


Abstract
Bacterial small regulatory RNAs (sRNAs) play a major role in the regulation of various cellular functions. Most sRNAs interact with mRNA targets via an antisense mechanism, modifying their translation and/or degradation. Despite considerable progresses in discovering sRNAs in Gram-positive bacteria, their functions, for the most part, are unknown. This is mainly due to difficulties in identifying their targets. To aid in the identification of sRNA targets in Gram-positive bacteria, we set up an in vivo method for fast analysis of sRNA-mediated post-transcriptional control at the 5΄ regions of target mRNAs. The technology is based on the co-expression of an sRNA and a 5΄ sequence of an mRNA target fused to a green fluorescent protein (GFP) reporter. The system was challenged on Staphylococcus aureus, an opportunistic Gram-positive pathogen. We analyzed several established sRNA–mRNA interactions, and in addition, we identified the ecb mRNA as a novel target for SprX2 sRNA. Using our in vivo system in combination with in vitro experiments, we demonstrated that SprX2 uses an antisense mechanism to prevent ecb mRNA translation initiation. Furthermore, we used our reporter assay to validate sRNA regulations in other Gram-positive organisms, Bacillus subtilis and Listeria monocytogenes. Overall, our method is broadly applicable to challenge the predicted sRNA–mRNA interactions in Gram-positive bacteria.
INTRODUCTION
Over the past decade, the discovery and functional identification of small RNA in bacteria has exploded. These sRNAs regulate many biological functions, and their specific expression throughout bacterial growth makes them an important class of macromolecules. They are usually non-coding and short (50–550 nt), and participate in the regulation of target gene expression. They have different modes of action: some interact with proteins to titrate them thus neutralizing them, while most interact with target mRNA (1). To date, the most discussed cases in the literature are sRNAs that interact by partial base-pairing with their mRNA targets near the ribosome binding site (RBS) via an antisense mechanism, in turn affecting the translation and/or stability of the mRNA targets (1).
Staphylococcus aureus is an opportunistic human and animal pathogen and the cause of nosocomial and community-acquired infections (2). Because of its remarkable adaptive capacity and resistance to multiple antibiotics, it can be a deadly infectious agent. The pathogenicity and success of S. aureus infections are due to the bacteria's capacity for efficient gene expression reprogramming in response to an ever-changing environment (3). Their sophisticated regulations are mediated by various effectors, including transcription factors, two-component systems, small signaling molecules, and sRNAs (1). Over 150 sRNAs have been discovered in S. aureus and are compiled in the SRD Staphylococcal regulatory RNA database (4) but so far the physiological functions of only a few of them have been demonstrated (5). Knowledge of the functions and mechanisms of action for each sRNA is mandatory for understanding their specific roles in S. aureus and other bacteria. A crucial step in this process is the determination of all the direct mRNA targets of a given sRNA. This identification is challenging, because sRNA interact with mRNA targets through limited and interrupted pairings. It is possible to determine a gene set whose expression is dependent on an sRNA by using high-throughput studies such as deep sequencing of RNA to monitor mRNA expression and two-dimensional difference gel electrophoresis (2D-DIGE) to monitor protein expression. Although the sRNA pulse expression methods permit the enrichment of the direct targets of the sRNA, it is difficult to discriminate the direct from the secondary targets in such high-throughput studies. There are different ways to narrow the identification of potential direct targets such as co-purifications of tagged sRNA complexes (6) or the use of in silico approaches, but these techniques produce many false positive (7,8). However, the in vivo evaluation of putative targets and the detection of direct targets remain critical issues.
In this study, we set up an in vivo technique for testing the predicted mRNA targets of the sRNAs expressed in S. aureus. We present a handy green fluorescent protein (GFP) reporter assay inspired by a system developed for Gram-negative bacteria (9). However to date, there were no easy genetic approaches available for Gram-positive bacteria. Here, we developed a technique suitable for Gram-positive bacteria, which allows a rapid and easy assessment in vivo of many targets identified by the high throughput studies. We validated the technique for different sRNAs expressed in S. aureus and also demonstrated that it can be used to study sRNA regulations from two other Gram positive bacteria, Bacillus subtilis and Listeria monocytogenes. Consequently, we expect our approach to be applicable for sRNA targets detected in many other Gram-positive bacteria. This study also revealed a new target for SprX2, an S. aureus sRNA that is involved in staphylococcal antibiotic resistance (10). Using our in vivo system, we show here that SprX2 downregulates the expression of the staphylococcal extracellular complement binding protein (Ecb). Mutational analysis shows a direct interaction between the first loop of SprX2 at the 5΄-end with the ecb mRNA RBS. This novel sRNA–mRNA interaction in S. aureus prevents ribosomal loading onto ecb mRNA, specifically inhibiting translation of the Ecb protein. The discovery of this novel mRNA target using our assay demonstrates its value in the search for novel mRNA targets of S. aureus sRNAs.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The strains used in this study are listed in Supplementary Table S1. The DH5-α strain of Escherichia coli was used for all cloning. The bacteria were grown at 37°C in Luria-Bertani broth (LB, Oxoid) or in LB supplemented with 50 μg/ml ampicillin. The S. aureus RN4220 strain was used to co-transform the target-gfp fusions and the sRNA-expressing vector. Cultures of these co-transformed S. aureus strains were grown 15 h at 37°C either in brain heart infusion broth (BHI, Oxoid) or on BHI agar plates. When necessary, the media were supplemented with 10 μg/ml of chloramphenicol and/or erythromycin.
Plasmid construction
Supplementary Table S2 lists all of the primers used. The tested mRNA targets and sRNAs from HG001 (NC_007795.1), W168 (NC_000964) and EDG-e (NC_003210) genomic DNA from S. aureus, B. subtilis and L. monocytogenes were amplified by PCR. To construct the sRNA-expressing vectors, the pRMC2 plasmid (11) was digested with PstI and NarI restriction enzymes, thus removing the tetracycline-inducible promoter. To create the pICS3 vector, the PstI and NarI digested pRMC2 DNA fragment was circularized by recombination with primer 1 (Supplementary Table S2). On 5΄ and 3΄ extremities, primer 1 contains sequences allowing recombination with PstI and NarI digested pRMC2 fragment. With its native promoters, sprD was first amplified with primer set 2–3 by PCR. The sprD mutated sequence were then amplified from plasmid pCN38-sprDmut (12) using the same primers. The resulting pICS3 vector, sprD, and sprDmut were then digested with PstI and NarI restriction enzymes then ligated overnight at 16°C with T4 DNA ligase (Invitrogen). All cloning experiments described below were done with a Gibson Assembly Master Mix (New England Biolabs). To construct the pICS3-sprX1 and pICS3-sprX2 vectors, we amplified the sprX1 and sprX2 sRNAs genes respectively with primers 4–5 and 6–7 by PCR. These DNA sequences include a predicted native promoter of SprX1 and SprX2. Using the same 6 and 7 primers, a sprX2 mutated sequence coding for RNA unable to interact with spoVG mRNA was amplified by PCR from the pCN38-sprX2mut plasmid (10). To express the SprX2mutL1 unable to interact with ecb mRNA two DNA fragments were amplified by PCR with primer sets 6,8 and 7,9 using HG001 genomic DNA. Then another PCR was performed using these two PCR fragments as a template and primers 6 and 7. The PCR product was recombined with pICS3 vector digested by PstI and NarI restriction enzymes. rnaIII, roxS (B. subtilis) and lhrA (L. monocytgenes) were amplified by PCR using primers sets 10–11, 12–13 and 14–15, respectively (primers 10, 12 and 14 include the sequence corresponding to the 41 nt-long PamiA promoter). Then PCR products were cloned onto the pICS3 vector creating pICS3-PamiA-rnaIII, pICS3-PamiA-roxS and pICS3-PamiA-lhrA vectors.
To prepare the target-gfp fusion vectors, we constructed a plasmid expressing the first 11 codons of sbi mRNA in translational fusion with gfp under the control of its native promoter (pCN33-Pendo-sbi-gfp). We amplified sbi and its promoter by PCR with primer set 16–17. sfgfp and the sbi transcriptional terminator (TT) were amplified by PCR using primer sets 18–19 and 20–21, respectively. Finally, we made pCN33-sbi-gfp vector by recombining (i) pCN33 vector digested by PstI and KpnI; (ii) the sbi PCR product; and (iii) the fragment comprising gfp and the transcriptional terminator of sbi. The EcoRV restriction site was added between sbi and gfp, thereby enabling the construction of the PtufA-sbi-gfp and PamiA-spoVG-gfp reporters.
To construct the pCN33-PtufA-sbi-gfp vector which expresses sbi under control of the PtufA promoter, we amplified the PtufA promoter by PCR with primer set 22–23. sbi was amplified by PCR with primer set 17–24 to overlap PtufA and gfp. The pCN33-Pendo-sbi-gfp plasmid was digested with PstI and EcoRV restriction enzymes to remove Pendo-sbi. The digested vector was recombined with the PtufA and sbi PCR products. The resulting pCN33-PtufA-sbi-gfp plasmid was used to clone the other targets controlled by PtufA promoter which were analyzed in this report. To do this, we amplified spoVG, gyrB, mapW, ecb, sucC (B. subtilis), ppnK (B. subtilis) and lmo0850 (L. monocytgenes) by PCR overlapping PtufA and gfp with primer sets 25–26, 27–28, 29–30, 31–32, 33–34, 35–36 and 37–38 respectively. These PCR products were recombined into pCN33-PtufA-sbi-gfp plasmid digested by the BglII and EcoRV restriction enzymes.
The construction of the pCN33-PamiA-spoVG-gfp vector, which expresses spoVG under control of the PamiA constitutive promoter, was done as follows: using primer set 39-26, we obtained the PCR product of spoVG overlapping gfp and PamiA. Using this PCR product and a primer 40 as templates a second PCR product was created using primers 41 and 26. This allowed us to construct a PamiA-spoVG fragment overlapping pCN33 at its 5΄ end and gfp at its 3΄ end. The pCN33-Pendo-sbi-gfp plasmid was digested by PstI and EcoRV restriction enzymes and recombined with the PamiA-spoVG PCR product. The resulting pCN33-PamiA-spoVG-gfp plasmid was used to clone gyrB and yabJ-spoVG under control of PamiA. gyrB was amplified by PCR using primers 42 and 28 and yabJ-spoVG was PCR amplified with primers 43 and 26. This resulted in two PCR products that could recombine with the pCN33-PamiA-spoVG-gfp digested by BglII and EcoRV restriction enzymes.
All cloning reactions were transformed by heat shock at 42°C into E. coli DH5-α. The plasmids were purified from overnight cultures (Miniprep Extraction Kit, Qiagen) and Sanger sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit, using a 3130xl capillary electrophoresis genetic analyzer (Applied Biosystems). S. aureus RN4220 strain was transformed with the purified plasmids, including pICS3 expressing the sRNA.
Fluorescent imaging on plates
RN4220 strains carrying each of the target-gfp fusions and the sRNA plasmids were streaked on BHI agar plates supplemented with 10 μg/ml chloramphenicol and erythromycin. After overnight growth at 37°C, colonies were scanned at 473 nm with a Typhoon FLA 9500 (GE Healthcare) using LPG filters. The same plates were used for comparing the fluorescence between strains expressing, or not, the sRNA being examined. Growth control was done by viewing the colonies with LPG filters at 532 nm in visible light.
Microplate fluorescence measurements
Overnight cultures of RN4220 strains carrying each of the pCN33 target-gfp fusions and each sRNA cloned into pICS3 were diluted to an OD600 of 0.1 in BHI. Triplicates of 150 μl cultures were placed in 96-well microtiter plates. Cultures were grown in a Synergy 2 Multi-Mode Reader (Biotek) at 37°C under continuous shaking. Every 10 min for 22 h, we measured absorbency at 600 nm and monitored fluorescence using a 485/20 nm excitation filter and a 528/20 nm emission filter (tungsten lamp). Experiments were performed with three independent cultures, and the average fluorescence and standard deviations were calculated on these triplicates.
Protein purifications and western blots
For the total protein extractions, cell pellets corresponding to 2 ml of culture at an OD600nm of 1 were resuspended into 0.2 ml of lysis buffer (10 mM Tris–HCl, pH 7.5, 20 mM NaCl, 1 mM EDTA, 5 mM MgCl2 and completed with EDTA-free protease inhibitor cocktail tablets (Roche) containing 0.1 mg/ml lysostaphin). Following incubation at 37°C for 10 min, Laemmli sample buffer was added (13). Samples were boiled for 5 min, separated by SDS-PAGE electrophoresis and transferred onto a hybond-P PolyVinyliDene Fluoride (PVDF) membrane (Amersham). For purifying the extracellular proteins, the supernatants were collected and precipitated with 10% trichloroacetic acid. The precipitates were washed with ice-cold acetone and loaded onto SDS-PAGE according to (13). GFP protein expression was visualized by anti-GFP antibodies (Roche) and, anti-rabbit IgG secondary antibodies (Jackson). Ecb protein expression was visualized by anti-Ecb antibodies (14) and, anti-rabbit IgG secondary antibodies (Jackson). Western blots were revealed using the Amersham ECL Plus detection Kit. Signals were visualized using LAS 4000 (GE Healthcare).
RNA isolation and northern blots
Total RNA were prepared as previously described (15). For SprD, SprX2 and RNAIII, northern blots were done using 10 μg total RNA as we have previously described (10). Specific 32P-labeled probes (the sequences are in Supplementary Table S2) were hybridized with membranes in ExpressHyb solution (Clontech) for 90 min at 37°C, washed, exposed, then scanned with a Typhoon FLA 9500 scanner (GE Healthcare). For the quantitative real-time PCR (qRT-PCR), cDNAs were prepared using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR experiments were performed using Power SYBR Green PCR Master Mix (Applied Biosystems) with the primers listed in Supplementary Table S2. Three independent experiments were performed, with independent RNA purifications. The hu gene was used for normalization.
Toeprint assays
The toeprint assays were performed as previously (12), with modifications. Annealing mixtures containing 0.2 pmol of ecb mRNA and 1 pmol of labeled primer 34 in a reaction buffer (100 mM Tris–HCl pH 7.5, 300 mM NH4Cl, 5 mM DTT) were incubated 2 min at 90°C, followed by 1 min incubation on ice. RNA refolding was performed in the presence of 10 mM MgCl2 for 10 min at room temperature. SprX2 and SprX2mutL1 RNAs were added at the specified concentrations before the addition of purified E. coli 70S ribosomes. The ribosomes were reactivated for 15 min at 37°C and diluted in presence of 1mM MgCl2. 1 pmol of 70S ribosomes were added in each assay and incubated 5 min at 37°C. The concentration of MgCl2 was adjusted to 10 mM. After 10 min at 37°C, 10 pmol of uncharged transfer RNAfMet (MP Biomedicals) was added and incubated 5 min at 37°C. Complementary DNAs were synthesized with 2 UI of AMV RT (Biolabs) for 15 min at 37°C. Reactions were ended by the addition of 15 μl of loading buffer II (Ambion). The cDNAs were separated in 8% denaturing polyacrylamide gel electrophoresis. Gels were dried and visualized using Typhoon FLA 9500 (GE Healthcare).
RESULTS
Designing the technology
We adapted a system for the in vivo study of the direct regulation of potential mRNA targets by sRNA expressed by S. aureus, a major Gram-positive human pathogen. Our method was inspired by a translational fusion system developed for Gram-negative bacteria (9). The technology is based on the co-expression from two compatible plasmids of an sRNA and a translational fusion made up of the tested target mRNA and gfp (green fluorescent protein) and allows studying sRNA-mediated translational control at and around the 5΄ region of a given target mRNA. Superfolder gfp (sfgfp) was used as a reporter gene for our investigations. sfgfp encodes a GFP variant favoring increased protein folding efficiency, thus higher fluorescence (16,17). To adapt the method to Gram-positive bacteria, we used two compatible E. coli and S. aureus shuttle vectors (Figure 1). The vectors are compatible due to their different replication origins (18). The target plasmid studied was a low-copy-number pCN33 vector (19) that carries a pT181 cop-wt repC replication origin and an erythromycin resistance cassette (ermC). Since some S. aureus strains are resistant to erythromycin, we also tested the use of a pCN36 vector carrying a tetracycline resistance cassette for the cloning of some fusion constructions (19) (Supplementary Figure S1). The target mRNA-gfp fusions were expressed under control of constitutive promoters. To control target gene expression, the use of a constitutive promoter allowed us to exclude the mRNA targets regulated at transcriptional level and to focus on the post-transcriptional regulations. The sRNA plasmid used was a high-copy-number pICS3 vector carrying a pC194 replication origin and cat chloramphenicol resistance (Figure 1). sRNA was expressed under control of either its native or constitutive promoters.
Figure 1.

Principles of the in vivo investigation of direct interactions between sRNA and their potential mRNA targets in Staphylococcus aureus. The potential targets are cloned into erythromycin (ermC)-resistant pCN33 vector as translational gfp fusions between BglII and EcoRV restriction enzyme sites. pCN33 is a low-copy-number vector having a pT181 replicon. The fusion is transcribed under control of a constitutive promoter (P.const). sRNA is cloned into pICS3, a high-copy-number vector conferring chloramphenicol resistance (cat) and having a pC194 replicon. In this case, the cloning occurs between the PstI and NarI restriction enzyme sites under control of their native (P.nat) or constitutive promoters. The impact of sRNA production on mRNA expression is monitored by GFP fluorescence measurements on BHI agar plates or during bacterial growth in liquid media in microplates using a BioTek microplate reader.
All in vivo studies of sRNA–mRNA target interactions were conducted in the S. aureus RN4220 strain because of its ease of transformation with plasmids purified from E. coli cells. RN4220 cells were initially transformed with the target plasmid, then either with the sRNA-expressing one or a control plasmid. There are several ways to monitor the fluorescence of the resulting transformants. BHI agar plates were used for qualitative investigations. Fluorescence quantification during bacterial growth in liquid culture was done using a microtiter plate-based assay (Figure 1) followed by subtraction of the natural fluorescence of S. aureus cells. Decreased fluorescence induced by sRNA expression is a sign of mRNA target translational repression. On the contrary, increased fluorescence indicates a positive regulation of the mRNA target by the sRNA.
Cloning and expression of the srna genes
Three sRNAs were cloned into pICS3: SprX2, (corresponding to the second copy of SprX in the HG001 strain) (10,15); SprD (20) and RNAIII (21). SRD sRNA gene identifiers (4) corresponding to the sRNA used in this study, are provided in Supplementary Table S3. They all have verified mRNA targets (10,12,22). Because SprX2 and SprD are expressed at all growth phases (12,15), we cloned them under control of their native promoters. In contrast, RNAIII expression varies widely during growth, starting very low at the beginning and accumulating during the exponential growth phase and beyond. To ensure high sRNA expression levels we therefore cloned rnaIII under control of a heterologous constitutive PamiA promoter (23).
Both sprX2 and sprD are located on pathogenicity islands originating from lysogenic phages (12,15). All phages were deleted from RN4220 strain (24,25), so there is no endogenous expression of these sRNAs, although RNAIII is expressed. We evaluated the expression levels of all these three sRNAs expressed by the pICS3 vector in RN4220, and compared the results with the endogenous levels in HG001. The expression levels of SprX2, SprD and RNAIII, expressed from plCS3 vectors, were stable during growth (Supplementary Figure S2). Moreover, the expression levels of three sRNAs were higher from the plCS3 vector than their endogenous expression in HG001 strain (Figure 2A and B). This shows that the vector used enables the high and constant expression of each studied sRNA, whether the controlling promoter was native or constitutive. This means that we were able to minimize the impact of endogenous sRNA on the regulations challenged by our assay.
Figure 2.
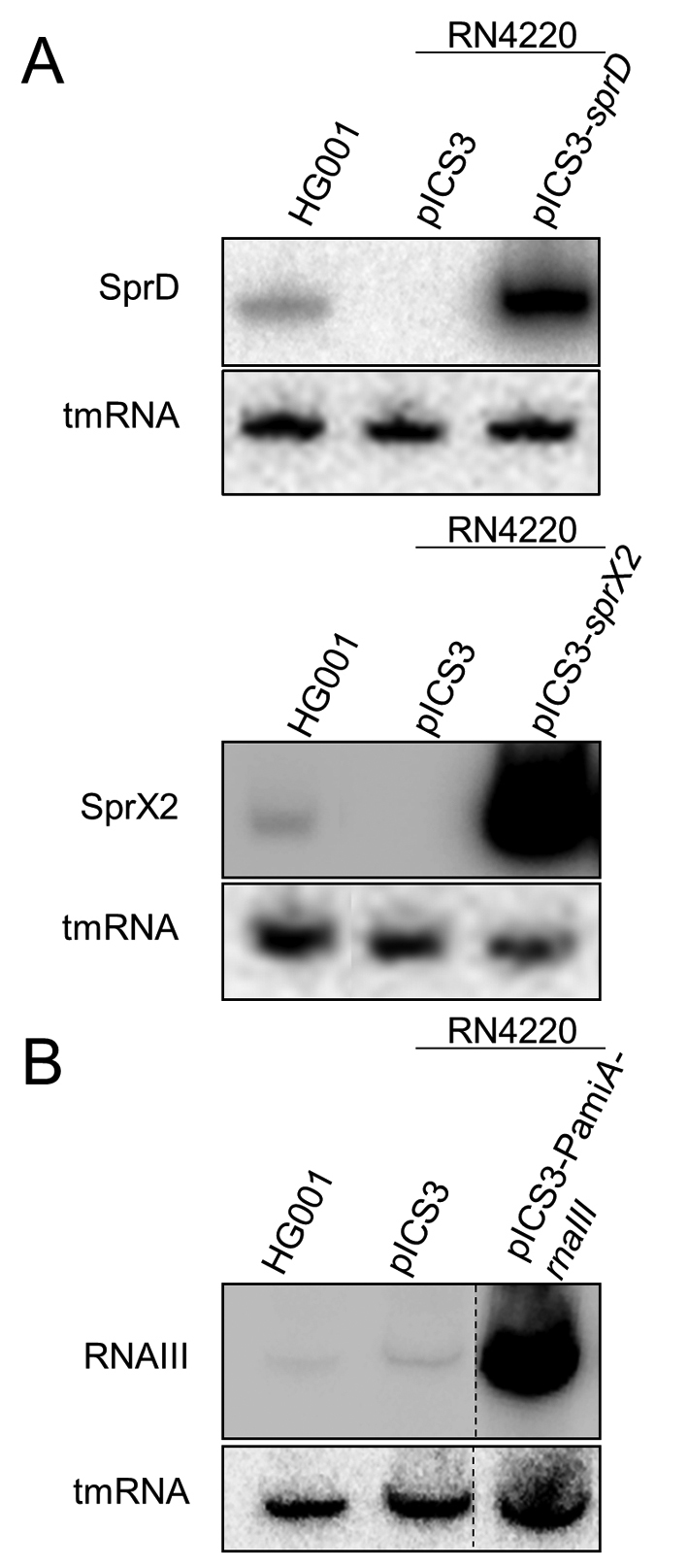
sRNA expression in Staphylococcus aureus cells. Northern blot analysis of sRNA expression in the RN4220 strain transformed with (A) pICS3-sprD, pICS3-sprX2 or (B) pICS3-PamiA-rnaIII. Expression levels were evaluated relative to the HG001 strain where SprD, SprX2 and RNAIII are endogenously expressed. Cultures were recovered at the exponential growth phase (OD600 ∼1.5). tmRNA was used as a loading control. Dotted line symbolizes the lanes that were cut out from the original gel.
Cloning and fluorescence analysis of translational fusions
Four mRNAs were studied: three known mRNA that are sRNA targets (spoVG, sbi and map); and the gyrB control gene (Figure 3A). The 5΄ mRNA portions of each target, including their 5΄ UTRs and the beginning of their coding sequences, were cloned as N-terminal translational fusions to gfp (Figure 3A). For the monocistronic sbi and mapW mRNA, the entire previously mapped 5΄ UTR was cloned (12,26). For the spoVG gene transcribed within an operon, two fusions were realized and tested. The first gfp fusion, named spoVG-gfp mRNA fusion, contains a shortened 5΄-UTR from spoVG. A second fusion, named yabJ-spoVG-gfp, had gfp fused to the full-length 5΄-domain of the yabJ-spoVG operon containing the first gene of the yabJ operon (Supplementary Figure S3A–C). For the gyrB gene transcribed within an operon, a sequence of ∼80 nt was cloned as an arbitrary 5΄ UTR. For three sRNA targets (sbi, spoVG and mapW mRNA), the cloned sequences contain known regions for pairing with respective sRNAs. The target genes were cloned using BglII and EcoRV restriction sites as N-terminal translational fusions with gfp under control of constitutive promoters (Figure 1). We tested two constitutive promoters: PamiA (23) and PtufA (27). PamiA is the promoter of the amiA gene in Streptococcus pneumoniae that encodes an oligopeptide binding precursor and it is constitutive in S. aureus (23). The PtufA promoter drives the expression of translation elongation factor G from S. aureus (27). The use of these two constitutive promoters allowed us to create several translational fusions with different mRNA transcription levels.
Figure 3.

Characterization of mRNA target-gfp fusions. (A) Overview of the gfp fusion plasmids used to set up and validate a double plasmid reporter system in Staphylococcus aureus cells. a, Gene names refer to the annotations in the S. aureus NCTC8325.4 genome (NC_007795.1). b, Number of nucleotides in the 5΄UTR of the cloned mRNA target. c, Number of nucleotides after each AUG start codon fused in-frame to gfp. d, Fluorescence levels of the fused target-gfp depending on the constitutive promoter used. (+) weak fluorescence emission; (++), intermediate fluorescence levels; (+++), high fluorescence; NT, untested combination. (B) Fluorescence of the mRNA-gfp fusions under control of the PamiA promoter or the PtufA promoter expressed from the pCN33 vector. The left image was obtained in the visible light and serves as a control for colony growth. (C) Western blot analysis of GFP fusion protein expression levels detected with monoclonal α-GFP antibodies. The same strains are shown here as in panel (B). Proteins were purified in post-exponential growth phase. The Coomassie blue coloration is used as a control for the amount of loaded proteins.
We first measured the fluorescence of each mRNA target-gfp fusion on BHI agar plates (Figure 3B). These levels varied according to the amount of target-GFP proteins detected (Figure 3C). Various fluorescence levels of mRNA-gfp fusion were observed (Figure 3A–C). As previously described in E. coli (28), the gfp fusion fluorescence was highly dependent on which mRNA was fused with gfp (Figure 3A and B). The fluorescence also varied according to which promoter was controlling the expression of the mRNA-gfp fusion. The expression of the same gyrB and spoVG genes driven by the PamiA promoter yielded higher protein levels (Figure 3C), which caused higher fluorescence emissions (Figure 3B) than that observed with the PtufA promoter. This indicates that the PamiA promoter is stronger than the PtufA one. We could not clone sbi or mapW-gfp fusions under control of the PamiA promoter, probably because the elevated expression of these fusion proteins is detrimental to bacterial viability.
S. aureus sRNA repression of mRNA-gfp translational fusions
We began by testing the putative non-specific regulations of each sRNA on GFP expression. The gyrB housekeeping gene was used as a negative control, since no interactions were predicted with any of the sRNAs being studied. SprX2, SprD and RNAIII expression did not induce any change in fluorescence levels for the PtufA-gyrB-gfp or the PamiA-gyrB-gfp fusions on plates (Figure 4A) or in liquid cultures (Figure 4B). This showed that overexpressing any of the three sRNAs did not induce non-specific regulations of mRNA targets using our technology.
Figure 4.

Verification of the targeting specificity. The putative regulatory control of each sRNA was tested using pCN33-gyrB-gfp fusion plasmid as negative control. Fluorescence was measured using two techniques. (A) Staphylococcus aureus strains carrying the pCN33-gyrB-gfp fusion plasmid under control of either PamiA or PtufA promoters co-transformed with pICS3-sprD, pICS3-sprX2, or pICS3-PamiA-rnaIII plasmids were grown on BHI agar plates. They were supplemented with chloramphenicol (10 μg/ml) and erythromycin (10 μg/ml). The left images were obtained by scanning fluorescence on plates. The right images were obtained using visible light and serve as colony growth controls. (B) The same strains as in panel (A) were also grown in liquid BHI medium on microplates. Growth (OD600nm) and fluorescence (using a 510 nm emission filter and a 460 nm excitation filter) were measured every 10 min over 24 h in a BioTek microplate reader.
We tested two sRNA-driven downregulations of target mRNAs in S. aureus which we had previously characterized: SprX2 which inhibits spoVG (10); and SprD, which inhibits sbi expression (12). Each interacts with the translation initiation signals of its mRNA target, preventing ribosome loading at the ribosomal binding site (RBS) and therefore inhibiting mRNA translation without inducing mRNA degradation. By using these two targets, we were able to challenge our system in vivo with sRNA regulation of targets that are encoded either by monocistronic (sbi) or bicistronic mRNA (spoVG). We also tested our in vivo reporter assay using SprX2 and SprD having mutations in their structural domains that are involved with their mRNA target interactions. These RNA were the same ones previously used by our group to study sRNA–mRNA interactions, and which have been shown to be unable to interact with and thus regulate their mRNA targets (10,12). The SprX2 mutated version, SprX2mut, contains mutations in the L2 loop involved in the regulation of SpoVG expression (10). The SprD mutated version, SprDmut, is deleted for the entire sbi mRNA interaction domain (nucleotides 35–70), which corresponds to the second stem-loop of SprD (12). The expression levels of both SprX2mut and SprDmut are comparable to the levels of respective non-mutated sRNA (10,12).
We obtained double transformants expressing gfp-fused targets and sRNA or their respective mutated versions. The fluorescence levels of the gfp fusions were then monitored in Petri dishes (Figure 5A) and in liquid cultures (Figure 5B). For both sRNA/mRNA target pairs (SprX-spoVG and SprD-sbi), we observed a significant decrease in fluorescence when the sRNA was expressed (Figure 5A and B). However, in the presence of the sRNA bearing mutations in the target-interacting domains, we saw a return to the control fluorescence levels (Figure 5A and B). Moreover, we verified the specificity of the sRNAs by testing SprD and SprX2 actions on unrelated mRNA-gfp fusions (SprD was tested with spoVG-gfp fusion and SprX2 with sbi-gfp fusion) (Supplementary Figure S3D). In all cases, co-expressing the unrelated sRNA resulted in fluorescence levels similar to the controls (Supplementary Figure S3D). Equivalent results were obtained when spoVG-gfp fusion was transcribed under control of PamiA or PtufA promoters, indicating that these promoters do not interfere in the sRNA–mRNA interaction. Similar effect of SprX2 on spoVG inhibition was observed either for the 5΄ shortened (Figure 5A–B) or for the full-length 5΄ versions of spoVG-gfp fusion (Supplementary Figure S3). Western blot analysis with anti-GFP antibodies confirmed that the changes in fluorescence were due to decreased levels of GFP fusion proteins (Figure 5C). SprX2 and SprD expression did not decrease the mRNA levels of gfp-fusion constructs (Figure 5D), confirming that the respective regulation of SpoVG and Sbi expression by SprX and SprD occurs at the translational level, as we have previously described (10,12).
Figure 5.

Negative in vivo mRNA regulation by two sRNAs in Staphylococcus aureus. Fluorescence decreased in cells expressing either spoVG-gfp or sbi-gfp fusions when they were co-expressed with SprX2 and SprD, their respective regulatory sRNAs. Fluorescence levels were restored when the sRNAs were mutated (SprX2mut and SprDmut) for the domains involved in the recognition with their mRNA targets. As in Figure 4, fluorescence levels were monitored on BHI agar plates (A) and in liquid cultures (B) during S. aureus growth. (C) Western blot analysis showing GFP fusion protein expression levels in each strain detected with monoclonal α−GFP antibodies. Samples were prepared from liquid cultures of strains from panels (A) and (B) in exponential growth phase. M, protein size (kDa). The Coomassie blue staining was used as a protein amount loading control. (D) qPCR analysis of the mRNA expression of gfp fusion using specific gfp primers. Same strains as in panel (C) were used.
Using our technology, sRNA–target interactions from other Gram-positive bacteria were also challenged. To test if we could use S. aureus as a host to assay sRNA regulations from other Gram-positive bacteria, we co-expressed in S. aureus RN4220 strain, two characterized sRNA–target pairs from B. subtilis and one from L. monocytogenes: RoxS sRNA inhibits sucC and ppnK expression in B. subtilis (29); while LhrA sRNA inhibits lmo0850 expression in L. monocytogenes (30). When expressed in S. aureus, both sRNAs are able to efficiently repress their corresponding targets (Supplementary Figure S4). Altogether, these experimental data show that our assay detects sRNA-mediated translational repression in S. aureus cells, and that our system can also be used for studying sRNA regulations of other Gram-positive bacteria.
Detection of positive gene regulation by an sRNA in S. aureus
Our procedure allows for detection of mRNA target repression by sRNAs, which is the mechanism most frequently described in bacteria. To test whether it could detect sRNA stimulation instead of repression, we examined a reported S. aureus example of positive regulation by an sRNA. RNAIII stimulates MapW protein production by directly interacting with mapW mRNA, thus increasing the mRNA levels (22,31). Whether this sRNA–mRNA interaction affects mapW translation was not reported. Using our system, we analyzed this activity. To do that, pCN33-PtufA-mapW-gfp vector was co-transformed in RN4220 with either pICS3-PamiA-rnaIII or a pICS3 control vector. In both Petri dishes and BHI liquid cultures, RNAIII expression stimulated fluorescence in the cells containing the pCN33-PtufA-mapW-gfp vector (Figure 6A and B). Western blot analysis demonstrated an increase in MapW-GFP fusion protein levels in the presence of RNAIII (Figure 6C), thus confirming the result obtained by the fluorescence assay. Analysis of the mapW-gfp fusion mRNA levels provided additional information, showing that RNAIII overexpression increases the levels of mRNA (Figure 6D). This therefore indicates that our assay also detected an increase in gene expression induced by the interaction of an sRNA and its mRNA target.
Figure 6.

Positive regulation of target mRNA expression by an sRNA in Staphylococcus aureus. Fluorescence levels of cells expressing the MapW-GFP fusion protein were increased in the presence of RNAIII expression under control of the PamiA promoter. Fluorescence was monitored on BHI agar plates (A) and in liquid cultures (B), as in Figure 4. (C) Western blot analysis detecting target protein expression levels using monoclonal α-GFP antibodies. Samples were taken from liquid cultures of cells in the exponential growth phase (OD600 ∼1.5) carrying both pCN33-PtufA-mapW-gfp and either pICS3 expressing RNAIII, or not. M, protein size (kDa). The Coomassie blue staining was used as a protein amount loading control. (D) qPCR analysis of mRNA-gfp fusion expression levels using specific gfp primers in the same strains as panel (C).
SprX2 specifically reduces Ecb protein expression by a direct interaction with the ecb mRNA
To confirm the soundness and utility of our in vivo system, we studied the well-known interaction between RNAIII and ecb mRNA (32). Extracellular complement-binding protein (Ecb) is a secreted protein that protects invading bacteria against the host immune system by binding and inhibiting the complement (31). ecb expression was shown to be controlled by RNAIII through antisense pairings with the ecb mRNA RBS, preventing its translation (32). We cloned a complete 5΄ UTR sequence (32) followed by 57 nts of the beginning of the ecb coding sequence, creating a translational fusion with gfp driven by the PtufA promoter. With our double plasmid system, we confirmed the effect of RNAIII on ecb expression (Figure 7). We assayed the putative effects of two other sRNAs already used in this study, SprD and SprX2. Interestingly, in the presence of SprX2 we noticed a significant and reproducible decrease in fluorescence emission in the cells expressing the Ecb-GFP fusion protein, while SprD had no effect on Ecb-GFP production (Figure 7). These results indicate that SprX2 inhibits ecb expression. To confirm the ecb regulation by SprX2, we evaluated the effect of SprX2 expression on endogenous Ecb protein levels in S. aureus RN4220 and HG001 strains (Supplementary Figure S5). Western blots show that SprX2 overexpression decreases Ecb protein amounts, confirming the regulation. Since in HG001 strain two copies of SprX are expressed simultaneously (Supplementary Figure S6A) we tested also the effect of SprX1 on ecb expression (Supplementary Figure S6). Similar to SprX2, SprX1 represses ecb expression.
Figure 7.

RNAIII and SprX2 lower Ecb expression in Staphylococcus aureus. The experiment was run on the Staphylococcus aureus RN4220 strain containing the pCN33-PtufA-ecb-gfp fusion plasmid co-transformed with pICS3, pICS3-sprD, pICS3-sprX2, or pICS3-PamiA-rnaIII plasmids. These samples were grown on BHI agar plates supplemented with both chloramphenicol (10 μg/ml) and erythromycin (10 μg/ml). The left image was obtained by scanning fluorescence emission on plates. The right image was collected in the visible light and serves as a bacteria growth control.
We went on to test whether the SprX2 effects depend on RNAIII expression, examining the effect of SprX2 on ecb-gfp expression in the strain deleted for rnaIII. For this purpose, the HG001 strain and its ΔrnaIII derivative (33) were transformed by pICS3 and pICS3-sprX2. Measurement of the fluorescence in the double transformants showed that SprX2 represses ecb expression in both wild-type HG001 and HG001ΔrnaIII strains (Supplementary Figure S7). These results indicate that SprX2 acts independently of RNAIII in the repression of ecb expression.
An interaction domain between SprX2 and ecb mRNA was predicted by intaRNA software (http://rna.informatik.uni-freiburg.de/IntaRNA/Input.jsp) to occur between the SprX2 L1 loop (nucleotides 20–32) and the 5΄ end of ecb mRNA, particularly on the Shine-Dalgarno (SD) sequence (16–29) (Figure 8A). To experimentally challenge this predicted interaction, we constructed a version of sprX2 containing mutations within loop L1 (sprX2mutL1) (Figure 8A), cloned it into pICS3 and then tested it in vivo. The expression level of SprX2mutL1 was comparable to that of SprX2 (Supplementary Figure S8). In Petri dishes and in liquid cultures, SprX2 strongly reduced the fluorescence emission of S. aureus cells containing pCN33-PtufA-ecb-gfp vector, but in the presence of SprX2mutL1, no fluorescence levels modifications were observed (Figure 8B and C). This in vivo result supports the interaction between the 5΄ end of ecb mRNA and the L1 loop of SprX2, as predicted in silico. We also tested the effect of SprX2 on ecb-gfp fusion mRNA levels by qPCR, demonstrating that they were not affected by SprX2 expression (Figure 8D). Western blot analysis with anti-GFP antibodies confirmed that the changes in fluorescence were due to decreased levels in the GFP fusion proteins (Figure 8E). This shows that SprX2 regulation of ecb occurs at a translational level, with no associated changes in mRNA levels.
Figure 8.

SprX2 reduces Ecb expression by preventing ribosomal loading onto the ecb mRNA RBS (A) Predicted base pairing between SprX2 and ecb mRNA by intaRNA software (http://rna.informatik.uni-freiburg.de/IntaRNA/Input.jsp). The predicted ΔG of the interaction was –11.5978 kcal/mol .The presumed SD sequence and AUG initiation codon are underlined. In SprX2, boxed nucleotides 5΄-CAUCUAUCCU-3΄ were mutated for 5΄-GUAGAAUGGA-3΄ to obtain SprX2mutL1. (B and C) Fluorescence extinction in cells expressing the ecb-gfp fusion when co-expressed with SprX2. Fluorescence levels were restored when the regulatory RNA was mutated in the domain predicted to pair with the ecb mRNA (SprX2mutL1). As in Figure 4, the fluorescence levels were monitored on BHI agar plates (B) and also on liquid cultures during growth (C). (D) qPCR analysis of ecb-gfp fusion mRNA expression using specific gfp primers in the same strains as used in panel (C) in exponential growth phase (OD600 ∼1.5). (E) Western blot analysis showing Ecb-GFP fusion protein expression levels in each strain detected by monoclonal α−GFP antibodies. Samples were prepared from liquid cultures of strains from panels (C) in exponential growth phase (E-phase, OD600 ∼1.5) and in stationary phase (S-phase, OD600 ∼7). The Coomassie blue staining was used as a protein amount loading control. (F) In vitro Toeprint assays on the ecb mRNA. These show that increasing concentrations of SprX2 (1, 2.5 and 5 pmol) prevent ribosomal loading onto the ecb mRNA and translation initiation, whereas identical concentrations of SprX2mutL1 do not. ‘+/–’, presence/absence of purified 70S ribosomes, SprX2, or SprX2mutL1; black arrow, toeprint. U, A, G and C refer to the nucleotide sequence of the ecb mRNA. (G) Quantification of the Toeprint signal on the ecb mRNA with either SprX2 or SprX2mutL1 from panel (F). Quantification was performed using ImageQuant software (GE Healthcare).
On the ecb mRNA, the potential SprX2 interaction domain is covered by the ribosomes during translation initiation. This implies that when it interacts with ecb mRNA, SprX2 should prevent ribosomal loading onto the ecb RBS. To test this hypothesis, toeprint assays were done. We formed a ternary initiation complex made of purified 70S ribosomes, initiator tRNAfMet, and ecb mRNA. A toeprint was detected 16 nts downstream from the AUG initiation codon of the ecb mRNA, indicating that the ribosome fixation blocked the elongation of reverse transcription (Figure 8F and G). SprX2 significantly reduced the toeprint in a concentration-dependent manner, indicating that SprX2 inhibits binding onto the ecb mRNA RBS in vitro. On the contrary, the toeprint does not vary in function of the SprX2mutL1 concentration, so SprX2 containing mutations in loop L1 fails to prevent ribosome loading onto the ecb mRNA RBS (Figure 8F and G). These results further support our in vivo data, showing that (i) SprX2 reduces Ecb-GFP fusion protein expression; and (ii) that the SprX2 L1 loop is essential for regulating ecb expression. Our system thus allowed us to detect a new direct mRNA target of S. aureus sRNA.
We provided experimental support that, in addition to RNAIII, another sRNA, SprX regulates ecb expression. We monitored the endogenous expression profiles of both sRNAs and compared them to the Ecb protein expression profile in HG001 strain (Figure 9). SprX2 expression is high at the beginning of growth and decreases at later stages of growth. RNAIII, however, is expressed at later growth stages, implying that at the beginning of growth ecb expression is controlled by SprX, while the regulation of ecb expression by RNAIII mainly occurs during the second part of growth. Interestingly, the peak of Ecb protein expression is situated at the exponential growth phase, corresponding to the lowest expression of both sRNAs (Figure 9B–C). Thus, we propose that the specific expression profiles of both sRNAs allow the precise regulating of ecb expression levels during bacterial growth and possibly also during host infection.
Figure 9.

SprX2, RNAIII and Ecb expression levels during S. aureus growth. (A) SprX2 and RNAIII expression profiles were monitored in HG001 strain during 8 h by northern blots using labeled DNA probes specific for each RNA. tmRNA was used as a loading control. (B) Western blot analysis of Ecb protein using specific anti-Ecb antibodies during growth (3–8 h). Extractions were performed on extracellular proteins. (C) Expression levels of the Ecb protein (green), RNAIII (yellow) and SprX2 (blue) in S. aureus strain during growth (gray). Quantifications of the Ecb protein is shown in arbitrary units (AU). The amounts of RNAIII and SprX2 are shown in arbitrary units (AU) and calculated relative to tmRNA.
DISCUSSION
Most known bacterial sRNAs having identified biological functions interact with their targets at the mRNA 5΄ end and around the ribosomal binding site, thus affecting mRNA translation and/or stability (34). In this study, we present a way to do rapid testing and validation of novel mRNA targets of Gram-positive bacterial sRNAs. The technology is based on the co-expression of the 5΄ sequence of a potential mRNA target fused to the gfp fluorescent reporter gene expressed, and the sRNA expressed from a second plasmid. This system allows for detection of a significant amount of sRNA-regulated mRNA targets. The advantage of the approach is that testing is done in vivo, so the regulation occurs in the presence of all the players necessary for the mRNA–sRNA interaction. Our method was validated using several already-known sRNA–mRNA regulations, and has been used to detect mRNA targets that are either negatively or positively regulated by sRNAs. Furthermore, our procedure can be used to test predicted interactions between sRNAs and targets that are encoded either mono- or polycistronically. The targets we tested were expressed under the control of constitutive promoters. This use of constitutive promoters enabled the avoidance of putative mRNA transcriptional regulations by the sRNA, leaving us free to investigate only post-transcriptional regulations and the direct mRNA targets of sRNA. Nevertheless, additional studies are required in order to determine whether the identified modifications occur during translational regulation and/or as part of mRNA target stability. The use of two different constitutive promoters (PtufA and PamiA) allowed us to check our method against varying mRNA expressions, and it proved to work well over a wide range of levels. The expression levels of sRNA and mRNA can be critical in specific cases to detect the regulation, then the use of constitutive or strong promoters might be an issue. We found that in S. aureus, PtufA induces a weaker expression of the fused mRNA targets than the PamiA promoter, enabling the cloning of mRNA sequences that when highly expressed are potentially toxic for the host bacteria.
Here, we were able to identify ecb, a novel direct mRNA target for sRNA SprX2. SprX has been shown to be expressed in most S. aureus strains (35), and in some strains there are even multiple copies (sprX1 and sprX2 in HG001; sprX1, sprX2 and sprX3 in Newman; (4)). We have previously shown that SprX shapes bacterial resistance to some antibiotics and downregulates the expression of spoVG encoding a DNA binding protein (10). ecb is transcribed as a monocistronic mRNA encoding an extracellular protein which affects bacterial virulence by binding to complement, in turn inhibiting its activation (31). Our double plasmid approach yielded proof that SprX2 downregulates ecb expression, and that this inhibition occurs at the post-transcriptional level. Furthermore, we produced in vivo confirmation of regions involved in the interaction between SprX2 and ecb mRNA, regions that had been predicted in silico.
ecb expression was previously shown to be repressed by another sRNA, RNAIII (32). Such RNAIII effects were observed in sprX-negative background using a strain deleted of prophages expressing SprX (21).
Using our technique, we show here that SprX2 also affects Ecb levels even without RNAIII (Supplementary Figure S7). Together, these results indicate that SprX2 and RNAIII independently control ecb expression. Regarding their specific expression profiles (Figure 9A–C), we assume that SprX probably sets the levels of ecb expression at the beginning of growth, whereas RNAIII intervenes at later stages to reduce the protein expression levels. Thus, in vivo Ecb synthesis is regulated during bacterial growth by the joint contribution of at least two sRNAs (Figure 9B). Examples of sRNA collaboration to regulate a shared target are known in both Gram-positive and negative bacteria, with both negative and positive regulations seen (36,37). In S. aureus, we have already reported a case wherein two sRNAs, SprD and RNAIII, control a virulence factor, Sbi (the Staphylococcal Binding Immunoglobulin protein) (33). During infection the expression of virulence factors such as Sbi and Ecb need to be tightly regulated, probably thus requiring multiple regulators. Interestingly, in addition to their dual sRNA regulatory control, there are other resemblances between Sbi and Ecb functions. Both are extracellular immune evasion factors that bind components of the complement (38,39). Both factors have similar expression profiles during growth with the highest expression corresponding to the exponential growth phase. sbi and ecb are both regulated by the SaeRS two-component system (40,41). Our present study demonstrated that both factors are regulated, at post-transcriptional level, by two sRNA. This dual sRNA control combines a common sRNA, RNAIII, in association with another sRNA, SprD or SprX, both expressed from the same immune evasion gene cluster of bacteriophage Φ3 (Φ3 IEC) (42). Altogether, this implies that these two immune evasion molecules could be controlled by identical external stimuli, most likely corresponding to the initial interactions with the host cells and tissues during the early stages of the infectious process.
We have done both in vitro and in vivo experiments to explore SprX2 downregulation of ecb expression. In fact, the regulatory RNAs SprX2 and RNAIII control ecb expression through a shared mechanism. Both sRNAs prevent translation initiation by antisense pairing at the RBS of ecb mRNA (Figure 8F) (32). For the RNAIII–ecb mRNA complex, the pairing involves 34 nts from RNAIII's 13 loop and the first ecb mRNA loop at the 5΄ end and includes its RBS (Figure 10A) (32). The SprX2–ecb mRNA pairing also involves the ecb mRNA RBS, but the interaction domain is smaller and includes only 13 nts of each RNA (Figure 10A). In the same way, the pairing between sbi mRNA and RNAIII is more extensive than that between sbi mRNA and SprD, and involves different structural domains (33). RNAIII is known to be a multifunctional sRNA with numerous direct molecular targets (Figure 10D). In order to coordinate binding and increase target specificity, RNAIII must use extended binding sites such as those in the ecb mRNA interaction, since RNAIII processes so many different mRNA targets. On the ecb mRNA, the sRNA interactions partly overlap (Figure 10A), implying that the sRNAs act independently on Ecb inhibition, and that their regulatory actions may be mutually exclusive. Interestingly, the interaction zone between SprX2 and ecb mRNA and that between SprX2 and another of its targets, spoVG mRNA, involves different sRNA structural domains (SprX2 L1 and L3 loops, respectively). This suggests that both mRNA targets could be regulated simultaneously by SprX2 (Figure 10C).
Figure 10.

SprX2 and RNAIII repress Ecb expression in Staphylococcus aureus. (A) Predicted base pairings between ecb mRNA and either RNAIII (32) or SprX2. The presumed SD sequence and AUG initiation codon are underlined. Nucleotides involved in the RNAIII interaction are yellow (43). (B) Schematic representation of RNAIII. The domain interacting with SprX is blue, that with ecb mRNA is red, and the domain coding for delta hemolysin (hld) is gray. (C) Schematic representation of SprX2. The domain interacting with RNAIII is yellow, red for ecb mRNA, and green for spoVG mRNA. (D) Model depicting the S. aureus regulatory network wherein two sRNAs, SprX2 and RNAIII, control Ecb expression.
Interestingly, interaction between RNAIII and the 5΄ region of Newman SprX1 has recently been reported. Furthermore, the nucleotide sequences of Newman SprX1 and HG001 SprX1 are identical (Supplementary Figure S9). This interaction involves the 5΄ region of SprX1, identical in SprX1 and HG001 SprX2 (Supplementary Figure S9), that pairs with the 5΄ region of RNAIII, at proximity from the hld coding sequence (43) (Figure 10B). Although the significance of this interaction is unclear, the authors propose that SprX1–RNAIII pairing might release the intramolecular base pairing between the RNAIII 5΄ and 3΄ ends, changing the secondary structure of RNAIII and allowing the ribosomal binding for effective hld translation. On the SprX sequence, the SprX1 region involved in the RNAIII interaction partially overlaps the SprX2 region involved in ecb mRNA binding (Figure 10A–C). Although additional study is needed to fully understand the collaboration and interactions between SprX and RNAIII, the current data support a functional relationship between the two.
In the last years, there has been growing interest in the regulatory functions of many bacterial sRNAs and especially those expressed by Gram-positive bacteria. However, since sRNAs were discovered in Gram-positive bacteria, functions have only been identified for a very few, in part because of the difficulties in identifying and validating sRNA–mRNA interactions in vivo. From this observation, our approach provides for a simple and accurate technique to study mRNA regulations by sRNA in Gram-positive bacteria. Our method can be used for rapid testing and validation of the many potential mRNA targets of bacterial sRNA in vivo. Since it is simple to use and very efficient, the technology we describe will be helpful in highlighting and understanding the specific roles of sRNAs in S. aureus. Furthermore, this approach provides an opportunity for implementing in Gram-positive bacteria high-throughput methods for screening for new sRNA targets as it was done recently in Gram-negative bacteria (44). We assayed with our approach the sRNA–target regulations from two other Gram-positive bacteria, B. subtilis and L. monocytogenes using S. aureus as a host. Furthermore, as both vectors used in our assay are transformable in other bacteria (45,46), our approach can be adapted for use with other Gram-positive bacteria.
Supplementary Material
ACKNOWLEDGEMENTS
We thank M. Kloc (University of Texas) for comments on the manuscript. The authors are grateful to Dr S.H. Rooijakkers (University of Utrecht, The Netherlands) for providing strains and the anti-Ecb antibodies, to Dr J. Vogel for providing us pXG10-SF vector, to Dr C. Condon (IBPC, Paris) and to Pr. P. Cossart (Institut Pasteur, Paris) for providing us respectively W168 Bacillus subtilis strain and BUG1600 Listeria monocytogenes DNA. We thank T. Mauro and C. Riffaud for technical assistance.
Footnotes
Present address: Alex Eyraud, Université de Sherbrooke, Department of Biochemistry, RNA Group, Sherbrooke, Québec J1E 4K8, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institut National de la Santé et de la Recherche Médicale (INSERM); University of Rennes1 (Défis scientifiques émergents); Lorraine Ivain was supported by a joint PhD fellowship from the Ministère de l’Enseignement Supérieur et de la Recherche and the Région Bretagne. Funding for open access charge: ANR (Agence Nationale de la Recherche) (sRNA-FIT project, grant number: DS0405).
Conflict of interest statement. None declared.
REFERENCES
- 1. Wagner E.G., Romby P.. Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv. Genet. 2015; 90:133–208. [DOI] [PubMed] [Google Scholar]
- 2. Lowy F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998; 339:520–532. [DOI] [PubMed] [Google Scholar]
- 3. Grundmann H., Aires-de-Sousa M., Boyce J., Tiemersma E.. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet. 2006; 368:874–885. [DOI] [PubMed] [Google Scholar]
- 4. Sassi M., Augagneur Y., Mauro T., Ivain L., Chabelskaya S., Hallier M., Sallou O., Felden B.. SRD: a Staphylococcus regulatory RNA database. RNA. 2015; 21:1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fechter P., Caldelari I., Lioliou E., Romby P.. Novel aspects of RNA regulation in Staphylococcus aureus. FEBS Lett. 2014; 588:2523–2529. [DOI] [PubMed] [Google Scholar]
- 6. Lalaouna D., Masse E.. Identification of sRNA interacting with a transcript of interest using MS2-affinity purification coupled with RNA sequencing (MAPS) technology. Genom. Data. 2015; 5:136–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tjaden B., Goodwin S.S., Opdyke J.A., Guillier M., Fu D.X., Gottesman S., Storz G.. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res. 2006; 34:2791–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pain A., Ott A., Amine H., Rochat T., Bouloc P., Gautheret D.. An assessment of bacterial small RNA target prediction programs. RNA Biol. 2015; 12:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Urban J.H., Vogel J.. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res. 2007; 35:1018–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eyraud A., Tattevin P., Chabelskaya S., Felden B.. A small RNA controls a protein regulator involved in antibiotic resistance in Staphylococcus aureus. Nucleic Acids Res. 2014; 42:4892–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corrigan R.M., Foster T.J.. An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid. 2009; 61:126–129. [DOI] [PubMed] [Google Scholar]
- 12. Chabelskaya S., Gaillot O., Felden B.. A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLoS Pathog. 2010; 6:e1000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227:680–685. [DOI] [PubMed] [Google Scholar]
- 14. Jongerius I., von Kockritz-Blickwede M., Horsburgh M.J., Ruyken M., Nizet V., Rooijakkers S.H.. Staphylococcus aureus virulence is enhanced by secreted factors that block innate immune defenses. J. Innate Immun. 2012; 4:301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bohn C., Rigoulay C., Chabelskaya S., Sharma C.M., Marchais A., Skorski P., Borezee-Durant E., Barbet R., Jacquet E., Jacq A. et al. Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res. 2010; 38:6620–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pedelacq J.D., Cabantous S., Tran T., Terwilliger T.C., Waldo G.S.. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006; 24:79–88. [DOI] [PubMed] [Google Scholar]
- 17. Corcoran C.P., Podkaminski D., Papenfort K., Urban J.H., Hinton J.C., Vogel J.. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol. Microbiol. 2012; 84:428–445. [DOI] [PubMed] [Google Scholar]
- 18. Novick R.P. Plasmid incompatibility. Microbiol. Rev. 1987; 51:381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Charpentier E., Anton A.I., Barry P., Alfonso B., Fang Y., Novick R.P.. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 2004; 70:6076–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pichon C., Felden B.. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc. Natl Acad. Sci. U.S.A. 2005; 102:14249–14254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Novick R.P., Ross H.F., Projan S.J., Kornblum J., Kreiswirth B., Moghazeh S.. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993; 12:3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y., Mu C., Ying X., Li W., Wu N., Dong J., Gao Y., Shao N., Fan M., Yang G.. RNAIII activates map expression by forming an RNA-RNA complex in Staphylococcus aureus. FEBS Lett. 2011; 585:899–905. [DOI] [PubMed] [Google Scholar]
- 23. Beard S.J., Salisbury V., Lewis R.J., Sharpe J.A., MacGowan A.P.. Expression of lux genes in a clinical isolate of Streptococcus pneumoniae: using bioluminescence to monitor gemifloxacin activity. Antimicrob. Agents Chemother. 2002; 46:538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kreiswirth B.N., Lofdahl S., Betley M.J., O’Reilly M., Schlievert P.M., Bergdoll M.S., Novick R.P.. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983; 305:709–712. [DOI] [PubMed] [Google Scholar]
- 25. Nair D., Memmi G., Hernandez D., Bard J., Beaume M., Gill S., Francois P., Cheung A.L.. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol. 2011; 193:2332–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harraghy N., Homerova D., Herrmann M., Kormanec J.. Mapping the transcription start points of the Staphylococcus aureus eap, emp, and vwb promoters reveals a conserved octanucleotide sequence that is essential for expression of these genes. J. Bacteriol. 2008; 190:447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joanne P., Falord M., Chesneau O., Lacombe C., Castano S., Desbat B., Auvynet C., Nicolas P., Msadek T., El Amri C.. Comparative study of two plasticins: specificity, interfacial behavior, and bactericidal activity. Biochemistry. 2009; 48:9372–9383. [DOI] [PubMed] [Google Scholar]
- 28. Urban J.H., Vogel J.. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res. 2007; 35:1018–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durand S., Braun F., Lioliou E., Romilly C., Helfer A.C., Kuhn L., Quittot N., Nicolas P., Romby P., Condon C.. A nitric oxide regulated small RNA controls expression of genes involved in redox homeostasis in Bacillus subtilis. PLoS Genet. 2015; 11:e1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nielsen J.S., Lei L.K., Ebersbach T., Olsen A.S., Klitgaard J.K., Valentin-Hansen P., Kallipolitis B.H.. Defining a role for Hfq in Gram-positive bacteria: evidence for Hfq-dependent antisense regulation in Listeria monocytogenes. Nucleic Acids Res. 2010; 38:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Amdahl H., Jongerius I., Meri T., Pasanen T., Hyvarinen S., Haapasalo K., van Strijp J.A., Rooijakkers S.H., Jokiranta T.S.. Staphylococcal Ecb protein and host complement regulator factor H enhance functions of each other in bacterial immune evasion. J. Immunol. 2013; 191:1775–1784. [DOI] [PubMed] [Google Scholar]
- 32. Boisset S., Geissmann T., Huntzinger E., Fechter P., Bendridi N., Possedko M., Chevalier C., Helfer A.C., Benito Y., Jacquier A. et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev/. 2007; 21:1353–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chabelskaya S., Bordeau V., Felden B.. Dual RNA regulatory control of a Staphylococcus aureus virulence factor. Nucleic Acids Res. 2014; 42:4847–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Waters L.S., Storz G.. Regulatory RNAs in bacteria. Cell. 2009; 136:615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bordeau V., Cady A., Revest M., Rostan O., Sassi M., Tattevin P., Donnio P.Y., Felden B.. Staphylococcus aureus regulatory RNAs as potential biomarkers for bloodstream infections. Emerg. Infect. Dis. 2016; 22:1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Papenfort K., Vanderpool C.K.. Target activation by regulatory RNAs in bacteria. FEMS Microbiol. Rev. 2015; 39:362–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guillier M., Gottesman S., Storz G.. Modulating the outer membrane with small RNAs. Genes Dev. 2006; 20:2338–2348. [DOI] [PubMed] [Google Scholar]
- 38. Burman J.D., Leung E., Atkins K.L., O'Seaghdha M.N., Lango L., Bernado P., Bagby S., Svergun D.I., Foster T.J., Isenman D.E. et al. Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein: indications of a novel mechanism of complement evasion by Staphylococcus aureus. J. Biol. Chem. 2008; 283:17579–17593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jongerius I., Garcia B.L., Geisbrecht B.V., van Strijp J.A., Rooijakkers S.H.. Convertase inhibitory properties of Staphylococcal extracellular complement-binding protein. J. Biol. Chem. 2010; 285:14973–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nygaard T.K., Pallister K.B., Ruzevich P., Griffith S., Vuong C., Voyich J.M.. SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J. Infect. Dis. 2010; 201:241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liang X., Yu C., Sun J., Liu H., Landwehr C., Holmes D., Ji Y.. Inactivation of a two-component signal transduction system, SaeRS, eliminates adherence and attenuates virulence of Staphylococcus aureus. Infect. Immun. 2006; 74:4655–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCarthy A.J., Lindsay J.A.. Staphylococcus aureus innate immune evasion is lineage-specific: a bioinfomatics study. Infect. Genet. Evol. 2013; 19:7–14. [DOI] [PubMed] [Google Scholar]
- 43. Kathirvel M., Buchad H., Nair M.. Enhancement of the pathogenicity of Staphylococcus aureus strain Newman by a small noncoding RNA SprX1. Med. Microbiol. Immunol. 2016; 205:563–574. [DOI] [PubMed] [Google Scholar]
- 44. Lee HJ, Gottesman S. sRNA roles in regulating transcriptional regulators: Lrp and SoxS regulation by sRNAs. Nucleic Acids Res. 2016; 44:6907–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ehrlich S.D., Niaudet B., Michel B.. Use of plasmids from Staphylococcus aureus for cloning of DNA in Bacillus subtilis. Curr. Top. Microbiol. Immunol. 1982; 96:19–29. [DOI] [PubMed] [Google Scholar]
- 46. Mojumdar M., Khan S.A.. Characterization of the tetracycline resistance gene of plasmid pT181 of Staphylococcus aureus. J. Bacteriol. 1988; 170:5522–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.