

Abstract
The impact of thyroid hormone (TH) on metabolism and energy expenditure is well established, but the role of TH in regulating nutritional sensing, particularly in the central nervous system, is only poorly defined. Here, we studied the consequences of hypothyroidism on leptin production as well as leptin sensing in congenital hypothyroid TRH receptor 1 knockout (Trhr1 ko) mice and euthyroid control animals. Hypothyroid mice exhibited decreased circulating leptin levels due to a decrease in fat mass and reduced leptin expression in white adipose tissue. In neurons of the hypothalamic arcuate nucleus, hypothyroid mice showed increased leptin receptor Ob-R expression and decreased suppressor of cytokine signaling 3 transcript levels. In order to monitor putative changes in central leptin sensing, we generated hypothyroid and leptin-deficient animals by crossing hypothyroid Trhr1 ko mice with the leptin-deficient ob/ob mice. Hypothyroid Trhr1/ob double knockout mice showed a blunted response to leptin treatment with respect to body weight and food intake and exhibited a decreased activation of phospho-signal transducer and activator of transcription 3 as well as a up-regulation of suppressor of cytokine signaling 3 upon leptin treatment, particularly in the arcuate nucleus. These data indicate alterations in the intracellular processing of the leptin signal under hypothyroid conditions and thereby unravel a novel mode of action by which TH affects energy metabolism.
Thyroid hormones (THs) are essential for the balance of metabolic processes. In humans, hyperthyroidism is accompanied by an overall increase in the metabolic rate whereas hypothyroidism results in reduced metabolic activity and decreased body temperature (1, 2). For metabolic functions, TH not only acts in well-established target organs such as liver, muscle, and adipose tissue but also in the central nervous system (CNS) where it is considered to affect central circuits regulating body weight (bw) and energy expenditure (3–6).
For integrating peripheral energy signals the hypothalamus represents an important area. In particular, the hypothalamic arcuate nucleus (Arc) located at the base of the third ventricle contains orexigenic Agouti-related peptide (AgRP)/neuropeptide Y (NPY) as well as anorexigenic proopiomelanocortin (POMC)/cocaine and amphetamine-regulated transcript-expressing neurons that receive information about the peripheral fat content via the adipocyte-derived hormone leptin (7). A second-order class of neurons is located in the paraventricular hypothalamic nucleus that integrates neuropeptidergic signals derived from the leptin receptor Ob-R expressing neurons in the Arc (8). Intriguingly, among these neurons are TRH-expressing neuroendocrine cells that respond to fasting with a decrease in TRH expression, thereby down-regulating the hypothalamus-pituitary-thyroid axis (8). The fasting-induced decrease in serum TH levels can be prevented by leptin administration, indicating that the activity of the hypothalamus-pituitary-thyroid axis is adapted to the size of peripheral energy stores (9).
Secondary to the neuroendocrine functions, hypothalamic TRH-expressing neurons exhibit widespread projections within the CNS and are thought to regulate food intake, locomotor activity, and thermogenesis (8, 10). Electrophysiological studies demonstrated modulatory activity of TRH on orexin/hypocretin (11, 12), histamine (13), and melanin-concentrating hormone-expressing neurons (14) in the hypothalamus. In rats, central application of TRH has repeatedly demonstrated to reduce food consumption (15, 16). Based on these observations, TRH has been classified as a neuropeptide with anorexigenic properties (17).
In rodents, action of TRH is mediated by two different, G protein-coupled TRH receptors (Trhr1 and Trhr2) of which only Trhr1 is found in the pituitary and therefore responsible for TRH action on thyrotrophs and lactotrophs (18). Trhr2 expression is restricted to the CNS, where it is predominantly expressed in cortical and thalamic areas. It thus may be involved in modulating higher sensory and cognitive functions (19). In comparison, Trhr1 is highly enriched in hypothalamic and visceral brain stem regions where it may mediate TRH effects on central pathways affecting appetite and energy expenditure. Notably, genome-wide association and replication studies identified Trhr1, the only TRH receptor present in humans, as a gene important for lean body mass (20). It is therefore conceivable that TH-induced alterations in TRH expression influence the central integration of the leptin signal via the activation of Trhr1.
In addition to this indirect mode of action, TH may also influence hypothalamic leptin signaling directly. TH receptor (TR) α and β isoforms exhibit a widespread expression in hypothalamic neurons (21, 22). Moreover, several studies have suggested that TH can indeed alter hypothalamic neuronal activity. In the Arc, TH has been shown to stimulate hypothalamic mitochondrial uncoupling specifically in NPY/AgRP-expressing neurons, thereby regulating fasting-induced rebound feeding (3). In the ventromedial hypothalamus, increased TH concentrations are associated with a down-regulation of AMP-activated protein kinase (AMPK) and consecutively with an activation of the sympathetic nervous system (5). Whether leptin receptor expression and signaling are direct targets of TH action has not been addressed yet.
Here, we report on the consequences of hypothyroidism and/or of an impaired TRH signaling on leptin production and hypothalamic leptin sensing by studying hypo- and euthyroid Trhr1 knockout (ko) mice as well as hypothyroid and euthyroid wild type (WT) animals expressing Trhr1. Our analysis revealed not only decreased leptin production under hypothyroid conditions but also striking alterations in hypothalamic leptin receptor expression and downstream signaling proteins. Our findings suggest that in mice, hypothyroidism, but not Trhr1 deficiency, is associated with a central resistance to leptin, thereby affecting bw and energy expenditure.
Materials and Methods
Animals
TRH receptor 1 (Trhr1) ko mice on a C57Bl/6N background were generated and genotyped as described previously (23). Trhr1/ob double knockout [dko] mice were obtained by mating Trhr1 ko mice with leptin-deficient ob/ob animals on a C57Bl6 background obtained from The Jackson Laboratory (Bar Harbor, Maine). Paired box transcription factor (Pax)-8 ko mice (24) were generated by mating heterozygous Pax8 animals on a mixed (C57Bl6/NMRI) background. Genotyping was performed as described elsewhere (25). Animal procedures were approved by the Animal Welfare Committee of the Medizinische Hochschule (Hannover, Germany) or by the Animal Welfare Committee of the Thüringer Landesamt für Lebensmittelsicherheit und Verbraucherschutz (TLLV Thüringen; Erfurt, Germany). Mice were kept at constant temperature (22°C) on a 12-hour light, 12-hour dark cycle and were provided with standard laboratory chow (2.98 kcal/g; Altromin, Hannover, Germany) and water ad libitum. In all experiments, only male mutant mice and WT littermates at the age of 3–5 months were analyzed, and only animals of the same genetic background were compared.
Hypothyroid Trhr1 ko and Trhr1/ob dko mice at the age of 3–4 weeks were rendered euthyroid by supplementing their drinking water with 0.01% BSA and thyroid hormones (Trhr1 ko: 4 ng/mL T3 and 120 ng/mL T4; Trhr1/ob dko: 4 ng/mL T3 and 60 ng/mL T4; Sigma-Aldrich, St. Louis, Missouri). Serum TH levels were monitored in orbital eye blood samples. Athyroid Pax8 ko animals were daily injected with 20 ng/g bw T4 sc for the first three postnatal weeks (26). After weaning, the animals received drinking water supplemented with 4 ng/mL T3, 120 ng/mL T4, and 0.01% BSA for 5 weeks. Animals were kept for additional 4 weeks on normal drinking water in order to render them hypothyroid.
To reduce TH serum levels in WT (C57Bl/6J background; purchased from Janvier, Paris France) and ob/ob mice, animals at the age of 8 weeks were supplied with drinking water supplemented with 0.02% 2-mercapto-1-methylimidazole, 1% sodium perchlorate, and 0.3% saccharin as a sweetener for 8 weeks until serum TH levels in orbital eye blood samples were close to the detection limit. Leptin-deficient (ob/ob and Trhr1/ob dko) mice and WT controls were injected twice daily (9:00 am and 5:00 pm) with recombinant mouse leptin (ip, 2 μg/g bw; PeproTech, Rocky Hill, New Jersey) for 3 consecutive days and killed 3 hours after the last injection. For determining leptin-induced alterations in food intake and bw, animals were first adapted to the injection-induced stress by injecting PBS ip for 5 days. Afterward, the animals were injected twice daily with leptin (ip, 2 μg/g bw) for 3 days. For phospho-signal transducer and activator of transcription (STAT)3 immunohistochemistry, the mice received a single leptin injection (ip, 2 μg/g bw) 45 minutes before they were killed by perfusion-fixation under deep anesthesia.
Indirect calorimetry
Indirect calorimetry was performed in a comprehensive laboratory animal monitoring system (Columbus Instruments, Columbus, Ohio), a computer-controlled open-circuit calorimetry system equipped with light beams to monitor animal activity. Studies were performed at 22°C, and animals were acclimatized to the chambers 24 hours before data collections.
Fat mass determination
In vivo body composition of mice was determined using the micro-CT scanner LaTheta LCT-100ATM (Zinsser Analytic, Frankfurt, Germany). Adipose tissue weights were computed using the common density factor of 0.92 g/cm3. Animals were scanned under deep anesthesia (5 μg/g midazolam, 0.5 μg/g medetomidine, and 2 μg/g fentanyl). In order to terminate the anesthesia, animals were injected with an antidote solution (2.5 μg/g atipamezol, 0.5 μg/g flumazenil, and 1.2 μg/g naloxon). Abdominal scans were obtained between vertebrae L5 and L6. Weights of visceral and sc fat were normalized to gram of total bw.
Determination of serum and tissue parameters
Serum T4 and T3 concentrations were determined by RIA. Tissues were rapidly frozen on dry ice and stored at −80°C. For determination of tissue T4 and T3 concentrations as well as type 2 iodothyronine deiodinase (Dio2) activities, hypothalami and brown adipose tissue (BAT) samples from five adult animals were pooled and processed as described previously (27). Leptin concentration in the serum was measured using a mouse Leptin ELISA Kit (Millipore Corp, Bedford, Massachusetts).
In situ hybridization (ISH)
Dissected brains were immediately frozen in dry ice-cooled isopentane and used for preparing cryostat sections. RNA probes were generated by in vitro transcription using [35S]UTP or digoxigenin (DIG)-11-UTP as substrate. For preparing RNA probes, a fragment corresponding to nt 1799–2437 of mouse Ob-R cDNA (GenBank accession number NM_146146), a fragment of mouse suppressor of cytokine signaling (SOCS)3 cDNA corresponding to nucleotide 1915-2427 (NM_007707), and a fragment of mouse TRH cDNA corresponding to nucleotide 202-733 (NM_009426), were generated by PCR and subcloned into the pGEM-T Easy Vector (Promega Corp, Madison, Wisconsin).
For ISH, frozen sections were air dried, followed by a 1-hour fixation in a 4% phosphate-buffered paraformaldehyde (PFA) solution (pH 7.4) and then permeabilized by incubation in 0.4% Triton-X 100/PBS for 10 min. Acetylation was carried out in 0.1 M triethanolamine (pH 8.0) containing 0.25% (vol/vol) acetic anhydride. Sections were dehydrated and then covered with hybridization mix containing cRNA probes diluted in hybridization buffer (50% formamide, 10% dextrane sulfate, 0.6 M NaCl, 10 mM Tris-HCl [pH 7.5], 1× Denhardt's solution, 100 μg/mL sonicated salmon sperm DNA, 1 mM EDTA, and 0.5 mg/mL t-RNA). 35S-labeled riboprobes were diluted in hybridization buffer to a final concentration of 5 × 104 cpm/μL. For nonradioactive ISH experiments, DIG-labeled riboprobes were diluted to a concentration of 5 ng/μL. Hybridization was performed overnight at 58°C. Slides were rinsed in 2× standard saline citrate (SSC) (0.3 M NaCl and 0.03 M sodium citrate, pH 7.0) and subsequently treated with ribonuclease A/T1 at 37°C for 30 minutes. Final washes were carried out in 0.2× SSC at 65°C for 1 hour. For detecting radioactive hybridization signals, the sections were dehydrated and then exposed to x-ray film (BioMax MR, Eastman Kodak Co, Rochester, New York) for 24–48 hours. Sections were dipped in Kodak NTB nuclear emulsion (Eastman Kodak) and stored at 4°C for 2 days for TRH, 3 weeks for Ob-R, and 6 weeks for SOCS3.
For nonradioactive ISH experiments, DIG-labeled riboprobes were diluted to a concentration of 5 ng/μL. Hybridization and posthybridization steps were carried out as described for radioactively labeled cRNA probes. After incubation in 0.2× SSC at 65°C, sections were rinsed with 150 mM NaCl, 100 mM Tris (pH 7.5) and then kept for 1 hour in a 10% milk powder blocking solution. The antidigoxigenin antibody conjugated with alkaline phosphatase (1:1000; Roche, Indianapolis, Indiana) was applied overnight at 4°C. Tissue sections were stained for 3–5 hours in a substrate solution containing nitroblue tetrazolium chloride (340 μg/mL; Sigma-Aldrich, St Louis, Missouri), 5-bromo-4-chloro-3-indoylphosphate (X-phosphate, 175 μg/mL; Sigma-Aldrich), 50 mM MgCl2, 100 mM NaCl, and 100 mM Tris (pH 9.5). Pictures were taken under bright-field illumination. Experiments carried out using the respective sense probes did not produce any specific hybridization signals.
Immunohistochemistry
Animals were deeply anesthetized by isoflurane and transcardially perfused with a 4% phosphate-buffered PFA (pH 7.4) solution. The brains were removed carefully and postfixed in the same fixative over night at 4°C. After cryoprotection in 30% sucrose, brains were frozen in isopentane cooled on dry ice. Coronal sections, 20 μm thick, were obtained using a cryostat and kept in PBS containing 0.02% sodium azide. Sections were carefully mounted on a positive-charged microscope slide and air dried, followed by a fixation step in 4% phosphate-buffered PFA.
Endogenous peroxidase activities were blocked by treating the sections with 1% NaOH/1% H2O2 in PBS for 20 minutes and with 0.3% glycine and 0.03% sodium dodecyl sulfate for 10 minutes each. Sections were fixed in methanol/acetone (1:1) for 10 minutes at room temperature (RT) and afterward washed in PBS. Sections were then incubated in 3% normal goat serum/0.2% Triton X-100/PBS for 1 hour at RT. P-STAT3 antibody (rabbit phospho-(Tyr705)-STAT3, 1:1000 in blocking solution; Cell Signaling Technology, Beverly, Massachusetts) was applied overnight at 4°C. Sections were washed three times in PBS before applying a biotinylated antirabbit IgG antibody (1:200 in blocking solution; Cell Signaling Technology) for 1 hour at RT. The slides were subsequently developed using avidin-conjugated horseradish peroxidase (Vectastain ABC Kit, Vector Laboratories, Burlingame, California) with 3,3′-diaminobenzidine as a substrate.
Laser-capture microdissection (LCM)
Frozen brains were cut into coronal 20-μm thick cryosections. In the area of the hypothalamic Arc (Bregma 3.9 mm), 24 consecutive sections were mounted on membrane slides (1.0 polyethylene naphthalate; Carl Zeiss, Jena, Germany). The sections were stained with a 0.1% (wt/vol) cresyl violet acetate solution for 5 minutes, dehydrated by successive washing in 95% and 100% isopropanol, Xylene, and dried on a heating plate at 37°C. Tissue covering the area of the Arc was dissected using an LCM microscope. Microdissected samples were collected in TRIzol reagent and subjected to RNA isolation using the RNeasy Micro Kit. Quality control of total RNA was performed with the Agilent Bioanalyzer 2100 (Agilent Technologies, Palo Alto, California). RNA was amplified with the MessageAmp II aRNA Amplification Kit.
Quantitative real-time PCR (qPCR)
Dissected tissues designated for qPCR analysis were rapidly frozen on dry ice and stored at −80°C. The RNeasy Lipid Tissue Mini Kit (QIAGEN, Hilden, Germany) was utilized to isolate total RNA from fat tissue whereas for all other tissues, the NucleoSpin RNA II Kit (Macherey-Nagel, Düren, Germany) was employed. Synthesis of cDNA was performed using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche). To exclude the presence of genomic DNA, one sample without reverse transcriptase was included as well. qPCR was performed using the iQ SYBR Green Supermix and the Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., Hercules, California). At least five samples per genotype were subjected to the analysis. As a housekeeping gene for normalization, cyclophilin D was used. The following primer pairs were designed to generate the PCR fragments at an annealing temperature of 55°C: AgRP: 5′-TGAGTTGTGTTCTGCTGTTG-3′ and 5′-TGAGGCCATTCAGACTTAGA-3′; cyclophilin D: 5′-GCAAGGATGGCAAGGATTGA-3′ and 5′-AGCAATTCTGCCTGGATAGC-3′; Dio2: 5′-GGAACAGCTTCCTCCTAGAT-3′ and 5′-GGTCTTCTCCGAGGCATAAT-3′; Leptin: 5′-CAGGGAGGAAAATGTGCTGGAG-3′ and 5′-CCGACTGCGTGTGTGAAATGTC-3′; Ob-R (all isoforms): 5′-ACAGTTCTGGCTGTCAATTC-3′ and 5′-GTGTCCAGGAAAGGATGAC-3′; Ob-Ra:5′-AAGTTGTTTTGGGACGATG-3′ and 5′-ATTGGGTTCATCTGTAGTGG-3′; Ob-Rb: 5′-GCACAAGGACTGAATTTCC-3′ and 5′-GTTCAGGCTCCAGAAGAAG-3′; Ob-Rc: 5′-CCTCTTGTGTCCTACTGCTC-3′ and 5′-GTGACCTTTTGGAAATTCAG-3′; POMC: 5′-TTCAGACCTCCATAGATGTGTGG-3′ and 5′-ATCTCCGTTGCCAGGAAACA-3′; PPARγ coactivator 1 α (PGC-1a): 5′-CAATGAATGCAGCGGTCTTA-3′ and 5′-GTGTGAGGAGGGTCATCGTT-3′; peroxisome proliferator-activated receptor (PPAR-g): 5′-TCGCTGATGCACTGCCTATG-3′ and 5′-GAGAGGTCCACAGAGCTGATT-3′; uncoupling protein (UCP)1: 5′-CGACTCAGTCCAAGAGTACTTCTCTTC-3′ and 5′-CCGGCTGAGATCTTGTTTC-3′; STAT3: 5′-TCTGCCTGGACCGTCTGGAA-3′ and 5′-CTGAACAGCTCCACGATCCT-3′.
Statistical analysis:
All data are presented as their mean value ± SEM. Statistical significance was determined by two-tailed Student's t test for comparing Pax8 WT and Pax8 ko, WT and WT+methimazole (MMI), ob and Trhr1/ob, ob/ob and ob+MMI, as well as one-way ANOVA for comparing WT, Trhr1 ko, and Trhr1+TH as well as ob/ob, Trhr1/ob, and Trhr1/ob+TH. For the statistical analysis of bw and food intake in response to leptin treatment (see Figure 5) repeated measures two-way ANOVA was applied. Differences that were considered significant are indicated as follows: *P < .05; **P < .01; and ***P < .001.
Figure 5.

Hypothyroid Animals Show a Blunted Response to Leptin Treatment. Hypothyroid Trhr1/ob dko mice (A), MMI-treated ob/ob mice (B), and euthyroid ob/ob mice were injected for 3 consecutive days with leptin. During this period as well as for additional 5 days of leptin withdrawal (W1–W5), daily food intake and bw were monitored. Changes are expressed as a percentage of baseline (day 0) weight and food intake. Whereas euthyroid ob/ob mice responded to the leptin injection with a robust decline in food intake and bw, the response was significantly less pronounced in hypothyroid Trhr1/ob dko and MMI-treated ob/ob mice. The effects of leptin treatment on bw and food intake were determined by two-way ANOVA with repeated measures. *P < .05; #P < .01; §P < .001.
Results
Determination of serum TH levels, bw, and food intake
Trhr1 ko mice exhibit a central hypothyroidism that is characterized by an approximately 50% decrease in serum T3 and T4 concentrations (23). In order to distinguish between effects related to a missing Trhr1 and consequences of the reduced serum TH levels, we rendered Trhr1 ko mice euthyroid by substituting them with TH (Figure 1A). As an additional animal model, we included Pax8 ko mice in our study that are born without a functional thyroid gland and therefore do not produce any TH endogenously. These athyroid animals were substituted with TH for the first 2 months and then rendered hypothyroid again (Figure 1A). Finally, we also included WT animals in our analysis that were treated for 8 weeks with MMI/perchlorate in order to render them hypothyroid. Determination of serum TH concentrations indeed revealed a pronounced reduction of both T4 and T3 serum concentrations compared with untreated control mice (Figure 1A).
Figure 1.

Serum TH Levels, bw, and Daily Food Intake in Adult Hypothyroid and Euthyroid Mice. WT, Trhr1 ko (R1 ko), TH-treated Trhr1 ko (R1 ko+TH), athyroid Pax8 ko mice, and WT animals rendered hypothyroid by MMI/perchlorate treatment were analyzed with respect to their serum TH levels (A), bw and daily food intake (B) at the age of 3–4 months. Due to the different genetic background, Pax8 WT animals (mixed C57Bl6/NMRI background) are heavier and exhibit a higher fat content than WT animals on either C57Bl6N (R1-WT) or C57Bl6J background (WT). Body weight was significant lower in hypothyroid animals when compared with background-matched control mice. Daily food intake normalized to bw was not altered. C, Visceral and sc white fat mass were determined with a micro-CT scanner and normalized to total bw. n = 8; *P < .05; **P < .01; ***P < .001.
Hypothyroid Trhr1 ko (C57Bl6/N background), Pax8 ko (mixed C57Bl6/NMRI background), and WT mice (C57Bl6/J background) exhibited a significant reduction in bw compared with the respective euthyroid littermates independent of the genotype and the background of the animals (Figure 1B). Daily food intake was in general lower in hypothyroid animals but not altered when normalized to bw (Figure 1B). Determination of fat content using a micro-CT scanner revealed a significant reduction in both visceral and sc fat mass in hypothyroid animals independent of the presence or absence of Trhr1 (Figure 1C). Analysis of oxygen consumption by indirect calorimetry showed increased values in hypothyroid Trhr1 ko mice whereas TH-treated Trhr1 ko mice displayed values similar to WT controls (Figure 2, A and B). Intriguingly, such alterations in oxygen consumption were not observed in MMI-treated WT animals (Figure 2B). Neither hypothyroidism nor Trhr1 deficiency had any influence on locomotor activity (Figure 2A).
Figure 2.
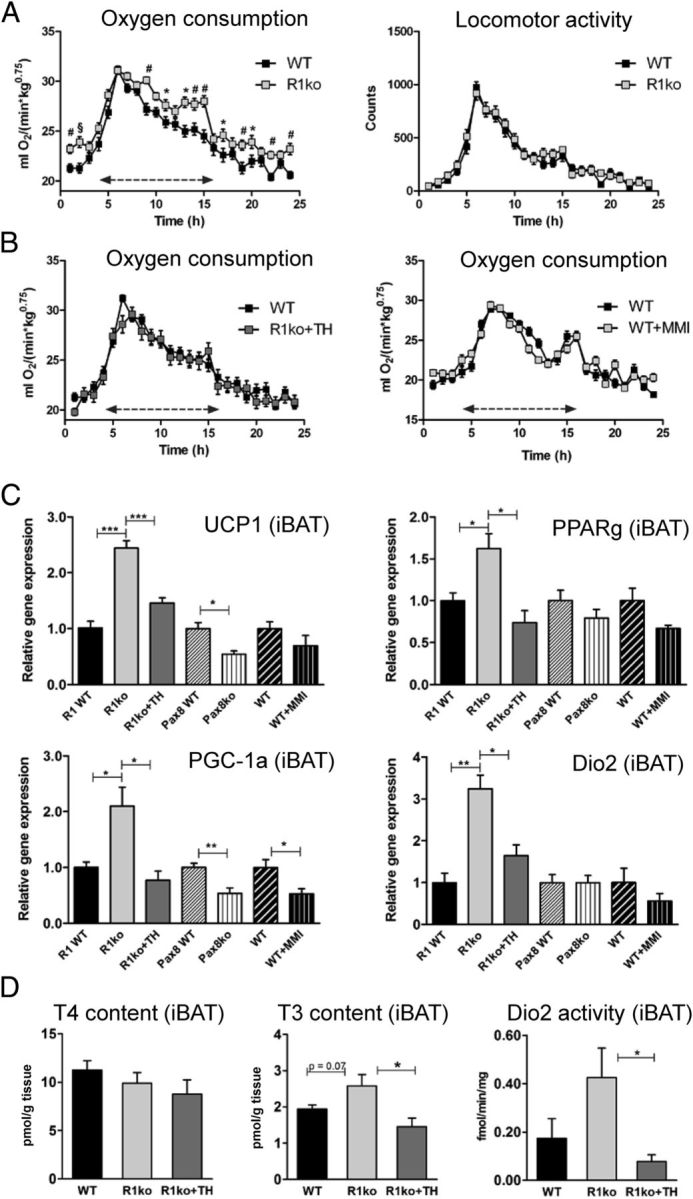
Increased Oxygen Consumption and BAT Activity in Hypothyroid Trhr1 ko Mice. Oxygen consumption and locomotor activity of hypothyroid Trhr1 ko, TH-treated Trhr1 ko, MMI-treated WT animals, and respective control mice was determined using a comprehensive laboratory animal monitoring system (CLAMS; Columbus Instruments). Double arrow marks the dark period. A, Hypothyroid Trhr1 ko mice exhibited a significant increased oxygen consumption but no alterations in locomotor activities. B, In contrast, TH-treated Trhr1 ko as well as MMI-treated WT animals exhibited normal oxygen consumption. n = 8, *P < .05; #P < .01; §P < .001. C, Expression levels of UCP1, PPARg, PGC-1a, and Dio2 were determined by qPCR in intercapsular BAT (iBAT) samples. Elevated expression levels for all four genes were found only in hypothyroid Trhr1 ko mice, suggesting that the BAT is in a highly activated state. D, In line with increased Dio2 expression, tissue T3 concentrations were found to be elevated in the BAT of hypothyroid Trhr1 ko mice whereas no alterations in T4 content were observed. n = 5, *P < .05; **P < .01; ***P < .001.
The increased oxygen consumption found in hypothyroid Trhr1 ko mice may be due to an enhanced activity of the BAT. In order to test this hypothesis, we collected intercapsular BAT samples from animals of all experimental groups and performed qPCR analysis. As depicted in Figure 2C, only hypothyroid Trhr1 ko mice exhibited a significant up-regulation of genes involved in thermogenesis such as UCP1, PGC-1a, and PPARg whereas MMI-treated WT mice and Pax8 ko mice rather showed a decrease in the respective transcript levels. Expression of Dio2, an important enzyme for local T4 to T3 conversion, was strongly induced in the BAT of hypothyroid Trhr1 ko mice but was altered neither in MMI-treated WT animals nor in Pax8 ko mice. Moreover, determination of tissue TH concentrations revealed elevated T3 levels in the BAT of hypothyroid Trhr1 ko mice whereas T4 tissue levels were not significantly altered (Figure 2D). Together these data indicate that specifically hypothyroid Trhr1 ko mice exhibit a hyperactive BAT and therefore show increased oxygen consumption.
Leptin expression is altered under hypothyroid conditions.
In view of the substantial differences in WAT mass found in all hypothyroid mouse models, we wondered whether leptin expression is altered under hypothyroid conditions as well. Indeed, as revealed by qPCR, Trhr1 ko mice exhibited a 2-fold decrease in leptin mRNA expression compared with WT and TH-treated Trhr1 ko mice (Figure 3A). A more pronounced reduction of about 76% was found in athyroid Pax8 ko mice. Even MMI treatment of WT animals resulted in a 48% reduction in leptin transcript levels. We further determined leptin serum levels using an ELISA kit. In agreement with the reduced leptin transcript levels and the reduced fat amount, leptin serum concentrations were found to be significantly lower in hypothyroid mice as well, with the strongest reduction of about 82% in athyroid Pax8 ko mice (Figure 3B). These findings indicate that in mice, hypothyroid conditions are associated with a decrease in leptin production.
Figure 3.

Hypothyroidism Compromises Leptin Production and Leptin Receptor Expression. Leptin transcript levels were determined in epidydimal white adipose tissues (WAT) by qPCR and found to be significantly decreased in all hypothyroid animal models (A). Determination of serum leptin levels by ELISA revealed significantly reduced leptin concentrations in all hypothyroid animals as well (B). *P < .05; **P < .01; ***P < .001. ISH studies revealed increased Ob-R transcript levels in the Arc of hypothyroid Trhr1 ko and athyroid Pax8 ko mice compared with euthyroid WT and TH-treated Trhr1 ko animals (C). In contrast, SOCS3 mRNA expression was strongly diminished in the ARC of hypothyroid animals (lower row). Scale bar, 100 μm.
Thyroid state of the hypothalamus in Trhr1 ko mice.
We next evaluated whether hypothalamic leptin signaling was altered in Trhr1-deficient animals. In order to assess the thyroid state of this brain area, hypothalamic T3 and T4 concentrations were determined in pools of five hypothalami per group (Supplemental Figure 1A published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). In the absence of Trhr1, mean hypothalamic T4 and T3 levels were reduced to 68% and 54% of WT values, respectively, whereas TH treatment was sufficient to normalize the local TH content in Trhr1 ko animals. As another indicator for the local thyroidal state, we analyzed TRH expression in T3-sensitive hypothalamic paraventricular nucleus neurons by radioactive ISH. TRH expression was markedly elevated in untreated Trhr1 ko mice and rather unaffected in TH-treated Trhr1 ko animals (Supplemental Figure 1B). Finally, we determined Dio2 mRNA levels in hypothalamic extracts and found elevated levels only in hypothyroid but not in TH-treated Trhr1 ko mice (Supplemental Figure 1C). These observations indicate that the TH treatment was successful in order to achieve an euthyroid state in the hypothalamus of Trhr1 ko mice.
Hypothyroidism influences hypothalamic leptin receptor (Ob-R) and SOCS3 expression
In order to assess the consequences of TH deficiency on leptin signaling, we studied the expression of leptin receptor Ob-R and its downstream target SOCS3 by radioactive ISH (Figure 3C). Signal intensities for Ob-R were markedly increased specifically in the Arc of Trhr1 ko mice as well as in athyroid Pax8 ko mice, whereas TH-treated Trhr1 ko mice showed similar signal intensities as WT animals. In comparison, mRNA expression levels of SOCS3 were visibly reduced in untreated Trhr1 ko and Pax8 ko mice but unaltered in TH-treated Trhr1 ko animals. These data suggest that hypothyroidism, but not Trhr1 deficiency, is associated with an increase in leptin receptor mRNA expression and a decrease in its downstream target SOCS3 specifically in the Arc region but not in other hypothalamic nuclei. These alterations may be caused by the low hypothalamic TH levels. Alternatively, these changes may rather reflect the decrease in serum leptin levels because leptin has been implicated to down-regulate the expression of its own receptor (28). In order to distinguish between the action of TH and the effects of leptin on Ob-R expression, we generated mouse mutants that, in addition to Trhr1, do not express leptin endogenously. For that purpose, we crossed leptin-deficient ob/ob mice with Trhr1 ko mice in order to obtain Trhr1/leptin dko animals. These Trhr1/ob dko mice were as obese and hyperphagic as ob/ob mice (Supplemental Figure 2A). However, in contrast to ob/ob animals, Trhr1/ob dko mice showed a 50% reduction in serum TH concentrations, indicating that Trhr1/ob dko mice exhibit the same decrease in serum TH as the Trhr1 ko mice (Supplemental Figure 2B). We also included TH-treated Trhr1/ob dko mice in our studies. This intervention was successful to normalize serum T3 levels whereas serum T4 was still slightly decreased compared with ob/ob mice.
Next, we treated ob/ob, Trhr1/ob, and TH-treated Trhr1/ob mice with leptin by injecting them twice daily with 2 μg/g bw ip for three consecutive days. Control animals received saline injections. Brains were collected 3 hours after the last injection and then subjected to ISH analysis. Independent of leptin, Ob-R signal intensities were markedly elevated in the Arc of Trhr1/ob dko mice (Figure 4A). In contrast, SOCS3-specific signals were close to the detection limit in the absence of leptin. Leptin treatment visibly induced SOCS3 expression in all leptin-deficient mouse mutants (Figure 4B). However, signal intensities were significantly lower in hypothyroid Trhr1/ob dko mice, suggesting an influence of the thyroidal state on the induction of SOCS3 expression.
Figure 4.

Analysis of Ob-R and SOCS3 Expression in Leptin-Deficient Animals. ISH studies were performed in order to analyze leptin receptor and SOCS3 expression in euthyroid ob/ob mice (ob), in hypothyroid mice deficient in Trhr1 and leptin (R1/ob) and TH-treated Trhr1/ob dko animals (R1/ob+TH) before and after the treatment of the animals for 3 days with leptin. Dark-field autoradiograms illustrate increased Ob-R-specific signals in the Arc of hypothyroid Trhr1/ob dko mice (A) whereas SOCS3 expression was strongly reduced in all leptin-deficient animals. Upon leptin injection, up-regulation of SOCS3 expression was markedly diminished in hypothyroid Trhr1/ob dko mice. C, Hypothalamic cryosections from leptin-treated ob/ob and Trhr1/ob dko mice were used for LCM experiments in order to collect specifically the tissue of the Arc for RNA isolation and qPCR analysis. Transcript levels of total Ob-R and of the short forms Ob-Ra and Ob-Rc were increased in leptin-treated hypothyroid Trhr1/ob dko mice compared with euthyroid ob/ob mice, whereas the levels of the long leptin receptor isoform Ob-Rb were not altered in hypothyroid animals. In contrast, SOCS3 mRNA expression was decreased in hypothyroid Trhr1/ob dko mice. *P < .05; **P < .01; ***P < .001.
In order to substantiate these findings, we again injected ob/ob and Trhr1/ob dko mice with leptin and prepared brains as described above. Hypothalamic area covering the Arc regions was identified by cresyl violet staining and collected using an LCM microscope. After RNA isolation and amplification, qPCR analysis was performed and revealed, in agreement with the ISH findings, a 1.9-fold increase in Ob-R transcript levels and a 2.3-fold down-regulation of SOCS3 mRNA expression in leptin-treated Trhr1/ob mice compared with leptin-treated ob/ob animals (Figure 4C).
The leptin receptor Ob-R gene produces several splice variants of which only the Ob-Rb form encodes the full leptin receptor protein and can activate the Jak-Stat pathway (29). Ob-Ra and Ob-Rc are shorter isoforms that are considered to be important for leptin transport and degradation. Because our ISH probe recognizes all isoforms, we performed qPCR in order to distinguish between the different splice variants. Indeed, after leptin treatment, transcript levels of the signaling isoform Ob-Rb were similar in ob/ob and Trhr1/ob dko mice, whereas expression of the shorter isoforms, Ob-Ra and Ob-Rc, was significantly higher in the hypothyroid Trhr1/ob dko mice (Figure 4C).
Hypothyroidism affects the physiologic response to leptin in terms of food intake and bw.
To which extent do these changes in Ob-R and SOCS3 expression affect the central integration of the leptin signal? To address this question, we again injected ob/ob and Trhr1/ob dko mice with leptin for 3 consecutive days and monitored the changes in bw and food intake during this period. Trhr1/ob dko mice responded to the leptin treatment with a decrease in food intake of about 54%, whereas ob/ob mice reacted to leptin application even more strongly and consumed only 13% of their normal daily food intake (Figure 5A). In line with these altered appetites, ob/ob mice lost 5% more bw upon leptin treatment than hypothyroid Trhr1/ob dko animals.
Next, we rendered leptin-deficient ob/ob mice hypothyroid by MMI/perchlorate treatment and then assessed the leptin response in this animal cohort. Upon leptin injection, MMI-treated ob/ob mice exhibited a 4.7% higher bw and 40% higher food intake than euthyroid ob/ob mice (Figure 5B), indicating that the reduced TH levels but not the absence of Trhr1 signaling compromise the physiologic response to leptin.
Hypothyroidism is associated with a reduced STAT3 activation upon leptin treatment
Phosphorylation of the signal transducer and activator of transcription 3 (STAT3) represents a central signaling event after activation of Ob-R that can be visualized by immunohistochemistry (30). Euthyroid and MMI-treated hypothyroid ob/ob mice, as well as hypothyroid Trhr1/ob dko and TH-treated Trhr1/ob dko animals, were injected with leptin or saline, respectively, and were subjected to perfusion fixation 45 minutes after the injection. Immunohistochemical stainings of hypothalamic cryosections with a phospho-STAT3 -specific antibody revealed the absence of any specific signal in all leptin-deficient animals (Figure 6A). Upon leptin treatment, pronounced phospho-STAT3-immunoreactive cells were detectable in all animals. However, MMI-treated ob/ob mice, as well as Trhr1/ob dko mice, showed markedly reduced signal intensities compared with the respective euthyroid controls. The attenuated up-regulation of phospho-STAT3 in the Arc was not due to a general reduction in STAT3 expression, because qPCR analysis of Arc-derived cDNA revealed similar transcript levels for STAT3 in leptin-treated ob/ob and Trhr1/ob dko mice (Figure 6B).
Figure 6.
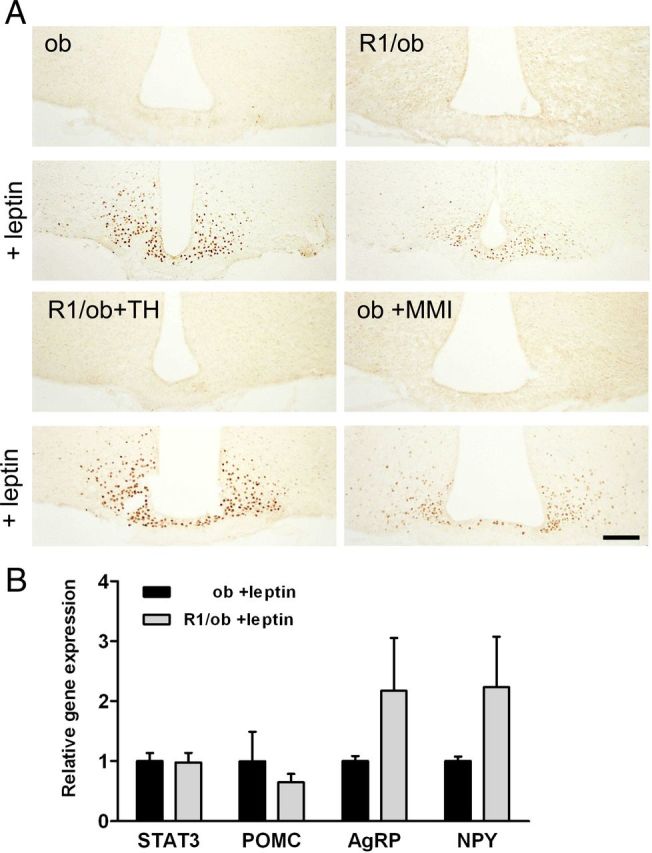
Hypothyroid Mice Show Diminished STAT-3 Activation upon Leptin Treatment. Phosphorylated STAT3 was visualized by immunohistochemistry in hypothalamic sections of hypothyroid Trhr1/ob dko and MMI-treated ob/ob mice compared with controls. Whereas euthyroid ob/ob and TH-treated Trhr1/ob dko mice showed a strong increase of phospho-STAT3 immunoreactivity upon leptin treatment, the elevation was less pronounced in hypothyroid Trhr1/ob dko and MMI-treated ob/ob mice (A). This diminished response was not due to alterations in STAT3 expression because qPCR analysis of STAT3 expression in the Arc revealed similar transcript levels in euthyroid ob/ob and hypothyroid Trhr1/ob dko mice (B). However, upon leptin treatment, transcript levels of POMC showed a slight decline and transcript levels of AgRP and NPY tended to be elevated in the arcuate nucleus of hypothyroid Trhr1/ob dko mice (B). Scale bar, 100 μm.
POMC and AgRP transcript levels are altered under hypothyroid conditions
Prominent targets of activated STAT3 are promoter regions in the AgRP, NPY, and POMC gene that respond to leptin with pronounced alterations in transcript levels. Leptin strongly up-regulates the POMC and down-regulates AgRP and NPY transcript levels. qPCR analysis revealed that in hypothyroid animals the response to leptin treatment was blunted. Hypothyroid Trhr1/ob dko mice exhibited higher NPY and AgRP and lower POMC expression levels compared with euthyroid ob/ob mice (Figure 6B). Overall, our findings indicate a resistance of neurons in the Arc to leptin treatment under hypothyroid conditions.
Discussion
A major aim of our study was to provide a first metabolic analysis of Trhr1 ko mice that exhibit central hypothyroidism (23). These mice presented a lean phenotype and an increase in oxygen consumption compared with WT littermates. We first suspected that these alterations are mainly caused by the central inactivation of Trhr1 because TRH has been implicated in the regulation of food intake and energy expenditure (8). Indeed, our analysis of the BAT revealed alterations in gene expression such as elevated UCP1 transcript levels, indicating a hyperactive state of this tissue. Such an activation could not be detected in Pax8 ko mice or MMI-treated WT animals and was also not reported in previous studies on hypothyroid mice kept at room temperature (31). Most strikingly, Dio2 was found to be highly up-regulated in hypothyroid but neither in TH-treated Trhr1 ko mice nor in the other hypothyroid mouse models used in this study. Likewise, only hypothyroid Trhr1 ko mice showed in the BAT-elevated T3 tissue concentration, a finding that may explain the rise in UCP1 expression. The seemingly hyperthyroid state of the BAT in an otherwise hypothyroid animal is intriguing and appears to be related, at least in part, to the lack of Trhr1 in the CNS. We speculate that a combination of central hypothyroidism and Trhr1 deficiency leads to an enhanced sympathetic tone that in turn stimulates Dio2 expression in the BAT (32). Further investigation using conditional TRH mouse mutants is expected to shed further light on this issue.
Concerning other metabolic parameters such as bw, food intake, and fat content we found the same changes in hypothyroid Trhr1 ko mice as in the other hypothyroid animal models, indicating that, in this respect, the lack of Trhr1 is of minor importance. In line with reported findings in a mouse mutant harboring dominant-negative mutations in the TRα1 gene (TRa1 R384C) (33), hypothyroid mice exhibited diminished circulating leptin levels. These alterations do not only reflect the overall reduction in the white fat content but may also be due to an extenuated leptin expression in adipose tissue. qPCR analysis indeed revealed reduced leptin transcript levels in epididymal white adipose tissue of hypothyroid animals (Figure 3). These findings are also fortified by previous reports (34, 35) demonstrating a stimulatory effect of T3 on leptin expression in primary 3T3L1 adipocytes. All these data advocate for a direct contribution of TH to the regulation of leptin expression.
Reduced circulating leptin levels are considered to be a potent stimulatory factor to increase food intake. TRa1 mutant mice are hyperphagic, a phenotype that has been discussed to be related to the low leptin serum levels (33). However, hypothyroid mice do not display increased food intake, suggesting that the reduction in circulating leptin levels is not properly sensed under hypothyroid conditions. This hypothesis prompted us to study the transcript levels of leptin receptor and its target genes in the hypothalamic Arc where prominent leptin target neurons reside. Our ISH analysis in fact revealed increased Ob-R and decreased SOCS3 transcript levels in the Arc of the hypothyroid Trhr1 ko and Pax8 ko mice whereas TH treatment led to a normalization of transcript levels (Figure 3C).
Binding of leptin to its receptor results in an activation of the JAK-STAT pathway. Within this signaling cascade, phosphorylation of STAT3 represents an important step as phospho-STAT3 enters the nucleus and stimulates the expression of leptin target genes such as SOCS3. Hence, the decreased SOCS3 mRNA levels that we observed in hypothyroid animals may reflect reduced intracellular leptin signaling as a consequence of the decreased leptin serum levels. In order to assess the impact of central hypothyroidism on intracellular leptin signaling, we generated leptin-deficient, hypothyroid Trhr1-deficient animals by crossing Trhr1 ko mice with leptin-deficient ob/ob and analyzed them before and after leptin application. In addition, we treated ob/ob mice with MMI/perchlorate and rendered them hypothyroid. As exptected, Trhr1/ob dko mice as well as MMI-treated ob/ob mice were as obese and hyperphagic as euthyroid ob/ob mice despite a significant reduction in TH values. When these animals were challenged by daily leptin injections, hypothyroid leptin-deficient mice lost significantly less weight and ate significantly more than the euthyroid ob/ob mice, indicating a partial resistance to the application of leptin (Figure 5). Moreover, hypothyroid Trhr1/ob dko mice, as well as MMI-treated ob/ob mice, showed reduced phospho-STAT3 activation and reduced up-regulation of SOCS3 expression in the Arc upon peripheral leptin injection (Figure 6). Finally, hypothyroid leptin-deficient animals showed increased AgRP and NPY and decreased POMC transcript levels after leptin stimulation. Taken together, these findings clearly indicate that hypothyroidism compromises the intracellular integration of leptin signaling specifically in the Arc.
The exact mechanism by which TH deficiency interferes with the intracellular leptin signaling, however, remains enigmatic. Hypothyroidism seemingly does not influence the expression levels of the long leptin receptor isoform Ob-Rb that is considered to be the major splice variant responsible for the activation of the JAK-STAT pathway (36) whereas expression of the shorter isoform Ob-Ra is highly elevated in the Arc of hypothyroid animals (Figure 4C). In contrast to Ob-Rb, Ob-Ra lacks the intracellular motifs required for the activation of the JAK-STAT-signal transduction pathway and has been suggested to be primarily involved in leptin clearance (37). It is therefore tempting to speculate that the diminished response of hypothyroid arcuate neurons towards leptin is at least partially due to an increased internalization and inactivation of leptin via the Ob-Ra receptor. Further studies would be needed to corroborate this hypothesis.
Currently, we also cannot exclude that TH deficiency impinges the activation of leptin signaling cascade by other ways. Hypothyroidism might affect the expression of proteins that modulate the interaction between Ob-R and JAK2 such as LPR1 (38). Moreover, other hypothalamic signaling pathways may be altered under hypothyroid conditions as well. As one prominent example, central application of T3 was shown to decrease the activation of hypothalamic AMPK, whereas hypothyroidism resulted in increased AMPKα1 levels and activity in the hypothalamus (5). Interestingly, an activated AMPK was recently identified as a potential negative regulator of leptin signaling (39). Thus, alterations in the cross talk between the JAK2/STAT3 pathway and the AMPK pathway may contribute to the blunted intracellular response toward leptin in the hypothyroid mouse mutants.
For more than a century, hypothyroidism in humans has been closely associated with obesity, and a central leptin resistance would fit well into this concept. However, hypothyroid mice are characterized by a lean phenotype, indicating that additional alterations in energy metabolism override any orexigenic effects due to a diminished leptin response. An increase in facultative thermogenesis has been suggested to be an important determinant for the lean phenotype of TRa1 R348C mutant mice because at room temperature but not at 30°C, these animals exhibit a highly activated brown adipose tissue due to an enhanced sympathetic outflow (33). Moreover, hypothyroid mice have been reported to show increased caloric intake and weight gain when the animals were acclimatized to thermoneutrality (31). We therefore speculate that the central leptin resistance comprises only one aspect of hypothyroidism-induced alterations in energy metabolism. Further studies with more sophisticated animal models are clearly needed to dissect central from peripheral consequences of hypothyroidism.
Acknowledgments
We thank Sabine Landmann (FLI, Jena) for excellent technical assistance and Karl Bauer (FLI, Jena) for helpful discussions.
This work was supported by the Leibniz Graduate School of Aging and Age Related Diseases.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AgRP
- Agouti-related peptide
- AMPK
- AMP-activated protein kinase
- Arc
- arcuate nucleus
- BAT
- brown adipose tissue
- bw
- body weight
- CNS
- central nervous system
- DIG
- digoxigenin
- Dio2
- type 2 iodothyronine deiodinase
- dko
- double knockout
- ISH
- in situ hybridization
- LCM
- laser-capture microdissection
- MMI
- methimazole
- NPY
- neuropeptide Y
- Ob-R
- leptin receptor
- PFA
- paraformaldehyde
- PGC-1a
- PPARγ coactivator 1 α
- POMC
- proopiomelanocortin
- PPARg
- peroxisome proliferator-activated receptor-γ
- qPCR
- quantitative real-time PCR
- RT
- room temperature
- SOCS
- suppressor of cytokine signaling
- SSC
- standard saline citrate
- STAT
- signal transducer and activator of transcription
- TH
- thyroid hormone
- Trhr1
- TRH receptor 1
- UCP
- uncoupling protein
- WAT
- white adipose tissue
- WT
- wild type.
References
- 1. Moller N , Nielsen S , Nyholm B , Porksen N , Alberti KG , Weeke J. Glucose turnover, fuel oxidation and forearm substrate exchange in patients with thyrotoxicosis before and after medical treatment. Clin Endocrinol (Oxf). 1996;44:453–459. [DOI] [PubMed] [Google Scholar]
- 2. Duntas LH. Thyroid disease and lipids. Thyroid. 2002;12:287–293. [DOI] [PubMed] [Google Scholar]
- 3. Coppola A , Liu ZW , Andrews ZB , et al. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007;5:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klieverik LP , Janssen SF , van Riel A , et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver. Proc Natl Acad Sci USA. 2009;106:5966–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lopez M , Varela L , Vazquez MJ , et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med. 2010;16:1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Varela L , Martinez-Sanchez N , Gallego R , et al. Hypothalamic mTOR pathway mediates thyroid hormone-induced hyperphagia in hyperthyroidism. J Pathol. 2012;227:209–222. [DOI] [PubMed] [Google Scholar]
- 7. Cowley MA , Smart JL , Rubinstein M , et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. [DOI] [PubMed] [Google Scholar]
- 8. Lechan RM , Fekete C. The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res. 2006;153:209–235. [DOI] [PubMed] [Google Scholar]
- 9. Legradi G , Emerson CH , Ahima RS , Flier JS , Lechan RM. Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus. Endocrinology. 1997;138:2569–2576. [DOI] [PubMed] [Google Scholar]
- 10. Wittmann G , Fuzesi T , Singru PS , Liposits Z , Lechan RM , Fekete C. Efferent projections of thyrotropin-releasing hormone-synthesizing neurons residing in the anterior parvocellular subdivision of the hypothalamic paraventricular nucleus. J Comp Neurol. 2009;515:313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hara J , Gerashchenko D , Wisor JP , Sakurai T , Xie X , Kilduff TS. Thyrotropin-releasing hormone increases behavioral arousal through modulation of hypocretin/orexin neurons. J Neurosci. 2009;29:3705–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gonzalez JA , Horjales-Araujo E , Fugger L , Broberger C , Burdakov D. Stimulation of orexin/hypocretin neurones by thyrotropin-releasing hormone. J Physiol. 2009;587:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parmentier R , Kolbaev S , Klyuch BP , et al. Excitation of histaminergic tuberomamillary neurons by thyrotropin-releasing hormone. J Neurosci. 2009;29:4471–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X , van den Pol AN. Thyrotropin-releasing hormone (TRH) inhibits melanin-concentrating hormone neurons: implications for TRH-mediated anorexic and arousal actions. J Neurosci. 2012;32:3032–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vijayan E , McCann SM. Suppression of feeding and drinking activity in rats following intraventricular injection of thyrotropin releasing hormone (TRH). Endocrinology. 1977;100:1727–1730. [DOI] [PubMed] [Google Scholar]
- 16. Suzuki T , Kohno H , Sakurada T , Tadano T , Kisara K. Intracranial injection of thyrotropin releasing hormone (TRH) suppresses starvation-induced feeding and drinking in rats. Pharmacol Biochem Behav. 1982;17:249–253. [DOI] [PubMed] [Google Scholar]
- 17. Horita A. An update on the CNS actions of TRH and its analogs. Life Sci. 1998;62:1443–1448. [DOI] [PubMed] [Google Scholar]
- 18. O'Dowd BF , Lee DK , Huang W , et al. TRH-R2 exhibits similar binding and acute signaling but distinct regulation and anatomic distribution compared with TRH-R1. Mol Endocrinol. 2000;14:183–193. [DOI] [PubMed] [Google Scholar]
- 19. Heuer H , Schafer MK , O'Donnell D , Walker P , Bauer K. Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J Comp Neurol. 2000;428:319–336. [PubMed] [Google Scholar]
- 20. Liu XG , Tan LJ , Lei SF , et al. Genome-wide association and replication studies identified TRHR as an important gene for lean body mass. Am J Hum Genet. 2009;84:418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cook CB , Kakucska I , Lechan RM , Koenig RJ. Expression of thyroid hormone receptor β 2 in rat hypothalamus. Endocrinology. 1992;130:1077–1079. [DOI] [PubMed] [Google Scholar]
- 22. Wallis K , Dudazy S , van Hogerlinden M , Nordstrom K , Mittag J , Vennstrom B. The thyroid hormone receptor α1 protein is expressed in embryonic postmitotic neurons and persists in most adult neurons. Mol Endocrinol. 2010;24:1904–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rabeler R , Mittag J , Geffers L , et al. Generation of thyrotropin-releasing hormone receptor 1-deficient mice as an animal model of central hypothyroidism. Mol Endocrinol. 2004;18:1450–1460. [DOI] [PubMed] [Google Scholar]
- 24. Mansouri A , Chowdhury K , Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet. 1998;19:87–90. [DOI] [PubMed] [Google Scholar]
- 25. Flamant F , Poguet AL , Plateroti M , et al. Congenital hypothyroid Pax8(−/−) mutant mice can be rescued by inactivating the TRα gene. Mol Endocrinol. 2002;16:24–32. [DOI] [PubMed] [Google Scholar]
- 26. Friedrichsen S , Christ S , Heuer H , et al. Regulation of iodothyronine deiodinases in the Pax8−/− mouse model of congenital hypothyroidism. Endocrinology. 2003;144:777–784. [DOI] [PubMed] [Google Scholar]
- 27. Reyns GE , Janssens KA , Buyse J , Kuhn ER , Darras VM. Changes in thyroid hormone levels in chicken liver during fasting and refeeding. Comp Biochem Physiol B Biochem Mol Biol. 2002;132:239–245. [DOI] [PubMed] [Google Scholar]
- 28. Mitchell SE , Nogueiras R , Morris A , et al. Leptin receptor gene expression and number in the brain are regulated by leptin level and nutritional status. J Physiol. 2009;587:3573–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gorska E , Popko K , Stelmaszczyk-Emmel A , Ciepiela O , Kucharska A , Wasik M. Leptin receptors. Eur J Med Res. 2010;15(suppl 2):50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vaisse C , Halaas JL , Horvath CM , Darnell JE , Stoffel M , Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–97. [DOI] [PubMed] [Google Scholar]
- 31. Ueta CB , Olivares EL , Bianco AC. Responsiveness to thyroid hormone and to ambient temperature underlies differences between brown adipose tissue and skeletal muscle thermogenesis in a mouse model of diet-induced obesity. Endocrinology. 2011;152:3571–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silva JE , Larsen PR. Potential of brown adipose tissue type II thyroxine 5′-deiodinase as a local and systemic source of triiodothyronine in rats. J Clin Invest. 1985;76:2296–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sjogren M , Alkemade A , Mittag J , et al. Hypermetabolism in mice caused by the central action of an unliganded thyroid hormone receptor α1. EMBO J. 2007;26:4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fain JN , Bahouth SW. Effect of tri-iodothyronine on leptin release and leptin mRNA accumulation in rat adipose tissue. Biochem J. 1998;332:361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoshida T , Monkawa T , Hayashi M , Saruta T. Regulation of expression of leptin mRNA and secretion of leptin by thyroid hormone in 3T3–L1 adipocytes. Biochem Biophys Res Commun. 1997;232:822–826. [DOI] [PubMed] [Google Scholar]
- 36. Fruhbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Uotani S , Bjorbaek C , Tornoe J , Flier JS. Functional properties of leptin receptor isoforms: internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes. 1999;48:279–286. [DOI] [PubMed] [Google Scholar]
- 38. Liu Q , Zhang J , Zerbinatti C , et al. Lipoprotein receptor LRP1 regulates leptin signaling and energy homeostasis in the adult central nervous system. PLoS Biol. 2011;9:e1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Su H , Jiang L , Carter-Su C , Rui L. Glucose enhances leptin signaling through modulation of AMPK activity. PLoS One. 2012;7:e31636. [DOI] [PMC free article] [PubMed] [Google Scholar]