

 A hexathymidine-DNA “hexT” tolerates acid and coinage metal catalysts, and can be ligated to coding DNA sequences.
A hexathymidine-DNA “hexT” tolerates acid and coinage metal catalysts, and can be ligated to coding DNA sequences.
Abstract
Libraries of DNA-tagged compounds are a validated screening technology for drug discovery. They are synthesized through combinatorial iterations of alternated coding and preparative synthesis steps. Thus, large chemical space can be accessed for target-based screening. However, the need to preserve the functionality of the DNA tag severely restricts the choice of chemical methods for library synthesis. Acidic organocatalysts, transition metals, and oxidants furnish diverse drug-like structures from simple starting materials, but cause loss of genetic information by depurination. A hexathymidine oligonucleotide, called “hexT” allows the chemist utilizing these classes of catalysts to access a potentially broad variety of structures in the initial step of library synthesis. We exploited its catalyst tolerance to efficiently synthesize diverse substituted β-carbolines, pyrazolines, and pyrazoles from readily available starting materials as hexT conjugates by acid- and Au(i)-catalysis, respectively. The hexT conjugates were ligated to coding DNA sequences yielding encoded screening libraries inspired by drug structures.
Introduction
DNA-encoded libraries (DELs) are mixtures of drug-like structures connected to individual DNA tags (1, Fig. 1A).1 DNA tagging allows for sampling of large compound collections, and identification of bioactive compounds from these collections by selection, followed by amplification and sequencing of the genetic information. Screening of DELs against numerous target proteins from different protein families has provided several bioactive compounds, among them valuable tool compounds for chemical biology, and even clinical candidates.1 Several DEL formats are actively used in drug discovery, namely DNA-recorded, DNA-templated, and DNA-routed libraries, DNA-encoded beads, and DNA-assembled fragment collections.2 DELs are synthesized by combinatorial strategies through alternated coding and synthesis steps.1,2 Crucially, the latter must not compromise the integrity of the DNA tag.3 Thus, only a tiny fraction of the rich inventory of organic chemistry was heavily utilized for DELs: carbonyl chemistry, C–C cross-coupling reactions, substitution of halides, and “click” reactions.4 These are appendage reactions that couple building blocks to appropriately functionalized structures. Due to the paucity in applicable synthesis methods DNA-encoded libraries show low variation in the connectivity of building blocks, they are heavily biased towards appendage diversity,5 and they require functionalized scaffolds to introduce topological diversity. Yet, the chemical space of bioactive compounds, synthetic compounds and natural products alike, contains a large variety of diversely substituted (hetero)cyclic structures.6 It has been argued that this structural wealth should be mirrored by screening libraries.7,8 An important access to structural diversity in compound libraries is the application of diverse synthesis methodology to build up molecules from simple starting materials. Currently, avenues to substituted (hetero)cyclic structures from simple starting materials are scarcely available for DEL synthesis: the Diels–Alder cycloaddition,9 condensation reactions,4c and a class of spirocycles were reported.10 These reports show a lively interest in expanding the chemistry toolbox for DELs.1,4 Prolific sources of drug-like structures are Brønsted acid-, and coinage metal-catalyzed reactions.11,12 Yet, they cause depurination,13,14 and thus require the chemically much more stable PNA for encoding.1c,15
Fig. 1. Outline of the oligothymidine-initiated DNA-encoded chemistry (TiDEC) strategy. (A) Schematic presentation of a DNA-tagged compound, the coloured shapes represent partial structures of the compound; (B) the TiDEC concept to DNA-encoded libraries: (a) heterocyclic chemistry; (b) isolation of hexT conjugates; (c) DNA ligation; (C) exemplary drugs with TiDEC-accessible scaffolds. Filled black circle denotes controlled pore glass (CPG) solid support; bold bond denotes connection of the hexT-DNA to the CPG solid support; wavy bond denotes 5′-amino-PEG(4)-linker; het-cycle: heterocycle.

We hypothesized that a hexathymidine DNA “hexT” should be stable enough to allow tapping into the fields of acid- and coinage metal-catalysis to convert hexT-conjugates of simple starting materials 2 into hexT-heterocycle conjugates 3. Furthermore, the short hexT oligonucleotide had to be recognized by T4 ligase to record the synthesis step with coding DNA sequences (encoded compounds 4, Fig. 1B, “TiDEC”, oligothymidine-initiated DNA-encoded chemistry).
In line with our hypothesis, we describe here the synthesis of hexT-conjugates of diverse substituted β-carbolines by the Brønsted acid-catalyzed Pictet–Spengler reaction,16 and the synthesis of hexT-conjugates of highly substituted pyrazol(in)es by a Au(i)-mediated cascade reaction17a from simple, readily available starting materials. These scaffolds can be found in numerous bioactive compounds, among them natural products and drugs (Fig. 1C). The hexT-heterocycle conjugates could subsequently be ligated to coding DNA sequences by T4 DNA ligation, and expanded to combinatorial libraries.
Results and discussion
Ligation of the hexT oligonucleotide to coding DNA sequences
For DNA tagging of the heterocycles we chose T4 ligation of 5′-phosphorylated duplex DNA sequences with tetramer overhangs. T4 ligation is a validated, frequently used method to encode large compound libraries.2b,c The DNA sequences were 5′-phosphorylated with polynucleotide kinase (PNK). Both Pictet–Spengler and the Au(i)-mediated cascade reaction efficiently provide heterocycle formation and introduction of a substituent in one step. Thus, the hexT conjugates I were to be ligated to two enzymatically 5′-phosphorylated duplex DNAs II and III in one step, too (Fig. 1B and 2A-b); see Fig. S1 in the ESI† for a more detailed description of the encoding strategy, all sequences are given in Tables S1 and S2†: sequence II contained a hexaadenine overhang, forward primer, and heterocycle code; sequence III contained the substituent code (Fig. 2A-b). The ligation products were then in turn ligated to sequences IV coding for a set of building blocks that concluded library synthesis (Fig. 2A-c). We established the encoding strategy with the fluorescein-labeled hexT-fluo that facilitated detection of the ligation products (Fig. 2B, S3–S6†). Ligation of the very short hexamer oligonucleotide hexT I to a much longer duplex DNA II (30/40 mer) expectedly results in only a small shift of the migration of the ligation product compared to the non-ligated duplex, when detected by DNA staining (Fig. 2B, left hand trace (a)). A fluorescence scan unambiguously confirmed successful ligation of the hexT oligonucleotide I to a duplex DNA II (Fig. 2B, right hand trace (a)), and, as we required for our library synthesis design (Fig. 1B), to two double stranded DNA sequences II and III in one step (Fig. 2B-b). The product of the latter ligation experiment was precipitated, and ligated to a third duplex DNA IV (Fig. 2B-c). Besides the expected product which was formed in high yields we observed minor formation of longer ligation products. The ligation product at 100 bp might be explained by ligation of two fluorescein-labeled duplex-DNAs. T4 ligase is known for tolerance of mismatches in overhangs.18 The encoding strategy was confirmed by several ligation experiments with hexT-heterocycle conjugates that demonstrated the viability of TiDEC for the synthesis of accordingly called tiDELs, oligothymidine-initiated DNA-encoded libraries (Fig. 2, S3–S11†).
Fig. 2. Ligation of DNA sequences to hexT-fluo. (A) Ligation schemes; grey segments denote primer regions; black, blue and light red segments denote coding sequences; red star: fluorescein; PNK: polynucleotide kinase; (B) detection of ligation products by DNA stain (left hand trace (a)) and fluorescence scan (right hand trace (a) and (b and c)).

Synthesis of hexT-β-carboline conjugates by acid-catalyzed Pictet–Spengler reaction
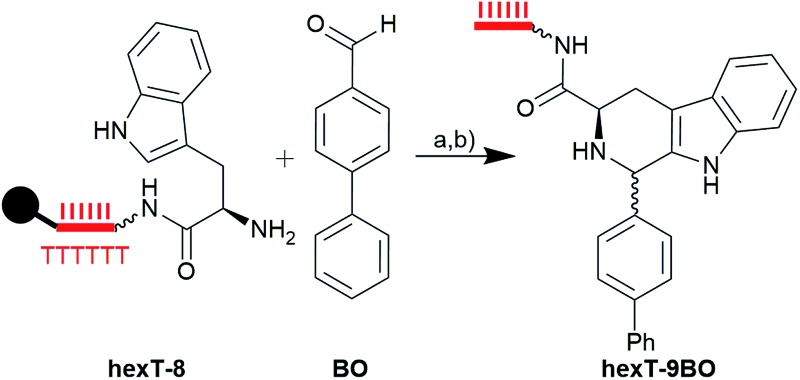
An essential structure in drug research is the β-carboline, constituting the core scaffold of the pharmacologically active natural product ajmalicine (5, Fig. 1C), the approved drug tadalafil (6, Fig. 1C), and drug candidates such as AZD9496 and the antimalarial NITD609. It can be accessed from tryptamines and aldehydes through the Brønsted acid catalyzed Pictet–Spengler reaction.16 We synthesized the controlled pore glass (CPG)-bound tryptophane conjugate hexT-8 (Fig. S12 and S13†), and tested several conditions (Table 1, Fig. S14†) to react hexT-8 with a 1000-fold excess of p-biphenylcarboxaldehyde BO (Table S3†) to the target β-carboline hexT-9BO.
Table 1. Exploration of reaction conditions for the synthesis of the hexT-β-carboline hexT-9BO; (a) see table; a (b) aq. NH3/aq. MeNH2. Wavy bond to hexT denotes 5′-amino-PEG(4)-linker, bold bond denotes connection to CPG solid support, filled circle denotes solid support.
| ||||
| Entry | Catalyst | Solvent | Time [h] | Conversion b [%] |
| 1 | TFA (1–3%) | CH2Cl2 | 18 | 100 |
| 2 | TFA (10%) | CH2Cl2 | 4 | 100 |
| 3 | TFA (10%) | CH2Cl2 | 18 | — c |
| 4 | TCA (1%) | CH2Cl2 | 18 | 100 |
| 5 | (PhO)2(HO)PO (1%) | CH2Cl2 | 18 | 80 |
| 6 | HCOOH (50%) | CH2Cl2 | 4 | 100 |
| 7 | BCl3 (1 N) | CH2Cl2 | 18 | — c |
| 8 | BF3 (1 N) | CH2Cl2 | 18 | — c |
| 9 | TFA (1%) | THF | 18 | 0 |
| 10 | TFA (1%) | Dioxane | 18 | 0 |
| 11 | TFA (1%) | DMF | 18 | 60 |
| 12 | TFA (1%) | DMF d | 18 | 0 |
| 13 | TFA (1%) | DMSO d | 18 | 0 |
| 14 | TFA (1%) | Toluene | 18 | 100 |
| 15 | TFA (1%) | C2H4Cl2 | 18 | 100 |
| 16 | TFA (1%) | MeCN | 18 | 100 |
a20 nmol hexT-8, 20 μmol BO, room temperature, dry solvent.
bEstimated by HPLC analysis of the crude product.
cDegradation of the DNA.
dSolvent contained trace amounts of water.
It was readily obtained with 1% of trifluoroacetic acid in dry non-polar solvents CH2Cl2, toluene, and C2H4Cl2, and in the polar, aprotic solvents DMF and acetonitrile overnight (Table 1, entries 1, 11, 14–16). The β-carboline was also formed with higher concentrations of TFA in CH2Cl2, (up to 3% overnight, or 10% for four hours), 1% of trichloroacetic acid in CH2Cl2, 1% of diphenyl phosphate in CH2Cl2, and 50% of formic acid in CH2Cl2. The product of the Pictet–Spengler reaction was confirmed by comparison with the reference ref-hexT-9BO (Fig. S15†). Further investigation of the reagent scope revealed that the Lewis acids BCl3 and BF3 in CH2Cl2, and overnight treatment with 10% of TFA in CH2Cl2 degraded the oligonucleotide.
In the library synthesis, we used 2% of TFA in CH2Cl2 which gave quantitative conversion of the tryptophane to the corresponding β-carboline with a 1000-fold excess of the carbonyl building blocks (aliquots of all reactions were analyzed by mass spectrometry). The Pictet–Spengler reaction revealed a broad scope with aliphatic, olefinic, heterocyclic, diverse aromatic aldehydes, and isatin furnishing 80 diversely substituted β-carbolines hexT-9A-9CB. The β-carboline displays a secondary amine, which lends itself to library expansion. However, in our hands this amine resisted amide bond formation efforts.4a Fortunately, the introduction of a Boc-protected 2-aminoethyl group at the secondary amine of the β-carboline by reductive amination was facile on solid support (Fig. S14†).4c All amine-substituted conjugates hexT-10A-10CB were cleaved from the solid support, and analyzed by analytical HPLC and MALDI-MS to ascertain their purity and identity. Indeed, 65 of the conjugates were obtained with a purity of >90%. The remainder were HPLC-purified. Inspection of the 15 products that had to be purified revealed that in all cases incomplete introduction of the aminoethyl group by reductive amination furnished a mixture of the β-carbolines hexT-9 and the C2-linker-substituted compounds hexT-10 (see Table S3†, and exemplary MALDI-MS spectra in the ESI†). The lipophilic Boc-group facilitated purification of the target compounds hexT-10 by reverse phase HPLC. Finally, the Boc-group of each an aliquot of every compound hexT-10 was removed with 10% of TFA in CH2Cl2 for six hours to provide 80 β-carbolines hexT-11A-11CB for further library synthesis (Fig. S14, Table S3†). These were again analyzed by MALDI-MS, which confirmed successful removal of the protective group, and stability of the hexT oligonucleotide under these reaction conditions.
Synthesis of hexT-pyrazol(in)e conjugates by a Au(i)-mediated cascade reaction
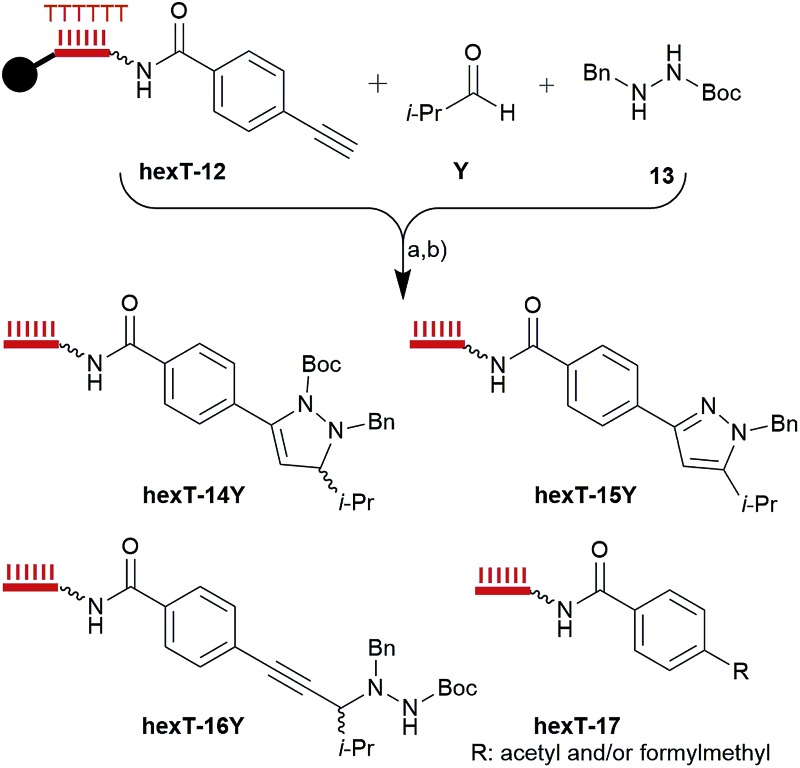
Many bioactive compounds, among them the former drug rimonabant (7, Fig. 1C), and the approved celecoxib represent highly substituted pyrazoles. A Au(i)-catalyzed cascade reaction provides access to the highly substituted pyrazolines from readily available alkynes, and azomethine imines that are formed in situ from aldehydes, and hydrazides (Table 2, Fig. S16†).17a Pyrazolines can in turn be oxidized to pyrazoles under mild conditions. The CPG-bound alkyne hexT-12, isobutyric aldehyde Y and hydrazide 13 served to investigate the accessibility of the hexT-pyrazoline conjugates hexT-14Y by Au(i) catalysis. Systematic exploration of reaction parameters yielded four distinct, isolatable products in different ratios (Table 2, see Table S4 in the ESI† for an expanded version of Table 2, and Fig. S17–S22†): the pyrazoline 14Y, the pyrazole 15Y, the propargylamine 16Y, and the hydration product 17.
Table 2. Exploration of reaction conditions for the synthesis of the hexT-pyrazol(in)es hexT-14Y and -15Y. (a) See table; a (b) aq. NH3/aq. MeNH2. Wavy bond to hexT denotes 5′-C6-amino-linker; bold bond denotes connection to CPG solid support; filled black circle denotes CPG solid support.
| |||||
| Entry | Conditions | Product ratio
b
[%] |
|||
| 14Y | 15Y | 16Y | 17 | ||
| 1 c | A, THF, 50 °C | — | — | — | — |
| 2 d | A, THF, 50 °C | 15 | — | 55 | — |
| 3 | A, THF, 50 °C | 50 | — | 25 | 10 |
| 4 | A, MeCN, 50 °C | 75 | — | 25 | — |
| 5 | A, C2H4Cl2, 50 °C | 40 | — | 10 | 50 |
| 6 | B, AcOH, 50 °C | 10 | 55 | 10 | — |
| 7 | A, MeCN, 25 °C | — | — | — | — |
| 8 | A, MeCN, 60 °C | 70 | 20 | — | — |
| 9 | B, AcOH, 60 °C | <5 | 90 | — | — |
| 10 e | B, C2H4Cl2, 50 °C | 85 | — | — | — |
| 11 e | C, C2H4Cl2, 50 °C | 85 | — | — | — |
| 12 | AgOTf, toluene, 50 °C | 40 | — | 60 | — |
a30 nmol hexT-12, 30 μmol Y, 30 μmol 13, 7.5 μmol A ([tris(2,4-di-tert-butylphenyl)phosphite]gold chloride)/AgSbF6, B (PPh3AuCl)/AgOTf, or C (chloro[1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]-gold(i))/AgOTf, 18 h.
bHPLC analysis of the crude, missing percentage to 100%: hexT-12.
c0.15 μmol A/AgSbF6.
d1.5 μmol A/AgSbF6.
e7.5 μmol AgOTf.
Performing the reaction with a 250 fold excess of the Au(i)catalyst A 17b versus hexT-12 at 50 °C yielded a 2 : 1 : 0.4 mixture of pyrazoline 14Y, propargylamine 15AJ, and the hydration product 17 (Table 2, entry 3). Reducing the excess of the catalyst reduced the yield of 14Y (entries 1 and 2),17a and lowering the temperature to 25 °C returned only starting material (entry 7). Changing the solvent had a profound effect on the product ratio: MeCN yielded a 3 : 1 mixture of 14Y/16Y (entry 4); 1,2-dichloroethane yielded a substantial amount of the hydration product 17 (entry 5); and running the reaction in glacial acetic acid with catalyst B we observed formation of pyrazole 15Y through ring closure, oxidation and Boc-group removal (entries 6 and 9). Finally, the triphenylphosphine- and N-heterocyclic carbene-based Au(i) catalysts B and C, respectively, yielded the pyrazoline as sole detectable product (entries 10 and 11).17a Ag(i), which reportedly afforded propargyl amines by a Mannich-type reaction,17a gave in our hands the ring-closed product 14Y and the propargyl amine 16Y in a 4 : 6 ratio, likely due to the high catalyst concentration (entry 12).
Prior to investigation of the scope of the reaction we synthesized reference compounds for the pyrazoline 14Y, the pyrazole 15Y, and the propargylamine 16Y (Fig. S31†) and coupled these to the hexT by amide formation. Comparison of these authentic samples with hexT-14Y-16Y by HPLC confirmed formation of the proposed products (Fig. S23 and S24†). The pyrazole hexT-15Y was also accessed in stepwise manner via synthesis of pyrazoline hexT-14Y, acid removal of its Boc-group to hexT-18Y, and oxidation with DDQ (Fig. 3 and S25†). Thus, by control of reaction conditions the Au(i)-mediated cascade reaction provides two densely substituted core scaffolds, namely pyrazolines and pyrazoles, as hexT-conjugates from the same reactants for encoded library synthesis, thereby adding diversity to the projected screening library in a highly efficient manner.
Fig. 3. Diverse routes to hexT-pyrazole 15Y; (a) B (see Table 2), AcOH, 60 °C, overnight; (b) aq. NH3/MeNH2, room temperature, 30 min; (c) A (see Table 2), MeCN, 50 °C, overnight; (d) DDQ/CH2Cl2; (e) 10% TFA/CH2Cl2. R: p-aminocarbonylphenyl; wavy bond to hexT denotes 5′-C6-amino-linker; bold bond denotes connection to CPG solid support; filled circle denotes solid support.

Several unsymmetrically substituted hydrazides furnished the target heterocycles, besides the Boc-group also an acetyl group was tolerated (Table S5†). The benzyl-substituent could be substituted with electron-donating, and -withdrawing groups. Because these hydrazides have to be synthesized de novo, we decided to diversify the scaffold with readily available aldehydes, and introduce a position for further substitution by the amine-displaying hydrazide 19 (Fig. S26, Table S5†). The synthesis of 23 pyrazolines hexT-20 and 48 pyrazoles hexT-21 succeeded with branched aliphatic, and substituted benzaldehydes (Tables S6 and S7†), whereas linear aliphatic, and several heteroaromatic aldehydes did not give the expected product. In the first case the reaction failed likely due to a lack of reactivity of the azomethine imine intermediate, in the second case likely due to poisoning of the metal catalyst. All pyrazol(in)es were HPLC-purified to provide authentic samples for the projected tiDEL (Fig. 4).
Fig. 4. Synthesis of tiDELs based on β-carbolines, and pyrazol(in)es. Reagents and conditions: (a) 2% TFA/CH2Cl2, room temperature, overnight; (b) NaCNBH4, 40 °C, overnight; (c) aq. NH3/MeNH2, room temperature, 30 min; (d) 10% TFA/CH2Cl2, 6 h; (e) A (see Table 2), MeCN, 50 °C, overnight; (f) B (see Table 2), AcOH, 60 °C, overnight; (g) T4 ligase, room temperature, overnight; (h) EDC, HOAt, DEAE sepharose, room temperature, overnight. R: p-aminocarbonylphenyl; wavy bond to hexT denotes 5′-amino-PEG(4)-linker.

Synthesis of the oligothymidine-initiated DNA-encoded libraries
For synthesis of the oligothymidine-initiated DNA-encoded libraries (tiDELs), pmol-aliquots of the in total 151 hexT-conjugates hexT-11, -20, and -21 were ligated in parallel to heterocycle-, and aldehyde-encoding DNA sequences with excellent yields (Fig. 4, S1 and S27A†). The ligation products were analyzed, and pooled by precipitation. The DNA pellet was then dissolved, split, and ligated to sequences containing the reverse primer, and genetic information for 96 carboxylic acids. These building blocks were taken from a previously published diversity set of carboxylic acids.18 We evaluated the reactivity of all members of this set with hexT-20Y or hexT-21Y, and selected those acids that gave the amide products with a yield of >80% (Table S8†).19
All encoded library constituents were transferred to DEAE sepharose in a 96 well plate, and reacted with the validated 96 carboxylic acids in parallel overnight.20 Washing steps removed the excess of reagents, and elution of the DNA-conjugates with a high-salt buffer, followed by pooling, and desalting provided a combined tiDEL with 14.496 compounds based on three scaffolds. The library was analyzed by gel electrophoresis, and PCR-amplified at two concentrations (Fig. S27B and C†). As ca. one percent of the library members contains the streptavidin binder desthiobiotin, the tiDEL was subjected to a streptavidin pull-down experiment versus a control DNA (Fig. S28†). PCR amplification indicated that a substantial part of the tiDEL was retained on the affinity matrix whereas only traces of the control DNA were detectable, suggesting the functionality of the tiDELs.
Conclusions
DNA-encoded libraries of small molecules are a validated technology for the identification of bioactive compounds.1–4 They have recently been proposed as an open source screening tool for biologists, because the infrastructure for screening these libraries consists of little more than a magnet and a PCR thermocycler, and importantly DNA sequencing to read out the screening results is nowadays affordable.21 Yet, expanding the diversity of DNA-encoded small molecule libraries has been recognized a major challenge in the field.1–4 We propose here a strategy to DNA-encoded libraries called TiDEC (oligothymidine-initiated DNA-encoded chemistry) that allows chemists to efficiently translate their creative synthesis methodology to these libraries, and thus make it accessible to biologists. Our herein described synthesis strategy relies on a hexathymidine DNA, hexT. HexT is recognized by T4 DNA ligase to record the synthesis with coding DNA, it is affordable in bulk, and displays a broad catalyst tolerance, e.g. acid and coinage metal catalysts that are well known to furnish diverse heterocycles – but are equally well known to cause DNA degradation. At the initial stage of library synthesis, the hexT-conjugates can be characterized by mass spectrometry and HPLC, and if necessary, be purified to provide authentic samples for library synthesis. Thus, also more complex reactions, such as multicomponent reactions that may give rise to side products can be utilized for DELs. We exploited the catalyst tolerance of the hexT to synthesize three exemplary drug scaffolds from simple, readily available starting materials as encodable hexT conjugates by acid catalysis, and a recently developed Au(i)-catalyzed transformation,17a respectively. Notably, the β-carbolines hexT-11 and the pyrazolines hexT-20 display a substituent through an sp3-hybridized carbon, thus adding in stereochemical diversity, a feature that is not provided by most synthesis methods used in DEL synthesis. All hexT conjugates were characterized; if necessary, purified to ensure fidelity; recorded by DNA ligation; and diversified by amide coupling to yield a 14.496-membered tiDEL (oligothymidine-initiated DNA-encoded library) for drug screening. Our TiDEC strategy holds promise to make several catalytic strategies11,12 available for DNA-recorded libraries, and thereby expand their chemical space. It is likely compatible with other DEL formats that are successfully used in drug research, too.1,2
Acknowledgments
This work was supported by the German Federal Ministry of Education and Research (BMBF) Grant 1316053.
Footnotes
References
- (a) Brenner S., Lerner R. A. Proc. Natl. Acad. Sci. U. S. A. 1992;89:5381–5383. doi: 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kleiner R. E., Dumelin C. E., Liu D. R. Chem. Soc. Rev. 2011;40:5707–5717. doi: 10.1039/c1cs15076f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Winssinger N. CHIMIA International Journal for Chemistry. 2013;67:340–348. doi: 10.2533/chimia.2013.340. [DOI] [PubMed] [Google Scholar]; (d) Franzini R. M., Neri D., Scheuermann J. Acc. Chem. Res. 2014;47:1247–1255. doi: 10.1021/ar400284t. [DOI] [PubMed] [Google Scholar]; (e) Salamon H., Klika Škopić M., Jung K., Bugain O., Brunschweiger A. ACS Chem. Biol. 2016;11:296–307. doi: 10.1021/acschembio.5b00981. [DOI] [PubMed] [Google Scholar]; (f) Goodnow Jr R. A., Dumelin C. E., Keefe A. D. Nat. Rev. Drug Discovery. 2017;16:131–147. doi: 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- (a) Mannocci L., Zhang Y., Scheuermann J., Leimbacher M., De Bellis G., Rizzi E., Dumelin C., Melkko S., Neri D. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17670–17675. doi: 10.1073/pnas.0805130105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Clark M. A., Acharya R. A., Arico-Muendel C. C., Belyanskaya A. L., Benjamin D. R., Carlson N. R., Centrella P. A., Chiu C. H., Creaser S. P., Cuozzo J. W., Davie C. P., Ding Y., Franklin G. J., Franzen K. D., Gefter M. L., Hale S. P., Hansen N. J., Israel D. I., Jiang J., Kavarana M. J., Kelley M. S., Kollmann C. S., Li F., Lind K., Mataruse S., Medeiros P. F., Messer J. A., Myers P., O'Keefe H., Oliff M. C., Rise C. E., Satz A. L., Skinner S. R., Svendsen J. L., Tang L., van Vloten K., Wagner R. W., Yao G., Zhao B., Morgan B. A. Nat. Chem. Biol. 2009;5:647–654. doi: 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]; (c) Hansen M. H., Blakskjaer P., Petersen L. K., Hansen T. H., Højfeldt J. W., Gothelf K. V., Hansen N. J. J. Am. Chem. Soc. 2009;131:1322–1327. doi: 10.1021/ja808558a. [DOI] [PubMed] [Google Scholar]; (d) Halpin D. R., Harbury P. B. PLoS Biol. 2004;2:1015–1021. doi: 10.1371/journal.pbio.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gartner Z. J., Tse B. N., Grubina R., Doyon J. B., Snyder T. M., Liu D. R. Science. 2004;305:1601–1605. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Li Y., Zhao P., Zhang M., Zhao X., Li X. J. Am. Chem. Soc. 2013;135:17727–17730. doi: 10.1021/ja409936r. [DOI] [PubMed] [Google Scholar]; (g) Melkko S., Scheuermann J., Dumelin C. E., Neri D. Nat. Biotechnol. 2004;22:568–574. doi: 10.1038/nbt961. [DOI] [PubMed] [Google Scholar]; (h) Reddavide F. V., Lin W., Lehnert S., Zhang Y. Angew. Chem., Int. Ed. 2015;54:7924–7928. doi: 10.1002/anie.201501775. [DOI] [PubMed] [Google Scholar]; (i) Daguer J.-P., Zambaldo C., Ciobanu M., Morieux P., Barluenga S., Winssinger N. Chem. Sci. 2015;6:739–744. doi: 10.1039/c4sc01654h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) MacConnell B., McEnaney P. J., Cavett V. J., Paegel B. M. ACS Comb. Sci. 2015;17:518–534. doi: 10.1021/acscombsci.5b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone M. L., Paegel B. M. ACS Comb. Sci. 2016;18:182–187. doi: 10.1021/acscombsci.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Franzini R. M., Samain F., Abd Elrahman M., Mikutis G., Nauer A., Zimmermann M., Scheuermann J., Hall J., Neri D. Bioconjugate Chem. 2014;25:1453–1461. doi: 10.1021/bc500212n. [DOI] [PubMed] [Google Scholar]; (b) Satz A. L., Cai J., Chen Y., Goodnow R., Gruber F., Kowalczyk A., Petersen A., Naderi-Oboodi G., Orzechowski L., Strebel Q. Bioconjugate Chem. 2015;26:1623–1632. doi: 10.1021/acs.bioconjchem.5b00239. [DOI] [PubMed] [Google Scholar]; (c) Luk K. C. and Satz A. L., in Handbook for DNA-Encoded Chemistry, ed. R. A. Goodnow Jr, Wiley, New York, 2014, pp. 67–98. [Google Scholar]; (d) Franzini R. M., Randolph C. J. Med. Chem. 2016;59:6629–6644. doi: 10.1021/acs.jmedchem.5b01874. [DOI] [PubMed] [Google Scholar]
- Feher M., Schmidt J. M. J. Chem. Inf. Comput. Sci. 2003;43:218–227. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- Shelat A. A., Guy R. K. Nat. Chem. Biol. 2007;3:442–446. doi: 10.1038/nchembio0807-442. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- Wetzel S., Bon R. S., Kumar K., Waldmann H. Angew. Chem., Int. Ed. 2011;50:10800–10826. doi: 10.1002/anie.201007004. [DOI] [PubMed] [Google Scholar]
- Buller F., Mannocci L., Zhang Y., Dumelin C. E., Scheuermann J., Neri D. Bioorg. Med. Chem. Lett. 2008;18:5926–5931. doi: 10.1016/j.bmcl.2008.07.038. [DOI] [PubMed] [Google Scholar]
- Tian X., Basarab G. S., Selmi N., Kogej T., Zhang Y., Clark M., Goodnow Jr R. A. Med. Chem. Commun. 2016;7:1316–1322. [Google Scholar]
- (a) Parmar D., Sugiono E., Raja S., Rueping M. Chem. Rev. 2014;114:9047–9153. doi: 10.1021/cr5001496. [DOI] [PubMed] [Google Scholar]; (b) Kampen D., Reisinger C. M., List B. Top. Curr. Chem. 2010;291:395–456. doi: 10.1007/978-3-642-02815-1_1. [DOI] [PubMed] [Google Scholar]
- (a) Gorin D. J., Sherry B. D., Toste F. D. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patil N. T., Yamamoto Y. Chem. Rev. 2008;108:3395–3442. doi: 10.1021/cr050041j. [DOI] [PubMed] [Google Scholar]; (c) Rudolph M., Hashmi S. K. Chem. Commun. 2011;47:6536–6544. doi: 10.1039/c1cc10780a. [DOI] [PubMed] [Google Scholar]
- Bannwarth W. Chimia. 1987;41:302–317. [Google Scholar]
- Hadjiliadis N. D. and Sletten E., Metal Complex–DNA Interactions, Wiley-Blackwell, New York, 2009. [Google Scholar]
- Chouikhi D., Ciobanu M., Zambaldo C., Duplan V., Barluenga S., Winssinger N. Chem.–Eur. J. 2012;18:12698–12704. doi: 10.1002/chem.201201337. [DOI] [PubMed] [Google Scholar]
- Stöckigt J., Antonchick A. P., Wu F., Waldmann H. Angew. Chem., Int. Ed. Engl. 2011;50:8538–8564. doi: 10.1002/anie.201008071. [DOI] [PubMed] [Google Scholar]
- (a) Suzuki Y., Naoe S., Oishi S., Fujii N., Ohno H. Org. Lett. 2012;14:326–329. doi: 10.1021/ol203072u. [DOI] [PubMed] [Google Scholar]; (b) Wagner B., Hiller W., Ohno H., Krause N. Org. Biomol. Chem. 2016;14:1579–1583. doi: 10.1039/c5ob02453f. [DOI] [PubMed] [Google Scholar]
- Harada K., Orgel L. E. Nucleic Acids Res. 1993;21:2287–2291. doi: 10.1093/nar/21.10.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klika Škopić M., Bugain O., Jung K., Onstein S., Brandherm S., Kalliokoski T., Brunschweiger A. Med. Chem. Commun. 2016;7:1957–1960. [Google Scholar]
- Halpin D. R., Lee J., Wrenn S. J., Harbury P. B. PLoS Biol. 2004;2:E175. doi: 10.1371/journal.pbio.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Brenner S., Lerner R. A. Angew. Chem., Int. Ed. 2017;56:1164–1165. doi: 10.1002/anie.201612143. [DOI] [PubMed] [Google Scholar]; (b) Yuen L. H., Franzini R. M. ChemBioChem. 2017;18:1–9. doi: 10.1002/cbic.201600567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.