

Abstract
MicroRNAs (miRNAs) are small (16–24 nucleotides) noncoding RNAs that negatively regulate gene expression. Growing evidence demonstrates that miRNAs participate in the regulation of numerous physiological and pathological processes. The clinical utility of the cell-type–specific miRNA expression profile (miRomics) has been directly demonstrated in molecular classification of tumor samples and in prediction of prognosis or therapeutic responsiveness. Identification of the relevant miRNAs and their targets requires both in silico and molecular biological methods. In this review, we summarize the methodological arsenal used in miRNA-related research, and through our own data on adrenal tumors, we present how miRNA could be integrated into omics-based networks. The expanding knowledge obtained from miRNA research may lead to the development of novel diagnostic and treatment modalities in future.
MicroRNAs (miRNAs) are short (∼19–25 nucleotides long), noncoding RNA molecules that posttranscriptionally regulate gene expression via RNA interference through binding of protein-coding mRNA (1). According to predictions, about 30%–50% of all protein-coding genes may be controlled by miRNAs (2, 3).
Due to next-generation sequencing data, the number of known miRNAs expands rapidly. In Release 18 of miRBase (available from November 2011) 18 226 hairpin precursor miRNAs (pre-miRNAs), and 21 643 mature miRNAs in 168 species have been included {ftp://mirbase.org/pub/mirbase/CURRENT/README}. For Homo sapiens, 1523 miRNA sequences have been described in this database (ftp://mirbase.org/pub/mirbase/CURRENT/README).
MiRNAs are transcribed from miRNA genes, which are estimated to represent about 1% of eukaryotic genomes (4). MiRNA genes can be located either within introns of protein-coding genes (miRtrons) or intergenicly (5). MiRNA genes can be found alone, but there are several regions where miRNAs encoding genes are clustered and expressed together (miRNA clusters). MiRNA coding genes are transcribed to the long primary transcript (primer miRNA) by RNA polymerase II (6). In the nucleus, primer miRNAs are processed by a ribonuclease III (Drosha)-containing microprocessor complex. Drosha asymmetrically cleaves both strands near the base of primary stem-loop into an approximately 60- to 70-nucleotide–long pre-miRNA. In addition to Drosha, this microprocessor complex contains an RNA-binding protein (DiGeorge syndrome critical region 8 or Pasha), characterized by the conserved motif Asp-Glu-Ala-Asp (D-E-A-D) box helicase p68 and p72, and nuclear ribonucleoproteins (7, 8). The hairpin structured pre-miRNA molecule is transported to the cytoplasm by Exportin-5 in a Ran GTP-dependent manner (9). Here pre-miRNAs processed by ribonuclease III (Dicer) and complexed with transactivation-responsive RNA-binding protein (TRBP). Dicer recognizes the 3′-end of pre-miR and cleaves it into an approximately 21-nucleotide–long miRNA:miRNAstar (miR:miR*) duplex (10, 11). The double-stranded miRNA duplex has 5′-phosphate and 2 nucleotides 3′-overhang. One strand of this RNA duplex (guide strand or matured miRNA) is incorporated into miRNA-induced silencing complex (miRISC), whereas the other strand (passenger strand or miR*) is mostly degraded (8, 12). However, in some cases, miR* could also be loaded into miRISC and function as a matured miRNA (13) The strand selection is based upon the thermodynamic properties of the duplex, and the strand with the less stability at the 5′-end is usually selected (14). The whole process presented above is called canonical miRNA processing (Figure 1). There are, however, 2 noncanonical steps in miRNA maturation. Some of the pre-miRs are transcribed from very short introns (called miRtrons) as a result of splicing and debranching and by this way bypass Drosha cleavage (15). In the cytoplasm, some pre-miRNAs could be cleaved at the 3′-arm by AGO2 (an argonaute protein that has endonuclease activity, which supports Dicer activity) resulting in additional intermediates called AGO2-cleaved pre-miRNA.
Figure 1.
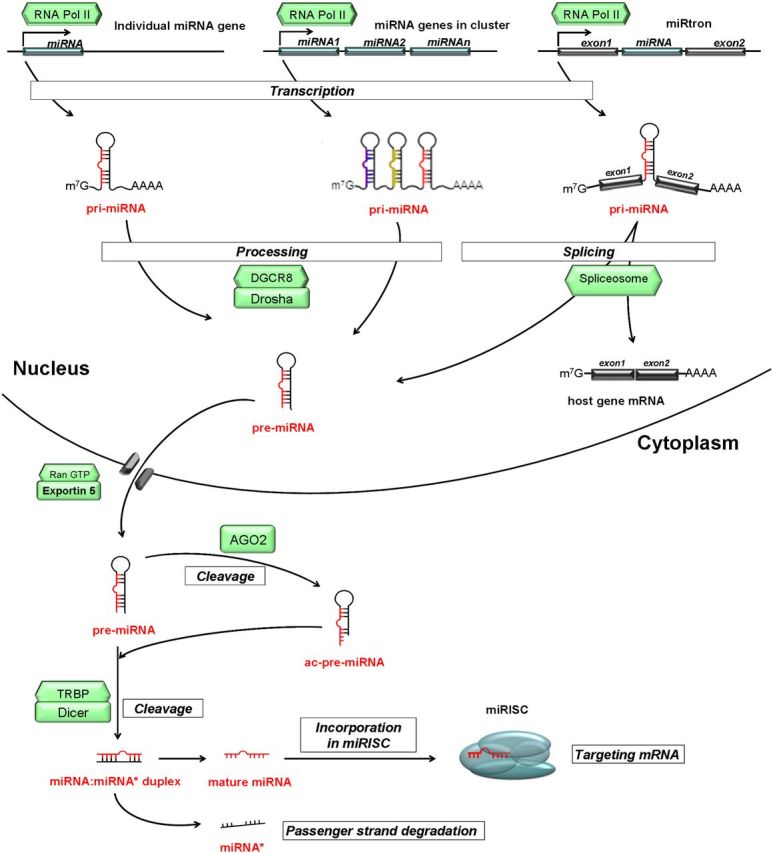
Schematic representation of biogenesis and function of miRNA. See details in the text. Italic characters indicate a process; green boxes indicate enzymes.
The single-stranded matured miRNA directly associates with argonaute proteins (in mammals, AGO1–4), which are core components of miRISC. In the miRISC, miRNA interacts with its 3′-untranslated region (UTR) mRNA target by base-pairing and causes repression of the expression of target genes. Although the seed region of miRNA is essential in this process, there is evidence that the central loop region is also involved (16). (In mature miRNAs, the seed region is defined as the consecutive stretch of ∼7 nucleotides starting from either the first or the second nucleotide at the 5′-end of the miRNA molecule [17].) Repression of the target genes occurs via 3 major processes: 1) mRNA cleavage by AGO2, 2) mRNA degradation by deadenylation, or 3) inhibition of different steps of translation processes (18–22). For AGO2-mediated cleavage of mRNA, a perfect or a nearly perfect complementarity is required between the mRNA and the seed sequence of the miRNA. In this case, AGO2 is able to cleave the mRNA leading to its degradation (18). In animals, the perfect complementarity is rare, whereas it is common in plants. In mRNA degradation through deadenylation, GW182, a 182-kDa glycine-tryptophan protein component of RISC, is required. The N terminus of GW182 interacts with AGO proteins, whereas its carboxyl-terminal region interacts with poly(A)-binding protein (PABP) and recruits deadenylase enzymes to the poly(A) tail of the mRNA molecule (23, 24). The mechanistic details of miRNA-mediated translational repression are not entirely understood. Due to the imperfect match between mRNA and miRNAs, RISC does not cleave the mRNA at the target site but causes translational suppression (19). Evidence suggests that the 7-methyl guanosine cap of mRNA is required for this process, and AGO2 is capable of binding to the 5′-cap of mRNA competing with eIF4E (a protein called eukaryotic translation initiation factor 4E), which is necessary for translation initiation (20, 21). In addition to binding to the 3′-UTR of protein encoding genes, miRNAs can also bind to the 5′-UTR or the coding sequences (25–27).
MiRNAs and the Endocrine System
MiRNAs have important interactions with the endocrine system at several levels. Hormones have been described as regulators of miRNA biogenesis. In human breast cancer cells, human endometrial stromal and myometrial smooth muscle cells, rat mammary gland, and mouse uterus, estrogens have been identified as regulators of miRNA biogenesis through regulation of the maturation process or as part of a negative autoregulatory feedback loop (28, 29). In addition to estrogens, GnRH (30), testosterone (31), and glucocorticoids (32) have also been implicated in the regulation of miRNA expression in cell-type–specific manner.
Like mRNA expression profile, the expression pattern of miRNAs (miRome) is highly tissue specific, so they can contribute to the cell type specificity of protein expression profiles (33, 34). Because miRNAs can influence many mRNAs, they can participate in the regulation of numerous physiological and pathological cellular processes including development (35), cell proliferation, differentiation (36), apoptosis (37), tumorigenesis (38), and hormone responsiveness (39). In regard to endocrine organs, it has been demonstrated that miRNAs regulate development of the pituitary gland by targeting Lef-1, a pivotal modulator of the expression of pituitary transcription factor 1 (Pit-1) (40), and in cooperation with cAMP-response element binding protein-1, it can affect the endocrine pancreas (41, 42) and the thyroid gland (43). In addition, Dicer1, the key enzyme in miRNA's biogenesis, is essential for the development of the female reproductive system (44), and Hawkins et al (45) showed that miRNAs have indispensable roles in the female and male reproductive tract (for a comprehensive review see Ref. 45).
Expression of miRNAs in Endocrine Tumors Has Been Explored With High-Throughput Technologies
The most important question related to the clinical properties of tumors is malignancy. Various high-throughput miRNA profiling methods have been introduced for identification of miRNAs that may have potential roles in various endocrine malignancies. Specific properties of miRNAs (their small size, sequence homology among miRNA family members, and cross-hybridization to their mRNA targets) have to be taken into account when their quantitative determination is planned. Several methods and various platforms have been developed for miRNA profiling, including Northern blot analysis, microarray analysis, and quantitative real-time PCR. In endocrine research, both commercially available platforms including microarrays, TaqMan chemistry-based low-density array (TLDA) cards and custom-made arrays have been tested (Table 1). Based on literature searches related to endocrine organs, miRNA expression profiles have been published for pituitary, endocrine pancreas, thyroid, and adrenal glands. However, comparison of miRNA expression of malignant and benign tumors is available only for thyroid and adrenal glands. MiRNA microarrays are miRNA-specific probe hybridization-based technologies allowing simultaneous profiling of all miRNAs identified at that time. The common technical limitations for arrays are related to the starting material (different platforms may need a larger amount of RNA isolated with dedicated reagents, and some of them need enrichment of small RNA fractions). Of the microarray platforms, the arrays using locked nucleic acid-modified oligonucleotide probes enhanced the binding affinity of the individual miRNA probes and significantly improved the specificity and sensitivity of miRNA detection. Another common platform used in these profiling experiments was the TLDA card (Life Technologies, Foster City, California). TLDA is a medium-throughput method using TaqMan chemistry for real-time PCR in a 384-well microfluidics cards. It is a rapid and reproducible methodology with a broad dynamic range compared with Northern blot or conventional RT-PCR when assessing RNA expression (46). Direct comparison of these assays failed to result in a clear conclusion regarding their superiority. Therefore, selection of the method used depends on facility and budget availability.
Table 1.
Differentially Expressed MiRNAs in Adrenal Carcinoma
| Number of Samples Analyzed | Platform | Differentially Expressed Validated MiRNAs | Ref. |
|---|---|---|---|
| 9 ACA, 4 ACC, 4 normal adrenal tissue | TLDA | Down-regulated miRs: miR-139-3p, miR-675, miR-335 | Schmitz et al, 2011 (48) |
| 4 ACA, 4 ACC, 4 CPA, 4 normal adrenal tissue | TLDA | Up-regulated miRs: miR-184, miR-503 | Tömböl et al, 2009 (49) |
| Down-regulated miRs: miR-511, miR-214 | |||
| 26 ACA, 22 ACC | Agilent miRNA microarray | Up-regulated miRs: miR-483-3p, miR-483-5p, miR-210, miR-21 | Özata et al, 2011 (50) |
| Down-regulated miRs: miR-195, miR-497, miR-1974 | |||
| 26 ACA, 10 ACC | miRCURY LNA arrays | Up-regulated miRs: miR-483-5p | Patterson et al, 2011 (51) |
| Down-regulated miRs: miR-100, miR-125b, miR-195 | |||
| 25 ACC, 5 normal (childhood ACC) | Agilent miRNA microarray | Up-regulated miRs: miR-99a, miR-100 | Doghman et al, 2010 (52) |
| 22 ACA, 27 ACC | miRCURY LNA arrays | Up-regulated miRs: miR-483-5p | Soon et al, 2009 (53) |
| Down-regulated miRs: miR-335, miR-195 |
Abbreviations: ACA, adrenocortical adenoma; ACC, adrenocortical carcinoma; CPA, cortisol-producing adrenocortical adenoma; LNA, locked nucleic acid.
Previous miRNA profiling studies were mainly descriptive, and they aimed at identifying differently expressed miRNAs in normal versus tumor or benign versus malignant tumor samples. Up-regulated miRNAs in tumor samples are frequently called oncomiRs, whereas down-regulated miRNAs are called tumor suppressor miRNAs. Here, it has to be mentioned that the results of a whole-genome miRNA profiling experiment must be validated by another independent technique. For this purpose, quantitative real-time PCR has been used almost exclusively. Both miRNA-specific TaqMan probes and SYBR Green-based methods are accepted. TaqMan using stem-loop structured reverse transcription primers for binding mature miRNA is a gold standard method for quantification of miRNA expression (47).
As an example, Table 1 summarizes the miRNA expression data obtained from adrenocortical tumor samples. Only the miR-483-5p has been identified as being overexpressed in adrenocortical cancer versus benign adrenal tumor or normal adrenal tissue in 3 independent studies (48–53). Another miRNA identified in 3 different studies (50, 51, 53), the miR-195 was down-regulated in adrenocortical cancer samples. Additional data are required to identify the explanation for this variability. It is possible that multiple factors, study design (comparing malignant with benign or malignant with normal, different sample cohorts, and even ethnic differences), starting material (fresh frozen samples or formalin-fixed paraffin-embedded tissues), and the profiling method (array versus RT-PCR, normalization steps, and reference transcripts) individually or together could influence the results.
Recently, deep sequencing has emerged as a promising novel method for direct evaluation of miRNAs. This technology has proved to be a suitable method not only for genome sequencing but also for miRNA profiling. No prior knowledge and no reference transcripts are needed. Theoretically, the direct comparison of sequences matching known miRNAs in tumor versus normal tissues could give the best evaluation of differentially expressed miRNAs. The main drawback of this technology is related to difficulties in the bioinformatics analysis. In addition to using dedicated algorithms for visualization of data from reads and mapping them against the transcriptome, the amount of data makes it necessary to use high-capacity computing facilities, including powerful hardware and dedicated software. Independently from the platform used, millions of short sequencing reads are generated. The raw data are encoded differently for their evaluation, and platform- dependent and -independent algorithms have been developed. It is worth noting that deep sequencing could also be applied for identification of novel miRNAs, but details of its bioinformatics would go beyond the scope of this article.
Unlike mRNA expression analysis, identification and validation of the most important miRNAs does not end at this step. Due to their diverse roles, further questions need to be clarified. Of these questions, identification of miRNA targets and their associated pathomechanism(s) are the most important. For identification of miRNA-related function, a combination of bioinformatics and molecular biological methods has been implemented. Bioinformatics analysis includes identification of potential miRNA targets and statistical analysis of cell- and context-dependent mRNA and miRNA expression profiles. Further perspective would be a high-throughput method for concomitant analysis of protein expression. The integrated analysis of miRNA-related experimental workflow is presented in Figure 2. In the following sections, we describe the complete workflow and, through our data obtained in adrenocortical cancer, we present how miRNAs can be integrated into a systems biology network.
Figure 2.

Schematic representation of high-throughput technologies, bioinformatics, and experimental tools used in miRNA-related research (orange indicates process; green, methods; blue, dedicated software; and purple, database). For example, miRNA expression profiles can be determined using high-throughput methods (labeled in green) or downloaded from depositories (purple). For statistical analysis, dedicated software packages, ie, GeneSpring or Bioconductor (marked in blue), can be used. The results of these analyses are further evaluated and complemented with inclusion of the outputs from target prediction algorithms. Target predictions can be performed both for miRNAs and for mRNAs/proteins revealed as differentially expressed between samples. Inclusion of the already published datasets, presented as Pubmed, together with the experimentally obtained data would further increase the chance for obtaining more relevant information. Having in hand all these datasets, the next step is the complex bioinformatics analysis, including pathway and network analysis (more detailed description can be found in text). The output from these analyses can be experimentally validated by evaluation of the direct interaction between miRNA-mRNAs and by studying the role of miRNAs through evaluation of the level or function of the targeted protein.
Bioinformatics Used in miRNA-Related Research
Target prediction algorithms
The first relevant question is to identify the miRNA targets. Most of the miRNA-mRNA target prediction algorithms are based on a search for perfect complementarity between the miRNA seed sequence and the mRNAs (54). Based on the GC content and size of the sequence, a perfect match of a 6-nucleotide–long seed in the whole genome can occur once in every 1.3 kilobases. Thus, 1 miRNA could have hundreds of targets theoretically. Due to the large number of factors that influence pairing, each of the predicted miRNA-mRNA target pairs needs to be experimentally verified because a simple high-throughput method to biologically validate miRNA targets does not exist.
Because miRNAs bind to the 3′-UTR, 5′-UTR, or even a coding region, target prediction must be performed on the entire sequence; however, the situation is very complex because 30% of protein-coding genes have no definitive 3′-UTR (55), and for instance, the Ensembl database uses cDNA and expressed sequence tag library sequences to identify these regions. Most target prediction algorithms scan miRNA seed sequences (positions 2–8) against all human 3′-UTR sequences (recently, 5′-UTR and coding sequences are also eligible in some algorithms) to identify perfect matches. Based on this alignment analysis, hundreds of potential targets could be established; however, false-positive results could be eliminated by examination of the level of cross-species conservation of seed pairing. Another possibility for selection is to investigate the stability of the mRNA-miRNA duplex and the free energy level of the binding (ΔG). For calculation of ΔG, several programs, web-based algorithms, eg, RNAFold, are available. A candidate target site is rejected if the ΔG value is higher than a predefined value, which may be dissimilar in different algorithms (56, 57). Increasing the ΔG could improve the specificity of the analysis. The most widely used target prediction algorithms are presented in Table 2.
Table 2.
Algorithms for Prediction of MiRNA-mRNA Interaction
| Name of the Method | Type of Algorithms Used | Ref. | Method Availability | Resource |
|---|---|---|---|---|
| Complementary | Stark et al, 2003 (100) | Online | http://www.russell.embl.de/miRNAs | |
| microRNA.org (miRanda) | Complementary | John et al, 2005 (101) | Online | http://www.microrna.org |
| microCosm (miRanda) | Complementary | Enright et al, 2003 (102) | Online | http://microrna.sanger.ac.uk |
| miRWalk | Complementary | Dweep et al, 2011 (103) | Online | http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html |
| TargetScan | Seed complementary | Grimson et al, 2007 (104) | Online | http://www.targetscan.org |
| TargetRank | Seed complementary and conservation | Nielsen et al, 2007 (105) | Online | http://hollywood.mit.edu/targetrank/ |
| MicroInspector | Complementarity and thermodynamics | Rusinov et al, 2005 (106) | Online | http://bioinfo.uni-plovdiv.bg/microinspector/ |
| PITA | Complementarity and thermodynamics | Kertesz et al, 2007 (107) | Online | http://genie.weizmann.ac.il/ |
| TargetSpy | Complementarity and thermodynamics | Sturm et al, 2010 (108) | Online | http://www.targetspy.org/ |
| DIANA microT | Thermodynamics | Kirakidou et al, 2004 (109) | Download | http://diana.cslab.ece.ntua.gr/ |
| PicTar | Thermodynamics | Krek et al, 2005 (110) | Online | http://pictar.mdc-berlin.de/ |
| RNAHybrid | Thermodynamics and statistical model | Rehmsmeier et al, 2004 (111) | Download | http://bibiserv.techfak.uni-bielefeld.de/rnahybrid |
| miRGen2+ | Baynesian inference | Huang et al, 2007 (112) | Mathlab Code | http://www.psi.toronto.edu/genmir |
| EIMMO | Bayesian phylogenetic model | Gaidatzis et al, 2007 (113) | Online | http://www.mirz.unibas.ch/ |
| MiTarget | Support vector machine | Kim et al, 2006 (114) | Online | http://cbit.snu.ac.kr/∼miTarget |
| miRDB (MiRtaget2) | Support vector machine | Wang et al, 2008 (115) | Online | http://mirdb.org |
| StaRmiR | Probabilistic tool based on secondary structure | Chan et al, 2005 (116) | Online | http://sfold.wadsworth.org/cgi-bin/starmir.pl |
| RNA22 | Pattern based | Miranda et al, 2006 (117) | Online | http://cbcsrv.watson.ibm.com/rna22.html |
| miracle | Based on RNA secondary structure | Online | http://miracle.igib.res.in/miracle/ | |
| Integrative target prediction algorithms | ||||
| MMIA (MicroRNA and mRNA integrated analysis) | Using 3 different algorithms (TargetScan, PicTar, and PITA) | Nam et al, 2009 (118) | Online | http://147.46.15.115/MMIA/mmia_main.html |
| miRTar | Using 4 different prediction programs (TargetScan, miRanda, PITA, and RNAHybrid) | Hsu et al, 2011 (119) | Online | http://mirtar.mbc.nctu.edu.tw/human/index.php |
| miRGen | Using 4 different prediction programs (microT, miRanda, PicTar, and TargetScanS) | Megraw et al, 2006 (120) | Online | http://www.diana.pcbi.upenn.edu/cgi-bin/miRGen/v3/Targets.cgi |
| GOmiR | Combining 4 different prediction programs (TargetScan, miRanda, RNAhybrid and PicTar) | Roubelakis et al, 2009 (121) | Download | http://www.bioacademy.gr/bioinformatics/projects/GOmir/#Introduction |
| MAMI (Meta miR target Inference) | Meta-prediction using 5 different algorhythms TargetScanS, miRandamicroT, miRtarget, and PicTar) | Online | http://mami.med.harvard.edu/david.html | |
| miRNAMap | Combining 3 prediction algorithms (miRanda, RNAhybrid, and TargetScan) | Hsu et al, 2006 (122) | Online | http://mirnamap.mbc.nctu.edu.tw/html/about.html |
| miRecords | Integrating 11 prediction algorithms (DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid, and TargetScan) | Xiao et al, 2009 (123) | Online | http://mirecords.biolead.org/index.php |
Integrative bioinformatics approaches
To identify the most relevant miRNAs and the related pathways, it is necessary to compare the miRNA expression data obtained using different platforms with the corresponding mRNA expression data. Combining miRNA and mRNA expression profiles from the same sample will further increase the specificity of the identification process. Incorporation of additional data retrieved either from publicly available databases (Ensembl, miRBase, etc), data repositories (Gene Expression Omnibus, GEO), or published datasets can further narrow the list of important miRNA-mRNA pairs. For these analyses, appropriate statistical methods are warranted.
An integrated approach is a commercially available software package called Gene Set Enrichment Analysis (GSEA). Gene Set Enrichment Analysis is a computational approach developed for the analysis of fine gene expression alterations. It was originally developed for analyses of mRNA expression. Mainly based on rank statistics, it determines whether a particular set of genes is over- or underrepresented in the samples compared (58). Inclusion of miRNA expression profiles could help the identification of the relevant miRNAs. This method is frequently used together with pathway analysis. For this purpose, a dedicated algorithm, the Ingenuity Pathways Analysis (IPA) Knowledge Base (Ingenuity Systems; Redwood City, California; www.ingenuity.com) has been developed. This approach is also commercially available, updated with current published datasets. By applying the Ingenuity Pathways Analysis software, gene sets can be functionally annotated, and biologically relevant pathways can be identified.
For example, in our studies on adrenal cancer, by analyzing all data of whole-genome microarray studies parallel with comparative genomic hybridization profiling available in different datasets, we have identified 3 major pathogenic pathways: cell cycle, retinoic acid signaling (including lipopolysaccharide/Toll like receptor 4 pathway), and complement system/antigen presentation (59). Extending this analysis with miRNA expression profiles may contribute to deepen our knowledge on adrenal tumorigenesis, and this may result in the identification of novel therapeutic targets (60). In the next part, we attempt to build an interacting network using our previous meta-analysis data complemented with 2 miRNAs that have been found to be common among studies.
Validation of the miRNA-Target mRNA Interaction
A possible way of investigating miRNA-mRNA interactions is to search for negative correlation between miRNA and mRNA expression (tissue-specific expression), which may strengthen target pairing (61, 62). However, the negative correlation between mRNA and miRNA expression is not exclusive because experimentally valid targets can be found even without changes in mRNA expression (63–65), further underlining the importance of the validation of individual miRNA-mRNA interactions. Reporter systems (eg, luciferase or green fluorescent protein) and direct assessment of the level of targeted protein after miRNA transfection using Western blot or ELISA are the most frequently used methods. In these experiments, selection of a cell line that expresses the candidate miRNA at a nondetectable or low level is recommended to minimize the effect of endogenously expressed miRNAs. Recently a high-throughput technique that allows the direct measurement of protein changes upon miRNA transfection (62, 66) is also available. This method is based on stable isotope labeling with amino acids in cell culture (SILAC) (67, 68). Using 2 different stable isotopes for pulse labeling (pSILAC) (heavy and medium heavy isotopes), it is possible to measure changes in protein production between 2 samples. Combining pSILAC with mass-spectrometry–based proteomics, Selbach et al (62) demonstrated that a single miRNA can repress the production of hundreds of proteins; however, the level of this repression was relatively low.
In addition to the direct measurement of protein changes after miRNA transfection or miRNA knockdown, another possibility for functional validation would be the evaluation of an antagomiR's effect on restoration of a candidate miRNA's effect. (AntagomiRs or anti-miRs are chemically engineered oligonucleotides which are used to silence endogenous miRNAs.) This approach will further validate and strengthen the interaction between miRNA and its target. In some cases, when the identified target protein involved in a signaling pathway is a kinase, a phosphatase, or an enzyme, it might be possible to directly test the function of the target proteins by phosphorylation/dephosphorylation assays or enzyme kinetic assays (69).
Our earlier work on evaluation of the role of miRNAs in pituitary tumorigenesis demonstrated that miRNAs through their targets influence TGFβ signaling and the dynamism of the cell cycle. Our workflow started with evaluation of miRNA expression profiles by high-throughput technologies followed by a complex bioinformatics analysis (target prediction, gene set enrichment analysis, and a comparison of each set of miRNA targets to all known Kyoto Encyclopedia of Genes and Genomes pathways). Each step was individually validated, ie, high-throughput profiling by quantitative RT-PCRs. Our results revealed that the significantly differentially expressed miRNAs between adenoma and normal pituitary tissues affected the TGFβ signaling pathway mainly through down-regulation of Smad3 (similar to the protein called mothers against decapentaplegic homolog in Drosophila). Interaction between the overexpressed and predicted miRNAs including miR-140 with Smad3 had also been experimentally validated (63). In addition, using the same high-throughput miRNA expression data, we identified that Wee1 kinase, which regulates the G2/M transition of the cell cycle, was down-regulated in nonfunctioning pituitary adenoma and GH-producing adenomas compared with normal tissues (64). Functional testing including in vitro luciferase reporter assays (Wee1 3′-UTR was cloned into luciferase vector and the miRNA-mRNA interaction was proved by site-directed mutagenesis) demonstrated that of the 5 predicted miRNAs, 3 indeed target Wee1 3′-UTR. In addition, evaluation of the Wee1 protein and mRNA levels after transfection of pre-miRNAs demonstrated that miR-155, miR-516a, and miR-128 significantly decreased the Wee1 protein (64). These data together underline that despite the bioinformatics limitations, valid mRNA-miRNA interactions and functional loops can be identified.
Integrating miRNAs into a systems biology approach
The recent technological revolution has led to the development of new high-throughput technologies resulting in the identification of genetic sequences of human and other genomes (genomics), gene expression profiles (transcriptomics), protein interactions (proteomics), and even metabolic alterations (metabolomics). A systems biology approach presents integration of these components into 1 network to decipher the complex biological background within 1 cell. Using datasets of adrenocortical cancer, we attempted to build a representative network for adrenocortical cancer including the 2 commonly identified miRNAs.
An integrated network for adrenocortical cancer
The basis of any interaction network is formed by data obtained either by an experimental work using high-throughput technologies evaluating whole genomes, transcriptomes, and proteomes or those that have been already published and deposited in databases. Relationships between different members can be predicted by computational algorithms and by including data obtained from model organisms (yeast 2-hybrid system for proteomics, Saccharomyces cerevisiae and Drosophila melanogaster for genomics and transcriptomics, and Caenorhabditis elegans for transcriptomics and miRomics). Experimentally validated individual interactions (DNA-protein, protein-protein, DNA-miRNA, and miRNA-DNA) retrievable from a literature search have to be also included into a network build (70). Including other regulatory levels, transcription factors, and miRNAs, integrating individual regulatory networks and complementing them with phenotypic data, a system-specific network can be built that may better represent the processes in its complexity (71–73). In addition, instead of presenting still images by linking different networks, the dynamism of certain processes can be modeled (70).
Any network can be characterized by its basic elements called nodes and edges (70). Depending on the network, nodes can represent a certain protein in a protein interacting network and edges represent physical interactions between them, whereas in a transcriptome network, nodes are transcription factors or transcription factor target genes and edges are physical connections between them. Integration of miRNA into such a network can be done similarly to the transcriptome network, keeping in mind that miRNAs negatively regulate gene/protein expression.
The first connections between proteins that might be called networks date back to 2005 (74, 75); however, the concept of interacting networks or a systems biology approach was introduced only in 2007 by Pujana et al (76). MiRNAs were introduced into biological networks 2 years ago (77, 78), and no data about miRNAs as members of a systems biology network have been published for the endocrine system yet. Therefore, the aim of our work was to integrate the 2 common miRNAs into our previous interacting network based on the transcriptome array obtained in adrenal cancer. This approach may allow us to analyze how these miRNAs might contribute to adrenal tumorigenesis.
In scale-free molecular networks, miRNAs and the target genes tend to be hubs, and usually the hub genes have more miRNA-binding sites (79). The betweenness measures the number of shortest paths going through a node; a node with high betweenness centrality is called a network bottleneck. Expression of the bottleneck proteins of a complex network is influenced by miRNAs because the target proteins of miRNAs have higher betweenness centrality than non-miRNA targets (80). If we take into consideration the large number of miRNA targets, a single miRNA could affect the expression of a large amount of downstream genes.
Modules are the main subcomponents of complex networks. Inside these modules, a higher density of links exists within a part of the network than outside of it. The expression of module genes can be regulated as a discrete entity, and the function or properties of a single module is separable from other ones (81).
Network motifs are basic structural units of gene regulatory networks, and these units occur more frequently than would be expected from random distribution. The so-called feed-forward loops are motifs, where a single node affects the expression of 2 others and another node also affects the third one. This structure often occurs when a transcription factor regulates the expression of miRNA and the target genes. In gene expression networks, regulated feedback loops are very frequently observed; in this case, 2 transcription factors mutually regulate their expression and an miRNA regulates both of them. Two main miRNA classes can be identified as the basis of the miRNA preference for network motifs. Class I miRNAs are mainly regulated by transcription factors, whereas class II miRNAs regulate transcription factors (82).
From the network context, the effects of miRNA expression changes seem to be very fundamental. It is well known that they are able to change the expression of hundreds of target genes, but the secondary effects of these changes in a complex network seem to be much more significant than the primary effects.
Network analysis of genes involved in adrenocortical tumorigenesis suggested the possible involvement of the underexpression of MYC oncogene in adrenocortical cancer (83). To complement this network with miRNA data, we collected the common miRNAs that are significantly differentially expressed in adrenal adenomas and carcinomas. The underexpression of hsa(human)-miR-195 and the overexpression of hsa-miR-483-5p were common in at least 3 independent studies (Table 1). To integrate these miRNAs into the network, we collected transcription factors regulating their expression using the TransmiR database (84) and their validated targets from the Tarbase database (85). All these data were integrated into the network by applying the Ingenuity Knowledge Database (86).
The merged network (Figure 3) was scale-free (power parameter, −1.21; R2, 0.84), and 10% of the highest-degree nodes were marked as hubs (hub). For network characterization, various algorithms were used: the vertex sort algorithm (87) for network hierarchy, mFinder software (88) for the identification of network motifs, and ModuLand software (89) for searching the network modules. Nodes of the transcription factor networks could be categorized into 3 main layers: 1) the top genes are the master regulators of the network because they should influence the whole network through their downstream targets; 2) core is the most abundant layer because it contains most of the HUBs and network motifs. The core layer plays a central role in the regulation of signal propagation. The bottom genes are located in the third layer, which directly regulates genes of effector molecules, or they are the effectors of the network (87).
Figure 3.

Interaction network complete with miRNAs in adrenocortical cancer. MiRNA-195 and MYC are hubs in this network having multiple inputs and outputs. In contrast, themiRNA-435-5p member of the top layer influences the expression of the other network members. However, the direct link between its target ATAD2 and MYC needs further clarification; hence, no information about the role of ATAD2 in adrenal carcinogenesis has been published. Green indicates underexpressed, pink/red indicates overexpressed, and white indicates molecules that have no assigned role in the adrenal gland. Lines represent experimentally proved connections between molecules.
Using the vertex sort algorithm, hsa-miR-483-5p has been identified as a member of the top layer. Therefore, overexpression of the hsa-miR-483-5p could highly influence the expression of the other network members in adrenocortical cancers, suggesting that hsa-miR-483-5p may be a class II miRNA. Another top layer gene, the early growth response 3 (EGR3) directly regulates the expression of hsa-miR-195, which was the member of the core layer and serves as a HUB in this network. The highest-degree node is the MYC protein that also directly regulates the expression of the hsa-miR-195. Most downstream targets of the hsa-miR-195 were localized in the bottom layer of the network, but several core member targets were identified. These results underline the significance of the hsa-miR-195 in the regulation of signaling processes in adrenal cancer. Due to the dominance of MYC, we have not found any significant modules or motifs in our network.
Of these deregulated miRNAs, hsa-miR-483-3p, hsa-miR-483-5p, and hsa-miR-195 was functionally tested. Using human adrenocortical cell line H295R, Özata and coworkers (50) demonstrated that down-regulation of hsa-miR-483-3p and increased expression of hsa-miR-195 reduced cell proliferation and induced cell death. Veronese et al. demonstrated that hsa-miR-483-3p located within intron 2 of the IGF2 gene, frequently overexpressed in adrenal cancer, directly targets the proapoptotic protein BBC3/PUMA (BCL2 binding component 3) (90). These data together suggest that in adrenocortical cancer, the overexpressed miR-483-3p and the underexpressed miR-195 contribute to carcinogenesis. These studies underline that bioinformatics evaluations extended with functional validation will result in identification of novel pathomechanisms, which may represent starting points for development of novel therapeutic approaches.
Future perspectives
Because miRNAs are very stable molecules within cells and body fluids, they represent ideal targets for biomarker discoveries. Certain miRNA or miRNA clusters have been suggested to be markers for various malignancies and for prognosis. Based on their tissue-specific expression, miRNAs can be used for classification and subclassification of samples hardly classifiable by routine immunohistochemistry.
MiRNAs are stable in plasma, suggesting that they may represent a novel class of biomarkers. Similar to chromogranin A, a neuroendocrine tumor-specific marker first used in immunohistochemistry analysis (91), certain miRNAs may follow this route, starting as a tissue-specific marker and ending as a useful marker, detectable in blood or other body fluids. The exact mechanism of the production and release of individual miRNAs in endocrine oncology is not yet known, but these processes may become elucidated shortly, considering the rapidly increasing number of publications showing that circulating miRNAs may serve as useful biomarkers. To date, circulating miRNAs have been used in evaluation of acute myocardiac infarction (92) and tissue damage (93) and proved to be valuable biomarkers for renal cell caner (94), breast cancer (95), and prostate cancer (96).
Based on their hormone-like properties and different production and acting sites, miRNAs have been suggested to be a novel class of hormones (hormomirs) by Selth et al (97). Indeed, similar to hormones acting through their receptors, miRNAs targeting various proteins may fulfill the hormone criteria, considering their presence in blood, and they are also potentially able to influence the function of a distant organ (98).
At this moment, it cannot be predicted how many of the miRNAs would serve as biomarkers for different diseases. Using high-throughput methodologies and evaluating datasets containing miRNA data through network approaches, a better understanding of the complexity and dynamic changes during physiological and pathophysiological processes can be achieved (99). A systems biology approach completed with small RNA-related mechanisms will help us to build disease models.
Acknowledgments
We acknowledge the financial support from the Hungarian Scientific Council Fund (ETT40/09), OTKA PD-100648, and TÁMOP-4.2.2.B-10/B-10/1-2010-0013. A.P. is a recipient of the Janos Bolyai Research Fellowship.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AGO2
- argonaute protein 2
- hsa
- human
- miRISC
- miRNA-induced silencing complex
- miRNA
- microRNA
- pre-miRNA
- precursor miRNA
- SILAC
- stable isotope labeling with amino acids in cell culture
- TLDA
- TaqMan chemistry-based low-density array
- UTR
- untranslated region.
References
- 1. Lagos-Quintana M , Rauhut R , Lendeckel W , Tuschl T. Identification of novel genes coding for small expressed. RNAs. Science. 2001;294:853–858. [DOI] [PubMed] [Google Scholar]
- 2. Chen K , Rajewsky N. Natural selection on human miRNA binding sites inferred from SNP data. Nat Genet. 2006;38:1452–1456. [DOI] [PubMed] [Google Scholar]
- 3. Lewis BP , Burge CB , Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. [DOI] [PubMed] [Google Scholar]
- 4. Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32(Database issue):D109–D11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krol J , Loedige I , Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. [DOI] [PubMed] [Google Scholar]
- 6. Lee Y , Kim M , Han J , et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee Y , Ahn C , Han J , et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415–419. [DOI] [PubMed] [Google Scholar]
- 8. Gregory RI , Chendrimada TP , Cooch N , Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123(4):631–640. [DOI] [PubMed] [Google Scholar]
- 9. Lund E , Guttinger S , Calado A , Dahlberg JE , Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. [DOI] [PubMed] [Google Scholar]
- 10. Zhang H , Kolb FA , Jaskiewicz L , Westhof E , Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118(1):57–68. [DOI] [PubMed] [Google Scholar]
- 11. Chendrimada TP , Gregory RI , Kumaraswamy E , et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436(7051):740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meister G , Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. [DOI] [PubMed] [Google Scholar]
- 13. Ghildiyal M , Xu J , Seitz H , Weng Z , Zamore PD. Sorting of Drosophila small silencing RNAs partitions microRNA* strands into the RNA interference pathway. RNA. 2010;16(1):43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khvorova A , Reynolds A , Jayasena SD. Functional siRNA and miRNAs exhibit strand bias. Cell. 2003;115:209–216. [DOI] [PubMed] [Google Scholar]
- 15. Ruby JG , Jan CH , Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448(7149):83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ye W , Lv Q , Wong CK , et al. The effect of central loops in miRNA:MRE duplexes on the efficiency of miRNA-mediated gene regulation. PLoS One. 2008;3:e1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sethupathy P , Megraw M , Hatzigeorgiou AG. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods. 2006;3(11):881–886. [DOI] [PubMed] [Google Scholar]
- 18. Zamore PD , Tuschl T , Sharp PA , Bartel DP. RNAi: double-staranded RNAdirects the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. [DOI] [PubMed] [Google Scholar]
- 19. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. [DOI] [PubMed] [Google Scholar]
- 20. Pillai RS , Bhattacharyya SN , Artus CG , et al. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. [DOI] [PubMed] [Google Scholar]
- 21. Kiriakidou M , Tan GS , Lamprinaki S , De Planell-Saguer M , Nelson PT , Mourelatos Z. An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell. 2007;129:1141–1151. [DOI] [PubMed] [Google Scholar]
- 22. Zinovyev A , Morozova N , Nonne N , Barillot E , Harel-Bellan A , Gorban AN. Dynamical modeling of microRNA action on the protein translation process. BMC Syst Biol. 2010;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eulalio A , Tritschler F , Izaurralde E. The GW182 protein family in animal cells: new insights into domains required for miRNA-mediated gene silencing. RNA. 2009;15(8):1433–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fabian MR , Sonenberg N , Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. [DOI] [PubMed] [Google Scholar]
- 25. Place RF , Li LC , Pookot D , Noonan EJ , Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sc. U S A. 2008;105:1608–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Orom UA , Nielsen FC , Lund AH. MicroRNA-10a binds the 5′-UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–471. [DOI] [PubMed] [Google Scholar]
- 27. Huang S , Wu S , Ding J , et al. MicroRNA-181a modulates gene expression of zinc finger family members by directly targeting their coding regions. Nucleic Acids Res. 2010;38:7211–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klinge CM. Estrogen regulation of MicroRNA expression. Curr Genomics. 2009;10(3):169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castellano L , Giamas G , Jacob J , et al. The estrogen receptor-α-induced microRNA signature regulates itself and its transcriptional response. Proc Natl Acad Sci U S A. 2009;106(37):15732–15737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuen T , Ruf F , Chu T , Sealfon SC. Microtranscriptome regulation by gonadotropin-releasing hormone. Mol Cell Endocrinol. 2009;302(1):12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Delić D , Grosser C , Dkhil M , Al-Quraishy S , Wunderlich F. Testosterone-induced upregulation of miRNAs in the female mouse liver. Steroids. 2010;75(12):998–1004. [DOI] [PubMed] [Google Scholar]
- 32. Smith LK , Shah RR , Cidlowski JA. Glucocorticoids modulate microRNA expression and processing during lymphocyte apoptosis. J Biol Chem. 2010;285(47):36698–36708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin SL , Chang D , Wu DY , Ying SY. A novel RNA splicing mediated gene silencing mechanism potential for genome evolution. Biochem Biophys Res Commun. 2003;310:754–760. [DOI] [PubMed] [Google Scholar]
- 34. Mattick JS , Makunin IV. Small regulatory RNAs in mammals. Hum Mol Genet. 2005;14(Spec No 1):R121–R132. [DOI] [PubMed] [Google Scholar]
- 35. Lee RC , Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. [DOI] [PubMed] [Google Scholar]
- 36. Chen JF , Mandel EM , Thomson JM , et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cimmino A , Calin GA , Fabbri M , et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ventura A , Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136(4):586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vreugdenhil E , Verissimo CS , Mariman R , et al. MicroRNA 18 and 124a down-regulate the glucocorticoid receptor: implications for glucocorticoid responsiveness in the brain. Endocrinology. 2009;150(5):2220–2228. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Z , Florez S , Gutierrez-Hartmann A , Martin JF , Amendt BA. MicroRNAs regulate pituitary development, and microRNA 26b specifically targets lymphoid enhancer factor 1 (Lef-1), which modulates pituitary transcription factor 1 (Pit-1) expression. J Biol Chem. 2010;285(45):34718–34728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lynn FC , Skewes-Cox P , Kosaka Y , McManus MT , Harfe BD , German MS. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes. 2007;56(12):2938–2945. [DOI] [PubMed] [Google Scholar]
- 42. Correa-Medina M , Bravo-Egana V , Rosero S , et al. MicroRNA miR-7 is preferentially expressed in endocrine cells of the developing and adult human pancreas. Gene Expr Patterns. 2009;9(4):193–199. [DOI] [PubMed] [Google Scholar]
- 43. Leone V , D'Angelo D , Ferraro A , et al. A TSH-CREB1-microRNA loop is required for thyroid cell growth. Mol Endocrinol. 2011;25(10):1819–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hong X , Luense LJ , McGinnis LK , Nothnick WB , Christenson LK. Dicer1 is essential for female fertility and normal development of the female reproductive system. Endocrinology. 2008;149(12):6207–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hawkins SM , Buchold GM , Matzuk MM. The roles of small RNA pathways in reproductive medicine. Mol Endocrinol. 2011;25(8):1257–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abruzzo LV , Lee KY , Fuller A , et al. Validation of oligonucleotide microarray data using microfluidic low-density arrays: a new statistical method to normalize real-time RT-PCR data. Biotechniques. 2005;38:785–792. [DOI] [PubMed] [Google Scholar]
- 47. Schmittgen TD , Lee EJ , Jiang J , et al. Real-time PCR quantification of precursor and mature microRNA. Methods. 2008;44:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmitz KJ , Helwig J , Bertram S , et al. Differential expression of microRNA-675, microRNA-139–3p and microRNA-335 in benign and malignant adrenocortical tumours. J Clin Pathol. 2011;64(6):529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tömböl Z , Szabó PM , Molnár V , et al. Integrative molecular bioinformatics study of human adrenocortical tumors: microRNA, tissue-specific target prediction, and pathway analysis. Endocr Relat Cancer. 2009;16(3):895–906. [DOI] [PubMed] [Google Scholar]
- 50. Özata DM , Caramuta S , Velázquez-Fernández D , et al. The role of microRNA deregulation in the pathogenesis of adrenocortical carcinoma. Endocr Relat Cancer. 2011;18(6):643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Patterson EE , Holloway AK , Weng J , Fojo T , Kebebew E. MicroRNA profiling of adrenocortical tumors reveals miR-483 as a marker of malignancy. Cancer. 2011;117(8):1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Doghman M , El Wakil A , Cardinaud B , et al. Regulation of insulin-like growth factor-mammalian target of rapamycin signaling by microRNA in childhood adrenocortical tumors. Cancer Res. 2010;70(11):4666–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Soon PS , Tacon LJ , Gill AJ , et al. miR-195 and miR-483-5p identified as predictors of poor prognosis in adrenocortical cancer. Clin Cancer Res. 2009;15(24):7684–7692. [DOI] [PubMed] [Google Scholar]
- 54. Lal A , Navarro F , Maher CA , et al. miR-24 inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′-UTR microRNA recognition elements. Mol Cell. 2009;35(5):610–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maziére P , Enright AJ. Prediction of microRNA targets. Drug Discovery Today. 2007;12:452–458. [DOI] [PubMed] [Google Scholar]
- 56. Wanatabe Y , Yachie N , Numata K , Saito R , Kanai A , Tomita M. Computational analysis of microRNA targets in Caenorhabditis elegans. Gene. 2006;365:2–10. [DOI] [PubMed] [Google Scholar]
- 57. Wang X , Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res. 2006;34:1646–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Subramanian A , Tamayo P , Mootha VK , et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Szabó PM , Tamási V , Molnár V , et al. Meta-analysis of adrenocortical tumour genomics data: novel pathogenic pathways revealed. Oncogene. 2010;29(21):3163–3172. [DOI] [PubMed] [Google Scholar]
- 60. Zsippai A , Szabó DR , Szabó PM , Tömböl Z , Bendes MR , Nagy Z , Rácz K , Igaz P. mRNA and microRNA expression patterns in adrenocortical cancer. Am J Cancer Res. 2011;1(5):618–628. [PMC free article] [PubMed] [Google Scholar]
- 61. Ruike Y , Ichimura A , Tsuchiya S , et al. Global correlation analysis of microRNA and mRNA expression profiles on human cell lines. J Hum Genet. 2008;53:515–523. [DOI] [PubMed] [Google Scholar]
- 62. Selbach M , Schwanhäusser B , Thierfelder N , Fang Z , Khanin R , Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. [DOI] [PubMed] [Google Scholar]
- 63. Pais H , Nicolas FE , Soond SM , et al. Analyzing mRNA expression identifies Smad3 as a microRNA-140 target regulated only at protein level. RNA. 2010;16(3):489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Butz H , Likó I , Czirják S , et al. Down-regulation of Wee1 kinase by a specific subset of microRNA in human sporadic pituitary adenomas. J Clin Endocrinol Metab. 2010;95(10):E181–E191. [DOI] [PubMed] [Google Scholar]
- 65. Gironella M , Seux M , Xie MJ , et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci U S A. 2007;104(41):16170–16175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Baek D , Villén J , Shin C , Camargo FD , Gygi SP , Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7:952–958. [DOI] [PubMed] [Google Scholar]
- 68. Ong SE , Blagoev B , Kratchmarova I , et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. [DOI] [PubMed] [Google Scholar]
- 69. Kuhn DE , Martin MM , Feldman DS , Terry AV , Nuovo GJ , Elton TS. Experimental validation of miRNA targets. Methods. 2008;44:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vidal M , Cusick ME , Barabási AL. Interactome networks and human disease. Cell. 2011;144(6):986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nibbe RK , Koyutürk M , Chance MR. An integrative -omics approach to identify functional sub-networks in human colorectal cancer. PLoS Comput Biol. 2010;6(1):e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saez-Rodriguez J , Alexopoulos LG , Epperlein J , et al. Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction. Mol Syst Biol. 2009;5:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fernández-Ramires R , Solé X , De Cecco L , et al. Gene expression profiling integrated into network modelling reveals heterogeneity in the mechanisms of BRCA1 tumorigenesis. Br J Cancer. 2009;101(8):1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rual JF , Venkatesan K , Hao T , et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437(7062):1173–1178. [DOI] [PubMed] [Google Scholar]
- 75. Stelzl U , Worm U , Lalowski M , et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. [DOI] [PubMed] [Google Scholar]
- 76. Pujana MA , Han JD , Starita LM , et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39:1338–1349. [DOI] [PubMed] [Google Scholar]
- 77. Volinia S , Galasso M , Costinean S , et al. Reprogramming of miRNA networks in cancer and leukemia. Genome Res. 2010;20:589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dong H , Luo L , Hong S , et al. Integrated analysis of mutations, miRNA and mRNA expression in glioblastoma. BMC Syst Biol. 2010;4:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liang H , Li WH. MicroRNA regulation of human protein protein interaction network. RNA. 2007;13(9):1402–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hsu CW , Juan HF , Huang HC. Characterization of microRNA-regulated protein-protein interaction network. Proteomics. 2008;8(10):1975–1979. [DOI] [PubMed] [Google Scholar]
- 81. Bonnet E , Tatari M , Joshi A , et al. Module network inference from a cancer gene expression data set identifies microRNA regulated modules. PLoS One. 2010;5(4):e10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yu X , Lin J , Zack DJ , Mendell JT , Qian J. 2008 Analysis of regulatory network topology reveals functionally distinct classes of microRNAs. Nucleic Acids Res. 2008;36(20):6494–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Szabó PM , Rácz K , Igaz P. Underexpression of C-myc in adrenocortical cancer: a major pathogenic event? Horm Metab Res. 2011;43(5):297–299. [DOI] [PubMed] [Google Scholar]
- 84. Wang J , Lu M , Qiu C , Cui Q. TransmiR: a transcription factor-microRNA regulation database. Nucleic Acids Res. 2010;38(Database issue):D119–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Vergoulis T , Vlachos I , Alexiou P , et al. Tarbase 6.0: capturing the exponential growth of miRNA targets with experimental support NAR. Nucleic Acids Res. 2012;40(Database issue):D222–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chang HS , Bae SM , Kim YW , et al. Comparison of diarsenic oxide and tetraarsenic oxide on anticancer effects: relation to the apoptosis molecular pathway. International journal of oncology. Int J Oncol. 2007;30(5):1129–1135. [PubMed] [Google Scholar]
- 87. Jothi R , Balaji S , Wuster A , et al. Genomic analysis reveals a tight link between transcription factor dynamics and regulatory network architecture. Mol Syst Biol. 2009;5:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kashtan N , Itzkovitz S , Milo R , Alon U. Efficient sampling algorithm for estimating subgraph concentrations and detecting network motifs. Bioinformatics. 2004;20(11):1746–1758. [DOI] [PubMed] [Google Scholar]
- 89. Kovács IA , Palotai R , Szalay MS , Csermely P. Community landscapes: an integrative approach to determine overlapping network module hierarchy, identify key nodes and predict network dynamics. PLoS One. 2010;5(9):e12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Veronese A , Lupini L , Consiglio J , et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010;70:3140–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. O'Toole D , Grossman A , Gross D , et al. Mallorca Consensus Conference participants; European Neuroendocrine Tumor Society. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: biochemical markers. Neuroendocrinology. 2009;90(2):194–202. [DOI] [PubMed] [Google Scholar]
- 92. Adachi T , Nakanishi M , Otsuka Y , et al. 2010 Plasma microRNA 499 as a biomarker of acute myocardial infarction. Clin Chem. 2010;56(7):1183–1185. [DOI] [PubMed] [Google Scholar]
- 93. Zhang Y , Jia Y , Zheng R , Guo Y , Wang Y , Guo H , Fei M , Sun S. Plasma microRNA-122 as a biomarker for viral-, alcohol-, and chemical-related hepatic diseases. Clin Chem. 2010;56:1830–1838. [DOI] [PubMed] [Google Scholar]
- 94. Redova M , Poprach A , Nekvindova J , et al. Circulating miR-378 and miR-451 in serum are potential biomarkers for renal cell carcinoma. J Transl Med. 2012;10(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. van Schooneveld E , Wouters MC , Van der Auwera I , et al. Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res. 2012;14(1):R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mitchell PS , Parkin RK , Kroh EM , et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Selth LA , Tilley WD , Butler LM. 2012 Circulating microRNAs: macro-utility as markers of prostate cancer? Endocr Relat Cancer. 2012;19(4):R99–R113. [DOI] [PubMed] [Google Scholar]
- 98. Zhang L , Hou D , Chen X , et al. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res. 2012;22(1):107–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zampetaki A , Willeit P , Drozdov I , Kiechl S , Mayr M. Profiling of circulating microRNAs: from single biomarkers to re-wired networks. Cardiovasc Res. 2012;93:555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Stark A , Brennecke J , Russell RB , Cohen SM. Identification of Drosophila microRNA targets. PLoS Biol. 2003;1(3):e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. John B , Enright AJ , Aravin A , et al. Human MicroRNA targets. PLoS Biol. 2005;3(7):e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Enright AJ , John B , Gaul U , Tuschl T , Sander C , Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dweep H , Sticht C , Pandey P , Gretz N. miRWalk database: prediction of possible miRNA binding sites by “walking” the genes of 3 genomes. J Biomed Inform. 2011;44(5):839–847. [DOI] [PubMed] [Google Scholar]
- 104. Grimson A , Farh KK , Johnston WK , Garrett-Engele P , Lim LP , Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nielsen CB , Shomron N , Sandberg R , Hornstein E , Kitzman J , Burge CB. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA. 2007;13:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rusinov V , Baev V , Minkov IN , Tabler M. MicroInspector: a web tool for detection of miRNA binding sites in an RNA sequence. Nucleic Acids Res. 2005;33(Web Server issue):W696–W700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kertesz M , Iovino N , Unnerstall U , Gaul U , Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39(10):1278–1284. [DOI] [PubMed] [Google Scholar]
- 108. Sturm M , Hackenberg M , Langenberger D , Frishman D. TargetSpy: a supervised machine learning approach for microRNA target prediction. BMC Bioinform. 2010;28(11):292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kiriakidou M , Nelson PT , Kouranov A , et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Krek A , Grun D , Poy MN , et al. Combinatorial miRNA target predictions. Nat Genet. 2005;37:495–500. [DOI] [PubMed] [Google Scholar]
- 111. Rehmsmeier M , Steffen P , Hochsmann M , Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Huang JC , Babak T , Corson TW , et al. Using expression profiling data to identify human microRNA targets. Nat Methods. 2007;4:1045–1049. [DOI] [PubMed] [Google Scholar]
- 113. Gaidatzis D , van Nimwegen E , Hausser J , Zavolan M. 2007 Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinformatics. BMC Bioinformatics. [Erratum (2007) 8(1):248] 2007;8(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kim SK , Nam JW , Rhee JK , Lee WJ , Zhang BT. miTarget: microRNA target gene prediction using a support vector machine. BMC Bioinformatics. 2006;7:411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chan CY , Lawrence CE , Ding Y. Structure clustering features on the Sfold Web server. Bioinformatics. 2005;21(20):3926–3928. [DOI] [PubMed] [Google Scholar]
- 117. Miranda KC , Huynh T , Tay Y , et al. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126(6):1203–1217. [DOI] [PubMed] [Google Scholar]
- 118. Nam S , Li M , Choi K , Balch C , Kim S , Nephew KP. MicroRNA and mRNA integrated analysis (MMIA): a web tool for examining biological functions of microRNA expression. Nucleic Acids Res. 2009;37(Web Server issue):W356–W362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hsu JB , Chiu CM , Hsu SD , et al. miRTar: an integrated system for identifying miRNA-target interactions in human. BMC Bioinformatics. 2011;12:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Megraw M , Sethupathy P , Corda B , Hatzigeorgiou AG. miRGen: a database for the study of animal microRNA genomic organization and function. Nucleic Acids Res. 2007;35(Database issue):D149–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Roubelakis MG , Zotos P , Papachristoudis G , et al. Human microRNA target analysis and gene ontology clustering by GOmir, a novel stand-alone application. BMC Bioinformatics. 2009;10(Suppl 6):S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hsu PW , Huang HD , Hsu SD , et al. miRNAMap: genomic maps of microRNA genes and their target genes in mammalian genomes. Nucleic Acids Res. 2006;34(Database issue):D135–D139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Xiao F , Zuo Z , Cai G , Kang S , Gao X , Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37(Database issue):D105–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]