

SUMMARY
Cell death is a fundamental biological phenomenon that is essential for the survival and development of an organism. Emerging evidence also indicate that cell death contributes to immune defense against infectious diseases. Pyroptosis is a form of inflammatory programed cell death pathway activated by human and mouse caspase-1, human caspase-4 and caspase-5, or mouse caspase-11. These inflammatory caspases are used by the host to control bacterial, viral, fungal or protozoan pathogens. Pyroptosis requires cleavage and activation of the pore-forming effector protein gasdermin D by inflammatory caspases. Physical rupture of the cell causes release of the pro-inflammatory cytokines IL-1β and IL-18, alarmins and endogenous danger-associated molecular patterns, signifying the inflammatory potential of pyroptosis. Here, we describe the central role of inflammatory caspases and pyroptosis in mediating immunity to infection and clearance of pathogens.
Keywords: Bacteria, caspase-1, caspase-4, caspase-5, caspase-11, cell death, gasdermin D, infection, inflammasomes, inflammation, inflammatory caspases, interferons, lysis, lytic, necrosis, necroptosis, pores, pyroptosis, viruses
INTRODUCTION
Programmed cell death pathways, including apoptosis, pyroptosis and necroptosis, are regulated by unique sets of host proteins that coordinate a variety of biological outcomes (1–6). Both apoptosis and pyroptosis are executed by caspases. Apoptosis is mediated by apoptotic caspases, which include caspase-2, caspase-3, caspase-6, caspase-7, caspase-8 and caspase-9 (7). Humans also express the apoptotic caspase family member caspase-10. Although apoptosis has generally been considered an immunologically silent process, emerging evidence indicates that apoptosis can be inflammatory when induced under certain conditions and has roles in the host defense against infection (3, 8, 9).
In contrast to apoptosis, pyroptosis is a form of necrotic and inflammatory programmed cell death induced by inflammatory caspases (6). The requirement of inflammatory caspases in executing pyroptosis distinguishes it from another necrotic and inflammatory form of programmed cell death called necroptosis (1, 10, 11), which is executed independently of caspases (Figure 1). Pyroptosis was initially observed in macrophages infected with Salmonella enterica serovar Typhimurium (S. Typhimurium) or Shigella flexneri and was thought to be apoptosis (12, 13). A study in 1998 found that cell death induced by S. flexneri was abolished in macrophages lacking the gene encoding caspase-1 [also known as interleukin-1β-converting enzyme (ICE)] (13). A similar study in 1999 also reported caspase-1-dependent cell death in macrophages infected with S. Typhimurium (14). These studies together provided important genetic evidence to pinpoint a role for caspase-1 in bacteria-induced cell death. Since previous studies have shown that caspase-1 can mediate proteolytic cleavage of the pro-inflammatory precursor cytokines pro-IL-1β and pro-IL-18 (15–18), the term pyroptosis was subsequently proposed in 2001 to ascribe the inflammatory nature to caspase-1-dependent cell death (19). The definition of pyroptosis has now been broadened to encompass cell death executed by most inflammatory caspases, namely human caspase-4, human caspase-5, and mouse caspase-11 (3).
Figure 1. Programed cell death pathways are regulated by different molecular components.
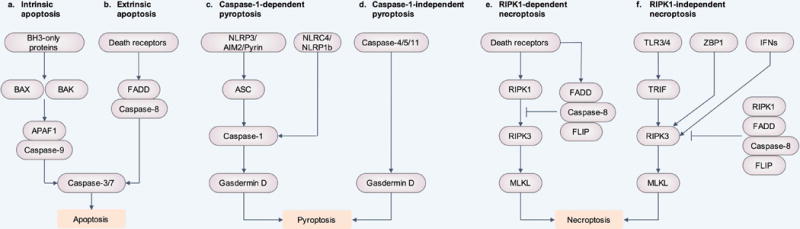
(a and b) Apoptosis can be activated via intrinsic and extrinsic pathways. (a) Intrinsic apoptosis requires BCL-2 homology domain 3 (BH3)-only proteins, which engages BAX and BAK activation. This leads to apoptosome assembly via APAF1 and caspase-9, resulting in the activation of the effectors caspase-3 and caspase-7, and apoptosis. (b) Extrinsic apoptosis requires death receptors to induce dimerization of caspase-8 or caspase-10 through the adaptor protein FADD. Active caspase-8 and caspase-10 cleave and activate caspase-3 and caspase-7, leading to apoptosis. (c and d) Pyroptosis can be induced via caspase-1, human caspase-4 and caspase-5, or mouse caspase-11. (c) The inflammasome sensors NLRP3, AIM2 and Pyrin require the inflammasome adaptor protein ASC in order to form a caspase-1-containing inflammasome complex. The inflammasome sensors NLRC4 and NLRP1b can directly bind to caspase-1 without ASC. Caspase-1 mediates cleavage of the substrate gasdermin D, generating an N-terminal fragment of gasdermin D that induces pyroptosis. (d) Human caspase-4 and caspase-5 or mouse caspase-11 directly cleave gasdermin D to induce pyroptosis. (e and f) Necroptosis can be induced via RIPK1 or independently of RIPK1. (e) Death receptors induce activation of RIPK1, RIPK3 and MLKL, leading to necroptosis. FADD, caspase-8, and the caspase-8 paralogue FLIP form a complex to inhibit RIPK1-dependent necroptosis. (f) TLR3 and TLR4 can directly recruit and activate RIPK3 via the adaptor protein TRIF, independently of RIPK1. In addition, ZBP1 (also known as DLM-1 and DAI) can bind and activate RIPK3. Interferons (IFNs) can also activate RIPK3. In this context, RIPK1, along with FADD, caspase-8, and FLIP inhibit RIPK3-dependent necroptosis.
With the exception of caspase-12, all inflammatory caspases are activated within an inflammasome (20–22). An inflammasome is a macromolecular protein complex composed of inflammasome-initiating sensors (NLRP1, NLRP3, NLRC4, AIM2 or pyrin) and inflammatory caspases, in the presence or absence of the inflammasome adaptor protein ASC (20, 23). Activation of inflammasome-associated inflammatory caspases drives cleavage of the pro-pyroptotic factor gasdermin D (24–26), generating an N-terminal fragment that oligomerizes to form pores on the host cell membrane and cause the lytic demise of the cell (27–31).
The field of inflammatory caspases has somewhat been confounded by studies showing that previously generated caspase-1-deficient mouse lines also lack caspase-11 (32, 33). In 1995, two groups each generated a caspase-1-deficient mouse line (called “ICE−/−” or “Casp1−/−”) using embryonic stem cells of the 129 background (34, 35). It was later revealed that these mouse lines carry a 129-associated inactivating passenger mutation in the caspase-11 locus (32). The close proximity of the caspase-1 and caspase-11 loci prevented segregation of these two proteins despite extensive backcrossing to the C57BL/6 background, essentially rendering these mice deficient in both caspase-1 and caspase-11 (32). In this review, we will refer previously-generated “ICE−/−” or “Casp1−/−” strains as Casp1−/−Casp11−/− (also known as Casp1−/−Casp11129mt/129mt) mice (32). Generation of new mutant mouse strains specifically lacking caspase-1 has been useful in propelling studies aiming to refine the biological functions of inflammatory caspases in health and disease (36–38). Here, we provide an overview on the functions and mechanisms of inflammatory caspases and pyroptosis in host defense against pathogens.
Molecular basis of pyroptosis and inflammasome activation
Pyroptosis is regulated via a caspase-1-dependent or caspase-1-independent mechanism (Figure 2). Caspase-1-independent pyroptosis is executed by human caspase-4, human caspase-5 or mouse caspase-11 (39). The morphological characteristics of caspase-1-dependent and caspase-1-independent pyroptosis are similar. Both are characterized by cell swelling, positivity for Annexin V and TUNEL staining, chromatin condensation and absence of DNA laddering (40–43). The mitochondria of pyroptotic cells also tend to lose membrane potential (42, 44). The terminal event is represented by rupture of the cell membrane, causing release of cytoplasmic contents of the cell, including pro-inflammatory cytokines, endogenous ligands, alarmins and other danger-associated molecular patterns (34, 35, 45–50).
Figure 2. Molecular basis of caspase-1-dependent pyroptosis and caspase-1-independent pyroptosis.
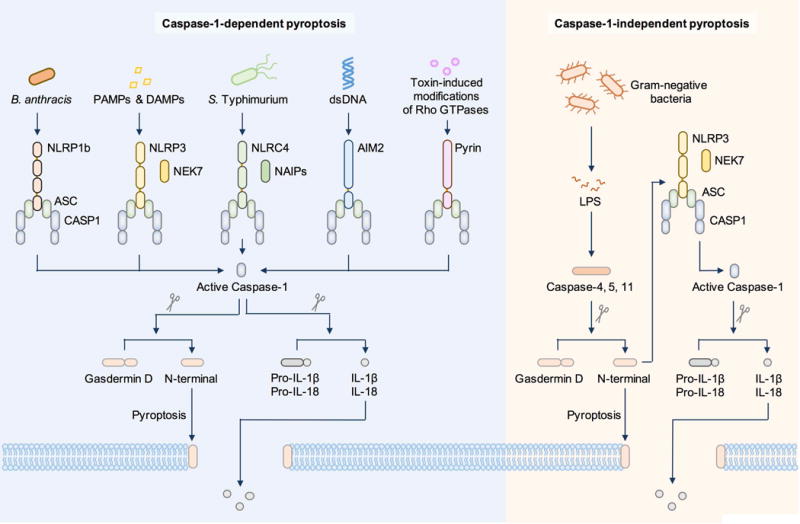
Caspase-1-dependent pyroptosis requires activation of the canonical inflammasomes. In this pathway, pathogen-associated molecular patterns or danger-associated molecular patterns activate their respective inflammasome sensors, including NLRP1b, NLRP3, NLRC4, AIM2 or Pyrin. Activation of the NLRP3 and NLRC4 inflammasomes requires the kinase NEK7 and ligand-binding NAIP proteins, respectively. Inflammasome sensors trigger recruitment of the inflammasome adaptor ASC and the cysteine protease caspase-1 into the same macromolecular complex. Caspase-1 directly cleaves gasdermin D and the precursor cytokines pro-IL-1β and pro-IL-18, initiating pyroptosis and maturation of IL-1β and IL-18, respectively. The 31-kDa cleaved N-terminal portion of gasdermin D forms pores on the host cell membrane to mediate the release of cytoplasmic contents. Caspase-1-independent pyroptosis requires activation of the non-canonical inflammasome. In this pathway, cytosolic LPS from Gram-negative bacteria is recognized by either caspase-4 or caspase-5 in human cells or by caspase-11 in mouse cells. These inflammatory caspases directly cleave gasdermin D and initiate pyroptosis. The N-terminal fragment also activates the NLRP3 inflammasome and caspase-1-dependent maturation of IL-1β and IL-18.
Both caspase-1-dependent and caspase-1-independent pyroptosis lead to the release of IL-1β and IL-18 – inflammatory hallmarks associated with inflammasome activation. IL-1β is a potent inducer of inflammation, vasodilation, and immune cell extravasation, but also has roles in shaping adaptive immune responses (51). IL-18 promotes interferon (IFN)-γ production in TH1 cells, NK cells and cytotoxic T cells, enhances the development of TH2 cells, and promotes local inflammation (52). The alarmins IL-1α and HMGB1 as well as other endogenous host molecules such as nuclear and mitochondrial DNA are also released by pyroptotic cells (34, 35, 47–50). Further studies have revealed that pyroptotic cells release ASC specks into the extracellular milieu to propagate inflammasome activation (53, 54). These signals together serve as triggers to initiate, amplify and perpetuate inflammation.
While caspase-1-dependent or caspase-1-independent pyroptosis are morphologically similar, there are notable differences. Caspase-1 is activated by inflammasome-initiating sensors on recognition of pathogen-associated molecular patterns or danger-associated molecular patterns (Figure 2). This mode of activation is called canonical inflammasome activation. The mouse NLRP1b and rat NLRP1 inflammasome sensors are activated following their cleavage by a lethal factor released by the Gram-positive bacterium Bacillus anthracis (55, 56). NLRP3 is activated by diverse pathogen-associated molecular patterns and danger-associated molecular patterns (57–59), facilitated by the kinase NEK7 (60–62). NLRC4 is activated by NAIP proteins; a set of NLRs which bind directly to flagellin or the inner rod or needle proteins of the Type III secretion system of bacteria (63–71). AIM2 is activated following direct binding to cytoplasmic dsDNA (72–75). Pyrin responds to bacterial toxin-induced modifications of Rho GTPases and requires microtubule assembly for its activation (76–82). Ultimately, these inflammasome sensors initiate the assembly of a caspase-1-containing inflammasome, licensing caspase-1 to directly cleave the precursor cytokines pro-IL-1β and pro-IL-18 (15–17, 83–87).
Caspase-1-independent pyroptosis is activated by non-canonical activation of the inflammasome (Figure 2). In this case, caspase-4, caspase-5 and caspase-11 are the apical activators, and, via their CARD domains directly recognize LPS from Gram-negative bacteria in the host cytoplasm (88–90). Caspase-4 has been suggested to be able to cleave pro-IL-1β and pro-IL-18 (91, 92), a finding which requires further confirmation. Caspase-11 cannot directly cleave pro-IL-1β and pro-IL-18 (93), but is able to induce pyroptosis independently of caspase-1.
Gasdermin D as a common executor of pyroptosis
All inflammasome-associated caspases directly cleave a 53-kDa substrate called gasdermin D (24–26). Cleavage of gasdermin D by these caspases generates a 31-kDa N-terminal fragment which initiates pyroptosis and a 22-kDa C-terminal fragment which has unknown functions (24, 25, 94). Further mechanistic studies revealed that the N-terminal fragment of gasdermin D associates with the inner leaflet of the cell membrane where it assembles into pores of 10–33 nm in diameter (Figure 2) (24–31). The N-terminal fragment of gasdermin D also drives activation of the NLRP3-dependent caspase-1 inflammasome (24, 25, 37), possibly requiring potassium efflux caused by gasdermin D-induced membrane pores (95). Gasdermin D can also damage and lyse bacteria directly, including Escherichia coli, Staphylococcus aureus and Bacillus megaterium protoplasts (27, 29). The proposed mechanism is that the N-terminal domain of gasdermin D binds to cardiolipin (a lipid found on the bacterial cell membrane) and oligomerize to form pores on the bacterial cell membrane (27, 29).
The physiological role of gasdermin D has only been examined in a mouse model of endotoxemia. Similar to mice lacking caspase-11, mice lacking gasdermin D are remarkably resistant to LPS-induced endotoxemia compared to wild-type mice (25). However, whether secretion of IL-1β and IL-18 in this model is dependent on gasdermin D remains unclear and further work is required to examine this issue.
Although gasdermin D induces pyroptosis, prolonged activation of the canonical inflammasome pathway by LPS plus ATP or flagellin leads to caspase-1-dependent and gasdermin D-independent pyroptosis (25). This finding suggests that other undefined caspase-1 substrates must also contribute to caspase-1-dependent pyroptosis (25). It is noteworthy to highlight that other members of the gasdermin family can also induce cell death, including mouse gasdermin A3, human gasdermin A, human gasdermin B, human gasdermin C and human and mouse DFNA5 (27, 96–98). Whether these proteins have a role in inflammasome signaling, pyroptosis or in host defense against pathogens remains to be determined.
Physiological roles of caspase-1 and caspase-11 during bacterial infection
Inflammatory caspases protect the host against a variety of bacterial pathogens (Tables 1 and 2). Because previously generated caspase-1-deficient mouse lines also lack caspase-11, the relative contribution of caspase-1 and caspase-11 in mouse models of bacterial infection requires further investigation. In addition, activation of inflammatory caspases leads to pyroptosis and secretion of IL-1β and IL-18, both of these functions could provide protection against pathogens.
Table 1.
The role of caspase-1 and caspase-11 in response to Gram-negative bacteria in mice.
| Mouse | Bacteria | Phenotype compared to wild-type mice |
|---|---|---|
|
Casp1−/− Casp11−/− |
Acinetobacter baumannii | Decreased IL-1β and IL-6 in the BALF, decreased lung pathology (188). |
| Burkholderia cepacia | No significant survival difference (115). | |
| Burkholderia pseudomallei | Reduced survival, increased bacterial burden in the spleen, lung and liver, decreased IL-1β, IL-18 and IFN-γ in BALF (104, 189, 190). | |
| Burkholderia thailandensis | Reduced survival (104, 115, 116), increased bacterial burden in the spleen, liver and MLN (116). | |
| Citrobacter rodentium | Increased body weight loss, bacterial burden in feces, and adaptive immune responses, shorter colon length, elevated pro-inflammatory cytokines, decreased IL-18 (191). | |
| Chromobacterium violaceum | Reduced survival, increased bacterial burden in the liver and spleen, increased macroscopic lesions and extensive neutrophil infiltration in the liver (115). | |
| Ehrlichia (lxodes ovatus ehrlichia) | No difference in survival, but higher bacterial burden in the liver (192). | |
| Francisella novicida | Increased pathogen burden in the liver, lung and spleen, reduced survival and IL-18 production (177, 178, 193). | |
| Francisella philomiragia | No survival difference (115). | |
| Francisella tularemia | Reduced survival and increased bacterial burden in lungs (194). | |
| Legionella bozemanii | Increased bacterial burden in the lungs (195). | |
| Legionella gratiana | Increased bacterial burden in the lungs (196). | |
| Legionella micdadei | Increased bacterial burden in the lungs (195). | |
| Legionella pneumophila | Increased bacterial burden in the lungs (195, 196). | |
| Legionella rubrilucens | Increased bacterial burden in the lungs (195). | |
| Salmonella Typhimurium | Increased pathogen burden in the liver, spleen, MLNs, and Peyer’s patches, and reduced survival, IL-18 production and bacterial uptake by neutrophils (99, 101–103, 197), increased bacterial burden in colonic mucosa but no significant difference of bacterial burden in the MLN and cecum (111), decreased IL-1β and IL-18 in the cecal tissue (92). | |
| Shigella flexneri | Reduced survival and increased bacterial burden in the lungs (198). | |
| Vibrio vulnificus | Reduced survival (199). | |
| Yersinia pestis | Increased survival and decreased symptoms of respiratory distress (200). | |
| Yersinia pseudotuberculosis | No difference in survival with WT Yersinia but reduced survival and increased bacterial burden in the spleen with ΔYopM (201, 202). | |
| LPS endotoxemia | Increased survival, reduced IL-1β and IL-18 production (32, 35, 47, 99, 122, 203). | |
| Casp1−/− (also known as Casp1Null) | Francisella novicida | Reduced survival and IL-18 production (175). |
|
Casp1−/− Casp11Tg* |
Legionella gratiana | Increased bacterial burden in the lungs (196). |
| Legionella pneumophila | Increased bacterial burden in the lungs (196, 204). | |
| Salmonella Typhimurium | Increased pathogen burden in the liver, spleen and MLNs and reduced bacterial uptake by neutrophils (102). | |
| LPS endotoxemia | No significant difference in survival, but reduced IL-1β and IL-18 production (32). | |
| Casp11−/− | Burkholderia pseudomallei | Reduced survival (104). |
| Burkholderia thailandensis | Reduced survival (104, 116), increased bacterial burden in the spleen, liver and MLN (116). | |
| Legionella gratiana | No difference in bacterial burden in the lungs (196). | |
| Legionella pneumophila | No difference in bacterial burden in the lungs (196). | |
| Listeria monocytogenes | No difference in survival, pathogen burden in the blood, liver, and spleen (205). | |
| Salmonella Typhimurium | Increased pathogen burden in the cecum and lumen (92), but not in the liver, spleen and MLNs (102). Reduced IL-18 production in the cecal tissue (92), no significant difference in bacterial burden in the colonic mucosa (111). | |
| LPS endotoxemia | Increased survival, reduced IL-1β and IL-18 production (32, 88, 89, 205, 206). |
Casp1−/−Casp11−/− mouse embryos microinjected with a bacterial artificial chromosome transgene encoding caspase-11, such that the mouse strain expresses caspase-11 to mimic a “Casp1−/−” mouse strain (32).
Table 2.
The role of caspase-1 and caspase-11 in response to Gram-positive and other non-Gram-negative bacteria in mice.
| Mouse | Bacteria | Phenotype compared to wild-type mice |
|---|---|---|
| Gram-positive bacteria | ||
| Casp1−/−Casp11−/− | Bacillus anthracis | Reduced survival and decreased IL-1β in the serum (207). |
| Listeria monocytogenes | Reduced survival, increased bacterial burden in the liver and spleen, decreased IL-18 and IFN-γ in the serum (208). | |
| Staphylococcus aureus | Reduced survival but no significant changes in bacterial burden (209). | |
| Streptococcus agalactiae (Group B Streptococcus) | Reduced survival and increased bacterial burden in the kidneys and blood (210). | |
| Streptococcus pneumonia | No difference in survival and in bacterial burdens in the lungs and blood (211). | |
| Gram-variable bacteria | ||
| Casp1−/−Casp11−/− | Mycobacterium tuberculosis | No difference in survival and bacterial burden in the lungs, liver and spleen (212). No difference in survival during the acute phase of infection (213). |
The differential contribution of inflammatory caspases and their substrates is best characterized in a murine model of salmonellosis. Earlier studies found that both IL-1β and IL-18 were important for the control of oral S. Typhimurium infection in the intestine, while IL-18 also controlled the infection at systemic sites (99, 100). The importance of inflammatory caspases in driving protection against S. Typhimurium infection is supported by studies showing that Casp1−/−Casp11−/− mice harbor increased bacterial load and that these mice succumb to infection more rapidly than wild-type mice (99, 101–103). The relative contribution between the two caspases has since been examined. In order to study the role of caspase-1 in a mouse model, Kayagaki and colleagues microinjected a bacterial artificial chromosome transgene encoding caspase-11 into Casp1−/−Casp11−/− mouse embryos to re-establish caspase-11 expression in this strain, generating a new strain known as Casp1−/−Casp11Tg (32). A subsequent study has shown that following orogastric infection with S. Typhimurium Casp1−/−Casp11Tg mice harbor more bacteria in the systemic organs than Casp1−/−Casp11−/− mice, and both strains have higher bacterial burden than wild-type mice (102). However, a similar number of bacteria was found between wild-type and Casp11−/− mice (102). These data would suggest that caspase-1, but not caspase-11, confers protection to S. Typhimurium infection. Moreover, caspase-11 is deleterious to the host in the absence of caspase-1.
A role for caspase-11 in Salmonella infection was revealed using a genetically engineered strain of S. Typhimurium (ΔsifA), which is unable to maintain integrity of the pathogen-containing vacuole and is susceptible to aberrant entry into the host cytoplasm. Caspase-11, but not canonical inflammasomes or IL-1β and IL-18, was shown to mediate clearance of S. Typhimurium ΔsifA in vivo (104). Additional studies have found that caspase-11 is required for the secretion of IL-18 in the intestinal tissue in response to S. Typhimurium infection and for controlling bacterial numbers in the cecum (92).
The importance of pyroptosis could also be partially inferred by studies showing that S. Typhimurium lacking both of its flagellin genes, fliC and fljB, has reduced capacity to induce inflammation and is attenuated in vivo, possibly because of its lack of motility and/or impaired activation of the flagellin-sensing NLRC4-caspase-1 inflammasome or TLR5 (105–108). A strain of S. Typhimurium which overexpresses flagellin due to lack of YdiV, the transcriptional repressor of the flagellin-encoding gene fliC, causes excessive pyroptosis in macrophages (109). This strain induces increased levels of IL-1β and TNF in the serum and fails to colonize the tissue in mice (109).
Pyroptosis releases intracellular bacteria residing within macrophages, including S. Typhimurium, Legionella pneumophila and Burkholderia thailandensis (110). These newly released bacteria can be phagocytosed and killed by neutrophils via a mechanism dependent on the production of reactive oxygen species but independent of IL-1β and IL-18 (110). Other studies have also reported that pyroptosis can clear bacterial infection even in the absence of IL-1α, IL-1β and IL-18 (92, 110, 111). More recent work proposed that viable bacteria can remain trapped within the cellular debris of pyroptotic macrophages called pore-induced intracellular traps (112). These pore-induced intracellular traps are efferocytosed and cleared by neutrophils (112). A more holistic view would be that both unbound and trapped bacteria are released by pyroptotic cells, both of which can be taken up by phagocytic cells in the tissue. An advantage of uptake of pyroptosis-released bacteria by neutrophils is that these host cells are relatively resistant to pyroptosis in response to S. Typhimurium infection and other inflammasome activators (113), indicating that neutrophils would be a suitable phagocytic cell type to clear residual bacteria in the tissue. Neutrophils can undergo pyroptosis in mice lacking the NADPH oxidase NOX2 infected with Pseudomonas aeruginosa (114), suggesting that pyroptosis can be activated to compensate for deficiency of another major antimicrobial pathway.
Another mechanism by which pyroptosis might clear bacteria is through extrusion of infected cells from the tissue. Enterocytes infected with S. Typhimurium undergo activation of the caspase-1 or caspase-11 inflammasome, resulting in physical extrusion of infected enterocytes from the intestine (92, 111). In addition to cell-type-specific roles for pyroptosis, organ-specific functions for pyroptosis have also been reported. The NLRC4-caspase-1 inflammasome is essential in mediating host protection to the ubiquitous environmental bacterium Chromobacterium violaceum (115). In this context, pyroptosis, but not IL-1β and IL-18 mediates bacterial clearance in the spleen, whereas both pyroptosis and IL-18-dependent NK cell responses are required in the liver (115). Caspase-1 can also act upstream of caspase-11 in the host defense against bacterial infection. Caspase-1 induces the production of IL-18, which then triggers the production of IFN-γ to prime caspase-11-mediated responses to clear B. thailandensis infection in mice (116). These data collectively indicate that pyroptosis and inflammatory cell death is generally protective against bacterial infection. In some cases, excessive inflammasome activation or pyroptosis is detrimental to the host. Unchecked inflammasome activation can either drive immunopathology in response to infection by Pseudomonas aeruginosa (117, 118) or impair the generation of CD8+ T-cell-mediated immunity to Listeria monocytogenes (119).
Caspase-4 and caspase-5 in bacterial infection
The caspase-11 homologs, human caspase-4 and caspase-5, also recognize LPS, induce activation of caspase-1, mediate cleavage of gasdermin D, and drive pyroptosis (24, 25, 90). Although both caspase-4 and caspase-5 have been shown to bind to LPS, the function of these inflammatory caspases is likely to be cell-type-specific and determined by the type of activators encountered by the cell. For example, caspase-4, but not caspase-5, was reported to drive cell death and IL-1β production in the human monocytic THP-1 cell line in response to LPS transfection (120, 121). Mouse bone-marrow-derived macrophages engineered to express human caspase-4 respond to LPS stimulation and release IL-1β and IL-18 (122). A further study has demonstrated that caspase-4 necessitated IL-1α release and cell death in primary human macrophages infected with S. Typhimurium, L. pneumophila or Yersinia pseudotuberculosis; however, caspase-4 was not required for IL-1β release in this setting (123). Other studies have found non-redundant functions between caspase-4 and caspase-5, with both caspases required for IL-1β secretion in human monocytes stimulated with LPS or in the THP-1 cell line infected with S. Typhimurium (120, 124).
Caspase-4 or caspase-5 can restrict the replication of L. pneumophila in human macrophages (125) and of S. Typhimurium in human colonic epithelial cells (92). The importance of these caspases is reflected by the presence of bacterial-encoded virulence factors that counteract the effect of these caspases. The effector protein NleF of enteropathogenic E. coli can bind the catalytic domain of caspase-4 and inhibit caspase-4-dependent IL-18 secretion in the human intestinal cell line Caco-2 (126). Enteropathogenic E. coli and enterohemorrhagic E. coli and their mouse relative C. rodentium encode effector proteins called NleB (127, 128) and NleH (129), which bind to components of the cell death pathway to suppress apoptosis and/or necroptosis.
To study the role of human caspase-4 in a mouse model, a transgene encoding human caspase-4 was introduced into Casp11−/− mice. Unlike Casp11−/− mice, Casp11−/− mice expressing human caspase-4 are protected from a lethal B. thailandensis infection (116). In an endotoxemia model, Casp11−/− mice are normally resistant to LPS-induced lethality; however, Casp11−/− mice expressing human caspase-4 are susceptible to LPS-induced lethality (122). These studies highlight the functional complementarity between human and mouse inflammatory caspases in infection and immunity.
Inflammatory caspases are required for cell-autonomous immunity to bacteria
Inflammatory caspases control antimicrobial cellular functions beyond pyroptosis and IL-1β and IL-18 release. Caspase-1 activation regulates phagosome maturation during both Gram-negative and Gram-positive bacterial infection (130, 131). Caspase-1 promotes fusion between vacuoles containing L. pneumophila and lysosomes (130). A further study has shown that caspase-1 regulates the pH of phagosomes containing Gram-positive bacteria such as Staphylococcus aureus, resulting in enhanced killing of the internalized pathogen in macrophages (131). Caspase-1 activation also reduces cellular stiffness to prevent excessive uptake of S. Typhimurium in macrophages, allowing the cell to control bacterial burden autonomously (132). A further study has shown that the catalytic activity of caspase-1 and caspase-11 is essential for dampening the intracellular growth of S. Typhimurium ΔsifA (133). In response to infection by L. pneumophila, caspase-11 regulates actin polymerization via cofilin to promote fusion between the vacuole containing L. pneumophila and lysosomes (125). The cell-autonomous functions of inflammatory caspases clearly expand their mechanistic repertoire beyond pyroptosis and cytokine production in anti-bacterial host defense.
Physiological roles of pyroptosis in viral, fungal and protozoan infection
The ability of inflammasome sensors to recognize a wide array of pathogen-associated molecular patterns suggests that inflammatory caspases are crucial for host defense against pathogens from virtually all domains of life. For example, both DNA and RNA viruses can activate the inflammasome and induce pyroptosis (134, 135). However, a deleterious role of pyroptosis in human immunodeficiency virus (HIV) infection has been reported. The DNA sensor IFI16 recognizes cytosolic viral DNA intermediates produced during HIV-1 infection in human macrophages or CD4+ T cells (136, 137). IFI16-mediated recognition of HIV-1 curtails virus replication in human macrophages (136), whereas this response drives pyroptosis in CD4+ T cells in lymphoid tissues via cell-to-cell transmission of HIV-1 (137–139). This phenomenon has been suggested to accelerate depletion of CD4+ T cells and progression to AIDS in humans (137, 138). Peripheral blood-derived CD4+ T cells do not undergo pyroptosis in response to HIV-1 infection owing to reduced levels of IFI16 expression and HIV-1 reverse transcripts in these cells (140). When co-cultured with lymphoid-derived CD4+ T cells, peripheral blood-derived CD4+ T cells become sensitized to HIV-1-induced pyroptosis (140), suggesting that pyroptosis is transmissible between different subsets of cells. Uninfected liver cells can also undergo caspase-1-dependent pyroptosis following infection of bystander cells with hepatitis C virus (HCV) (141).
Studies in mice have revealed the importance of caspase-1 and caspase-11 against infection by influenza A virus and West Nile virus (Table 3) (142). The influenza A virus activates the NLRP3 inflammasome (143–146). Indeed, Casp1−/−Casp11−/− mice are more susceptible to infection by influenza A virus (143, 144, 147), and produce less IL-1β and IL-18 in the lungs, and have diminished lung functions and increased viral titers compared to wild-type mice (143, 147). However, Casp1−/−Casp11−/− mice are as resistant as Mx1 congenic mice on the C57BL/6 background after infection with influenza A virus but the absence of Casp1 and Casp11 provides protection from lethality in Tlr7−/−Mavs−/− mice (148). Individual roles for caspase-1 and caspase-11 have not been deciphered in vivo. Influenza A viruses and pathogens other than Gram-negative bacteria do not carry LPS, the pathogen-associated molecular pattern that activates caspase-11. However, it is still too early to presume that caspase-11 has no role in the host defense against microbial agents other than Gram-negative bacteria in vivo. Indeed, a role for both caspase-1 and caspase-11 has been observed in host defense against the fungal pathogen Aspergillus fumigatus, a pathogen which do not carry LPS (36, 149). A. fumigatus activates the NLRP3 inflammasome in human THP-1 cells and both the AIM2 and NLRP3 inflammasomes in mouse bone-marrow-derived dendritic cells and the lung tissue (149, 150). This caspase-1-dependent response is crucial for the generation of protective cytokines IL-1β and IL-18 in a mouse model of aspergillosis (149). Interestingly, Casp11−/− mice are also more susceptible to A. fumigatus-induced mortality compared with wild-type, but succumb to infection with a delayed kinetics compared with mice lacking caspase-1 or both caspase-1 and caspase-11 (36). How caspase-11 is activated or conferred protection during A. fumigatus infection is not known. It is possible that activation of caspase-11 might induce actin-mediated phagosomal killing in order to control A. fumigatus dissemination in vivo (6, 125, 151). Caspase-1-dependent release of IL-18 induces production of IFN-γ, which might provide a priming signal for caspase-11 to control aspergillosis in vivo (149). A recent study has suggested that in addition to LPS, caspase-11 recognizes host-derived oxidized phospholipids, a form of danger-associated molecular pattern that can induce IL-1β release in dendritic cells without triggering pyroptosis (152). Other than A. fumigatus, increased susceptibility of Casp1−/−Casp11−/− mice to the fungal pathogens Candida albicans and Paracoccidioides brasiliensis, compared with wild-type mice has also been reported (Table 4) (153–156). In macrophages, pyroptosis induced by C. albicans requires the development of fungal hyphae and/or neutralization of the phagosome (157, 158). However, the precise physiological role of pyroptosis in fungal infection has not been investigated.
Table 3.
The role of caspase-1 and caspase-11 in response to viral infection in mice.
| Mouse | Virus | Phenotype compared to wild-type mice |
|---|---|---|
| Casp1−/−Casp11−/− | Encephalomyocarditis virus | No difference in survival (214). |
| Influenza A virus | Reduced survival (143, 144, 147), decreased IL-1β, IL-18, TNF, IL-6, KC, MIP-2 in the BALF, decreased neutrophils and monocytic dendritic cells in the BALF, diminished respiratory function (143), decreased IFN-γ producing CD4+ and CD8+ T cells, reduced nasal IgA, increased pulmonary viral titer (147). No difference in survival and body weight change in Mx1 sufficient host (148). | |
| Murine gamma-herpesvirus 68 (MHV68) | No difference in viral burden in the lungs (215). | |
| Vesicular stomatitis virus | No difference in survival (214). | |
| West Nile virus | Reduced survival (142). |
Table 4.
The role of caspase-1 and caspase-11 in response to fungal infection in mice.
| Mouse | Fungus | Phenotype compared to wild-type mice |
|---|---|---|
| Casp1−/−Casp11−/− | Aspergillus fumigatus | Increased lung damage and hemorrhage, fungal dissemination, and reduced survival (36, 149). |
| Candida albicans | Reduced survival and increased fungal burden in the kidneys, diminished TH1/TH17 responses (153–155). | |
| Paracoccidioides brasiliensis | Reduced survival, increased lung damage and hemorrhage, and fungal dissemination (156). | |
| Casp1−/− (also known as Casp1Null) | Aspergillus fumigatus | Reduced survival (36). |
| Casp11−/− | Aspergillus fumigatus | Reduced survival (36). |
The biological relevance of inflammatory caspases extends to studies on protozoan parasites, but the specific contribution of pyroptosis is unknown (Table 5). Opposing roles of inflammatory caspases can be observed during leishmaniasis, which appears to depend on the background of the mouse strains and the species of protozoa involved. Casp1−/−Casp11−/− mice on the C57BL/6 background infected with Leishmania amazonensis develop lesions of larger size and harbor increased parasite burden in ear, lymph node and spleen compared with wild-type mice (159). Casp1−/−Casp11−/− mice on the BALB/c background infected with Leishmania major have less footpad swelling and parasite burden owing to increased IFN-γ and reduced IL-4 and IL-5 production (160).
Table 5.
The role of caspase-1 and caspase-11 in response to protozoan infection in mice.
| Mouse | Protozoan | Phenotype compared to wild-type mice |
|---|---|---|
| Casp1−/−Casp11−/− | Leishmania amazonensis | Increased lesion size and parasite burden in ear, lymphnode and spleen (159). |
| Leishmania major | Decreased footpad swelling, decreased pathology score and parasite burden in the footpads, decreased IL-1β and IL-18 in the footpads, decreased IL-4, IL-5 and increased IFN-γ (160), decreased infiltration of CD11b+ and PMN cells in the ear (216). | |
| Plasmodium berghei | No difference in survival when infected with sporozoites or iRBCs and no difference in parasitemia level (217, 218). | |
| Toxoplasma gondii | Reduced survival and decreased IL-1β and IL-18 in the serum (161). | |
| Trypanosoma cruzi | Reduced survival, higher parasitism in the heart, spleen and blood, higher heart injury, decreased IL-1β (162, 163), decreased production of NO from splenocytes (163). | |
| Casp11−/− | Toxoplasma gondii | Increased survival, decreased clinical score, altered immune responses, increased brain cysts and neuroinflammation during late stages of disease (164). |
Casp1−/−Casp11−/− mice are also susceptible to infection by the protozoa Toxoplasma gondii and Trypanosoma cruzi, a phenotype associated with reduced IL-1β or IL-18 production (161–163). A further study demonstrates that in response to infection with T. gondii, mice lacking caspase-11 alone have reduced levels of local and systemic cytokines during the acute phase, whereas inflammation is elevated in the brain during the chronic phase of infection (164). Further investigations are required to unveil individual functions of inflammatory caspases in protozoan infections.
IFN signaling and IFN-inducible proteins as critical regulators of pyroptosis and inflammasome activation
IFN signaling is a central modulator of pyroptosis induced by pathogens (165). Extracellular LPS from Gram-negative bacteria is recognized by TLR4, initiating a signaling cascade requiring the TLR adaptor TRIF (102, 166–169). TRIF is crucial in inducing the production of type I IFNs. Type I IFNs bind the heterodimeric type I IFN receptor composed of the subunits IFNAR1 and IFNAR2, and drive transcription of hundreds of IFN-stimulated genes (102, 167). Type I IFN signaling also induces caspase-11, an essential component that activates the non-canonical inflammasome pathway and pyroptosis. Reactive oxygen species and/or the Cpb1-C3-C3aR complement pathway can upregulate the expression of caspase-11 by operating downstream of the TLR4 and type I IFN signaling pathway (170, 171).
The IFN-inducible network of proteins include IFN-inducible GTPases, such as the 47-kD immunity-related GTPases (IRGs) and the 65-kD guanylate-binding proteins (GBPs) (165). GBPs rupture the pathogen-containing vacuole encasing Gram-negative bacteria, allowing the bacteria and their LPS to enter the cytoplasm for sensing by caspase-11 (Figure 3) (172–174). Once the bacteria are in the cytoplasm, IRGB10 further disrupts the structural integrity of the bacteria, enhancing liberation of LPS in the cytoplasm and accessibility by caspase-11 (175). Delivery of LPS into the cytoplasm can also be achieved by bacterial outer membrane vesicles (176), but whether IFN signaling is involved in cytoplasmic entry of bacterial outer membrane vesicles has not been investigated.
Figure 3. Type I IFN signaling regulates pathogen-induced inflammasome activation and pyroptosis.
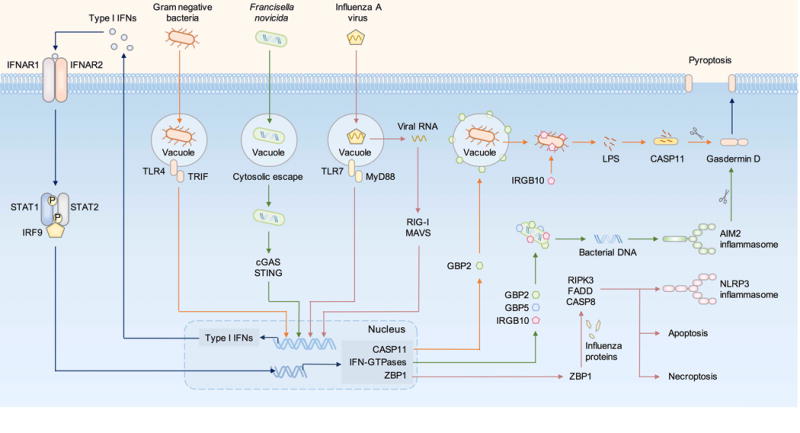
LPS from Gram-negative bacteria is recognized by TLR4, inducing TRIF-dependent type I IFN production. Francisella novicida is a cytosolic bacterium which is recognized by the DNA sensor cGAS, inducing STING-dependent type I IFN production. RNA from influenza A virus triggers the RNA sensors TLR7 via the adaptor MyD88 and RIG-I via the adaptor MAVS, both inducing production of type I IFNs. The type I IFN signaling pathway is activated via the transcription factors STAT1, STAT2 and IRF9, leading to upregulation of caspase-11, IFN-inducible GTPases, including guanylate-binding proteins (GBPs) and Immunity-related GTPases (IRGs), and other IFN-inducible proteins including the sensor ZBP1. GBP2 ruptures the vacuole containing Gram-negative bacteria, mediating the liberation of LPS into the cytoplasm for recognition by caspase-11. IRGB10 further disrupts Gram-negative bacteria to increase LPS accessibility for detection by caspase-11. GBP2, GBP5 and IRGB10 directly target the bacterial membrane of cytosolic-dwelling F. novicida, exposing its DNA for sensing by AIM2. Activation of the caspase-11-NLRP3 inflammasome in response to Gram-negative bacteria and activation of the AIM2 inflammasome in response to F. novicida lead to pyroptosis via gasdermin D. ZBP1 recognizes proteins from the influenza A virus and induce pyroptosis, necroptosis, and apoptosis via RIPK3, FADD and caspase-8.
Activation of pyroptosis induced by the AIM2 inflammasome in response to Francisella novicida infection also requires type I IFN signaling (177–183). This pathway is initiated by the DNA sensor cGAS upon detection of F. novicida, inducing STING-dependent production of type I IFNs (177, 183, 184). Type I IFNs potentiate expression of GBPs and IRGs via the transcription factor IRF1 (175, 177). GBP2, GBP5 and IRGB10 co-operate synergistically to rupture F. novicida that have entered the cytoplasm, resulting in the exposure of F. novicida DNA for sensing by AIM2 (175, 177, 178).
In addition to bacterial infection, type I IFN signaling contributes to cell death induced by influenza A virus. Type I IFN signalling mediates upregulation of the innate immune sensor ZBP1 (also known as DLM-1 or DAI) (146). ZBP1 recognizes the influenza A virus nucleoprotein and RNA polymerase subunit PB1, and mediates activation of the NLRP3 inflammasome, necroptosis and apoptosis via the kinase RIPK3, caspase-8 and FADD (146). These findings were recently confirmed by another group (185, 186), providing further evidence to underscore the importance of the type I IFN–ZBP1 axis in driving influenza virus-induced cell death (Figure 3).
In addition to type I IFN signaling, IFN-γ can prime caspase-11 expression and activation of the inflammasome in macrophages (172, 178). A further study has shown that IFN-γ rather than type I IFNs is necessary to drive induction of caspase-11 responses in mice infected with B. thailandensis (116). IFN-γ is partially required for the upregulation of caspase-11 protein in the colon tissue of mice during DSS-induced colitis (187). These studies collectively highlight a role for both type I and type II IFN signaling pathways in the activation of inflammasomes and pyroptosis.
Conclusions and future perspectives
Host cells are often invaded by intracellular pathogens that are capable of replicating in the pathogen-containing vacuoles or in the cytoplasm. Cell death directly removes these replicative niches exploited by the pathogens. Infectious agents released from dying cells are exposed to extracellular immune defense and are often taken up by other immune cells for killing. In addition, pyroptosis releases cytoplasmic contents from the dying host cells, thereby providing potent signals to initiate an inflammatory cascade. Local inflammation leads to recruitment and priming of immune cells, ultimately contributing to clearance of pathogen from the host.
Our understanding of the molecular mechanisms governing activation and execution of pyroptosis has evolved substantially over the years. Issues associated with the loss of caspase-11 in previously generated caspase-1–deficient mouse strains are being addressed with newly generated single knockout mouse strains (36–38), providing the scientific community new genetic tools to redefine the biological functions of caspase-1 and caspase-11. The identification of gasdermin D as a substrate of inflammatory caspases and a pore-forming protein opens up new avenues by which the function of pyroptosis can be specifically dissected in health and disease. The susceptibility profile of mice lacking gasdermin D or other gasdermin proteins to infectious agents have not been investigated. Direct comparison between mice lacking gasdermin D, mice lacking caspase-1 and/or caspase-11 and mice lacking IL-1β and IL-18 in response to a range of infectious agents would unequivocally reveal the unique and overlapping contributions of these components in the anti-microbial host defense. Given gasdermin D is a terminal effector of pyroptosis and is thought to be a major pathway in clearing intracellular pathogens, it is reasonable to speculate that mice lacking gasdermin D would be susceptible to a range of infectious agents, phenocopying the susceptibility profile seen in mice lacking caspase-1 and/or caspase-11. Further investigations into the relationship between inflammatory caspases and pyroptosis in innate immunity will uncover novel signaling components that can be targeted and translated to prevent or treat infections in human patients.
Acknowledgments
Grant Support
Work from our laboratory is supported by the US National Institutes of Health (AI101935, AI124346, AR056296 and CA163507 to T.D.K.) and the American Lebanese Syrian Associated Charities (to T.D.K.).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nature reviews Molecular cell biology. 2016 doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 2.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nature reviews Molecular cell biology. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 3.Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nature reviews Immunology. 2016;16:7–21. doi: 10.1038/nri.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annual review of immunology. 2015;33:79–106. doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blander JM. A long-awaited merger of the pathways mediating host defence and programmed cell death. Nature reviews Immunology. 2014;14:601–618. doi: 10.1038/nri3720. [DOI] [PubMed] [Google Scholar]
- 6.Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26:R568–R572. doi: 10.1016/j.cub.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 7.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nature reviews Molecular cell biology. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 8.Davidovich P, Kearney CJ, Martin SJ. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biological chemistry. 2014;395:1163–1171. doi: 10.1515/hsz-2014-0164. [DOI] [PubMed] [Google Scholar]
- 9.Creagh EM. Caspase crosstalk: integration of apoptotic and innate immune signalling pathways. Trends in immunology. 2014;35:631–640. doi: 10.1016/j.it.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Vercammen D, Beyaert R, Denecker G, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. The Journal of experimental medicine. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holler N, Zaru R, Micheau O, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature immunology. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 12.Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci U S A. 1996;93:9833–9838. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilbi H, Moss JE, Hersh D, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. The Journal of biological chemistry. 1998;273:32895–32900. doi: 10.1074/jbc.273.49.32895. [DOI] [PubMed] [Google Scholar]
- 14.Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci U S A. 1999;96:2396–2401. doi: 10.1073/pnas.96.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kostura MJ, Tocci MJ, Limjuco G, et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci U S A. 1989;86:5227–5231. doi: 10.1073/pnas.86.14.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Black RA, Kronheim SR, Sleath PR. Activation of interleukin-1 beta by a co-induced protease. FEBS letters. 1989;247:386–390. doi: 10.1016/0014-5793(89)81376-6. [DOI] [PubMed] [Google Scholar]
- 17.Ghayur T, Banerjee S, Hugunin M, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 18.Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 19.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–114. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 20.Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunological reviews. 2015;265:6–21. doi: 10.1111/imr.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell. 2016;165:792–800. doi: 10.1016/j.cell.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 23.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 24.Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 25.Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 26.He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell research. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 28.Aglietti RA, Estevez A, Gupta A, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sborgi L, Ruhl S, Mulvihill E, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. The EMBO journal. 2016 doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, He WT, Hu L, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell research. 2016;26:1007–1020. doi: 10.1038/cr.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 33.Kang SJ, Wang S, Hara H, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. The Journal of cell biology. 2000;149:613–622. doi: 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 35.Li P, Allen H, Banerjee S, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 36.Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, Kanneganti TD. Generation of a caspase-1–deficient mouse line to study the differential roles of caspase-1 and caspase-11 inflammasomes. Sci Rep. 2017 doi: 10.1038/srep45126. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Case CL, Kohler LJ, Lima JB, et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A. 2013;110:1851–1856. doi: 10.1073/pnas.1211521110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu B, Jin C, Li HB, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354:765–768. doi: 10.1126/science.aaf7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stowe I, Lee B, Kayagaki N. Caspase-11: arming the guards against bacterial infection. Immunological reviews. 2015;265:75–84. doi: 10.1111/imr.12292. [DOI] [PubMed] [Google Scholar]
- 40.Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci U S A. 2008;105:4312–4317. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cellular microbiology. 2006;8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 42.Fernandes-Alnemri T, Wu J, Yu JW, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. The Journal of cell biology. 2016;213:617–629. doi: 10.1083/jcb.201602089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimada K, Crother TR, Karlin J, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mariathasan S, Newton K, Monack DM, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 46.England H, Summersgill HR, Edye ME, Rothwell NJ, Brough D. Release of interleukin-1alpha or interleukin-1beta depends on mechanism of cell death. The Journal of biological chemistry. 2014;289:15942–15950. doi: 10.1074/jbc.M114.557561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamkanfi M, Sarkar A, Vande Walle L, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. Journal of immunology. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eigenbrod T, Park JH, Harder J, Iwakura Y, Nunez G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. Journal of immunology. 2008;181:8194–8198. doi: 10.4049/jimmunol.181.12.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nature medicine. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 50.Dombrowski Y, Peric M, Koglin S, et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med. 2011;3:82ra38. doi: 10.1126/scitranslmed.3002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joosten LA, Netea MG, Dinarello CA. Interleukin-1beta in innate inflammation, autophagy and immunity. Seminars in immunology. 2013;25:416–424. doi: 10.1016/j.smim.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 52.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Frontiers in immunology. 2013;4:289. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franklin BS, Bossaller L, De Nardo D, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nature immunology. 2014;15:727–737. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baroja-Mazo A, Martin-Sanchez F, Gomez AI, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nature immunology. 2014;15:738–748. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 55.Levinsohn JL, Newman ZL, Hellmich KA, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS pathogens. 2012;8:e1002638. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hellmich KA, Levinsohn JL, Fattah R, et al. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PloS one. 2012;7:e49741. doi: 10.1371/journal.pone.0049741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 58.Kanneganti TD, Ozoren N, Body-Malapel M, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 59.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 60.He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–357. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi H, Wang Y, Li X, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nature immunology. 2016;17:250–258. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. The Journal of biological chemistry. 2016;291:103–109. doi: 10.1074/jbc.C115.700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao Y, Yang J, Shi J, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 65.Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A. 2013;110:14408–14413. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. Cutting Edge: Mouse NAIP1 Detects the Type III Secretion System Needle Protein. Journal of immunology. 2013;191:3986–3989. doi: 10.4049/jimmunol.1301549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Franchi L, Amer A, Body-Malapel M, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nature immunology. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 68.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nature immunology. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 69.Miao EA, Mao DP, Yudkovsky N, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kortmann J, Brubaker SW, Monack DM. Cutting Edge: Inflammasome Activation in Primary Human Macrophages Is Dependent on Flagellin. Journal of immunology. 2015;195:815–819. doi: 10.4049/jimmunol.1403100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE. Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol Cell. 2014;54:17–29. doi: 10.1016/j.molcel.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burckstummer T, Baumann C, Bluml S, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nature immunology. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 75.Roberts TL, Idris A, Dunn JA, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 76.Gavrilin MA, Abdelaziz DH, Mostafa M, et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. Journal of immunology. 2012;188:3469–3477. doi: 10.4049/jimmunol.1102272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 78.Aubert DF, Xu H, Yang J, et al. A Burkholderia Type VI Effector Deamidates Rho GTPases to Activate the Pyrin Inflammasome and Trigger Inflammation. Cell host & microbe. 2016;19:664–674. doi: 10.1016/j.chom.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 79.Van Gorp H, Saavedra PH, de Vasconcelos NM, et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc Natl Acad Sci U S A. 2016;113:14384–14389. doi: 10.1073/pnas.1613156113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nature immunology. 2016;17:914–921. doi: 10.1038/ni.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Masters SL, Lagou V, Jeru I, et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med. 2016;8:332ra345. doi: 10.1126/scitranslmed.aaf1471. [DOI] [PubMed] [Google Scholar]
- 82.Gao W, Yang J, Liu W, Wang Y, Shao F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci U S A. 2016;113:E4857–4866. doi: 10.1073/pnas.1601700113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Auron PE, Webb AC, Rosenwasser LJ, Mucci SF, Rich A, Wolff SM, Dinarello CA. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci U S A. 1984;81:7907–7911. doi: 10.1073/pnas.81.24.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.March CJ, Mosley B, Larsen A, et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature. 1985;315:641–647. doi: 10.1038/315641a0. [DOI] [PubMed] [Google Scholar]
- 85.Ushio S, Namba M, Okura T, et al. Cloning of the cDNA for human IFN-gamma-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. Journal of immunology. 1996;156:4274–4279. [PubMed] [Google Scholar]
- 86.Okamura H, Tsutsi H, Komatsu T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 87.Gu Y, Kuida K, Tsutsui H, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1 beta converting enzyme. Science. 1997;275:206–209. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- 88.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kayagaki N, Wong MT, Stowe IB, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 90.Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 91.Fassy F, Krebs O, Rey H, et al. Enzymatic activity of two caspases related to interleukin-1beta-converting enzyme. European journal of biochemistry/FEBS. 1998;253:76–83. doi: 10.1046/j.1432-1327.1998.2530076.x. [DOI] [PubMed] [Google Scholar]
- 92.Knodler LA, Crowley SM, Sham HP, et al. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses against Enteric Bacterial Pathogens. Cell host & microbe. 2014;16:249–256. doi: 10.1016/j.chom.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang S, Miura M, Jung Y, et al. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. The Journal of biological chemistry. 1996;271:20580–20587. doi: 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- 94.Agard NJ, Maltby D, Wells JA. Inflammatory stimuli regulate caspase substrate profiles. Molecular & cellular proteomics: MCP. 2010;9:880–893. doi: 10.1074/mcp.M900528-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ruhl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. European journal of immunology. 2015;45:2927–2936. doi: 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- 96.Shi P, Tang A, Xian L, et al. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. The Biochemical journal. 2015;468:325–336. doi: 10.1042/BJ20150204. [DOI] [PubMed] [Google Scholar]
- 97.Lin PH, Lin HY, Kuo CC, Yang LT. N-terminal functional domain of Gasdermin A3 regulates mitochondrial homeostasis via mitochondrial targeting. Journal of biomedical science. 2015;22:44. doi: 10.1186/s12929-015-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nature communications. 2017;8:14128. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Franchi L, Kamada N, Nakamura Y, et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nature immunology. 2012;13:449–456. doi: 10.1038/ni.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lara-Tejero M, Sutterwala FS, Ogura Y, et al. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. The Journal of experimental medicine. 2006;203:1407–1412. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Man SM, Hopkins LJ, Nugent E, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A. 2014;111:7403–7408. doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Aachoui Y, Leaf IA, Hagar JA, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iqbal M, Philbin VJ, Withanage GS, et al. Identification and functional characterization of chicken toll-like receptor 5 reveals a fundamental role in the biology of infection with Salmonella enterica serovar typhimurium. Infect Immun. 2005;73:2344–2350. doi: 10.1128/IAI.73.4.2344-2350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schmitt CK, Ikeda JS, Darnell SC, et al. Absence of all components of the flagellar export and synthesis machinery differentially alters virulence of Salmonella enterica serovar Typhimurium in models of typhoid fever, survival in macrophages, tissue culture invasiveness, and calf enterocolitis. Infect Immun. 2001;69:5619–5625. doi: 10.1128/IAI.69.9.5619-5625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stecher B, Hapfelmeier S, Muller C, Kremer M, Stallmach T, Hardt WD. Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar Typhimurium colitis in streptomycin-pretreated mice. Infect Immun. 2004;72:4138–4150. doi: 10.1128/IAI.72.7.4138-4150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vijay-Kumar M, Wu H, Jones R, et al. Flagellin suppresses epithelial apoptosis and limits disease during enteric infection. The American journal of pathology. 2006;169:1686–1700. doi: 10.2353/ajpath.2006.060345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stewart MK, Cummings LA, Johnson ML, Berezow AB, Cookson BT. Regulation of phenotypic heterogeneity permits Salmonella evasion of the host caspase-1 inflammatory response. Proc Natl Acad Sci U S A. 2011;108:20742–20747. doi: 10.1073/pnas.1108963108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nature immunology. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sellin ME, Muller AA, Felmy B, et al. Epithelium-Intrinsic NAIP/NLRC4 Inflammasome Drives Infected Enterocyte Expulsion to Restrict Salmonella Replication in the Intestinal Mucosa. Cell host & microbe. 2014;16:237–248. doi: 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 112.Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. The Journal of experimental medicine. 2016;213:2113–2128. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, Schroder K. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell reports. 2014;8:570–582. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 114.Ryu JC, Kim MJ, Kwon Y, et al. Neutrophil pyroptosis mediates pathology of P. aeruginosa lung infection in the absence of the NADPH oxidase NOX2. Mucosal immunology. 2016 doi: 10.1038/mi.2016.73. In Press. [DOI] [PubMed] [Google Scholar]
- 115.Maltez VI, Tubbs AL, Cook KD, et al. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity. 2015;43:987–997. doi: 10.1016/j.immuni.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aachoui Y, Kajiwara Y, Leaf IA, et al. Canonical Inflammasomes Drive IFN-gamma to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell host & microbe. 2015;18:320–332. doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. The Journal of clinical investigation. 2013;123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Faure E, Mear JB, Faure K, et al. Pseudomonas aeruginosa type-3 secretion system dampens host defense by exploiting the NLRC4-coupled inflammasome. American journal of respiratory and critical care medicine. 2014;189:799–811. doi: 10.1164/rccm.201307-1358OC. [DOI] [PubMed] [Google Scholar]
- 119.Theisen E, Sauer JD. Listeria monocytogenes-Induced Cell Death Inhibits the Generation of Cell-Mediated Immunity. Infect Immun. 2017;85 doi: 10.1128/IAI.00733-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baker PJ, Boucher D, Bierschenk D, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. European journal of immunology. 2015;45:2918–2926. doi: 10.1002/eji.201545655. [DOI] [PubMed] [Google Scholar]
- 121.Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. European journal of immunology. 2015;45:2911–2917. doi: 10.1002/eji.201545523. [DOI] [PubMed] [Google Scholar]
- 122.Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O, Buxbaum JD. A critical role for human caspase-4 in endotoxin sensitivity. Journal of immunology. 2014;193:335–343. doi: 10.4049/jimmunol.1303424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Casson CN, Yu J, Reyes VM, et al. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1421699112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nature communications. 2015;6:8761. doi: 10.1038/ncomms9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Akhter A, Caution K, Abu Khweek A, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. 2012;37:35–47. doi: 10.1016/j.immuni.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pallett MA, Crepin VF, Serafini N, et al. Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal immunology. 2016 doi: 10.1038/mi.2016.77. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pearson JS, Giogha C, Ong SY, et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature. 2013;501:247–251. doi: 10.1038/nature12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li S, Zhang L, Yao Q, et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature. 2013;501:242–246. doi: 10.1038/nature12436. [DOI] [PubMed] [Google Scholar]
- 129.Hemrajani C, Berger CN, Robinson KS, Marches O, Mousnier A, Frankel G. NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc Natl Acad Sci U S A. 2010;107:3129–3134. doi: 10.1073/pnas.0911609106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Amer A, Franchi L, Kanneganti TD, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. The Journal of biological chemistry. 2006;281:35217–35223. doi: 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- 131.Sokolovska A, Becker CE, Ip WK, et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nature immunology. 2013;14:543–553. doi: 10.1038/ni.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Man SM, Ekpenyong A, Tourlomousis P, et al. Actin polymerization as a key innate immune effector mechanism to control Salmonella infection. Proc Natl Acad Sci U S A. 2014;111:17588–17593. doi: 10.1073/pnas.1419925111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thurston TL, Matthews SA, Jennings E, et al. Growth inhibition of cytosolic Salmonella by caspase-1 and caspase-11 precedes host cell death. Nature communications. 2016;7:13292. doi: 10.1038/ncomms13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. European journal of immunology. 2016;46:269–280. doi: 10.1002/eji.201545839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lupfer C, Kanneganti TD. The expanding role of NLRs in antiviral immunity. Immunological reviews. 2013;255:13–24. doi: 10.1111/imr.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jakobsen MR, Bak RO, Andersen A, et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci U S A. 2013;110:E4571–4580. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science. 2014;343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Doitsh G, Galloway NL, Geng X, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Galloway NL, Doitsh G, Monroe KM, Yang Z, Munoz-Arias I, Levy DN, Greene WC. Cell-to-Cell Transmission of HIV-1 Is Required to Trigger Pyroptotic Death of Lymphoid-Tissue-Derived CD4 T Cells. Cell reports. 2015;12:1555–1563. doi: 10.1016/j.celrep.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Munoz-Arias I, Doitsh G, Yang Z, Sowinski S, Ruelas D, Greene WC. Blood-Derived CD4 T Cells Naturally Resist Pyroptosis during Abortive HIV-1 Infection. Cell host & microbe. 2015;18:463–470. doi: 10.1016/j.chom.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kofahi HM, Taylor NG, Hirasawa K, Grant MD, Russell RS. Hepatitis C Virus Infection of Cultured Human Hepatoma Cells Causes Apoptosis and Pyroptosis in Both Infected and Bystander Cells. Sci Rep. 2016;6:37433. doi: 10.1038/srep37433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ramos HJ, Lanteri MC, Blahnik G, et al. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS pathogens. 2012;8:e1003039. doi: 10.1371/journal.ppat.1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thomas PG, Dash P, Aldridge JR, Jr, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Allen IC, Scull MA, Moore CB, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kanneganti TD, Body-Malapel M, Amer A, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. The Journal of biological chemistry. 2006;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 146.Kuriakose T, Man SM, Malireddi RK, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Science immunology. 2016;1:aag2045. doi: 10.1126/sciimmunol.aag2045. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. The Journal of experimental medicine. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pillai PS, Molony RD, Martinod K, et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. 2016;352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Karki R, Man SM, Malireddi RK, Gurung P, Vogel P, Lamkanfi M, Kanneganti TD. Concerted Activation of the AIM2 and NLRP3 Inflammasomes Orchestrates Host Protection against Aspergillus Infection. Cell host & microbe. 2015;17:357–368. doi: 10.1016/j.chom.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Said-Sadier N, Padilla E, Langsley G, Ojcius DM. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PloS one. 2010;5:e10008. doi: 10.1371/journal.pone.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Caution K, Gavrilin MA, Tazi M, et al. Caspase-11 and caspase-1 differentially modulate actin polymerization via RhoA and Slingshot proteins to promote bacterial clearance. Sci Rep-Uk. 2015;5 doi: 10.1038/srep18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zanoni I, Tan Y, Di Gioia M, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352:1232–1236. doi: 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.van de Veerdonk FL, Joosten LA, Shaw PJ, et al. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. European journal of immunology. 2011;41:2260–2268. doi: 10.1002/eji.201041226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell host & microbe. 2009;5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mencacci A, Bacci A, Cenci E, et al. Interleukin 18 restores defective Th1 immunity to Candida albicans in caspase 1-deficient mice. Infect Immun. 2000;68:5126–5131. doi: 10.1128/iai.68.9.5126-5131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ketelut-Carneiro N, Silva GK, Rocha FA, Milanezi CM, Cavalcanti-Neto FF, Zamboni DS, Silva JS. IL-18 triggered by the Nlrp3 inflammasome induces host innate resistance in a pulmonary model of fungal infection. Journal of immunology. 2015;194:4507–4517. doi: 10.4049/jimmunol.1402321. [DOI] [PubMed] [Google Scholar]
- 157.Uwamahoro N, Verma-Gaur J, Shen HH, et al. The pathogen Candida albicans hijacks pyroptosis for escape from macrophages. mBio. 2014;5:e00003–00014. doi: 10.1128/mBio.00003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vylkova S, Lorenz MC. Phagosomal Neutralization by the Fungal Pathogen Candida albicans Induces Macrophage Pyroptosis. Infect Immun. 2017;85 doi: 10.1128/IAI.00832-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lima-Junior DS, Costa DL, Carregaro V, et al. Inflammasome-derived IL-1beta production induces nitric oxide-mediated resistance to Leishmania. Nature medicine. 2013;19:909–915. doi: 10.1038/nm.3221. [DOI] [PubMed] [Google Scholar]
- 160.Gurung P, Karki R, Vogel P, Watanabe M, Bix M, Lamkanfi M, Kanneganti TD. An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. The Journal of clinical investigation. 2015;125:1329–1338. doi: 10.1172/JCI79526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gorfu G, Cirelli KM, Melo MB, et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio. 2014;5 doi: 10.1128/mBio.01117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Silva GK, Costa RS, Silveira TN, et al. Apoptosis-associated speck-like protein containing a caspase recruitment domain inflammasomes mediate IL-1beta response and host resistance to Trypanosoma cruzi infection. Journal of immunology. 2013;191:3373–3383. doi: 10.4049/jimmunol.1203293. [DOI] [PubMed] [Google Scholar]
- 163.Goncalves VM, Matteucci KC, Buzzo CL, et al. NLRP3 controls Trypanosoma cruzi infection through a caspase-1-dependent IL-1R-independent NO production. PLoS Negl Trop Dis. 2013;7:e2469. doi: 10.1371/journal.pntd.0002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Coutermarsh-Ott SL, Doran JT, Campbell C, Williams TM, Lindsay DS, Allen IC. Caspase-11 Modulates Inflammation and Attenuates Toxoplasma gondii Pathogenesis. Mediators Inflamm. 2016;2016:9848263. doi: 10.1155/2016/9848263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Man SM, Place DE, Kuriakose T, Kanneganti TD. Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. Journal of leukocyte biology. 2016;101:143–150. doi: 10.1189/jlb.4MR0516-223R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gurung P, Malireddi RK, Anand PK, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. The Journal of biological chemistry. 2012;287:34474–34483. doi: 10.1074/jbc.M112.401406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rathinam VA, Vanaja SK, Waggoner L, et al. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. The Journal of biological chemistry. 2002;277:41624–41630. doi: 10.1074/jbc.M207852200. [DOI] [PubMed] [Google Scholar]
- 169.Sander LE, Davis MJ, Boekschoten MV, et al. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474:385–389. doi: 10.1038/nature10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lupfer CR, Anand PK, Liu Z, Stokes KL, Vogel P, Lamkanfi M, Kanneganti TD. Reactive Oxygen Species Regulate Caspase-11 Expression and Activation of the Non-canonical NLRP3 Inflammasome during Enteric Pathogen Infection. PLoS pathogens. 2014;10:e1004410. doi: 10.1371/journal.ppat.1004410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Napier BA, Brubaker SW, Sweeney TE, et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. The Journal of experimental medicine. 2016;213:2365–2382. doi: 10.1084/jem.20160027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Meunier E, Dick MS, Dreier RF, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. 2014;509:366–370. doi: 10.1038/nature13157. [DOI] [PubMed] [Google Scholar]
- 173.Pilla DM, Hagar JA, Haldar AK, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A. 2014;111:6046–6051. doi: 10.1073/pnas.1321700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Finethy R, Jorgensen I, Haldar AK, et al. Guanylate Binding Proteins Enable Rapid Activation of Canonical and Noncanonical Inflammasomes in Chlamydia-Infected Macrophages. Infect Immun. 2015;83:4740–4749. doi: 10.1128/IAI.00856-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Man SM, Karki R, Sasai M, et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell. 2016;167:382–396 e317. doi: 10.1016/j.cell.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, Rathinam VA. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell. 2016;165:1106–1119. doi: 10.1016/j.cell.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Man SM, Karki R, Malireddi RK, et al. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nature immunology. 2015;16:467–475. doi: 10.1038/ni.3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Meunier E, Wallet P, Dreier RF, et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nature immunology. 2015;16:476–484. doi: 10.1038/ni.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. The Journal of experimental medicine. 2007;204:987–994. doi: 10.1084/jem.20062665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Fernandes-Alnemri T, Yu JW, Juliana C, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nature immunology. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cole LE, Santiago A, Barry E, et al. Macrophage proinflammatory response to Francisella tularensis live vaccine strain requires coordination of multiple signaling pathways. Journal of immunology. 2008;180:6885–6891. doi: 10.4049/jimmunol.180.10.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nature immunology. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jones JW, Kayagaki N, Broz P, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. Journal of immunology. 2015;194:3236–3245. doi: 10.4049/jimmunol.1402764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Thapa RJ, Ingram JP, Ragan KB, et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell host & microbe. 2016;20:674–681. doi: 10.1016/j.chom.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Nogusa S, Thapa RJ, Dillon CP, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell host & microbe. 2016;20:13–24. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Oficjalska K, Raverdeau M, Aviello G, et al. Protective Role for Caspase-11 during Acute Experimental Murine Colitis. Journal of immunology. 2014;194:1252–1260. doi: 10.4049/jimmunol.1400501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kang MJ, Jo SG, Kim DJ, Park JH. NLRP3 inflammasome mediates IL-1beta production in immune cells in response to Acinetobacter baumannii and contributes to pulmonary inflammation in mice. Immunology. 2016 doi: 10.1111/imm.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS pathogens. 2011;7:e1002452. doi: 10.1371/journal.ppat.1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Breitbach K, Sun GW, Kohler J, et al. Caspase-1 mediates resistance in murine melioidosis. Infect Immun. 2009;77:1589–1595. doi: 10.1128/IAI.01257-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liu Z, Zaki MH, Vogel P, et al. Role of inflammasomes in host defense against Citrobacter rodentium infection. The Journal of biological chemistry. 2012;287:16955–16964. doi: 10.1074/jbc.M112.358705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yang Q, Stevenson HL, Scott MJ, Ismail N. Type I interferon contributes to noncanonical inflammasome activation, mediates immunopathology, and impairs protective immunity during fatal infection with lipopolysaccharide-negative ehrlichiae. The American journal of pathology. 2015;185:446–461. doi: 10.1016/j.ajpath.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. The Journal of experimental medicine. 2005;202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Periasamy S, Le HT, Duffy EB, Chin H, Harton JA. Inflammasome-Independent NLRP3 Restriction of a Protective Early Neutrophil Response to Pulmonary Tularemia. PLoS pathogens. 2016;12:e1006059. doi: 10.1371/journal.ppat.1006059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pereira MS, Morgantetti GF, Massis LM, Horta CV, Hori JI, Zamboni DS. Activation of NLRC4 by flagellated bacteria triggers caspase-1-dependent and -independent responses to restrict Legionella pneumophila replication in macrophages and in vivo. Journal of immunology. 2011;187:6447–6455. doi: 10.4049/jimmunol.1003784. [DOI] [PubMed] [Google Scholar]
- 196.Cerqueira DM, Pereira MS, Silva AL, Cunha LD, Zamboni DS. Caspase-1 but Not Caspase-11 Is Required for NLRC4-Mediated Pyroptosis and Restriction of Infection by Flagellated Legionella Species in Mouse Macrophages and In Vivo. Journal of immunology. 2015;195:2303–2311. doi: 10.4049/jimmunol.1501223. [DOI] [PubMed] [Google Scholar]
- 197.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. The Journal of experimental medicine. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sansonetti PJ, Phalipon A, Arondel J, et al. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity. 2000;12:581–590. doi: 10.1016/s1074-7613(00)80209-5. [DOI] [PubMed] [Google Scholar]
- 199.Toma C, Higa N, Koizumi Y, et al. Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins and TLR/nucleotide-binding oligomerization domain-mediated NF-kappa B signaling. Journal of immunology. 2010;184:5287–5297. doi: 10.4049/jimmunol.0903536. [DOI] [PubMed] [Google Scholar]
- 200.Sivaraman V, Pechous RD, Stasulli NM, Eichelberger KR, Miao EA, Goldman WE. Yersinia pestis activates both IL-1beta and IL-1 receptor antagonist to modulate lung inflammation during pneumonic plague. PLoS pathogens. 2015;11:e1004688. doi: 10.1371/journal.ppat.1004688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell host & microbe. 2012;12:799–805. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zheng Y, Lilo S, Mena P, Bliska JB. YopJ-induced caspase-1 activation in Yersinia-infected macrophages: independent of apoptosis, linked to necrosis, dispensable for innate host defense. PloS one. 2012;7:e36019. doi: 10.1371/journal.pone.0036019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Netea MG, Fantuzzi G, Kullberg BJ, et al. Neutralization of IL-18 reduces neutrophil tissue accumulation and protects mice against lethal Escherichia coli and Salmonella Typhimurium endotoxemia. Journal of immunology. 2000;164:2644–2649. doi: 10.4049/jimmunol.164.5.2644. [DOI] [PubMed] [Google Scholar]
- 204.Caution K, Gavrilin MA, Tazi M, et al. Caspase-11 and caspase-1 differentially modulate actin polymerization via RhoA and Slingshot proteins to promote bacterial clearance. Sci Rep. 2015;5:18479. doi: 10.1038/srep18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mueller NJ, Wilkinson RA, Fishman JA. Listeria monocytogenes infection in caspase-11-deficient mice. Infection and Immunity. 2002;70:2657–2664. doi: 10.1128/IAI.70.5.2657-2664.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 207.Moayeri M, Crown D, Newman ZL, et al. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS pathogens. 2010;6:e1001222. doi: 10.1371/journal.ppat.1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, Nakanishi K, Flavell RA. Roles of caspase-1 in Listeria infection in mice. Int Immunol. 2004;16:335–343. doi: 10.1093/intimm/dxh041. [DOI] [PubMed] [Google Scholar]
- 209.Hanamsagar R, Aldrich A, Kielian T. Critical role for the AIM2 inflammasome during acute CNS bacterial infection. Journal of neurochemistry. 2014;129:704–711. doi: 10.1111/jnc.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Costa A, Gupta R, Signorino G, et al. Activation of the NLRP3 inflammasome by group B streptococci. Journal of immunology. 2012;188:1953–1960. doi: 10.4049/jimmunol.1102543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Albiger B, Dahlberg S, Sandgren A, et al. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cellular microbiology. 2007;9:633–644. doi: 10.1111/j.1462-5822.2006.00814.x. [DOI] [PubMed] [Google Scholar]
- 212.McElvania Tekippe E, Allen IC, Hulseberg PD, et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PloS one. 2010;5:e12320. doi: 10.1371/journal.pone.0012320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mayer-Barber KD, Barber DL, Shenderov K, et al. Caspase-1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. Journal of immunology. 2010;184:3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rajan JV, Rodriguez D, Miao EA, Aderem A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol. 2011;85:4167–4172. doi: 10.1128/JVI.01687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Cieniewicz B, Dong Q, Li G, et al. Murine Gammaherpesvirus 68 Pathogenesis Is Independent of Caspase-1 and Caspase-11 in Mice and Impairs Interleukin-1beta Production upon Extrinsic Stimulation in Culture. J Virol. 2015;89:6562–6574. doi: 10.1128/JVI.00658-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Charmoy M, Hurrell BP, Romano A, et al. The Nlrp3 inflammasome, IL-1beta, and neutrophil recruitment are required for susceptibility to a nonhealing strain of Leishmania major in C57BL/6 mice. European journal of immunology. 2016;46:897–911. doi: 10.1002/eji.201546015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kordes M, Matuschewski K, Hafalla JC. Caspase-1 activation of interleukin-1beta (IL-1beta) and IL-18 is dispensable for induction of experimental cerebral malaria. Infect Immun. 2011;79:3633–3641. doi: 10.1128/IAI.05459-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Reimer T, Shaw MH, Franchi L, et al. Experimental cerebral malaria progresses independently of the Nlrp3 inflammasome. European journal of immunology. 2010;40:764–769. doi: 10.1002/eji.200939996. [DOI] [PMC free article] [PubMed] [Google Scholar]