


Abstract
Whole transcriptome sequencing (RNA-seq) has become a standard for cataloguing and monitoring RNA populations. One of the main bottlenecks, however, is to correctly identify the different classes of RNAs among the plethora of reconstructed transcripts, particularly those that will be translated (mRNAs) from the class of long non-coding RNAs (lncRNAs). Here, we present FEELnc (FlExible Extraction of LncRNAs), an alignment-free program that accurately annotates lncRNAs based on a Random Forest model trained with general features such as multi k-mer frequencies and relaxed open reading frames. Benchmarking versus five state-of-the-art tools shows that FEELnc achieves similar or better classification performance on GENCODE and NONCODE data sets. The program also provides specific modules that enable the user to fine-tune classification accuracy, to formalize the annotation of lncRNA classes and to identify lncRNAs even in the absence of a training set of non-coding RNAs. We used FEELnc on a real data set comprising 20 canine RNA-seq samples produced by the European LUPA consortium to substantially expand the canine genome annotation to include 10 374 novel lncRNAs and 58 640 mRNA transcripts. FEELnc moves beyond conventional coding potential classifiers by providing a standardized and complete solution for annotating lncRNAs and is freely available at https://github.com/tderrien/FEELnc.
INTRODUCTION
The development of high-throughput RNA sequencing (RNA-seq) has revealed the presence of many RNAs in different organisms such as mammals (1–3), insects (4,5) and plants (6,7). Particularly, whole transcriptome sequencing sheds light on the pervasive transcription of the genomes with messenger RNAs (mRNAs) only representing a small fraction of the genome, outnumbered by a vast repertoire of small (miRNAs, snRNAs…) and long non-coding RNAs (lncRNAs) (1). LncRNAs, basically defined as transcripts longer than 200 nucleotides and without any protein-coding capabilities, have been involved in many aspects of normal and pathological cells. From the pioneer discovery of the Xist lncRNA involved in X chromosome inactivation in placental females (8) to the more recent links between lncRNAs and cancers (9,10), lncRNAs have emerged as key actors of the cell machinery with diverse modes of action such as gene expression regulation, control of translation or imprinting.
Following RNA sequencing, the computational reconstruction of transcripts models either by genome-guided (11,12) or de novo assembly (13) usually produces tens of thousands of known and novel transcript models. Among this wealth of assembled transcripts, it remains crucial to annotate the different classes of RNAs and especially to distinguish protein-coding from non-coding RNAs. To this aim, several bioinformatic tools have been developed in order to compute a coding potential score (hereafter termed CPS) used to discriminate the coding status of the RNA gene models. Broadly, they can be divided into programs using sequence alignments, either between species (14) or alignments to protein databases (15), and alignment-free software (16–18). The alignment-dependent methods, although very specific in terms of performance, are often very time- and resource-consuming. For instance, the PhyloCSF program requires a multispecies sequence alignment to predict the likelihood of a sequence to be a conserved protein-coding transcript based on the evolution of the codon substitution frequencies (14). It is thus dependent on the quality of the input alignments and may also be biased toward misclassifying species-specific or lowly conserved coding and non-coding transcripts (19). In contrast, the alignment-free methods compute a CPS only depending on intrinsic features of the input RNA sequences. One of the main features is given by the length of the longest open reading frame (ORF) (20,21) since a transcript harboring a long ORF will most likely be translated into a protein. However, the definition of the longest ORF can vary between programs, especially when it involves the strict inclusion of either or both start and stop codons. This is particularly important to model since some transcripts from reference annotations and/or newly assembled transcripts are not full-length. For instance, the number of protein-coding transcripts in the human EnsEMBL (v83) annotation (22) lacking a start codon or a stop codon is 7677 (∼10%) and 16 649 (∼25%), respectively. A complementary feature to discriminate mRNAs from non-coding RNAs is the relative frequency of oligonucleotides or k-mer (where k denotes the size of the oligonucleotide). Some tools already use k-mer frequencies but are often limited to one and/or small k-mers (generally k ≤ 6), whereas longer k-mers could help resolve ambiguities by taking into account lncRNA-specific repeats or spatial information (23,24). Finally, common to all methods is the lack of an explicit modeling and cut-off definition for ‘non-model’ organisms (25), for which it can be crucial to train the programs with species-specific data and to automatically derive a CPS cut-off which provides better discriminative power.
Here, we present FEELnc, for FlExible Extraction of LncRNAs, a new tool to annotate lncRNAs from RNA-seq assembled transcripts. FEELnc is an all-in-one solution from the filtering of non-lncRNA-like transcript models, to the computation of a coding potential score and the formalization of the definition of the lncRNA classes. Based on a relaxed definition of ORFs and a very fast analysis of small and large k-mer frequencies (from k = 1 to 12), the program implements an alignment-free strategy using Random Forests (26) to classify lncRNAs and mRNAs. We benchmarked FEELnc and five existing programs (PhyloCSF (14), CPC (15), CPAT (16), PLEK (17) and CNCI (18)) using known sets of lncRNAs annotated in multiple organisms (GENCODE for human and mouse (27) and NONCODE for other species (28)), and showed that FEELnc performance metrics outperformed or are similar to state-of-the-art programs. We developed FEELnc to be used on ‘non-model’ organisms for which no set of lncRNAs is available by deriving species-specific lncRNA models from mRNA sequences and automatically computing the CPS cut-off that maximizes classification performances. FEELnc also allows users to provide their own specificity thresholds in order to annotate high-confidence sets of lncRNAs and mRNAs and to define a class of transcripts with ambiguous status. Finally, as part of the LUPA consortium (29), we produced 20 RNA sequencing data sets from 16 different canine tissues and applied FEELnc on the reconstructed models to annotate 10 374 lncRNA and 58 640 mRNA new transcripts from known and novel loci. We also classified lncRNAs into 5033 long intergenic non-coding RNAs (lincRNAs) and 5341 genic sense or antisense lncRNAs based on the FEELnc classifier module. The number of lncRNA transcripts detected by our data considerably expands the canine genome annotation providing an extended resource which will help deciphering genotype to phenotype relationships (30).
MATERIALS AND METHODS
Data set
For the sake of reproducibility, all data sets and scripts used to generate the benchmark files are available in Supplementary Data.
Human long non-coding and protein-coding genes were obtained from the manually curated GENCODE version 24 annotation (EnsEMBL v83 corresponding to the GRCh38 human genome assembly) selecting the long non-coding gene biotypes ‘lincRNA’ and ‘antisense’, and ‘protein_coding’ for coding genes. From each of this set, 10 000 transcripts were extracted and further divided into two sets of 5000 transcripts, that are used for the learning and the testing steps, denoted HL and HT data sets, respectively. Importantly, only one transcript per locus was extracted for all biotypes in order not to create a bias by introducing two isoforms of the same gene in both the HL and HT sets. For mouse, we used the GENCODE version M4 annotation (EnsEMBL v79) and derived the learning and testing sets in the same way as for human (denoted ML and MT). Due to the lower number of GENCODE lncRNAs annotated in mouse compared to human, each file contains ∼2000 lncRNAs and 5000 mRNAs. For ‘non-model organisms’, lncRNAs belonging to the lincRNA and antisense classes (NONCODE codes 0001 and 1000, respectively) were downloaded from the latest version of the NONCODE database (NONCODE 2016) (28) while mRNAs were retrieved from the EnsEMBL database (v84). A summary of the number of mRNAs/lncRNAs per species is available in Supplementary Table S1.
Whole transcriptome sequencing of dog RNA samples (n = 20) was performed by the LUPA consortium. These biological samples, corresponding to 16 unique tissues and 7 breeds, were obtained from the ‘Cani-DNA CRB’ biobank at the University Rennes1, CNRS-IGDR, France (http://dog-genetics.genouest.org), the biobank at University of Copenhagen, Denmark, the biobank at University of Helsinky, Finland and the Vetsuisse Biobank at University of Bern, Switzerland. The dog owners consented to the use of the data for research purposes anonymously. RNAs were extracted from tissues using the NucleoSpin RNA kit (Macherey–Nagel) according to the manufacturer's instructions. Polyadenylated RNAs were captured by oligo-dT beads and RNA-seq libraries were constructed via the Illumina TruSeq™ Stranded mRNA Sample Preparation Kit. Sequencing was done in paired-end and stranded fashion on the HiSeq2000 platform using v3 chemistry (TruSeq PE Cluster Kit v3-cBot-HS, TruSeq SBS Kit v3-HS, TruSeq SR Cluster Kit v3-cBot-HS) to a depth of about 50 million reads per tissue (Supplementary Table S2). The RNA-seq data are available in the short read archive (SRA) under NCBI bioproject PRJNA327075 and SRA accession SRP077559.
FEELnc filter module (FEELncfilter)
The first FEELnc module aims at identifying non-lncRNA transcripts from the reconstructed transcript models given by genome-guided transcriptome assemblers such as Cufflinks (11) or more recently StringTie (12). To achieve this goal, FEELnc flags every assembled transcript that overlaps any exon of the reference annotation in sense. To deal with the plethora of input models inherent to high-depth RNA-seq experiments, the comparison of transcript intervals is parallelized through the Parallel:ForkManager Perl module. Importantly, FEELnc allows the user to parameter the percentage of overlap and also the transcript biotype (e.g. ‘protein_coding’ or ‘pseudogene’) to be considered from the reference annotation. Indeed, transcripts matching protein-coding exons should be flagged as they likely indicate novel mRNA isoforms. FEELncfilter also filters out short transcripts (default 200 nt) and can deal with single-exon transcripts depending on whether the protocol used to construct libraries is stranded or not. For instance, the module allows the removal of intergenic single-exon models as they may correspond to mapping artifacts due to repeat sequences and for which the checking of the consensus splice sites could not be assessed (27).
FEELnc coding potential module (FEELnccodpot)
FEELnccodpot predictors
The second FEELnc module aims at computing a coding potential score given the assembled sequences following transcriptome reconstruction. To deal with the incompleteness of ORF annotation where both the reference and the reconstructed transcripts may not be full-length, FEELnc computes all ORFs and annotates five ORF types from the stricter ‘type 0’ which corresponds to the longest ORF having both a start and a stop codon, to the more relaxed ‘type 4’ that is the whole input RNA sequence (see Supplementary Data for detailed description of the ORF types). Because the size of the protein-coding ORF is generally correlated with the length of the input RNA sequence, we used the ORF coverage, i.e. the proportion of the transcript size covered by an ORF, as the first predictor to discriminate mRNAs/lncRNAs in the FEELnc model.
The second predictor of FEELnc relies on the computation of the multi k-mer frequencies between mRNAs and lncRNAs. Biases in nucleotide frequencies and codon usage have already been described in the literature as important discriminative features between coding and non-coding RNAs (21,31). Within the framework of FEELnc, we developed an extremely fast and exact k-mer counter called KIS (for K-mer in short) that relies on the open-source GATB library (32). For example, KIS can compute all 6-mers (hexamers) and 12-mers of the human GRCh38 genome assembly (∼3 billions k-mers) in ∼2 min and 2 min 50 s, respectively (on a linux RedHat station with one core Intel(R) Xeon(R) CPU X5550 @ 2.67GHz). Due to the high speed of KIS, a major contribution of this work was to be able to combine different lists of k-mers, including longer k-mers, in order to better discriminate lncRNAs from mRNAs. Specifically, we assigned a score for each sequence 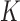 (e.g. TGC) of size k (e.g. 3) based on the occurrence of this sequence in each predicted mRNA ORF sequence and lncRNA whole sequence from the learning data set. This score
(e.g. TGC) of size k (e.g. 3) based on the occurrence of this sequence in each predicted mRNA ORF sequence and lncRNA whole sequence from the learning data set. This score  , similar to the one used in Claverie et al. in (33), is computed for each
, similar to the one used in Claverie et al. in (33), is computed for each 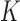 of size
of size 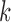 as follows:
as follows:
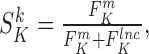 with
with 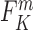 and
and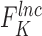 the observed frequencies of
the observed frequencies of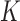 in mRNA ORFs and in lncRNA sequences for the two learning sets, respectively. Note that these k-mer profiles are made on a subset of the learning set (10% by default) and the transcripts used to compute the k-mer profiles are removed from the random forest model in order to avoid overfitting.
in mRNA ORFs and in lncRNA sequences for the two learning sets, respectively. Note that these k-mer profiles are made on a subset of the learning set (10% by default) and the transcripts used to compute the k-mer profiles are removed from the random forest model in order to avoid overfitting.
Once the k-mer profiles are made, i.e. all 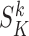 have been computed for all k-mer, FEELnc associates a k-mer score
have been computed for all k-mer, FEELnc associates a k-mer score 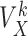 for each remaining ORFs
for each remaining ORFs  as follows:
as follows:
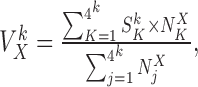 with
with 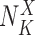 the number of occurrences of
the number of occurrences of  in the ORF
in the ORF  and
and 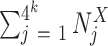 the total number of k-mer of size k. Using this scoring method, a k-mer score is associated to each sequence for each k-mer size selected in the model.
the total number of k-mer of size k. Using this scoring method, a k-mer score is associated to each sequence for each k-mer size selected in the model.
FEELnc coding potential also uses the total RNA sequence length as a predictor of the model since lncRNAs have been shown to be significantly shorter than mRNAs (34,35). For illustration purposes, a distribution of the FEELnc predictor scores with ‘type 3’ ORF (i.e. longest ORF having either or both a start and a stop codon) and multi k-mer scores with k in {1, 2, 3, 6, 9, 12} is given in Supplementary Figure S1 for the 5000 mRNAs and 5000 lncRNAs of the HL data set.
Random forest classification and optimized coding potential cut-offs
The aforementioned predictor scores are incorporated into a machine learning method—Random Forest (RF) (26)—that computes a coding potential score (CPS) for each input training transcripts. As also shown by others with respect to lncRNAs annotation, RF often outperforms other machine learning techniques especially due to the random sampling of features to build the ensemble of trees (36,37). In addition, our RF model which is based on the randomForest R package (38), can deal with imbalanced training set by down-sampling the majority class (most likely mRNAs in many organisms). In fact, the CPS in our RF model corresponds to the proportion of all trees (500 trees by default) which ‘vote’ for the input sequence to be coding or non-coding. A proportion close to 0 will indicate a non-coding RNA and close to 1 an mRNA.
To define an optimal CPS cut-off, FEELnc automatically extracts CPS that maximizes both sensitivity (Sn) and specificity (Sp) (see performance section) based on a 10-fold cross-validation. Using the ROCR package (39), FEELnc provides users with a two-graph ROC curve in order to display the performances of the model and to visualize the optimal CPS (Figure 1 A).
Figure 1.

FEELnccodpot and FEELncclassifier description. (A) Two graph ROC curves for automatic detection of optimized CPS threshold and user specificity threshold, the latter defining two conservative sets of lncRNAs and mRNAs and a class of transcripts with ambiguous biotypes termed TUCp (Transcripts of Unknown Coding potential). B) Sub classification of intergenic and genic lncRNA/transcripts interactions by the FEELncclassifier module.
Even if this approach aims at providing the highest performances, it could sometimes misclassify coding and/or non-coding transcripts whose CPS is closed to the optimized threshold (40). To take this into account, FEELnc allows fixing two minimal specificity cut-offs for lncRNAs and mRNAs (this approach is termed ‘two cut-offs’). This naturally leads to the annotation of two high-confident classes of lncRNAs and mRNAs and also to the definition of a third class of ambiguous transcripts (i.e. transcripts whose the CPS is between the two cut-offs) that are named TUCp (34,40) for Transcripts of Unknown Coding potential (Figure 1A).
FEELnc without long non-coding training set
One issue when using machine learning algorithms is the requirement of both a positive and negative sets (here mRNA and lncRNA) to train the model. While the former is often available for most organisms, the latter is usually not especially for non-model organisms (25). To model non-coding RNAs in the absence of a true set of lncRNAs, we assessed three strategies called intergenic, shuffle and cross-species. As DNA composition varies between species, a first naive approach consists in extracting decoy sequences from the genome of interest to model species-specific non-coding sequences. More precisely, we extracted random intergenic sequences of length L (L is given by the distribution of the mRNA sizes) as the non-coding training set. The shuffle strategy employs a more sophisticated method which is based on the idea that lncRNAs are derived from ‘debris’ of protein-coding genes (41,42), as exemplified by the Xist lncRNA that emerged from the disruption of the mRNA gene Lnx3 (43). To this end, we shuffled mRNA sequences from the reference annotation using the Ushuffle program (44), while preserving a given k-mer frequency of the input sequences. Note, however, that shuffling sequences and preserving frequencies for one fixed k-mer does not constrain the frequencies of the other k-mers. In addition, when k increases, it is possible that Ushuffle could not permute some input sequences because of the constraint to preserve the given k-mer frequencies. Finally, the cross-species strategy makes use of lncRNA sets annotated in other species to extract non-coding predictors and train the RF model. For the latter strategy, we trained the FEELnc model using human mRNAs and species-specific lncRNAs (NONCODE lncRNA catalogues being available in 13 different species). For all strategies, we assessed the performance on the HT data sets.
Performance evaluation
We evaluated the performance of FEELnc and five other state-of-the-art programs: CNCI (version 2 Feb 28, 2014) (18), CPC (version 0.9-r2) (15), CPAT (version 1.2.1) (16), PhyloCSF (version 20121028-exe) (14) and PLEK (version 1.2) (17), by computing classical performance metrics:
- Sensitivity (Sn) or True Positive Rate = 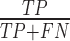 ;
;
- Specificity (Sp) or True Negative Rate =  ;
;
- Precision (Prec) or Positive Predicted Value =  ;
;
- Accuracy (Acc) =  ;
;
With TP: True Positive, TN: True Negative, FP: False Positive and FN: False Negative.
In addition, we used two complementary metrics in order to capture the global performance of the tools in a single measure (45):
- F-score =  , which is a statistic measuring the harmonic mean of precision and sensitivity;
, which is a statistic measuring the harmonic mean of precision and sensitivity;
- MCC (Matthews Correlation Coefficient) = 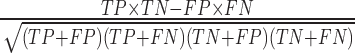 , which is particularly useful when the two classes are of very different sizes (which is often the case for mRNAs and lncRNAs in non-model organisms) and which could be seen as a correlation coefficient between the true classes and the predicted classes (46).
, which is particularly useful when the two classes are of very different sizes (which is often the case for mRNAs and lncRNAs in non-model organisms) and which could be seen as a correlation coefficient between the true classes and the predicted classes (46).
For each performance metric, we considered lncRNAs as the negative class and mRNAs as the positive class. Note that, as a binary classification, the mRNA specificity corresponds to the lncRNA sensitivity (and conversely). Moreover, for the CPAT and PLEK programs, which allow training their models, we used species-specific set of mRNAs/lncRNAs for training (called CPATtrain and PLEKtrain). Nevertheless, contrary to FEELnc, PLEK and CPAT required us to a priori extract the CDS of the mRNA input file to learn the coding parameters. For CPATtrain, we referred to the optimal CPS cut-off mentioned on their website (16) to discriminate between coding and non-coding RNAs. A detailed description of the command lines used to run each program is given in Supplementary Data.
FEELnc classifier module (FEELncclassifier)
Given a known reference annotation, it is essential to classify newly annotated lncRNAs based on their closest annotated transcripts. It will potentially guide researchers towards functional annotation and relationships between lncRNAs and their annotated partners (lncRNA/mRNA pairs for instance). For this purpose, the FEELnc classifier module (FEELncclassifier) employs a sliding window strategy (whose length is fixed by the user) around each lncRNAs to report all the reference transcripts located within the window. Not only does the FEELnc classifier annotate lincRNAs and antisense lncRNAs but it also formalizes the definition of lncRNA subclasses with respect to annotated transcripts (Figure 1B). First, these rules involve the direction (sense or antisense) and the type of interactions (genic or intergenic). Then, within each type of interaction, a subtype level allows to narrow down the classification (e.g. divergent for lincRNAs or containing for genic lncRNAs). Finally, a location level is added informing about the position of the lncRNA with respect to the annotated transcripts (e.g. upstream for lincRNA or exonic for genic lncRNAs). In addition, the FEELncclassifier can be used with all transcript biotypes (e.g. short ncRNAs such as snoRNAs) from a reference annotation and therefore is capable of annotating lncRNAs that are host genes for short RNAs (47) (hence the ‘genic sense exonic’ class).
Because one lncRNA could belong to different classes depending on which reference transcript is considered, our approach reports all interactions within the defined window and defines a best partner transcript using the following priorities: for genic lncRNAs, the exonic class has priority over the intronic class and the intronic over the containing, while for lincRNAs, the nearest reference transcript is selected.
Reads mapping and transcript model reconstruction of canine RNA-seq samples
The processing of RNA-seq reads from the mapping of the reads to the transcript model reconstruction was performed using standard bioinformatic pipeline (48). Such genome-guided transcript model reconstruction has already been validated as for instance in the canine genome (49). Briefly, the mapping of the canine RNA-seq reads was done using the STAR v2.5.0a program (50) while Cufflinks v2.2.1 (11) was used to reconstruct transcript models for each sample separately using the dog genome annotation by EnsEMBL and by the Broad (49) as a guide. Finally, the cuffmerge tool, from the Cufflinks package, was used to compute a single consensus file with all reconstructed transcript models (all command lines and parameters used for each tool are also provided in Supplementary Data).
RESULTS
FEELnc modules to annotate lncRNAs
Starting from assembled transcripts and a reference annotation, the FEELnc pipeline is composed of three independent modules to classify and annotate lncRNAs (Figure 2). The FEELncfilter module filters out input transcript models reconstructed via a genome-guided assembly strategy that do not correspond to potential novel lncRNA candidates. Due to its ability to take into account reference transcript biotypes (See Methods), the module allows to keep novel transcript models overlapping other referenced ncRNAs for instance, as these models may correspond to long non-coding RNAs that are host genes for small RNAs (51). After the FEELncfilter module, the remaining transcripts are thus candidates to be new lncRNAs or mRNAs.
Figure 2.

General overview of the FEELnc pipeline. The FEELnc filter module (FEELncfilter) identifies newly assembled RNA-seq transcripts and removes non-lncRNA transcripts. The FEELnc coding potential module (FEELnccodpot) computes a coding potential score (CPS) and automatically defines the optimal CPS score cut-off to discriminate lncRNAs versus mRNAs (and eventually TUCPs). The FEELnc classifier module (FEELncclassifier) annotates lncRNA classes based on RNA partners from the reference annotation.
The second module (FEELnccodpot) computes a coding potential score for every candidate transcript based on a RF model trained with several predictors such as ORF coverage, multi k-mer frequencies and RNA sizes (see Methods for details about predictors). Using the gold-standard GENCODE human learning set (HL) for training, we evaluated the performance of FEELnc on 5000 lncRNAs and 5000 mRNAs of the test data set with respect to the five ORF types and multi k-mer combinations. Remarkably, we observed that ‘type 1’ and ‘type 3’ ORFs, which extract the longest ORF even in the absence of stop codon, consistently display better achievements (mean MCC = 0.816) than ‘type 0’ and ‘type 2’ ORFs (mean MCC = 0.67 and 0.68, respectively) whichever combination of k-mers is considered (Supplementary Figure S2). In addition, the multi k-mer strategy improves the performance of the program with a MCC performance starting at 0.80 when only using 6-mers but reaching 0.85 with a combination of {1, 2, 3, 6, 9, 12}-mers (with a fixed ORF type 3, Supplementary Figure S2).
In addition to measuring performance, we conducted several evaluations to assess the robustness of the FEELnc predictions. First, we showed that FEELnc was not biased by unbalanced or low numbers of transcripts in the input training set with sensitivity and specificity values higher than 0.9 using only 400 mRNAs and lncRNAs (Supplementary Figure S3). Second, FEELnc performed similarly or better than other methods to classify very small or long mRNAs and lncRNAs (Supplementary Figure S4). Third, in order to model incomplete RNA reconstructions, we removed the 10%, 25% and 50% of either 5΄-end or 3΄-end of transcript sequences from the HT data set. Even if FEELnccodpot performance decreased with increasing degradation percentages (MCCs = 0.827, 0.749 and 0.53 for 10%, 25% and 50%, respectively), it performed better than other tested methods (Supplementary Figure S5). Fourth, we observed similar high performance with mouse GENCODE data set ML and MT (see materials) composed of 5000 mRNAs and 2000 lncRNAs where FEELnc achieves 0.938 in sensitivity, 0.941 in specificity and an MCC of 0.856. Finally, among the manually curated list of 35 ‘well-characterized’ lncRNAs from Chen et al. (52), FEELnc correctly classifies 33 (95%) of them as non-coding. The two discordant lncRNAs are PWRN1, which has a CPS (0.374) just above the optimized cut-off (0.372), and FIRRE, which exhibits the highest CPS (0.58 when the median CPS for the 35 lncRNAs is 0.088). This high coding potential score for FIRRE lncRNA can be explained by a large and complete ORF (546 nt, i.e. 58% of the total RNA sequence) and a high level of sequence similarity with the FAM195A protein-coding gene.
The third FEELnc module (FEELncclassifier) formalizes the annotation of lncRNAs based on neighboring genes in order to predict lncRNA functions and RNA partners (see classes in methods). To illustrate the outcome of the FEELncclassifier, we applied it on the human EnsEMBL v83 annotation composed of 24 659 lncRNAs (‘lincRNA’ and ‘antisense’ biotypes) and 79 901 mRNA transcripts (‘protein_coding’ biotype). For instance, FEELncclassifier annotates 5544 lncRNAs as ‘intergenic antisense upstream’ which correspond to divergent lincRNAs (i.e. transcribed in head to head orientation with the RNA partner) among which 28.3% (n = 1572) are less than 1kb from their mRNA partner Transcription Start Sites (TSSs). This class directly pinpoints to lincRNAs potentially sharing a bi-directional promoter with their mRNA partners (53,54). On the other hand, 408 lincRNAs located less than 5kb from their mRNA partner, belong to the ‘sense intergenic upstream’ class and may correspond to dubious lncRNAs that are actually 5΄UTR extensions of the neighboring protein-coding RNAs. FEELncclassifier also annotates 5006 lncRNAs in the ‘antisense exonic’ class as potential candidates for complementary interactions with the mRNA transcribed in opposite direction (55,56).
Benchmarking FEELnc and existing tools
We next compared the performance of FEELnc with five state-of-the-art programs either alignment-free (CPAT, CNCI and PLEK) or alignment-based (PhyloCSF and CPC). Similarly to FEELnc, we used the balanced human learning data set (HL) composed of 5000 lncRNAs and 5000 mRNAs (see Methods) to construct the models for CPAT and PLEK (denoted CPATtrain and PLEKtrain). We also used default pre-built models for PLEK and CPAT although some of the transcripts from the human GENCODE data set test file (HT) could have been used for building these models. For all programs, performance metrics were calculated according to the HT data set. This showed that FEELnc had the highest classification power (AUC, i.e. Area Under the Curve value = 0.97) compared to the others tools as illustrated by the ROC curves (Figure 3A). Accordingly, FEELnc displays the highest sensitivity (0.923) and the second highest specificity (0.915) among all tools while PLEK displays the highest specificity (0.985) and precision (0.981). In general, alignment-based methods have lower classification metrics than alignment-free programs as they usually depend on the quality of the input cross-species alignments or the completeness of species-specific protein databases. Finally, FEELnc also shows the highest classification accuracy (0.919), F-score (0.919) and MCC values (0.838) indicating that it performs well on the human GENCODE data set in comparison to other tools (Table 1).
Figure 3.

FEELnc performance against coding potential tools and with shuffle, intergenic and cross-species approaches. (A) ROC curve analysis of FEELnc versus coding potential tools based on GENCODE human data set (HT). (B) Empirical cumulative distribution of FEELnccodpot feature scores with the true set of human lncRNAs (‘lncRNA’) in comparison with the ‘shuffle’ and ‘intergenic’ methods. (C) FEELnccodpot MCC values tested on human HT set and trained using human mRNAs and species-specific NONCODE lncRNAs (cross-species). The x-axis represents the time of speciation between human and NONCODE species as given in (69). Species abbreviations are the following: Atha: Arabidopsis; Btau: Cow; Cele: Nematode; Dmel: Fly; Drer: Zebrafish; Ggal: Chicken; Ggor: Gorilla; Hsap: Human; Mdom: Opossum; Mmul: Rhesus; Mmus: Mouse; Oana: Platypus; Pabe: Orangutan; Ptro: Chimpanzee; Rnor: Rat.
Table 1. Tools performance on the GENCODE human data sets. Bold-underlined values correspond to the highest values of each metrics. CPAT_train and PLEK_train correspond to program versions trained with the HL data set. Programs are sorted by MCC values.
| HUMAN data set | Program | Sensitivity | Specificity | Precision | Accuracy | F-score | MCC |
|---|---|---|---|---|---|---|---|
| FEELnc | 0.923 | 0.915 | 0.916 | 0.919 | 0.919 | 0.838 | |
| CPAT | 0.899 | 0.924 | 0.922 | 0.912 | 0.910 | 0.823 | |
| CPAT_train | 0.920 | 0.901 | 0.903 | 0.910 | 0.911 | 0.821 | |
| CNCI | 0.829 | 0.979 | 0.975 | 0.904 | 0.896 | 0.817 | |
| PLEK | 0.732 | 0.985 | 0.981 | 0.858 | 0.838 | 0.741 | |
| PhyloCSF | 0.906 | 0.802 | 0.820 | 0.854 | 0.861 | 0.712 | |
| PLEK_train | 0.582 | 0.960 | 0.936 | 0.770 | 0.718 | 0.584 | |
| CPC | 0.699 | 0.739 | 0.728 | 0.719 | 0.713 | 0.438 |
We further investigated the performance on the mouse data sets composed of 2000 lncRNAs and 5000 mRNAs in order to replicate the analysis in another organism using an unbalanced data set. For this benchmark, we included the same programs except PhyloCSF due to the labor-intensive task to extract input cross-species multiple alignments. Again, FEELnc displays the highest classification accuracy, F-score and MCC (Table 2) while CPC shows the best specificity (0.992) and precision (0.996) despite a weak sensitivity (0.744). As in human, the CPAT program performs well even if we consider both the re-trained and prebuilt models. Interestingly, CPAT used with the trained mouse model has only slightly better performance than used with the human prebuilt model. This suggests that within-species training achieves relatively few performance gains compared to cross-species training with a closely related model species.
Table 2. Program performances on the GENCODE mouse data sets. Bold-underlined values correspond to the highest values of each metrics. Programs are sorted by MCC values.
| MOUSE data set | Program | Sensitivity | Specificity | Precision | Accuracy | F-score | MCC |
|---|---|---|---|---|---|---|---|
| FEELnc | 0.938 | 0.941 | 0.976 | 0.939 | 0.956 | 0.856 | |
| CPAT_train | 0.950 | 0.880 | 0.952 | 0.930 | 0.951 | 0.828 | |
| CPAT | 0.892 | 0.960 | 0.982 | 0.911 | 0.935 | 0.806 | |
| CNCI | 0.857 | 0.972 | 0.987 | 0.890 | 0.918 | 0.772 | |
| CPC | 0.744 | 0.992 | 0.996 | 0.815 | 0.852 | 0.667 | |
| PLEK | 0.710 | 0.913 | 0.954 | 0.768 | 0.814 | 0.564 | |
| PLEK_train | 0.630 | 0.891 | 0.936 | 0.704 | 0.753 | 0.470 |
Finally, we assessed the computational time of each program including the time required for computing the model for training-based tools on a linux RedHat station (Intel(R) Xeon(R) CPU X5550 @ 2.67GHz). FEELnc took ∼46 min to classify the 10 000 human lncRNAs and mRNAs while PLEK was the fastest (6 min as compared to ∼10 h when we trained its model) and CPC the longest (∼2 days; with default parameters).
Annotating lncRNAs without a species-specific training set of lncRNAs
In the absence of a species-specific lncRNAs set, machine learning strategies require to simulate non-coding RNA sequences to train the model. In order to evaluate the intergenic and shuffle strategies on the human training sets (see Materials and Methods), we computed their predictor scores in comparison with the true set of 5000 HL lncRNAs. For the shuffle strategy, it is essential to determine a priori which given k-mer frequencies should be preserved by Ushuffle to maximize classification accuracy. We thus shuffled HL mRNA sequences for different sizes of k and showed that preserving 7-mer frequencies gave the best MCC values on the HT set while sustaining a high number of permuted sequences (Supplementary Figure S6). We then compared the intergenic versus shuffle-derived lncRNAs and observed that the cumulative distribution of FEELnc predictor values for the shuffle strategy tended to be closer to the one observed in the true set of human lncRNAs compared to the intergenic approach (Figure 3B). This result was confirmed by assessing performance of these two strategies on the HT data set where the shuffle method outperformed the intergenic approach (MCCs = 0.768 versus 0.646) as compared to true set of lncRNAs (MCC = 0.846) (Supplementary Figure S7).
As described above, FEELnc can be used in a stringent mode in order to distinguish high-confidence sets of lncRNAs and mRNAs. To this end, we also applied the ‘two cut-offs’ option for both strategies with increasing specificity thresholds (0.93, 0.96, 0.99 for both mRNAs and lncRNAs) as compared to the automatic optimal CPS cut-off defined previously (Supplementary Figure S7). With these cut-offs, we also observed higher performances for the shuffle approach (MCCs = 0.823, 0.881 and 0.943) versus intergenic (MCCs = 0.646, 0.654 and 0.785) (Supplementary Figure S7). Moreover, the greater interest of the shuffle approach could be appreciated by the lower variability between sensitivity and specificity metrics as defined within the FEELnc methodology compared to the intergenic approach (Supplementary Figure S7).
In order to directly evaluate FEELnc performance for ‘non-model’ organisms, we used the FEELnc shuffle strategy where the protein-coding predictors were learnt on species-specific mRNAs and the non-coding predictors on species-specific mRNAs shuffled by Ushuffle (with preserved 7-mer frequencies). All tests were further assessed on the catalogues of lncRNAs annotated in the NONCODE database. We also compared the performance with CNCI knowing that NONCODE uses both CNCI and matching protein-coding coordinates from RefSeq database to remove all ncRNAs annotated as protein-coding. In Supplementary Table S3, we showed that FEELnc without annotated lncRNAs achieved good classification metrics in many species (MCCs ranging from 0.70 for rat to 0.903 for cow) and even better than CNCI in five diverse species (nematode, fly, gorilla, orangutan and rhesus macaque).
As a third approach to model non-coding sequences, we sought to use cross-species lncRNAs for learning FEELnc model parameters. To this end, NONCODE lncRNAs from 13 diverse organisms were used to serve as a proxy for modeling human lncRNAs, and evaluation metrics were then performed on the HT data set. As expected, FEELnc performance is negatively correlated with respect to the evolutionary distance between human and NONCODE species (Spearman rho = −0.85; P-value = 5.6e-05) with MCC values of 0.374 when using Caenorhabditis elegans lncRNAs to 0.823 with chimpanzee lncRNAs (Figure 3C and Supplementary Table S4). This probably reflects the variability in term of lncRNA sequence conservation (and thus in non-coding k-mer frequencies) between human and NONCODE species (35,52). Interestingly, the shuffle strategy showed a MCC of 0.748, which corresponds to the performance obtained with species that diverged about 100 million years ago. This indicates that it constitutes an interesting approach when no lncRNAs from closely related species are available.
Application to identify an extended catalog of canine lncRNAs
Within the framework of the LUPA consortium (29,57), we performed 20 whole transcriptome sequencing experiments of 16 canine tissues (Supplementary Table S1). After QC, ∼1, 3 billions reads were mapped onto the CanFam3 genome assembly using the STAR mapper (50). Cufflinks (11) was further used to reconstruct the transcript models in each tissue separately guided by a consensus reference annotation given by the Broad (49) and EnsEMBL v83 (22) annotations (called CanFam3.1). Then, the cuffmerge tool (Cufflinks package) merged the tissue samples files into a single GTF file containing 211 794 transcript models of more than 50 000 gene models. In order to annotate new transcribed loci, we used the FEELncfilter module to flag all transcripts which overlap exons (in sense) from the reference annotation, single exonic intergenic transcripts and transcripts with a size below 200 nt. A total of 5523 remaining candidate transcripts were then analyzed by the FEELnccodpot module. The ‘two cut-offs’ option was run with a minimal specificity threshold fixed at 0.93 in both biotypes. These cut-offs allow leveraging the number of ambiguous transcripts (TUCps) while optimizing classification specificity (see Materials and Methods). This analysis identified 3822 novel lncRNA transcripts, 477 new mRNA transcripts and 884 TUCps.
Because we also aimed at developing a comprehensive annotation of novel transcript isoforms, we used FEELnccodpot, with the exact same parameters as before, on the subset of assembled models overlapping (but not included in) the current CanFam3.1 annotation (n = 69 602) (See Supplementary Methods for details). In addition, since a novel transcript isoform could merge two or more genes from the reference annotation, we defined rules to avoid the merging of transcripts having different biotypes by removing these incompatible transcripts (see Supplementary Methods for details). This resulted in the annotation of 67 312 transcripts with more mRNA isoforms (58 163) compared to lncRNAs (6552) and TUCps (2597).
Combining the results of these analyses with the reference annotation, the new canine annotation that we called CanFam3.1-plus, includes a total of 36 237 loci (with 3145 new loci) and 189 114 transcripts (with 10 374 lncRNA and 58 640 mRNA newly annotated transcripts) (Supplementary Table S5).
This study improves the dog genome annotation in several aspects. First, we extended the number of mRNA transcripts by 50% and doubled the number of annotated lncRNA transcripts. Second, we found that novel lncRNA and mRNA transcripts were longer in terms of number of exons, CDS/UTRs and RNA sizes compared to CanFam3.1 transcripts suggesting a more complete reconstruction of their gene structures (Supplementary Table S6). Also, the number of isoforms per gene locus has been expanded to ∼2.2 and ∼7.2 for lncRNAs and mRNAs, respectively. Third, using STAR and RSEM (58) programs to quantify transcripts expression levels, we found that 86% of novel lncRNAs have a TPM (transcript per million) value higher than 0.5 in at least one of the 20 tissues highlighting a robust set of new lncRNAs. Finally, among the novel canine lncRNA genes, ∼15% are also found as non protein-coding genes in the human GENCODE annotation by using the EnsEMBL compara EPO alignments (59). The CanFam3.1-plus now annotates, for instance, three CAncer Susceptibility Candidates lincRNAs (CASC lincRNAs) such as the CASC9 lincRNA located on canine chr29:23,554,585-23,605,371 and involved in esophageal squamous cell carcinoma (60). Other examples include the well-described MALAT1 cancer-associated lincRNA (61) which was considered as an unclassified non-coding transcript in CanFam3.1 and the IFNG-AS antisense lncRNA involved in T-cell differentiation (62).
Finally, by employing the FEELncclassifier on all CanFam3.1-plus lncRNA transcripts (see Supplementary Table S4), we annotated 8209 lincRNAs. Among them, 1279 are located at a distance smaller than 5 kb and transcribed in a divergent orientation from their neighbor mRNA which could suggest potential canine bi-directional promoters (54). For genic lncRNAs, FEELncclassifier identified 5085 antisense exonic lncRNAs as possible candidates for sense-antisense regulation by sequence complementarity (55). The CanFam3.1-plus annotation constitutes a new resource that will help identifying lncRNA candidates for understanding genotype to phenotype relationships.
DISCUSSION
In this study, we designed a new program to identify and annotate lncRNAs called FEELnc for FlExible Extraction of Long non-coding RNAs. Using the gold-standard GENCODE annotation in human and mouse (27), we showed that FEELnc performs well to discriminate long non-coding versus protein-coding RNAs. Most probably, FEELnc includes predictors (multi k-mer frequencies and ORF coverage) that are general enough to capture all lncRNAs classes whereas alignment-based methods will be biased toward misclassifying species-specific transcripts or coding transcripts that are not referenced in peptide databases. The integration of a Random Forest-based model in FEELnc contributes to the good achievements of the tool because of the intrinsic properties of randomly sub-sampled features thereby encompassing diverse lncRNA characteristics. In recent years, major advances have been made in the machine learning field (63) allowing the programs to cope with thousands of parameters simultaneously. Thus, it would thus be of great value to investigate deep learning approaches to coding and non-coding RNA annotations.
The contribution of FEELnc is not only limited to provide high classification performance metrics on models organisms since the tool is also accompanied by several modules and options that enable fine-tuning and precisely adjusting lncRNA annotations for any species of interest. To our knowledge, it is the first tool that allows users to annotate conservative sets of lncRNAs and mRNAs by automatically fixing their own specificity thresholds (40). Second, FEELnc can be used for any given species even in the absence of a lncRNA training set due to the possibility to model species-specific lncRNAs. For instance, the shuffle strategy only requires species-specific protein-coding sequences and it is thus suitable for analyses without a reference genome assembly, which is still the case for many non-model organisms. Third, we expect the FEELnc classifier module to be of great interest to researchers in order to automatically annotate novel lncRNAs thus directly providing candidate pairs of lncRNA and mRNA partners to be investigated for experimental validations.
For non-model organisms, we have shown that FEELnc performs similarly to CNCI although the benchmark was done on NONCODE lncRNAs that were a priori filtered by the CNCI tool. As for human and mouse where the manually curated GENCODE annotation is considered as a standard, this stressed the importance of also defining gold-standard sets of lncRNAs/mRNAs to correctly evaluate programs for ‘non-model’ organisms. This is envisaged within the framework of collaborative projects such as FAANG, the Functional Annotation of Animal Genomes project (30).
Although the purpose of FEELnc is not to assemble transcripts from RNA-seq data, the program relies on the correct modeling of transcripts. For weakly expressed mRNAs, the corresponding reconstructed models might not be full-length and thus may also introduce a bias for the FEELnc sequence length predictors. However, the benchmarking on shortened sequences (Supplementary Figure S5) showed that FEELnc performs well even if 25% of either transcript-end sequences are removed. With the availability of very long reads from third generation sequencing technologies (64), the issue will not so much consist in annotating full-length transcripts but rather in taking into account the high error rate inherent to these technologies, which could lead to the misannotation of the correct ORF. Using a specific FEELnc option, preliminary tests on degraded transcript sequences showed that it raised good evaluation metrics (Sn = 0.87, Prec = 0.78) when the computation of multi k-mer frequencies is done on the entire transcript sequence (i.e independently of the ORF annotation) and the ORF coverage is removed from the predictors (see Supplementary Table S7).
Finally, we illustrated the usefulness of FEELnc on the dog transcriptome for which 20 RNA-seq samples were sequenced in the frame of the LUPA consortium. The biological relevance of this expanded canine resource can be illustrated by the increased transcript and CDS sizes as well as higher exon numbers. Although this improved annotation will facilitate the identification of genotype to phenotype associations, it will still need further investigations given the plethora of expressed biotypes beside lncRNAs and mRNAs. For instance, one could consider annotating transcribed (processed or unprocessed) pseudogenes or enhancer RNAs by using FEELnc as a multiclass classifier instead of a binary classifier (e.g. coding versus non-coding). Indeed, pseudogenes that recently derived from a protein-coding gene should harbor multi k-mer frequencies similar to their parent mRNAs but without long ORFs. Their CPS would then be gathered in an intermediate class between lncRNAs and mRNAs. For instance, FEELnc identifies that the FIRRE lncRNA has an intermediary CPS score of 0.58 that is supported by a relatively long ORF of 152 AA and significant percentage of sequence similarity with the FAM195 protein-coding genes. Interestingly, it also appeared that the fourth exon of FIRRE is almost completely embedded in the MCRIP2P1 pseudogene, the latter deriving from FAM195 mRNA. This highlights the potential use of FEELnc to annotate pseudogene-derived lncRNAs (65,66) for which the CPS score should be in between lncRNAs and mRNAs. Similarly, enhancer RNAs defined as non-coding transcripts derived from enhancer elements (67,68) should harbor small ORFs and specific patterns of k-mers corresponding to their transcription factor binding sites, which would then be caught by FEELnc predictors.
Altogether, FEELnc provides a standardized and exhaustive protocol to identify and annotate lncRNAs.
AVAILABILITY
FEELnc is implemented in Perl and R (and KmerInShort in C++) and is available throw a github: https://github.com/tderrien/FEELnc. A UCSC track hub for the extended canine annotation CanFam3.1-plus is available using the following URL:
http://tools.genouest.org/data/tderrien/canFam3.1p/annotation/trackhub/canfam3.1p_trackhub/hub.txt with transcripts from the original CanFam3.1 annotation in blue, new isoforms from CanFam3.1 genes in green and transcripts from CanFam3.1-plus genes in red. A guideline is available in the Supplementary Data for running FEELnc analyses with or without a reference genome.
ACCESSION NUMBERS
The RNA-seq data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under NCBI bioproject PRJNA327075 and SRA accession SRP077559.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the Cani-DNA CRB for providing RNA samples for the project (http://dog-genetics.genouest.org), and the biobank at University of Copenhagen. They are grateful to referring veterinarians, all dog owners and breeders for sample collection and also colleagues from the European LUPA consortium (www.eurolupa.org), especially Anne-Sophie Lequarré and Marilou Ramos for coordinating the program. We also thank FEELnc users and more particularly Kevin Muret, Sandrine Lagarrigue, Mark Cock and Sarah Djebali. Sequencing was performed at the Broad Institute's Genomics Platform. Finally, we are grateful to Rory Johnson and Jacques Nicolas for useful discussions and the GenOuest Bioinformatics core facility (www.genouest.org) for storing sequencing data, hosting the Cani-DNA website and for the use of the bioinformatic cluster to analyze the data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The 7th PCRD ‘Health programs’ LUPA consortium [FP7, GA: 201370]; French National Research Agency in the frame of the ‘Investing for the Future’ program [ANR-11-INBS-0003 to The ‘Cani-DNA’ biobank, which is part of the CRB-Anim infrastructure] (https://www.crb-anim.fr); European Young Investigator Award from European Science Foundation as well as a Consolidator Award from the European Research Council [to K.L.T.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. et al. Landscape of transcription in human cells. Nature. 2012; 489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pervouchine D.D., Djebali S., Breschi A., Davis C.A., Barja P.P., Dobin A., Tanzer A., Lagarde J., Zaleski C., See L.-H. et al. Enhanced transcriptome maps from multiple mouse tissues reveal evolutionary constraint in gene expression. Nat. Commun. 2015; 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carninci P., Kasukawa T., Katayama S., Gough J., Frith M.C., Maeda N., Oyama R., Ravasi T., Lenhard B., Wells C. et al. The transcriptional landscape of the mammalian genome. Science. 2005; 309:1559–1563. [DOI] [PubMed] [Google Scholar]
- 4. Brown J.B., Boley N., Eisman R., May G.E., Stoiber M.H., Duff M.O., Booth B.W., Wen J., Park S., Suzuki A.M. et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014; 512:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Legeai F., Derrien T.. Identification of long non-coding RNAs in insects genomes. Curr. Opin. Insect Sci. 2015; 7:37–44. [DOI] [PubMed] [Google Scholar]
- 6. Paytuví Gallart A., Hermoso Pulido A., Anzar Martínez de Lagrán I., Sanseverino W., Aiese Cigliano R.. GREENC: a Wiki-based database of plant lncRNAs. Nucleic Acids Res. 2016; 44:D1161–D1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li L., Eichten S.R., Shimizu R., Petsch K., Yeh C.-T., Wu W., Chettoor A.M., Givan S.A., Cole R.A., Fowler J.E. et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014; 15:R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brockdorff N., Ashworth A., Kay G.F., McCabe V.M., Norris D.P., Cooper P.J., Swift S., Rastan S.. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 1992; 71:515–526. [DOI] [PubMed] [Google Scholar]
- 9. Prensner J.R., Chinnaiyan A.M.. The Emergence of lncRNAs in Cancer Biology. Cancer Discov. 2011; 1:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leucci E., Vendramin R., Spinazzi M., Laurette P., Fiers M., Wouters J., Radaelli E., Eyckerman S., Leonelli C., Vanderheyden K. et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016; 531:518–522. [DOI] [PubMed] [Google Scholar]
- 11. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., Couger M.B., Eccles D., Li B., Lieber M. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013; 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin M.F., Jungreis I., Kellis M.. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011; 27:i275–i282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kong L., Zhang Y., Ye Z.Q., Liu X.Q., Zhao S.Q., Wei L., Gao G.. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007; 35:W345–W349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L., Park H.J., Dasari S., Wang S., Kocher J.-P., Li W.. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013; 41:e74–e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li A., Zhang J., Zhou Z.. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014; 15:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., Liu Y., Chen R., Zhao Y.. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013; 41:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dewey C.N., Pachter L.. Evolution at the nucleotide level: the problem of multiple whole-genome alignment. Hum. Mol. Genet. 2006; 15:R51–R56. [DOI] [PubMed] [Google Scholar]
- 20. Brent M.R. How does eukaryotic gene prediction work. Nat. Biotechnol. 2007; 25:883–885. [DOI] [PubMed] [Google Scholar]
- 21. Blanco E., Parra G., Guigó R.. Using geneid to identify genes. Curr. Protoc. Bioinformatics. 2007; 4:3. [DOI] [PubMed] [Google Scholar]
- 22. Yates A., Akanni W., Amode M.R., Barrell D., Billis K., Carvalho-Silva D., Cummins C., Clapham P., Fitzgerald S., Gil L. et al. Ensembl 2016. Nucleic Acids Res. 2016; 44:D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johnson R., Guigo R.. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA. 2014; 20:959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zucchelli S., Fasolo F., Russo R., Cimatti L., Patrucco L., Takahashi H., Jones M.H., Santoro C., Sblattero D., Cotella D. et al. SINEUPs are modular antisense long non-coding RNAs that increase synthesis of target proteins in cells. Front. Cell. Neurosci. 2015; 9:1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tagu D., Colbourne J.K., Nègre N.N.. Genomic data integration for ecological and evolutionary traits in non-model organisms. BMC Genomics. 2014; 15:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Breiman L. Random Forests. Mach. Learn. 2001; 45:5–32. [Google Scholar]
- 27. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y., Li H., Fang S., Kang Y., Wu W., Hao Y., Li Z., Bu D., Sun N., Zhang M.Q. et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016; 44:D203–D208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lequarré A.-S., Andersson L., André C., Fredholm M., Hitte C., Leeb T., Lohi H., Lindblad-Toh K., Georges M.. LUPA: a European initiative taking advantage of the canine genome architecture for unravelling complex disorders in both human and dogs. Vet. J. 2011; 189:155–159. [DOI] [PubMed] [Google Scholar]
- 30. Andersson L., Archibald A.L., Bottema C.D., Brauning R., Burgess S.C., Burt D.W., Casas E., Cheng H.H., Clarke L., Couldrey C. et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015; 16:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bussotti G., Raineri E., Erb I., Zytnicki M., Wilm A., Beaudoing E., Bucher P., Notredame C.. BlastR–fast and accurate database searches for non-coding RNAs. Nucleic Acids Res. 2011; 39:6886–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Drezen E., Rizk G., Chikhi R., Deltel C., Lemaitre C., Peterlongo P., Lavenier D.. GATB: Genome Assembly & Analysis Tool Box. Bioinformatics. 2014; 30:2959–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Claverie J.M., Sauvaget I., Bougueleret L.. K-tuple frequency analysis: from intron/exon discrimination to T-cell epitope mapping. Methods Enzymol. 1990; 183:237–252. [DOI] [PubMed] [Google Scholar]
- 34. Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L.. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011; 25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Achawanantakun R., Chen J., Sun Y., Zhang Y.. LncRNA-ID: Long non-coding RNA IDentification using balanced random forests. Bioinformatics. 2015; 31:3897–3905. [DOI] [PubMed] [Google Scholar]
- 37. Lertampaiporn S., Thammarongtham C., Nukoolkit C., Kaewkamnerdpong B., Ruengjitchatchawalya M.. Identification of non-coding RNAs with a new composite feature in the hybrid random forest ensemble algorithm. Nucleic Acids Res. 2014; 42:e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liaw A., Wiener M.. Classification and regression by randomForest. R. News. 2002; 2:18–22. [Google Scholar]
- 39. Sing T., Sander O., Beerenwinkel N., Lengauer T.. ROCR: visualizing classifier performance in R. Bioinformatics. 2005; 21:3940–3941. [DOI] [PubMed] [Google Scholar]
- 40. Mattick J.S., Rinn J.L.. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015; 22:5–7. [DOI] [PubMed] [Google Scholar]
- 41. Ponting C.P., Oliver P.L., Reik W.. Evolution and functions of long noncoding RNAs. Cell. 2009; 136:629–641. [DOI] [PubMed] [Google Scholar]
- 42. Ulitsky I. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat. Publishing Group. 2016; 17:601–614. [DOI] [PubMed] [Google Scholar]
- 43. Duret L., Chureau C., Samain S., Weissenbach J., Avner P.. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science. 2006; 312:1653–1655. [DOI] [PubMed] [Google Scholar]
- 44. Jiang M., Anderson J., Gillespie J., Mayne M.. uShuffle: a useful tool for shuffling biological sequences while preserving the k-let counts. BMC Bioinformatics. 2008; 9:192–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baldi P., Brunak S., Chauvin Y., Andersen C.A., Nielsen H.. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000; 16:412–424. [DOI] [PubMed] [Google Scholar]
- 46. Powers D.M.W. Evaluation: from precision, recall and F-measure to ROC, informedness, markedness and correlation. J. Mach. Learn. Technol. 2011; 2:37–63. [Google Scholar]
- 47. Askarian-Amiri M.E., Crawford J., French J.D., Smart C.E., Smith M.A., Clark M.B., Ru K., Mercer T.R., Thompson E.R., Lakhani S.R. et al. SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA. 2011; 17:878–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Djebali S., Wucher V., Foissac S., Hitte C., Corre E., Derrien T.. Bioinformatics pipeline for transcriptome sequencing analysis. Methods Mol. Biol. 2017; 1468:201–219. [DOI] [PubMed] [Google Scholar]
- 49. Hoeppner M.P., Lundquist A., Pirun M., Meadows J.R.S., Zamani N., Johnson J., Sundström G., Cook A., FitzGerald M.G., Swofford R. et al. An Improved Canine Genome and a Comprehensive Catalogue of Coding Genes and Non-Coding Transcripts. PLoS One. 2014; 9:e91172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lykke-Andersen S., Chen Y., Ardal B.R., Lilje B., Waage J., Sandelin A., Jensen T.H.. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev. 2014; 28:2498–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen J., Shishkin A.A., Zhu X., Kadri S., Maza I., Guttman M., Hanna J.H., Regev A., Garber M.. Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol. 2016; 17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sigova A.A., Mullen A.C., Molinie B., Gupta S., Orlando D.A., Guenther M.G., Almada A.E., Lin C., Sharp P.A., Giallourakis C.C. et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carrieri C., Cimatti L., Biagioli M., Beugnet A., Zucchelli S., Fedele S., Pesce E., Ferrer I., Collavin L., Santoro C. et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. 2012; 491:454–457. [DOI] [PubMed] [Google Scholar]
- 56. He Y., Vogelstein B., Velculescu V.E., Papadopoulos N., Kinzler K.W.. The antisense transcriptomes of human cells. Science. 2008; 322:1855–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vaysse A., Ratnakumar A., Derrien T., Axelsson E., Rosengren Pielberg G., Sigurdsson S., Fall T., Seppala E.H., Hansen M.S.T., Lawley C.T. et al. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 2011; 7:e1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Herrero J., Muffato M., Beal K., Fitzgerald S., Gordon L., Pignatelli M., Vilella A.J., Searle S.M.J., Amode R., Brent S. et al. Ensembl comparative genomics resources. Database (Oxford). 2016; 2016:bav096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hao Y., Wu W., Shi F., Dalmolin R.J., Yan M., Tian F., Chen X., Chen G., Cao W.. Prediction of long noncoding RNA functions with co-expression network in esophageal squamous cell carcinoma. BMC Cancer. 2015; 15:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gutschner T., Hämmerle M., Eissmann M., Hsu J., Kim Y., Hung G., Revenko A., Arun G., Stentrup M., Gross M. et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013; 73:1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hu G., Tang Q., Sharma S., Yu F., Escobar T.M., Muljo S.A., Zhu J., Zhao K.. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat. Immunol. 2013; 14:1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. LeCun Y., Bengio Y., Hinton G.. Deep learning. 2015; 521:436–444. [DOI] [PubMed] [Google Scholar]
- 64. Sharon D., Tilgner H., Grubert F., Snyder M.. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013; 31:1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Milligan M.J., Lipovich L.. Pseudogene-derived lncRNAs: emerging regulators of gene expression. Front Genet. 2014; 5:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Johnsson P., Ackley A., Vidarsdottir L., Lui W.-O., Corcoran M., Grandér D., Morris K.V.. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 2013; 20:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ørom U.A., Shiekhattar R.. Long noncoding RNAs usher in a new era in the biology of enhancers. 2013; 154:1190–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ren B. Transcription: enhancers make non-coding RNA. 2010; 465:173–174. [DOI] [PubMed] [Google Scholar]
- 69. Hedges S.B., Marin J., Suleski M., Paymer M., Kumar S.. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 2015; 32:835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.