 150 beats/min), a respiratory rate of 30 counts/min, and a blood pressure of 90/60 mmHg in the supine position. Glasgow coma scale score was 8. On day 2, she had intractable vomiting with altered level consciousness and irritability. She was hemodynamically unstable and developed metabolic acidosis. The patient revealed that deep icterus (over skin and sclera) and multiple ecchymotic patches of varying sizes ranging from 0.5 to 5 cm were seen over extremities. In addition, there was bilateral periorbital swelling with a chemosed conjunctiva. Nasogastric tube was inserted, and the aspirate revealed the presence of blood (75 ml) which gave us a suspicion of a gastroduodenal ulcer. Sonographic imaging of the abdomen revealed fatty liver with GB wall edema with mild ascites. CT revealed multiple parenchymal hemorrhages of the liver, breach in the continuity of wall of second part of the duodenum with associated pneumoperitoneum. Based on the above findings, a diagnosis of acute FHF secondary to toxic hepatitis with a complication of duodenal perforation following a rodenticide poisoning was made. Metabolic acidosis was corrected by sodium bicarbonate infusion. Central line insertion and endotracheal intubation were done. The patient was treated with ventilatory support. Her falling blood pressure was managed with IV fluids, vasopressors, and inotropes.
The clinical effects of acute poisoning with YP have classically been divided into three stages. The initial gastrointestinal (GI) stage is characterized by vomiting, nausea, diarrhea, and abdominal pain, which occur within the first 24 h after ingestion. Laboratory tests are almost normal during this period. During this stage, sudden death may occur; it may be that the ingestion of a very large amount of fireworks can directly result in cardiovascular arrhythmia and collapse within the first 24 h. Cardiac failure may also occur because of fluid and electrolyte loss due to vomiting or diarrhea in addition to cardiac toxicity with repeated electrocardiograms demonstrating dysrhythmias, widened QRS complexes, or depression of ST segments, and early death is generally due to cardiac dysrhythmias secondary to electrolyte abnormalities such as hypocalcemia and hyperkalemia.[6] The second stage (1–4 days), which may last for several days, is essentially a symptom-free period, but liver enzyme levels become elevated, and toxic hepatitis begins to spread. The third stage can end in acute liver failure and acute renal failure with metabolic derangements, encephalopathy, coagulopathy, arrhythmia, cardiogenic shock, and abnormal liver tests. Central nervous system effects include changes in mental status such as confusion, psychosis, hallucinations, and coma. In short, this is a multiorgan failure; the third stage occurred between 4 and 7 days. Patients progress to the third stage because of the systemic effects of high-dose phosphorus after it has been absorbed. If liver transplantation, which is the only treatment during the last stage, is not performed, death is inevitable.
Garlic odor, mucosal burns, and phosphorescent vomitus or feces occurred in only a small percentage of cases.[7] The treatment should include the removal of phosphorus and general supportive measures only during the first and second stages. In the past, acute poisoning management involved removal of the toxin with gastric lavage and potassium permanganate (1:5000), which oxidizes phosphorus into relatively less toxic phosphoric acid and phosphates.[3] The administration of potassium permanganate is not safe and is no longer used in the treatment of phosphorus poisoning.
Studies have shown no clear benefit from N-acetyl cysteine, corticosteroids, or exchange transfusion.[8] She recovered within 3 days after giving N-acetyl cysteine (20 h regimen) which has shown clinical benefit in the presenting case. Her INR improved from 2.98 to 1.0 within three-dimensional and stabilized thereafter. The regimen of N-acetyl cysteine used was a loading dose of 150 mg/kg IV over 1 h, followed by 50 mg/kg IV over 4 h and 100 mg/kg IV over 16 h. This treatment regimen may be proposed for use by clinical toxicologists for acute white phosphorus poisoning. Careful monitoring of hepatic and renal function and management of their failure is required. There is no antidote for yellow phosphorous poisoning. Liver transplantation may be lifesaving in candidates with acute hepatic failure.[9]
Abdominal ultrasonography with Doppler can demonstrate fatty changes of the liver, solid-organ injury, hepatic outflow obstruction, and/or biliary tract obstruction. Contrast-enhanced CT of the abdomen is the investigation of choice to demonstrate parenchymal hemorrhages of the liver as well as their resolution and for the evaluation of bowel which can demonstrate intestinal perforation with pneumoperitoneum[10] as noted in our patient.
The presented patient allegedly ingested YP, which is used as a rodenticide in households and agricultural farms. The toxicological properties were consistent with the known pharmacological effects of YP poisoning such as nausea, vomiting, jaundice, pitting edema, oliguria, high pulse rate, and low blood pressure which are signs of hepatic, renal, and cardiovascular system compromise. In our patient, the first manifestation of intoxication was restlessness, irritability, drowsiness, along with primary GI symptoms such as multiple episodes of vomiting, abdominal pain, hematemesis, and developed jaundice on the second day of admission with progression to FHF evidenced as parenchymal hemorrhages of the liver. She had duodenal perforation subsequently. Similar to the reports of Fernandez and Canizares, 1995, patients with YP poisoning mainly present with acute hepatic failure, coagulopathy, and deranged liver function, early elevations in transaminase, alkaline phosphatase, derangement in PT, and associated metabolic acidosis and was witnessed in our patient. She was treated with IV Vitamin K, IV fluids, IV antibiotics, and fresh frozen plasma and was discharged on day 15 after bleeding parameters were stabilized. It is evident that large-scale use of other anticoagulant rodenticide such as warfarin has led to the development of resistance among rodents which lead the use of ratol more prominent.
CONCLUSION
Prevention strategies by restricting access to this poison can be one of the best methods to avoid complications. General public as well as clinicians should be made aware of the lethality of inorganic phosphorus. Reducing deaths from poisoning will require interventions to both reduce the incidence of harmful behavior and to improve medical management of acute poisoning.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Brent J, Wallace KL, Burkhart KK. Phosphorus. In: Brent J, Wallace KL, Burkhart KK, Phillips SD, Donovan JW, editors. Critical Care Toxicology – Diagnosis and Management of the Critically Poisoned Patient. Philadelphia, PA: Elsevier Mosby; 2005. pp. 851–61. [Google Scholar]
- 2.Tenenbein M. Position statement: Whole bowel irrigation. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol. 1997;35:753–62. doi: 10.3109/15563659709162571. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez OU, Canizares LL. Acute hepatotoxicity from ingestion of yellow phosphorus-containing fireworks. J Clin Gastroenterol. 1995;21:139–42. doi: 10.1097/00004836-199509000-00015. [DOI] [PubMed] [Google Scholar]
- 4.Ates M, Dirican A, Ozgor D, Aydin C, Isik B, Ara C, et al. Living donor liver transplantation for acute liver failure in pediatric patients caused by the ingestion of fireworks containing yellow phosphorus. Liver Transpl. 2011;17:1286–91. doi: 10.1002/lt.22384. [DOI] [PubMed] [Google Scholar]
- 5.Mauskar A, Mehta K, Nagotkar L, Shanbag P. Acute hepatic failure due to yellow phosphorus ingestion. Indian J Pharmacol. 2011;43:355–6. doi: 10.4103/0253-7613.81500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Talley RC, Linhart JW, Trevino AJ, Moore L, Beller BM. Acute elemental phosphorus poisoning in man: Cardiovascular toxicity. Am Heart J. 1972;84:139–40. doi: 10.1016/0002-8703(72)90318-3. [DOI] [PubMed] [Google Scholar]
- 7.McCarron MM, Gaddis GP, Trotter AT. Acute yellow phosphorus poisoning from pesticide pastes. Clin Toxicol. 1981;18:693–711. doi: 10.3109/15563658108990295. [DOI] [PubMed] [Google Scholar]
- 8.Marin GA, Montoya CA, Sierra JL, Senior JR. Evaluation of corticosteroid and exchange-transfusion treatment of acute yellow-phosphorus intoxication. N Engl J Med. 1971;284:125–8. doi: 10.1056/NEJM197101212840303. [DOI] [PubMed] [Google Scholar]
- 9.Akman SA, Cakir M, Baran M, Arikan C, Yuksekkaya HA, Tumgor G, et al. Liver transplantation for acute liver failure due to toxic agent ingestion in children. Pediatr Transplant. 2009;13:1034–40. doi: 10.1111/j.1399-3046.2008.01119.x. [DOI] [PubMed] [Google Scholar]
- 10.Maniatis V, Chryssikopoulos H, Roussakis A, Kalamara C, Kavadias S, Papadopoulos A, et al. Perforation of the alimentary tract: Evaluation with computed tomography. Abdom Imaging. 2000;25:373–9. doi: 10.1007/s002610000022. [DOI] [PubMed] [Google Scholar]