

Abstract
CRH is a key regulator of neuroendocrine, autonomic, and behavioral response to stress. CRH-stimulated CRH receptor 1 (CRHR1) activates ERK1/2 depending on intracellular context. In a previous work, we demonstrated that CRH activates ERK1/2 in limbic areas of the mouse brain (hippocampus and basolateral amygdala). ERK1/2 is an essential mediator of hippocampal physiological processes including emotional behavior, synaptic plasticity, learning, and memory. To elucidate the molecular mechanisms by which CRH activates ERK1/2 in hippocampal neurons, we used the mouse hippocampal cell line HT22. We document for the first time that ERK1/2 activation in response to CRH is biphasic, involving a first cAMP- and B-Raf–dependent early phase and a second phase that critically depends on CRHR1 internalization and β-arrestin2. By means of mass-spectrometry–based screening, we identified B-Raf–associated proteins that coimmunoprecipitate with endogenous B-Raf after CRHR1 activation. Using molecular and pharmacological tools, the functional impact of selected B-Raf partners in CRH-dependent ERK1/2 activation was dissected. These results indicate that 14-3-3 proteins, protein kinase A, and Rap1, are essential for early CRH-induced ERK1/2 activation, whereas dynamin and vimentin are required for the CRHR1 internalization-dependent phase. Both phases of ERK1/2 activation depend on calcium influx and are affected by calcium/calmodulin-dependent protein kinase II inactivation. Thus, this report describes the dynamics and biphasic nature of ERK1/2 activation downstream neuronal CRHR1 and identifies several new critical components of the CRHR1 signaling machinery that selectively controls the early and late phases of ERK1/2 activation, thus providing new potential therapeutic targets for stress-related disorders.
The 41-amino acid peptide CRH plays a key role in the regulation of neuroendocrine, autonomic, and behavioral response to stress (1, 2). Hypothalamic CRH mediates basal and stress-induced responses through hypothalamic-pituitary-adrenal axis activation, whereas central extrahypothalamic CRH modulates a wide range of adaptations, including anxiety-like behaviors (3, 4). Dysregulation of the CRH system is implicated in human stress-related affective disorders, mainly anxiety and depression (2, 5–7).
CRH exerts its actions by activating 2 distinct G protein-coupled receptors (GPCRs), CRH receptor 1 (CRHR1), and CRH receptor 2 (CRHR2), that display not only specific expression patterns throughout the brain but also different ligand affinities: CRH is a high-affinity ligand for CRHR1 and binds poorly to CRHR2 for which other ligands such as urocortin II and III have higher affinity (8, 9).
A large number of preclinical and clinical studies have clearly described the involvement of CRHR1 in the onset and maintenance of stress-related disorders (6, 10–13). In this regard, conditional CRHR1-knockout mice have provided interesting information: although mice lacking the CRHR1 in all principal neurons of the forebrain show reduced anxiety-related behavior (11), a recent work that analyzed behavioral effects of CRHR1 deletions in neurotransmitter-specific neurons shows that the receptor exerts different functions (anxiogenesis vs anxiolysis) acting on different neuronal populations (14). These novel results suggest that transitions from physiological to pathological states may be regulated by a delicate balance of the CRH/CRHR1 system.
In most systems, activated CRHRs bind the Gαs protein, resulting in adenylyl cyclase (AC) stimulation with the consequent increase in intracellular cAMP (reviewed in Ref. 15). However, CRHR1 downstream signaling is far more complex than previously thought.
In fact, recent reports indicate that CRHR1 is able to activate different intracellular pathways depending on the cellular context (12, 16–29). One group of signaling molecules that can be activated by CRHR1 depending on the specific cellular circumstances is the family of MAPKs ERK1/2. Different results in terms of ERK1/2 activation by CRH have been reported. In particular cellular systems, CRH increases cAMP intracellular levels but is ineffective in activating ERK1/2 (20–22). On the other hand, under different cellular circumstances, CRH is able to trigger a MAPK signal transduction pathway via cAMP (23–26, 29).
In vivo studies demonstrated that MAPKs ERK1/2 are activated by acute intracerebroventricular administration of CRH in specific limbic areas such as hippocampus and basolateral amygdala. These brain structures are functionally involved in environment information processing and behavioral adaptation to stress (12). However, other regions with high CRHR1 expression levels such as cortex, hypothalamic nuclei, and the central nucleus of the amygdala did not show CRH-mediated ERK1/2 activation (12). The fact that CRH injection triggers such a specific activation pattern suggests that a molecular and functional link between CRHR1 and downstream MAPKs may be restricted to these brain structures (19).
ERK1/2 is widely distributed through the central nervous system (CNS) and is highly expressed in limbic brain areas such as the hippocampus, prefrontal cortex, and basolateral amygdala (30, 31) where it acts as a central regulator of molecular processes that mediate synaptic plasticity, learning and memory processes, and emotional behavior (32–34). In addition, several studies in rodents indicate that stress can change levels of activated ERK1/2 (35, 36), and pharmacological manipulations of the ERK1/2 pathway affect different parameters in stress-coping behavior and anxiety tests (37–39). Because a number of CNS disorders in humans are closely related to the ERK1/2 activation cascade, attention remains focused on the underlying actions of these molecules (33).
Given that the signal transduction pathways leading to ERK1/2 activation by CRH depend on cellular context and that in hippocampal neurons the mechanisms involved are not known, we carried out a molecular analysis of CRHR1 signaling pathways in HT22 cells, a mouse hippocampal cell line. Our results reported in this work demonstrated that these cells are a valuable tool to understand molecular aspects of CRH signaling, because this in vitro model proved to be useful to perform molecular and cellular experiments that would be more complex, difficult, and sometimes even unfeasible, in vivo. Although several studies using non-neuronal cellular models demonstrated ERK1/2 activation by CRHR1, the involvement of at least 2 distinct activation mechanisms (one cAMP-dependent and the other one CRHR1 internalization-dependent) involving different signaling pathways has not been reported to date and may be specific to the HT22 cellular environment. Several studies using this immortalized mouse hippocampal cell line consider this as a suitable model of hippocampal neurons (40–48). Thus, this is the first report of molecular components and mechanisms involved in ERK1/2 activation by CRH mediated through CRHR1 in a cell model that embrace a hippocampal cellular context. We identified critical components of the CRHR1 signaling machinery that selectively controls the early and late phases of ERK1/2 activation. These findings may have important implications for our understanding of the functional connections of the main signaling pathways controlling CRH actions.
Materials and Methods
Cell culture and generation of cell lines stably expressing CRHR1
HT22 mouse hippocampal cells were cultured as described (49). The lack of bona fide commercial antibodies that recognize CRHR1 with specificity and sensitivity (14) prompted us to use a c-Myc-CRHR1 construct to generate stable HT22-CRHR1 clones. The use of a mouse monoclonal anti–c-Myc-antibody allowed us to perform the immunocytochemical (see Figure 2) and the flow cytometry experiments (see Figure 7B and Supplemental Figure 2, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) shown in this work. The mouse CRHR1 expression vector pcDNA3-c-Myc-mCRHR1 (14) was kindly provided by Dr Jan Deussing (Max Planck Institute of Psychiatry, Munich, Germany). pcDNA3-c-Myc-mCRHR1 was used to transfect HT22 cells using Lipofectamine reagent (Life Technologies, Grand Island, New York) according to the manufacturer's instructions. For the generation of cell lines stably expressing CRHR1 (HT22-CRHR1), transfected cells were grown in DMEM (Life Technologies) in the presence of G418 (800 μg/ml; Life Technologies) to select for transfected cells. G418 surviving clones were subcultured and maintained in DMEM supplemented with 5% (vol/vol) fetal calf serum (FCS), 4mM l-glutamine (Sigma, St Louis, Missouri), 10 mM HEPES (Sigma), 2.2 mg/ml NaHCO3, 100 U/ml penicillin (Life Technologies), 100 mg/ml streptomycin (Life Technologies), and 200 μg/ml G418 (Life Technologies) at 37°C in a humidified 5% CO2 incubator. CRHR1 expression levels in the stably transfected cell lines was assessed by Western blot using a mouse monoclonal anti c-Myc-antibody (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, California). Thirteen positive clones of HT22-CRHR1 were obtained. All the experiments were performed at least 2 times with 2 individual clones (clones 2 and 3) with different expression levels of c-Myc-CRHR1. Data from a representative clone (clone 2) are shown. Using HT22-CRHR1 clones increased sensitivity in the study by ensuring that experimental conditions were homogeneous (same CRHR1 levels in all the experiments).
Figure 2.

pERK1/2 and c-Myc–CRHR1 subcellular distribution induced by CRH in HT22-CRHR1 cells. pERK1/2 and c-Myc–CRHR1 distribution was monitored over different time points after 100 nM CRH stimulation by indirect double immunofluorescence using specific antibodies against pERK1/2 (green) and c-Myc (red) as described in Materials and Methods. Cell nuclei were stained with the DNA-specific dye propidium iodide (PI) (blue). pERK1/2 and CRHR1 colocalization appears as yellow in the merged images. Scale bar, 20 μM.
Figure 7.

CRHR1 internalization and β-arrestin2 requirement for CRH-induced ERK1/2 activation. A and D, HT22-CRHR1 cells were transfected with 600 ng dynamin-K44A (A) or 600 ng β-arr (319–418) (D) as described in Materials and Methods. Cells were stimulated with 100 nM CRH for the indicated time points and pERK1/2 and total ERK1/2 were determined by Western blot. Signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes of CRH stimulation under control conditions ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05; ** P < .01 compared with control treatment (empty vector) at the same time point. Statistical analysis was performed using a one-way ANOVA. B, HT22-CRHR1 cells were pretreated for 30 minutes with vehicle, PAO (2.5 μM), or MDC (100 μM) and stimulated with 100 nM CRH for 30 minutes. Using specific antibodies against c-Myc, tagged-receptor distribution was monitored by indirect immunofluorescence in fixed and permeabilized HT22-CRHR1 cells (red). c-Myc–CRHR1 internalization was quantified by flow cytometry, labeling cell surface with c-Myc antibody. HT22 cells were used as control. A representative flow cytometry experiment is shown. c-Myc–CRHR1 internalization quantifications are expressed as the percentage of maximum receptor on cell surface (nonstimulated HT22-CRHR1 cells) ± SEM from 3 independent experiments. ** P < .01 compared with nonstimulated HT22-CRHR1 cells. Statistical analysis was performed using a one-way ANOVA followed by post hoc Scheffé's test. C, HT22-CRHR1 cells were pretreated for 30 minutes with vehicle, PAO (2.5 μM), or MDC (100 μM) and stimulated with 100 nM CRH for 30 minutes. pERK1/2 and total ERK1/2 were determined by Western blot. Signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as mean fold increase compared with basal ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05 compared with basal. Statistical analysis was performed using a one-way ANOVA followed by post hoc Scheffé's test. E, HT22-CRHR1 cells were transfected with 100 nM scramble or 100 nM β-arrestin2 siRNA as described in Materials and Methods. After 18 hours in Opti-MEM, cells were stimulated with 100 nM CRH for different times and pERK1/2 and total ERK1/2 were determined by Western blot. Signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes of stimulation in control conditions ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05; ** P < .01 compared with control treatment (scramble siRNA) at the same time point. Statistical analysis was performed using a one-way ANOVA. In the inset is shown a Western blot of β-arrestin2 and total ERK1/2 as a loading control from a representative experiment.
Small interfering RNA transfection
Chemically synthesized, double-stranded small interfering RNAs (siRNAs) with 21-nucleotide duplex RNA and 2-nucleotide 3′-dTdT overhangs were obtained from Invitrogen in deprotected and desalted form. The siRNA sequences targeting mouse β-arrestin1, β-arrestin2, and B-Raf were 5′-AAAGCCUUCUGUGCUGAGAAC-3′ (positions 732–752), 5′-AAGGACCGGAAAGUGUUCGUG-3′ (positions 226–246), and 5′-AAGUGGCAUGGUGAUGUGGCA-3′ (positions 1702–1722), respectively. For vimentin silencing, siRNA was from a commercial source (Santa Cruz sc-29523). A nonsilencing RNA duplex (5′-CUUACGCUGAGUACUUCGATT-3′) was used as a control. For siRNA experiments, HT22-CRHR1 cells (40% confluence) on 100-mm dishes were transfected with 100 nM siRNA in Opti-MEM (Life Technologies), using 24 μl of Lipofectamine 2000 Reagent (Life Technologies), according to the manufacturer's instructions, and 24 hours later, cells were split into either 40-mm or 6-well dishes and the experiments performed 72 hours after transfection.
Transient DNA transfections and plasmids
The expression vectors dynamin-K44A, β-arrestin (319–418) (kindly provided by Dr J. Benovic, Department of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, Pennsylvania), Rap1A-17N and Ras-17N (kindly provided by Dr O. Coso, Laboratorio de Fisiología y Biología Molecular, Universidad de Buenos Aires, Buenos Aires, Argentina) were used. HT22-CRHR1 cells grown in 100-mm dishes (50% confluent) were transiently transfected with plasmids (6 μg) using 36 μl of Lipofectamine reagent combined with 24 μl of PLUS Reagent (Life Technologies) following the manufacturer's instructions, and 24 hours later, cells were split into either 40-mm or 6-well dishes and experiments performed 48 hours after transfection. The plasmid expressing the dominant-negative (DN)–14-3-3η (R56A, R60A), (kindly provided by Dr A. S. Shaw, Department of Pathology and Immunology, Washington University School of Medicine, St Louis, Missouri)A. S. was used at the amounts indicated (see Figure 6).
Figure 6.

Involvement of B-Raf partners in the early CRH-induced ERK1/2 activation. A, HT22-CRHR1 cells were transfected with increasing amounts of DN–14-3-3η as indicated. Cells were stimulated with 100 nM CRH for 3 minutes. pERK1/2 (top panel), total ERK1/2 (middle panel), and total 14-3-3 proteins (endogenous and transfected) were determined by Western blot. Note that c-Myc–DN–14-3-3ηs have slower gel mobility compared with endogenous 14-3-3s. B, HT22-CRHR1 cells were pretreated for 30 minutes with 10 μM H89 and stimulated with 100 nM CRH for the indicated time points. Controls were preincubated with vehicle. pERK1/2 and total ERK1/2 were determined by Western blot; signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes of stimulation in the absence of inhibitor ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05; ** P < .01 compared with vehicle plus CRH treatment at the same time point. Statistical analysis was performed using a one-way ANOVA. C, HT22-CRHR1 cells were transfected with 600 ng RapN17A, 600 ng RasN17A, or the empty vector. Cells were stimulated with 100 nM CRH for the indicated time points, and pERK1/2 and total ERK1/2 were determined by Western blot. Signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes of stimulation in control conditions ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05; ** P < .01 compared with control treatment (empty vector) at the same time point. Statistical analysis was performed using a one-way ANOVA followed by post hoc Scheffé's test.
Ligand stimulation, drugs, and pharmacological inhibitors
HT22 and HT22-CRHR1 cells on 6-well or 40-mm plates were starved for 18 hours in Opti-MEM before stimulation or drug pretreatments. Cells were stimulated with human/rat CRH (H-2435; Bachem, Torrance, California) or with platelet-derived growth factor (PDGF) (01-305; Millipore, Billerica, Massachusetts) at the concentrations and time points indicated in the figures. After incubations, cells were washed with ice-cold PBS and maintained in ice. When CRHR1 antagonist DMP696, calcium chelator 1,2-Bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) (5 μM) (B6769; Life Technologies), extracellular EGTA (2 mM) or pharmacological inhibitors were used, cells were pretreated with the drugs (or vehicle) 30 minutes before stimulation. CRHR1-specific antagonist DMP696 (5 μM) was a generous gift from Dr Hausch (Max Planck Institute of Psychiatry, Munich, Germany). Receptor internalization was inhibited by pretreatment with phenylarsine oxide (2.5 μM; Sigma) or monodansylcadaverine (100 μM; Sigma). Adenylate cyclase, protein kinase A (PKA), calcium/calmodulin-dependent protein kinase (CaMKII), phospholipase C (PLC), and MAPK kinase (MEK)1/2 were inhibited with 2′,5′-dideoxyadenosine (2′,5′–ddA) (50 μM; Calbiochem, La Jolla, California), H89 (10 μM; Calbiochem), KN-62 (10 μM; Calbiochem), U-73122 (5 μM; Sigma), and UO126 (10 μM; Calbiochem), respectively. All pretreatments and stimulations were carried out at 37°C in a humidified incubator at 5% CO2.
Preparation of cellular extracts and immunoblotting
After treatments, cells were washed with ice-cold PBS and lysed directly in Laemmli sample buffer. Whole-cell lysates were sonicated, heated to 95°C for 5 minutes, and cooled on ice. Samples were resolved by SDS-PAGE and transferred onto nitrocellulose membranes (Millipore) for immunoblotting. Membranes were blocked in TBS-Tween 20 (0.05%) containing 5% milk at room temperature for 1 hour under shaking and probed overnight at 4°C with the primary antibodies listed below, dissolved in the same blocking solution unless it is explicitly defined. Membranes were incubated with the corresponding secondary antibodies, and antibody binding was detected by enhanced chemiluminescence (ECL; Pierce, Rockford, Illinois) using an LAS-1000 imaging system (Fujifilm, Tokyo, Japan).
The following antibodies were used: anti–c-Myc, anti–phospho-ERK1/2 (pERK1/2), anti–pan-14-3-3, antivimentin, anti–total-B-Raf (Santa Cruz Biotechnology); anti–phospho-B-Raf Ser44, anti–total-ERK1/2, anti-phospho-CaMKII Thr286 (Cell Signaling, Danvers, Massachusetts); anti–β-arrestin (BD Transduction Laboratories, Lexington, Kentucky). To normalize the phosphorylated isoforms of ERK1/2 and B-Raf to their total expression level, after stripping, membranes were incubated with total ERK1/2 or with total B-Raf antibodies. Immunoreactive signals were quantified by densitometry using ImageJ software (http://rsbweb.nih.gov/ij/, National Institutes of Health).
Immunoprecipitation assays
HT22-CRHR1 cells were grown to 70% confluence and serum starved with Opti-MEM (Gibco) 18 hours before being stimulated with 100 nM CRH. After treatments, cells were washed with ice-cold PBS and lysed for 2 h at 4°C with modified RIPA buffer (50 mM Tris [pH 7.4], 100 mM NaCl; 1 μM EDTA; 1% Nonidet P-40) containing a cocktail of protease inhibitors (Roche Molecular Biomedicals, Indianapolis, Indiana). To maximize the recovery of the B-Raf–interacting proteins, three 100-mm dishes were used for each condition. Immunoprecipitation assays were performed at 4°C overnight with rabbit anti–B-Raf antibodies raised against an epitope corresponding to amino acids 12–156 mapping at the N terminus of B-Raf (1 μg) (sc-9002; Santa Cruz Biotechnology). Immunoprecipitates were collected using protein A/G plus agarose (Santa Cruz Biotechnology). A preclearing step with a commercial polyclonal nonimmune rabbit antibody (sc-2345; Santa Cruz Biotechnology) and protein A/G plus agarose (Santa Cruz Biotechnology) was included before performing B-Raf immunoprecipitation to prevent nonspecific protein binding to the anti–B-Raf rabbit polyclonal antibody. Immunoprecipitates were washed once with modified RIPA buffer and once with cold PBS and resuspended in Laemmli sample buffer. After heating at 95°C for 5 minutes, the samples were centrifuged briefly and the supernatants were run by SDS-PAGE. After electrophoresis, proteins were stained by Coomassie Brilliant Blue following the manufacturer's protocol (Bio-Rad, Hercules, California) and scanned with the densitometer GS-800 using the PDQuest software (Bio-Rad).
Proteomic analysis by liquid chromatography-electrospray ionization-tandem mass spectrometry
Gel lanes were cut in 23 equal slices and in-gel tryptic digestion was performed as previously described (49). The extracted peptides were lyophilized and dissolved in 10 μl 0.1% formic acid aqueous solution (Merck, Darmstadt, Germany). Five microliters were then loaded onto an in-house packed fused silica nano RP-C18 column (0.075 mm × 20 cm) using 5 μm inner diameter packing material (Maisch, Monheim, Germany). The nano column was washed with 0.1% formic acid for 10 minutes and eluted with a gradient of 95% acetonitrile/0.1% formic acid from 2% to 10% in 2 minutes, from 10% to 45% in 45 minutes at a flow rate of 200 nl/min using a nano LC-2D system (Eksigent, Dublin, California) coupled online to an LTQ-Orbitrap mass spectrometer equipped with a nano spray source (Thermo Fisher Scientific, Bremen, Germany). The mass spectrometer was operated in the positive-ion mode, with ion spray voltage of 2 kV, applying a data-dependent automatic scan switch between mass spectrometry (MS) and MS/MS acquisition. Full scans were recorded in the Orbitrap mass analyzer at a mass range of mass to charge ratio of 380–1600 at resolution of 60,000 (mass to charge ratio of 400) in profile mode. The MS/MS analysis of the 3 most intense peptide ions for each scan was recorded in the LTQ mass analyzer in centroid mode.
Database searching
The identification of the proteins was achieved by searching against SwissProt version 15.3 database (uniprot29.05.09), Mus musculus as taxonomy specie, trypsin as enzyme, and allowing one missed cleavage site. The methionine oxidation and cysteine carboxyamidomethylation were set as dynamic and static modifications, respectively. The mass accuracies of the precursor and the fragment ions were set 20 ppm and 0.7 Da, respectively. For the peptide identification, the MS/MS spectra were searched with the support of Mascot version 2.2 (50). The results with a significance level over 95% were accepted. The protein identifications were considered confident when at least 2 peptides were contained. The proteins reported in this work were identified in 2 independent experimental replicates.
Immunofluorescence and confocal microscopy
Cells were seeded on glass coverslips and grown until 50% confluency. After treatments, cells were washed with ice-cold PBS and fixed with 4% paraformaldehyde in PBS. Next, cells were permeabilized with 0.01% Triton X-100/PBS. After a 5-minute wash with PBS, nonspecific binding was reduced by incubating coverslips with 1% FCS in PBS for 1 hour. Primary antibodies were diluted 1:200 and incubated overnight at 4°C. After washing with PBS, cells were incubated with a 1:200 dilution of Alexa Fluor 647- or 488-conjugated secondary antibodies (Life Technologies) for 45 minutes at room temperature. When double staining was carried out, both primary and secondary antibodies were incubated together. Controls without primary antibody were performed for each experiment. Coverslips were washed with PBS and mounted in Vectashield mounting media with or without propidium iodide (Vector Laboratories, Inc, Burlingame, California) on microscope slides. The slides were examined with an oil immersion objective (×60) using an FV300 BX61 laser-scanning confocal fluorescence microscope (Olympus, Center Valley, Pennsylvania) using the appropriate lasers and filters and a confocal slice thickness of 0.45 μm. Images were acquired with the Fluoview analyzer program. For each treatment, between 15 and 20 individual cells were randomly selected and examined. No fluorescence was observed in cells treated with secondary Alexa-Fluor antibodies only. All images were acquired with the same confocal settings to ensure uniformity detection for each cell, including pinhole size, detector gain, offset, and laser power.
Flow cytometry assays
For surface staining, HT22-CRHR1 cells on 100-mm plates were starved for 18 hours in Opti-MEM before stimulation or drug pretreatments. Cells were stimulated with 100 nM CRH for 30 minutes. When pharmacological inhibitors were used, cells were pretreated with the drugs (or vehicle) 30 minutes before stimulation. After incubations, cells were washed and harvested in ice-cold PBS, and 3.0 × 105 HT22-CRHR1 cells of each treatment were incubated with 1% FCS in ice-cold PBS for 1 hour in ice water to reduce nonspecific binding. Subsequently, cells were incubated with 2 μg anti-c-Myc for 4 hours in ice water. After washing with PBS, cells were stained with a 1:100 dilution of antimouse Alexa Fluor 488-conjugated secondary antibody (Life Technologies) for 1 hour in ice water. Controls staining parental HT22 cells were performed for each experiment. Flow cytometry data were acquired on a FACSAriaII (Becton Dickinson, San Jose, California). Data were analyzed using the FlowJo software (Tree Star Inc).
cAMP determination
HT22-CRHR1 cells were grown to 80% confluence in 12- or 24-well plates and serum starved with Opti-MEM for 18 hours. For concentration-response experiments, cells were incubated for 5 minutes with 500 μM 3-isobutyl-1-methylxanthine and exposed to CRH at different concentrations for 10 minutes. To determine EC50 of CRH-induced cAMP production, data were adjusted with a 3-parameter concentration-response curve with GraphPad Prism software. Where indicated, cells were pretreated for 30 minutes with the selective AC inhibitor 2′,5′-ddA. Then, cells were subjected to ethanol extraction followed by 15 minutes centrifugation at 3000g. Supernatants were dried and resuspended in 50 mM Tris-HCl buffer (pH 7.4). cAMP content was determined by means of competition with [3H]cAMP for PKA as described previously (51) and expressed as the percentage of stimulation relative to maximal response. The standard curve was performed using 8 cAMP amounts ranging from 0.1 to 90 pmol. Duplicate samples in at least 3 independent experiments were analyzed.
Calcium imaging
All experiments were performed at room temperature (20°C–24°C). Cells in 40-mm petri dishes were loaded for 30 minutes in darkness with Fluo-4 AM 6 μM (Life Technologies) and Pluronic F-127 0.14% (Life Technologies) in a Ca2+ buffer (125 mM NaCl, 5 mM KCl, 0.4 mM CaCl2, 1 mM MgSO4, 5 mM NaHCO3, 1 mM Na2HPO4, 10 mM glucose, 20 mM HEPES [pH 7.4]) and then placed on the stage of a fluorescence BX-FLA Olympus microscope. Cells were illuminated with a USH-I 02DH mercury lamp (USHIO) and imaged using a ×40 water immersion objective and a cooled CCD Quantix camera (Photometrix). A filter block containing a 460- to 490-nm excitation filter (band pass), a 505-nm dichroic mirror, and a 515-nm barrier filter (high pass) was used. Exposure times ranged from 100 to 300 milliseconds, and frames were taken every 0.4 seconds. Images were acquired with the Axon Imaging Workbench version 2.1 program and analyzed with Image J version 1.29v (National Institutes of Health, Bethesda, Maryland). Calcium imaging data are presented as ΔF/Fo, where Fo is the resting fluorescence (before stimulation) and ΔF is the peak change in fluorescence from resting levels. For quantification, fluorescence was measured in 4 cells per visual field and at least 3 fields randomly selected in each experiment. Data are presented as the average ± SEM from 3 independent experiments.
Statistics
Each experiment was performed at least 3 independent times. The results are presented as the mean ± SEM of each measurement. Data were tested for homogeneity, and comparisons between treatment means were performed using 1-way ANOVA (GraphPad Prism version 5 for Windows; GraphPad Software, San Diego California) followed by post hoc Scheffé's test when more than 2 means were compared (StatView, SAS Institute Inc, Cary, North Carolina). Statistically significant differences are indicated.
Results
CRHR1 induces biphasic ERK1/2 activation in a CRH-stimulated hippocampal cell line
Given our previous findings that established a specific pattern of ERK1/2 activation in vivo by CRH in the mouse brain, involving limbic areas such as hippocampus and amygdala (12), we decided to carry out a molecular analysis of the signaling components and mechanisms involved. We first analyzed the effects of CRH in terms of ERK1/2 activation in mouse hippocampal neuronal HT22 cells. In these cells, unlike CRHR2, CRHR1 mRNA is detected after 18 hours of serum starving (data not shown). As shown in Figure 1A, CRH induces a biphasic ERK1/2 activation, with a first peak as early as 3–6 6 minutes after stimulation with 100 nM CRH, followed by a decline to about basal levels at minute 12. Remarkably, a second phase of ERK1/2 phosphorylation was observed at minute 20 (∼30% of maximal response) and showed sustained ERK1/2 phosphorylation levels for at least 60 minutes (Figure 1A). Stable overexpression of an N-terminal Myc-tagged murine CRHR1 in HT22 cells (HT22-CRHR1) augments the overall ERK1/2 activation ∼2-fold at early time points and up to 3-fold in the second phase (Figure 1B). However, the overall pattern of ERK1/2 activation is identical for both endogenous and exogenous receptor expression. In addition, control kinetics of ERK1/2 activation performed with vehicle showed that pERK1/2 levels in the absence of CRH do not change during the time course of the experiment (Supplemental Figure 1A).
Figure 1.

Time- and concentration-dependent ERK1/2 activation via CRHR1 in HT22 cells. A and B, pERK1/2 and total ERK1/2 were determined by Western blot in HT22 (A) or HT22-CRHR1 (B) cells stimulated with 100 nM CRH for the indicated time points. C and D, pERK1/2 and total ERK1/2 were determined by Western blot in HT22 (C) or HT22-CRHR1 (D) cells stimulated with 100 nM CRH for the indicated time points after preincubation in the presence of vehicle or the CRHR1 antagonist DMP696 (5 μM). A–D, Signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes of stimulation in the absence of antagonist ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05; ** P < .01 with respect to vehicle plus CRH treatment at the same time point. Statistical analysis was performed using a 1-way ANOVA. E, HT22-CRHR1 cells were stimulated with CRH at the indicated concentrations for 3 minutes (left panel) or 40 minutes (right panel). pERK1/2 and total ERK1/2 were determined by Western blot. Signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximal pERK1/2 response obtained at 3 minutes (left panel) or 40 minutes (right panel) ± SEM from 3 independent experiments. To determine EC50 for CRH-induced ERK1/2 phosphorylation, data were adjusted with a 3-parameter dose-response curve with GraphPad Prism software. EC50 at 3 minutes was 6.02−11 M (95% confidence interval of EC50 at 3 minutes, 2.03−11 M to 1.79−10 M). EC50 at 40 minutes was 7.16−10 M (95% confidence interval of EC50 at 40 minutes, 2.31−10 M to 2.22−9 M).
To further validate the role of CRHR1 in mediating ERK1/2 activation in both parental and HT22-CRHR1 cells, we examined the time course of CRH-induced pERK1/2 in the presence or absence of DMP696, a well-defined selective CRHR1 antagonist. As shown in Figure 1, C and D, 5 μM DMP696 pretreatment abolished ERK1/2 activation in both the early phase (3–6 minutes) and the late phase (20–60 minutes), indicating that CRH-induced ERK1/2 activation is mediated by CRHR1. In this line, the concentration-response curves, in which pERK1/2 levels were saturated at high concentrations of hormone, also demonstrate a receptor-mediated effect of CRH on ERK1/2 phosphorylation for both phases of CRH response (Figure 1E).
Subcellular distribution of activated ERK1/2 and CRHR1 in CRH-stimulated cells
Using indirect immunofluorescence confocal microscopy, we examined the spatiotemporal distribution of pERK1/2 and CRHR1 in HT22-CRHR1 cells after CRH stimulation (Figure 2). Under basal conditions, a very low immunoreactive signal of pERK1/2 (pERK1/2 immunoreactivity) was homogeneously distributed throughout the cell. As expected, most of the Myc-tag immunoreactivity corresponding to CRHR1 was detected in the plasma membrane. However, appreciable CRHR1 signal was also found intracellularly, likewise it was previously observed in cortical neurons transfected with tagged forms of CRHR1 (52). Treatment of the cells with 100 nM CRH promoted CRHR1 internalization (Figure 2 and Supplemental Figure 2), and a perinuclear accumulation was rapidly observed over time (Figure 2). After agonist stimulation, pERK1/2 immunoreactivity increased with a comparable kinetic to that detected by Western blot (Figures 1, A and B and 2). At the early time points of CRH exposure, a small amount of pERK1/2 immunoreactivity concentrated in the cell surface and colocalized with CRHR1 (Figure 2, 3 minutes). Noteworthy, most of CRH-activated ERK1/2 was evidenced as discrete granular accumulations that were persistently retained in the cytoplasm and cell surface without significant translocation into the nucleus. This pERK1/2 distribution was clearly different from the predominant nuclear location observed in HT22-CRHR1 cells stimulated for 5 minutes with PDGF, another ERK1/2-activating stimulus (Figure 3). These results indicate that a specific intracellular pERK1/2 distribution pattern is generated in response to CRH in HT22-CRHR1 cells.
Figure 3.

pERK1/2 subcellular distribution induced by CRH and PDGF in HT22-CRHR1 cells. HT22-CRHR1 cells were stimulated with 100 nM CRH for 3 minutes or 10 ng/ml PDGF for 5 minutes. pERK1/2 distribution was monitored by indirect immunofluorescence using specific antibodies (green) as described in Materials and Methods. Cell nuclei were stained with the DNA-specific dye propidium iodide (PI) (blue). Colocalization appears as cyan in the merged images. Scale bar, 20 μM.
cAMP on CRH-induced ERK1/2 activation: critical role of B-Raf
Based on previous studies demonstrating that CRHR1 activation by CRH triggers ERK1/2 activation downstream of cAMP (23–26, 29), we decided to determine whether in the HT22 cellular context, CRH-induced ERK1/2 phosphorylation is a consequence of an increase in cAMP levels. To that end, we first examined the role of AC in ERK1/2 activation in terms of CRH signaling. Pretreatment of HT22-CRHR1 cells with the AC inhibitor 2′,5′-ddA significantly reduced CRH ability to activate ERK1/2 at the early phase (∼40%), showing no effect on late time points (Figure 4A). As expected, the maximum cAMP activation induced by CRH (after 3 minutes of 100 nM CRH) is blocked in cells pretreated with 50 μM 2′,5′ddA (Figure 4B). Interestingly, the EC50 of early-phase CRH-induced pERK (∼0.1 nM, Figure 1E, left panel) is in the same range of the EC50 of CRH-induced cAMP (∼0.5 nM, data not shown) in HT22-CRHR1 cells. These results demonstrate that AC and its product, cAMP, are determining factors for the initial peak increase in CRH-induced ERK1/2 activation.
Figure 4.

Role of AC and specific requirement of B-Raf for early CRH-induced ERK1/2. A and B, HT22-CRHR1 cells were stimulated with 100 nM CRH for the indicated time points after preincubation in the presence of vehicle or the AC inhibitor 2′,5′-ddA (50 μM) for 30 minutes. pERK1/2 and total ERK1/2 (A) were determined by Western blot; signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes of stimulation in control conditions ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05; ** P < .01 with respect to vehicle plus CRH treatment at the same time point. Statistical analysis was performed using a one-way ANOVA. cAMP levels (B) were determined by means of competition with [3H]cAMP for PKA as described in Materials and Methods. Results are expressed as the percentage of maximum cAMP levels obtained at 3 minutes of stimulation ± SEM from 3 independent experiments. ** P < .01 compared with the basal (time 0 minutes) in the presence of vehicle. Statistical analysis was performed using a one-way ANOVA followed by post hoc Scheffé's test. C and D, HT22-CRHR1 cells were transfected with 100 nM scramble or 100 nM B-Raf siRNA as described in Materials and Methods. Cells were stimulated with 100 nM CRH (C) or 10 ng/ml PDGF (D) for the indicated time points and pERK1/2 and total ERK1/2 were determined by Western blot. Signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes (C) or 5 minutes (D) of stimulation in control conditions ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05; ** P < .01 compared with control treatment (scramble siRNA) at the same time point. Statistical analysis was performed using a one-way ANOVA. E, Western blot of B-Raf and glyceraldehyde 3-phosphate dehydrogenase as a loading control from a representative siRNA experiment. F, pB-Raf (top panel), pERK1/2 (middle panel), and total B-Raf (bottom panel) in HT22-CRHR1 cells stimulated with 100 nM CRH at different time points were determined by Western blot. A representative Western blot from 3 independent experiments is shown.
In cells in which cAMP stimulates ERK activity, B-Raf constitutes the main activator of the cascade Raf/MEK/ERK (53). B-Raf is the MAPKKK (MAP kinase kinase kinase) predominantly expressed in the nervous system and is highly expressed in HT22 hippocampal cells, as we previously demonstrated (49). Therefore, we decided to investigate the role of this MAPKKK in ERK1/2 activation by CRH. We analyzed the time course of CRH-induced pERK1/2 after depleting cellular levels of B-Raf by transfecting HT22-CRHR1 cells with a siRNA specifically directed against this protein. Silencing ∼85% of B-Raf expression (Figure 4E) led to a significant decrease of ERK1/2 activation (∼50%) on the early phase but had no effect on the late phase, indicating that B-Raf is involved in early CRH-induced ERK1/2 activation (Figure 4C). It is worth mentioning that, in contrast to the effects observed using CRH, PDGF-stimulated ERK1/2 activation in the same cells was not affected by B-Raf silencing (Figure 4D).
B-Raf Ser446 phosphorylation is essential for its MAPKKK activity (54, 55). We examined by Western blot the phosphorylation status of B-Raf Ser446 (pB-Raf) after CRH stimulation. Interestingly, this B-Raf modification was transient in HT22-CRHR1 cells (Figure 4F). pB-Raf was detected after 1 minute of CRH exposure, preceding in time the peak of early-phase CRH-induced pERK1/2 (3 minutes) and rapidly returned to basal levels after 3–6 minutes.
Taken together, these results suggest that active B-Raf kinase is required to elicit the early phase of pERK1/2. Moreover, there is a critical and specific role of B-Raf in mediating early ERK1/2 activation by CRH in HT22-CRHR1 cells.
Identification of B-Raf partners showing a role in CRH signaling
In a recent work, we described the B-Raf interactome in HT22 cells under nonstimulating conditions (49). Given the critical role of this MAPKKK in mediating ERK1/2 activation by CRH in HT22-CRHR1 cells and that proteins do not act in isolation to fulfill their diverse functions, we performed B-Raf immunoprecipitation followed by MS in CRH-treated cells with the aim of identifying B-Raf partners with a potential role in CRH signaling in terms of ERK1/2 activation (Figure 5). Coimmunoprecipitated proteins with endogenous B-Raf obtained from lysates of CRH-treated HT22-CRHR1 cells were resolved by SDS-PAGE and subjected to in-gel proteolytic digestion. The resulting proteolytic peptides were analyzed by liquid chromatography-electrospray ionization-tandem MS, and the identification of sequences was performed by the use of Mascot algorithm. At first, those proteins with a potential role in cellular signaling were selected by extracting and mining biological relationships from biomedical literature. Finally, a list of B-Raf partners whose function in CRH-induced ERK1/2 activation has been confirmed in this work was created (Table 1).
Figure 5.
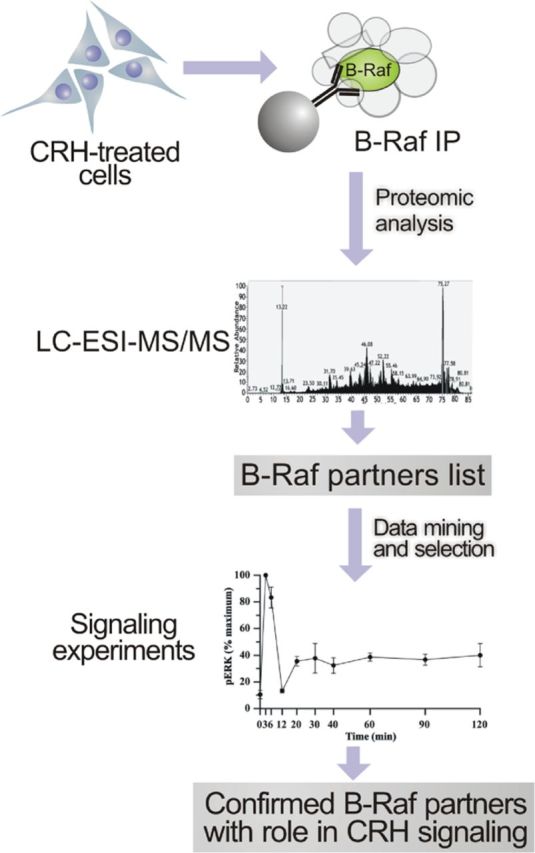
Identification of B-Raf partners with a role in CRH signaling. A schematic representation of the approach used to identify B-Raf partners with a role in CRH signaling is depicted (see text). The list of B-Raf partners whose function in CRH-induced ERK1/2 activation has been confirmed in this work is shown in Table 1.
Table 1.
B-Raf Partners Identified From CRH-Stimulated HT22-CRHR1 Cells Whose Role in Signaling Is Described in This Work
| Protein | Entry Name | Molecular Mass, kDa |
|---|---|---|
| 14-3-3 | ||
| β/α | 1433B | 28 |
| ϵ | 1433E | 29 |
| η | 1433F | 28 |
| γ | 1433G | 28 |
| σ | 1433S | 28 |
| θ | 1433T | 28 |
| ζ/δ | 1433Z | 28 |
| PKA (catalytic subunit α) | PRKACA | 40 |
| Rap1a | RAP1A | 21 |
| MEK1 | Map2K1 | 21 |
| Dynamin-1 | DYN1 | 98 |
| Vimentin | VIME | 54 |
| CaMKII | ||
| α | Camk2a | 54 |
| δ | Camk2d | 56 |
The 14-3-3 proteins interact with many different cellular phosphoproteins and play a central role in the activation process of Raf kinases (56). We identified the 7 mammalian isoforms of these proteins in association with B-Raf (Table 1). Homodimeric and heterodimeric forms of 14-3-3 associate with B-Raf (57). To evaluate the biological function of 14-3-3 in the B-Raf–dependent phase of CRH-induced ERK1/2 activation, we used the double arginine mutant forms (R56A and R60A) of 14-3-3η (DN–14-3-3η). Because DN–14-3-3 forms generate inactive heterodimers with endogenous 14-3-3 monomers (58) and taking into account that 14-3-3 proteins are highly abundant in CNS, different amounts of DN–14-3-3η were transfected in HT22-CRHR1 cells. Although 0.2 μg of DN–14-3-3η did not show changes in pERK1/2 signal in response to 3 minutes of stimulation with 100 nM CRH, 0.6 μg of plasmid substantially reduced ERK1/2 phosphorylation. Furthermore, the higher the amount of transfected DN–14-3-3η, the higher the inhibition of CRH-induced ERK1/2 activation (Figure 6A). Therefore, we conclude that 14-3-3 proteins, previously identified as B-Raf partners (57), are essential at early time points of ERK1/2 activation induced by CRH in HT22-CRHR1 cells.
Previous work in corticotrophs demonstrated that a rapid CRH/CRHR1-triggered ERK1/2 activation takes place downstream of PKA and B-Raf (24). In addition, B-Raf activation via PKA was also previously described in PC12 cells used as aneuronal model (59). In this work, we identified the catalytic subunit of the PKA in a complex with B-Raf (Table 1). To evaluate the dependence of ERK1/2 activation on PKA activity, we examined the time course of CRH-induced pERK1/2 in the presence or absence of H89, a selective PKA inhibitor. In line with the observed effects of inhibiting AC and B-Raf, 10 μM H89 pretreatment reduced most of the ERK1/2 activation on the early phase (∼60%) but had no significant effects on the late time points, indicating that PKA is the principal target of the second-messenger cAMP involved in early CRH-induced ERK1/2 activation (Figure 6B).
Several studies have demonstrated that the small G protein Rap1, like Ras, activates the MEK/ERK signaling pathway through activation of B-Raf (60). A Rap1/B-Raf complex as a consequence of PKA activation has been previously identified in PC12 cells (59). In CRH-treated HT22-CRHR1 cells, we found B-Raf in association with Rap1 (Table 1). To investigate the role of these 2 members of small G proteins, we examined the effect of a DN mutant of Rap1 (Rap1A-17N) or a DN form of Ras (Ras-17N) on CRH-induced ERK1/2 activation in HT22-CRHR1 cells. ERK1/2 phosphorylation after CRH stimulation was significantly inhibited (∼50%) either by Rap1A-17N or by Ras-17N only in the early phase (Figure 6C). These data indicate that Rap1 and Ras are also involved in the activation of ERK1/2 induced by CRH at early time points.
MEK1/2 are dual-specificity kinases that phosphorylate the tyrosine and threonine residues on ERK1/2 required for their activation. MEK1/B-Raf interaction was previously described (61). Because MEK1 was detected in B-Raf immunoprecipitates from CRH-treated cells, we analyzed the effect of UO126, a selective MEK1/2 inhibitor, on CRH-induced ERK1/2 activation. As expected, no pERK1/2 signal was detected when HT22-CRHR1 cells were pretreated with the MEK1/2 inhibitor (Supplemental Figure 1B).
Taken together, these results indicate that B-Raf and specific identified partners (14-3-3 proteins, PKA, Rap1, and MEK1/2) are essential players of early time points of CRH-induced pERK1/2.
CRHR1 internalization and β-arrestins are required for CRH-induced ERK1/2 activation
Clathrin-mediated endocytosis involves recruitment of clathrin to forming pits and the scission of vesicles from the plasma membrane induced by the GTPase dynamin. We identified dynamin as a B-Raf partner in CRH-treated cells (Table 1). To assess a potential role of dynamin in CRH-induced pERK1/2, HT22-CRHR1 cells were transfected with dynamin DN mutant (dynamin-K44A), an effective inhibitor of clathrin-mediated endocytosis. In this case, interestingly, we found that dynamin-K44A transfection significantly reduced ERK1/2 activation of the late phase (∼40%) compared with control cells (Figure 7A).
Several studies have demonstrated that, at least in certain cell types, GPCR internalization is required for activation of downstream signaling cascades including those leading to ERK1/2 phosphorylation (18, 62, 63). Given that dynamin is involved in CRH-induced ERK1/2 activation of the late phase (Figure 7A) and a link between CRHR endocytosis and ERK1/2 activation has been described in peripheral cell line expression systems (18), we investigated the relationship between CRHR1 internalization and ERK1/2 activation in our neuronal model HT22-CRHR1 cells. To that end, we evaluated the effects of different endocytosis inhibitors on CRH-induced ERK1/2 activation. Pretreatment of cells with 2.5 μM phenylarsine oxide (PAO), a general endocytosis inhibitor, or with 100μΜ monodansylcadaverine (MDC), a specific blocker of clathrin-mediated endocytosis, significantly reduced CRHR1 internalization after 30 minutes of agonist stimulation (Figure 7B). Interestingly, impaired CRHR1 endocytosis was accompanied by a decrease to nearly basal levels in ERK1/2 activation after 30 minutes of CRH (Figure 7C).
β-arrestins specifically target GPCRs for internalization by clathrin-coated vesicles and have also been described as mediators of diverse signaling pathways within the cell, including ERK1/2 activation (63). We investigated the role of β-arrestin1 and β-arrestin2 in CRH-induced ERK1/2 activation in HT22-CRHR1 cells. We first analyzed the effect of the clathrin-binding domain of β-arrestin β-arr (319–418), which functions as a DN by preventing β-arrestin interaction with receptors. β-Arr (319–418) expression significantly decreased pERK1/2 in the late phase of CRH stimulation (∼50%) (Figure 7D). To evaluate the contribution of each β-arrestin isoform, we analyzed the time course of CRH-induced pERK1/2 after depleting cellular levels of β-arrestin1 or β-arrestin2 by alternatively transfecting cells with a siRNA specifically directed against each isoform. Silencing β-arrestin1 expression had no significant effect on the magnitude of the pERK1/2 signal after CRH treatment (Supplemental Figure 3A). In contrast, silencing β-arrestin2 expression reduced CRH-induced ERK1/2 activation on the early phase and led to an almost complete elimination of pERK1/2 signal from the late phase (∼75%) (Figure 7E). As expected, depleting both β-arrestin isoforms simultaneously produced a similar reduction in ERK1/2 activation to β-arrestin2 siRNA-pretreated HT22-CRHR1 cells (Supplemental Figure 3B). These results demonstrate that β-arrestin2 plays an active and critical role in the late phase of ERK1/2 activation by CRH.
In an effort to further understand the mechanisms of CRH signal transduction, we analyzed the time course of CRH-induced pERK1/2 after depleting cellular levels of vimentin, a B-Raf partner detected in this work (Table 1) and also previously described as a component of the B-Raf interactome in HT22 cells (49). Interestingly, transfecting siRNA specifically directed against vimentin led to an inhibition of ERK1/2 activation in the late phase of CRH stimulation (∼55%) (Figure 8A) with a similar pattern to that observed when receptor internalization is altered (compare Figure 7, A, D, and E, with Figure 8A). To discard a general and nonspecific ERK1/2 inhibition caused by low cellular levels of vimentin, we analyzed the B-Raf–independent PDGF-stimulated ERK1/2 activation pattern in HT22-CRHR1 vimentin-depleted cells. Silencing vimentin had no effect on ERK1/2 activation by PDGF (Figure 8B). These results confirm that vimentin plays a key role in ERK1/2 activation specifically elicited by CRH in mouse hippocampal HT22-CRHR1 cells.
Figure 8.
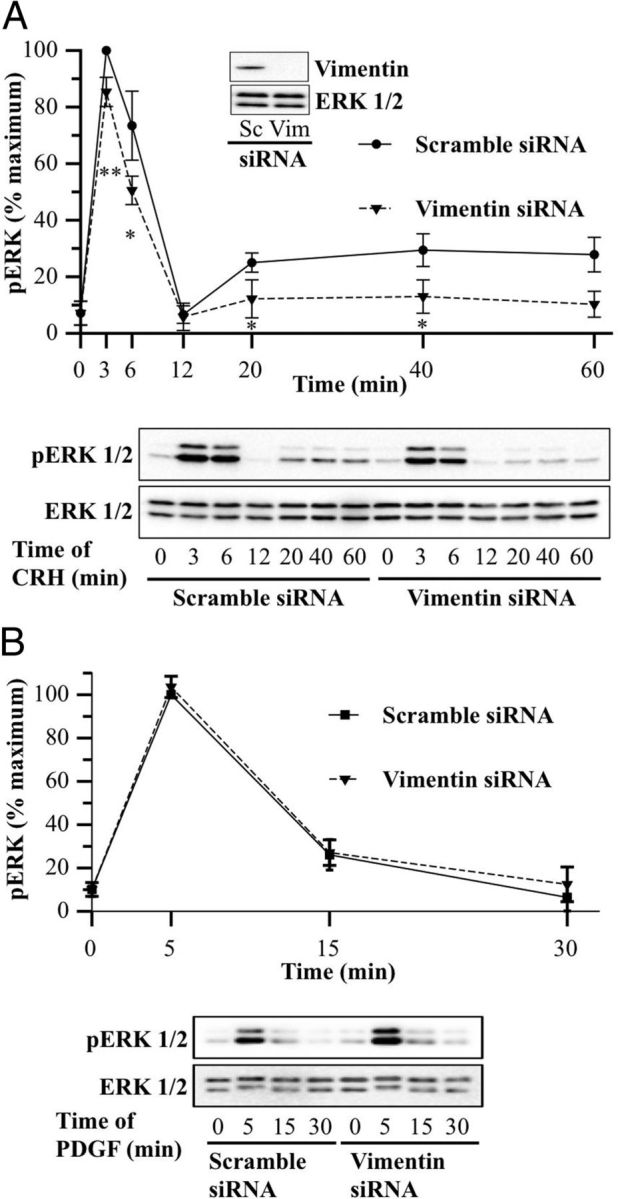
Vimentin is required for the late phase of CRH-induced ERK1/2. A and B, HT22-CRHR1 cells were transfected with 100 nM scramble or 100 nM vimentin siRNA as described in Materials and Methods. Cells were stimulated with 100 nM CRH (A) or 10 ng/ml PDGF (B) for different times. pERK1/2 and total ERK1/2 were determined by Western blot. Signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes (A) or 5 minutes (B) of stimulation in control conditions ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 and total ERK1/2 from a representative experiment. * P < .05; ** P < .01 compared with control treatment (scramble siRNA) at the same time point. Statistical analysis was performed using a one-way ANOVA. The inset in A shows a Western blot of vimentin and ERK1/2 as a loading control from a representative experiment.
Taken together, these findings provide evidence that CRH-induced ERK1/2 activation of the late phase in HT22-CRHR1 cells requires CRHR1 internalization, β-arrestin2, and vimentin expression.
Calcium in CRH signaling: role of CaMKII
Although CRHR1 mainly couples to the stimulatory Gαs protein, CRH receptors may also stimulate transient calcium (Ca2+) mobilization in certain cell types, depending on the cellular background, by PLC activation (27, 28). On the other hand, it was also described that CRH may elicit calcium entry through L-type voltage-dependent calcium channels (24, 64, 65). Because we identified CaMKII, the major calcium downstream effector, as a B-Raf partner (Table 1), we investigated the effect of CRH on intracellular calcium in HT22-CRHR1 cells. As shown in Figure 9A, CRH elevates intracellular calcium levels very shortly after stimulation (∼6 seconds). We therefore determined whether the cytoplasmic calcium signal after application of CRH depends on calcium influx from the extracellular environment. To this end, intracellular calcium was monitored in HT22-CRHR1 cells stimulated with 100 nM CRH in presence of 2 mM of the calcium chelator EGTA, to achieve depletion of extracellular calcium. In the presence of EGTA, agonist stimulation failed to induce an increase in intracellular calcium. Although cell-impermeable EGTA treatment in this assay may also induce depletion of inositol triphosphate-sensitive intracellular calcium stores, 5 μM U-73122, a potent inhibitor of PLC, did not change the measured intracellular calcium triggered by CRH (Figure 9B).
Figure 9.

Calcium and CaMKII are involved in both phases of CRH-induced ERK1/2 activation. A, HT22-CRHR1 cells were loaded with Fluo-4 AM, as described in Materials and Methods, and a time course of intracellular calcium in single HT22-CRHR1 cells in response to 100 nM CRH was performed. Fluorescence was determined as described in Materials and Methods. Results are presented as the peak change in fluorescence from resting levels ± SEM from 3 independent experiments. B, HT22-CRHR1 cells were loaded with Fluo-4 AM, as described in Materials and Methods, and cells were pretreated with EGTA (2 mM), U-73122 (5 μM), or vehicle for control for 30 minutes. Fluorescence was determined as described in Materials and Methods. Results are presented as the percentage of calcium response (peak change in fluorescence from resting levels) obtained at control conditions ± SEM from 3 independent experiments. ** P < .01 compared with the control (vehicle plus CRH). Statistical analysis was performed using a one-way ANOVA followed by post hoc Scheffé's test. C–E, HT22-CRHR1 cells were pretreated for 30 minutes with 10 μM KN-62 (C), 5 μM BAPTA-AM (D), 2 mM EGTA (E), or 5 μM U-73122 (E) for 30 minutes and stimulated with 100 nM CRH for the indicated times. Controls were preincubated with vehicle. pERK1/2, total ERK1/2, and pCaMKII were determined by Western blot. pERK1/2 and ERK1/2 signals were quantified by densitometry as described in Materials and Methods, and each value of pERK1/2 was normalized to total ERK1/2. Results are expressed as the percentage of maximum pERK1/2 obtained at 3 minutes of stimulation in the absence of inhibitor ± SEM from 3 independent experiments. Bottom panels show Western blots of pERK1/2 (top), total ERK1/2 (middle), and pCaMKII (bottom) from a representative experiment. * P < .05; ** P < .01 compared with vehicle plus CRH treatment at the same time point. Statistical analysis was performed using a one-way ANOVA followed by post hoc Scheffé's test.
Calcium triggers a variety of cellular responses including MAPK signaling. In corticotrophs, ERK1/2 activation by CRH involves L-type voltage-dependent calcium channel-dependent and calcium-independent mechanisms (24). Because calcium channel regulation by cAMP/PKA is a major effector mechanism for neurotransmitters and neuromodulators in hippocampal neurons (66), and calcium effects are mediated by CaMKII, we decided to analyze the role of this serine/threonine kinase identified in this work as a B-Raf partner (Table 1). To that end, we examined the time course of pERK1/2 in the presence or absence of KN-62, a selective CaMKII inhibitor. Interestingly, 10 μM KN-62 pretreatment significantly reduced pERK1/2 levels after CRH treatment in both phases (∼35% of early and ∼55% of late), indicating that CaMKII activity is required for CRH-induced ERK1/2 activation at early and at late time points (Figure 9C).
Upon activation, CaMKII phosphorylates a threonine residue (Thr286) in the autoinhibitory domain of the neighboring subunit of the CaMKII multimer (67). This phosphorylation prevents the autoinhibitory domain from reassociating with the kinase domain and is used as a reporter for CaMKII activation within cells. We examined by Western blot the phosphorylation status of CaMKII Thr286 (pCaMKII) after CRH stimulation. Phosphorylated CaMKII (pCaMKII) was detected early after CRH treatment and throughout the late phase of ERK1/2 activation. As expected, pCaMKII levels were severely diminished when HT22-CRHR1 cells were pretreated with KN-62 (Figure 9C). The CaMKII phosphorylation pattern and the inhibition of CRH-stimulated ERK1/2 by KN-62 suggest that an active CaMKII is required in both phases of ERK1/2 phosphorylation induced by CRH in HT22-CRHR1 cells.
Because calcium/calmodulin stimulation of CaMKII activity is important for ERK1/2 activation, then chelation of intracellular calcium should also affect pERK1/2 levels induced by CRH. HT22-CRHR1 cells were pretreated with cell-permeable calcium chelator BAPTA-AM (5 μM), and CRH-induced pERK1/2 was analyzed. Similar to the effect observed with the CaMKII inhibitor KN-62, calcium chelation affected ERK1/2 activation at early and late time points of CRH stimulation (Figure 9D). Accordingly, pCaMKII levels were also reduced in HT22-CRHR1 cells pretreated with BAPTA-AM before CRH stimulation (Figure 9D).
Therefore, we explored the main source of calcium involved in CRH-induced ERK1/2 activation. pERK1/2 levels were analyzed after 100 nM CRH stimulation in the absence or presence of 2 mM EGTA (no extracellular calcium), or 5 μM U-73122 (no PLC-dependent intracellular calcium mobilization). As shown in Figure 9D, HT22-CRHR1 cells depleted of total calcium (BAPTA-AM) showed reduced ERK1/2 activation compared with control cells. Similar results were observed when cells were depleted of extracellular calcium with EGTA (Figure 9E). Notably, CRH-induced pERK levels were similar compared with control cells, unlike glutamate-induced ERK1/2 activation that was significantly reduced when cells were preincubated with U-73122 (Supplemental Figure 4), demonstrating that CRH activation of this MAPK is independent of PLC activity (Figure 9E). In this line, the calcium chelator BAPTA-AM also affected ERK1/2 activation induced by CRH at lower concentrations (1 nM), revealing that calcium still plays a role in CRH signaling at suboptimal concentrations for Gαq coupling to CRHR1 (68) in HT22-CRHR1 cells (Supplemental Figure 5).
Taken together, these results demonstrate that CRH elevates intracellular calcium levels imported from an extracellular source and that calcium-mediated activation of CaMKII is necessary to activate ERK1/2 by CRH in HT22-CRHR1 cells.
Discussion
In this work, we describe the molecular components and cellular mechanisms involved in ERK1/2 activation by CRH in neuronal hippocampal HT22 cells. We document for the first time that ERK1/2 activation in response to CRH is biphasic in these cells (Figure 1A). This is a novel CRH-induced ERK1/2 phosphorylation pattern because a number of studies in non-neuronal cellular models did not show different waves of ERK1/2 activation (24–26, 29), and it may reflect a specific CRH response in a neuronal context. Our group and others have used AtT-20 cells to study molecular aspects of CRH signaling in the pituitary, where cAMP is involved in ERK1/2 activation induced by CRH. Moreover, our previous work demonstrated that ERK1/2 activation in corticotrophs is implicated in the expression of POMC (24). However, a study of CRH signaling in neurons of specific brain areas like the hippocampus had not been performed to date. Our results showing the involvement of at least 2 distinct activation mechanisms (one cAMP-dependent and other CRHR1 internalization-dependent) for CRH-induced ERK1/2 activation are not found in AtT-20 cells (24, 25) and may be specific to the cellular environment. Biphasic ERK1/2 activation involving different signaling pathways has been previously demonstrated for other 7 transmembrane domain receptors, although the biological basis of the differential cellular responses and its physiological consequences are far from being understood (69, 70). Our data indicate that the mechanisms and molecular components underlying the first early and second late activation of ERK1/2 are distinct. The mechanisms and molecular players reported in this work are summarized in the model depicted in Figure 10.
Figure 10.

Molecular mechanisms involved in biphasic ERK1/2 activation triggered by CRH-activated CRHR1 in hippocampal neuronal cells. Early phase is depicted on the left. Upon CRHR1 activation by CRH, the coupled Gαs protein leads to AC stimulation with a consequent increase of intracellular cAMP levels and PKA activation. In addition, CRH also triggers rapid calcium influx and activation of CaMKII. Association of PKA-activated small G proteins Rap1 and Ras to B-Raf induces conformational changes and stimulation of this MAPKKK. Phosphorylated (activated) B-Raf is represented in a complex with the interacting proteins whose role in CRH signaling is described in this work. B-Raf serine/threonine kinase activity triggers sequential phosphorylation and activation of MEK1/2 and ERK1/2 of the early phase. Late phase is depicted on the right. Shortly after stimulus, activated dynamin (Dyn) and vimentin, identified as B-Raf partners, become involved in clathrin-coated pits containing CRHR1/β-arrestin2 complexes. Binding of β-arrestin2 to agonist-occupied CRHR1 triggers receptor internalization and the assembly of a MAPK activation complex resulting in ERK1/2 activation of the late phase. Activated CaMKII is also involved in this sustained phase, suggesting its potential role in CRHR1-internalization–dependent signaling. Activated (phosphorylated) ERK1/2 (pERK1/2) generated from both mechanisms is mainly retained in the cytoplasm with very low amounts translocated to the nucleus. Cellular mechanisms and physiological consequences of the distribution of CRH-activated ERK1/2 remain still unknown.
pERK1/2 in CRH-stimulated HT22-CRHR1 cells was primarily detected at the cell surface and in the cytoplasm, with very low amounts translocated to the nucleus (Figures 2 and 3). Although ERK1/2 are common effectors of multiple signaling pathways, this pattern of activated ERK1/2 subcellular distribution was CRH-specific in these hippocampal neuronal cells and clearly different from that obtained using PDGF as stimulus. In HT22-CRHR1 cells, the pERK1/2 localization pattern triggered by CRH is consistent with a contribution of 2 pathways: a G protein-mediated pERK1/2 that is distributed in variable amounts in the cytoplasm and nucleus and a long-lasting β-arrestin–mediated pERK1/2 that is localized in membrane and cytoplasmic vesicles (71). Interestingly, immunocytochemistry and confocal analysis detected extranuclear pERK1/2 in all the specific mouse brain areas in which intracerebroventricular administered CRH activates the MAPKs (12).
In our system, internalization of the CRHR1 appears to be rapid (Figure 2) as previously observed in transfected HEK293 and primary cortical neurons (18, 52). In vivo studies in rodents have documented a relationship between CRHR1 internalization and stress in specific brain areas. Transgenic mice in which CRH overexpression was restricted to limbic structures showed that acute stress resulted in a significant reduction of CRHR1 expression in the amygdala (72), and evidence of CRHRs stress-induced trafficking was reported in rat locus coeruleus and dorsal raphe (73, 74).
Different ERK1/2 activation mechanisms downstream of CRHR1 have been described depending on the cellular model analyzed (12, 16–21, 23, 24). Classically, activated CRHRs bind the Gαs protein and activate AC, leading to an increase in intracellular cAMP. In this work, we demonstrated that AC is involved in CRH-induced ERK1/2 activation in the early phase but seems dispensable for the late ERK1/2 activation. A crucial player linking cAMP to ERK1/2 activation is the MAPKKK B-Raf. Our results provide evidence for a critical role of this MAPKKK in early ERK1/2 activation induced by CRH in HT22-CRHR1 cells. Notably, B-Raf provides specificity to CRH signaling because B-Raf silencing experiments (Figure 4, C and D) showed that this MAPKKK is not required for PDGF-activated ERK1/2, demonstrating that 2 different stimuli (CRH vs PDGF), leading to the activation of same effectors (pERK1/2), signal through different molecular components in the same cells.
Surprisingly, late ERK1/2 activation was not affected by B-Raf silencing. Activation of the ERK1/2 signaling cascade by CRHR1 is a complex process that may involve multiple mechanisms and molecular components. Sustained MAPK activation mediated by β-arrestin requires the assembly of a MAPKKK-MAPKK-MAPK-β-arrestin signaling complex (75, 76). In recent years, the fact that Raf proteins operate as homodimers and heterodimers became evident (77, 78). Although B-Raf is the most active MAPKKK, it may not be the only MAPKKK involved in CRH/CRHR1-activated ERK1/2. That scenario may explain the apparent independence of B-Raf of the late phase of CRH-induced pERK1/2 (Figure 4C) as well as the incomplete blockage of ERK1/2 activation in cells transfected with B-Raf siRNA. Moreover, the particular situation generated by silencing B-Raf may lead to a compensation response of the MAPKKK activity by other Raf family members. Indeed, it has been reported that conditional ablation of B-Raf in neuronal precursors in mice leads to increased Raf-1 recruited to MEK1 in oligodendrocytes (79). Furthermore, a recent study using single B-Raf and Raf-1 knockouts in mice showed that these 2 Raf proteins can compensate for each other in certain Raf signaling processes (80).
In our previous work, we characterized the B-Raf interactome in this hippocampal neuronal cell line under basal conditions, using MS-based proteomic analysis, as a starting point to understand MAPK pathways in neurons (49). Given the role of B-Raf in CRH signaling in HT22-CRHR1 cells, we performed a similar analysis to identify B-Raf–associated proteins in CRH-stimulated cells. To avoid biochemical artifacts from overexpression studies, B-Raf immunoprecipitations were performed under endogenous B-Raf expression conditions. Table 1 shows a list of selected B-Raf partners whose role in ERK1/2 stimulation by CRH was analyzed in the present study. The functional analysis performed on this subset of identified B-Raf partners suggests that other biomolecular interactions detected would also play a role in CRH signaling and will be the focus of future experiments.
B-Raf biological activity was found to be dependent on 14-3-3s binding (81). Our results indicate that 14-3-3 proteins are essential at B-Raf–dependent time points of ERK1/2 activation by CRH. However, because 14-3-3s play a central role in the activation process of all Raf kinases (56), the late phase of CRH-induced pERK1/2 is also expected to be regulated by 14-3-3 proteins. In line with this hypothesis, when HT22-CRHR1 cells were transfected with DN–14-3-3, both phases of ERK1/2 activation were affected (Supplemental Figure 6).
The catalytic subunit of PKA was identified as a B-Raf partner in HT22-CRHR1 cells treated with CRH (Table 1). Previous work in corticotrophs demonstrated that a rapid CRH/CRHR1-triggered ERK1/2 activation takes place downstream of the PKA (24). Our results suggest that, in neuronal cells, a similar pathway operates in the early-detected ERK1/2 activation triggered by CRH. However, although many of the cellular effects induced by cAMP require PKA activation, ERK1/2 phosphorylation could also proceed via alternative mechanisms (82). In a recent work, it was demonstrated that exchange protein activated by cAMP can also mediate CRH-induced ERK1/2 signaling in AtT-20 corticotrophs (25). That possibility may explain incomplete blockage of ERK1/2 activation in the early phase of H89-pretreated cells. Future investigations analyzing the role of exchange proteins activated by cAMP in ERK1/2 activation by CRH in hippocampal cells should be performed.
We found that 2 members of the Ras family of small G proteins, Rap1 and Ras, play a role in CRH-induced ERK1/2 activation at early time points. These results are consistent with the fact that B-Raf, unlike Raf-1, can be activated by these 2 related small G proteins via PKA-dependent mechanisms (83). CRH stimulation of the MAPK pathway involving Rap1 was previously described in corticotrophs (24). In the mouse hippocampus, both Rap1 and Ras link increments in intracellular cAMP to ERK1/2 activation, and interestingly, Rap1 is required to activate a specific membrane-associated pERK1/2 pool implicated in regulation of learning and memory (83, 84).
It is noteworthy that short times of CRH stimulation also identified molecular associations whose role was evident in the late phase without showing a remarkable contribution to the first phase of ERK1/2 activation, in which B-Raf is a key player. Besides its role as a MAPKKK, proteins of the Raf family behave as scaffolds interacting with a complex network of numerous proteins that are also engaged in dynamic multimolecular complexes. Some molecular components may be detected with B-Raf at a time that may not reflect its precise stage of action. Molecular interactions established at some point in the cell at a basal and/or stimulated situation may operate independently of each other in a specific signaling phase. One may hypothesize that an interaction between B-Raf and a given partner may be necessary for its function in a later stage. That may explain the identification of dynamin-1 and vimentin as B-Raf partners in our studies.
The intermediate filament protein vimentin is a target of a number of kinases. Vimentin was reported to interact with pERK1/2 and to protect these activated MAPKs from dephosphorylation in vitro (85). In our work, vimentin was detected in B-Raf immunoprecipitates at basal and after CRH stimulation. The association of vimentin and vimentin kinases with Raf-1 has been described, and vimentin phosphorylation was found to be dependent on activated Raf-1 (86). B-Raf may act similarly to Raf-1 and regulate vimentin function by promoting specific phosphorylations that are required for vimentin activation. The β-arrestin2–dependent (late) phase of pERK1/2 triggered by CRH may require activated vimentin that was generated at early time points of CRH stimulation. Interestingly, vimentin was found to potently stimulate the dynamin GTPase activity (87) and was identified, as well as dynamin-1, as a β-arrestin2–interacting partner (88).
Our results demonstrate that the late phase of CRH-induced ERK1/2 activation depends on receptor internalization. In addition to leading to an almost complete blockage of the late phase, alteration of receptor internalization also partially affected the early phase of CRH-induced ERK1/2 activation. Because internalization of the CRHR1 initiates shortly after stimulus in HT22-CRHR1 cells, an overlap of ERK1/2 activation by both mechanisms may contribute to the pERK1/2 signal detected at the initial time points analyzed, although the G protein-dependent pathway is the principal mechanism that activates ERK1/2 at the early phase.
The internalization of many GPCRs occurs primarily via clathrin-coated pits involving proteins of the arrestin family with a crucial role, β-arrestins 1 and 2 (89). Notably, in HEK293 cells and primary cortical neurons transfected with CRHR1, the CRH-activated receptor was found to interact selectively with β-arrestin2 (52). Indeed, we observed that when HT22-CRHR1 cells are depleted of β-arrestin2, the second phase of CRH-induced ERK1/2 activation is affected. These findings suggest that controlling the fate of internalized CRHR1 (recycling to the cell surface or lysosomal degradation) might represent a new level of regulation of CRHR1 signaling. The study of these new mechanisms of signaling regulation as well as the temporal aspects of receptor resensitization and turnover will provide important insight in our understanding of CRHR1 signaling in neurons and should be further explored in the future. Specifically, unraveling the mechanism of how CRHR1 internalizes might result in unprecedented targets that can be used to reduce pathologically relevant enhancement of CRH/CRHR1 signaling.
CRH signaling through Ca2+ has been documented in many cellular systems. Previous reports have shown that CRHR1 stimulates calcium mobilization in HEK-293 cells but not in human neuroblastoma SK-N-MC cells, demonstrating once again the relevance of the cellular context (27, 28). In this line, we confirmed that CRH does not stimulate calcium mobilization in the neuronal HT22-CRHR1 cells, but instead, CRH stimulates the ion influx from the extracellular environment, as previously observed (24) for AtT-20 corticotrophs (Figure 9B).
The cross talk between calcium and ERK1/2 pathways has been studied in detail (90, 91). Our results demonstrate that calcium influx and CaMKII are essential to activate the B-Raf–mediated (early) and the β-arrestin–mediated (late) phase of CRH-induced ERK1/2 activation (Figure 9). CaMKII association with Raf kinases regulating their activity is already known (90). In our system, CaMKII is a B-Raf partner and may be affecting early pERK1/2 levels by regulating its MAPKKK activity. However, a CaMKII role in the late phase of ERK1/2 activation may be different. CaMKII is known to bind to β-arrestins (88), and an essential role for β-arrestin in mediating CaMKII signaling after β1-adrenoreceptor stimulation has been shown (92). Interestingly, in the same line, the interaction of calmodulin, a critical factor needed for CaMKII activation, with the 5-HT2C receptor has been found to be necessary for β-arrestin–dependent ERK1/2 signaling (93). In our system, CaMKII may be part of a complex with β-arrestins that could translocate to agonist-occupied CRHR1 on the plasma membrane and trigger β-arrestin2–dependent (late phase) ERK1/2 activation induced by CRH.
In conclusion, in this work, we describe for the first time a comprehensive study on signaling mechanisms and molecular components involved in CRH-induced ERK1/2 activation in neuronal cells. Our results provide novel evidence that CRH activates ERK1/2 through different pathways involving different intracellular molecules that may become potential targets to develop therapies for affective disorders such as depression and anxiety. Given the importance of modulating CRHR1 responses in pathological situations, our approach constitutes a basis to design therapies for disorders caused by an impaired CRH/CRHR1 system.
Acknowledgments
We thank J. Deussing for providing pcDNA-c-Myc-mCRHR1 plasmid, J. Benovic for providing dynamin-K44A and β-arrestin (318-418) plasmids, O. Coso for providing Rap1A-17N and Ras-17N plasmids, A. S. Shaw for providing DN 14-3-3η plasmid, C. Shayo for sharing plasmids and reagents, and C. Davio for advice and reagents for experiments of cAMP measurements.
This work was supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT-Argentina, PICT-Max-Planck 06:205 and 10:2791) to S.S. and C.W.T., the Max Planck Society, the University of Buenos Aires (UBA) and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) (National Research Council, Argentina). A National Alliance for Research on Schizophrenia and Depression (NARSAD) young Investigator award also supported D.R. J.J.B. is a graduate student of UBA, supported by a fellowship from CONICET.
Disclosure Summary: The authors declare no conflict of interest.
Footnotes
- AC
- adenylyl cyclase
- BAPTA-AM
- 1,2-Bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid tetrakis(acetoxymethyl ester)
- CaMKII
- calcium/calmodulin-dependent protein kinase
- CNS
- central nervous system
- CRHR1
- CRH receptor 1
- CRHR2
- CRH receptor 2
- 2′,5′–ddA
- 2′,5′-dideoxyadenosine
- DN
- dominant-negative
- FCS
- fetal calf serum
- GPCR
- G protein-coupled receptor
- MAPKKK
- MAP kinase kinase kinase
- MDC
- monodansylcadaverine
- MEK
- MAPK kinase
- MS
- mass spectrometry
- PAO
- phenylarsine oxide
- PDGF
- platelet-derived growth factor
- pERK
- phospho-ERK
- PKA
- protein kinase A
- PLC
- phospholipase C
- siRNA
- small interfering RNA.
References
- 1. Vale W , Spiess J , Rivier C , Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. [DOI] [PubMed] [Google Scholar]
- 2. de Kloet ER , Joels M , Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. [DOI] [PubMed] [Google Scholar]
- 3. Smith SM , Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci. 2006;8:383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gallagher JP , Orozco-Cabal LF , Liu J , Shinnick-Gallagher P. Synaptic physiology of central CRH system. Eur J Pharmacol. 2008;583:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arborelius L , Owens MJ , Plotsky PM , Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. [DOI] [PubMed] [Google Scholar]
- 6. Holsboer F. The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. J Psychiatr Res. 1999;33:181–214. [DOI] [PubMed] [Google Scholar]
- 7. Holsboer F , Ising M. Stress hormone regulation: biological role and translation into therapy. Annu Rev Psychol 2010;61:81–109, C1–C11. [DOI] [PubMed] [Google Scholar]
- 8. Grigoriadis DE. The corticotropin-releasing factor receptor: a novel target for the treatment of depression and anxiety-related disorders. Expert Opin Ther Targets. 2005;9:651–684. [DOI] [PubMed] [Google Scholar]
- 9. Holsboer F , Ising M. Central CRH system in depression and anxiety: evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol. 2008;583:350–357. [DOI] [PubMed] [Google Scholar]
- 10. Smith GW , Aubry JM , Dellu F , et al. . Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. [DOI] [PubMed] [Google Scholar]
- 11. Muller MB , Zimmermann S , Sillaber I , et al. . Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci. 2003;6:1100–1107. [DOI] [PubMed] [Google Scholar]
- 12. Refojo D , Echenique C , Muller MB , et al. . Corticotropin-releasing hormone activates ERK1/2 MAPK in specific brain areas. Proc Natl Acad Sci U S A. 2005;102:6183–6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang XD , Chen Y , Wolf M , et al. . Forebrain CRHR1 deficiency attenuates chronic stress-induced cognitive deficits and dendritic remodeling. Neurobiol Dis. 2011;42:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Refojo D , Schweizer M , Kuehne C , et al. . Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333:1903–1907. [DOI] [PubMed] [Google Scholar]
- 15. Bonfiglio JJ , Inda C , Refojo D , Holsboer F , Arzt E , Silberstein S. The corticotropin-releasing hormone network and the hypothalamic-pituitary-adrenal axis: molecular and cellular mechanisms involved. Neuroendocrinology. 2011;94:12–20. [DOI] [PubMed] [Google Scholar]
- 16. Hillhouse EW , Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev. 2006;27:260–286. [DOI] [PubMed] [Google Scholar]
- 17. Dautzenberg FM , Hauger RL. The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci. 2002;23:71–77. [DOI] [PubMed] [Google Scholar]
- 18. Punn A , Levine MA , Grammatopoulos DK. Identification of signaling molecules mediating corticotropin-releasing hormone-R1α-mitogen-activated protein kinase (MAPK) interactions: the critical role of phosphatidylinositol 3-kinase in regulating ERK1/2 but not p38 MAPK activation. Mol Endocrinol. 2006;20:3179–3195. [DOI] [PubMed] [Google Scholar]
- 19. Arzt E , Holsboer F. CRF signaling: molecular specificity for drug targeting in the CNS. Trends Pharmacol Sci. 2006;27:531–538. [DOI] [PubMed] [Google Scholar]
- 20. Teli T , Markovic D , Hewitt ME , Levine MA , Hillhouse EW , Grammatopoulos DK. Structural domains determining signalling characteristics of the CRH-receptor type 1 variant R1β and response to PKC phosphorylation. Cell Signal. 2008;20:40–49. [DOI] [PubMed] [Google Scholar]
- 21. Grammatopoulos DK , Randeva HS , Levine MA , Katsanou ES , Hillhouse EW. Urocortin, but not corticotropin-releasing hormone (CRH), activates the mitogen-activated protein kinase signal transduction pathway in human pregnant myometrium: an effect mediated via R1α and R2β CRH receptor subtypes and stimulation of Gq-proteins. Mol Endocrinol. 2000;14:2076–2091. [DOI] [PubMed] [Google Scholar]
- 22. Cao J , Cetrulo CL , Theoharides TC. Corticotropin-releasing hormone induces vascular endothelial growth factor release from human mast cells via the cAMP/protein kinase A/p38 mitogen-activated protein kinase pathway. Mol Pharmacol. 2006;69:998–1006. [DOI] [PubMed] [Google Scholar]
- 23. Cibelli G , Corsi P , Diana G , Vitiello F , Thiel G. Corticotropin-releasing factor triggers neurite outgrowth of a catecholaminergic immortalized neuron via cAMP and MAP kinase signalling pathways. Eur J Neurosci. 2001;13:1339–1348. [DOI] [PubMed] [Google Scholar]
- 24. Kovalovsky D , Refojo D , Liberman AC , et al. . Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol Endocrinol. 2002;16:1638–1651. [DOI] [PubMed] [Google Scholar]
- 25. Van Kolen K , Dautzenberg FM , Verstraeten K , et al. . Corticotropin releasing factor-induced ERK phosphorylation in AtT20 cells occurs via a cAMP-dependent mechanism requiring EPAC2. Neuropharmacology. 2010;58:135–144. [DOI] [PubMed] [Google Scholar]
- 26. Huising MO , van der Meulen T , Vaughan JM , et al. . CRFR1 is expressed on pancreatic beta cells, promotes beta cell proliferation, and potentiates insulin secretion in a glucose-dependent manner. Proc Natl Acad Sci U S A. 2010;107:912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dautzenberg FM , Gutknecht E , Van der Linden I , Olivares-Reyes JA , Durrenberger F , Hauger RL. Cell-type specific calcium signaling by corticotropin-releasing factor type 1 (CRF1) and 2a (CRF2(a)) receptors: phospholipase C-mediated responses in human embryonic kidney 293 but not SK-N-MC neuroblastoma cells. Biochem Pharmacol. 2004;68:1833–1844. [DOI] [PubMed] [Google Scholar]
- 28. Gutknecht E , Van der Linden I , Van Kolen K , Verhoeven KF , Vauquelin G , Dautzenberg FM. Molecular mechanisms of corticotropin-releasing factor receptor-induced calcium signaling. Mol Pharmacol. 2009;75:648–657. [DOI] [PubMed] [Google Scholar]
- 29. Wu SV , Yuan PQ , Lai J , et al. . Activation of Type 1 CRH receptor isoforms induces serotonin release from human carcinoid BON-1N cells: an enterochromaffin cell model. Endocrinology. 2011;152:126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ortiz J , Harris HW , Guitart X , Terwilliger RZ , Haycock JW , Nestler EJ. Extracellular signal-regulated protein kinases (ERKs) and ERK kinase (MEK) in brain: regional distribution and regulation by chronic morphine. J Neurosci. 1995;15:1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Benedetto B , Hitz C , Holter SM , Kuhn R , Vogt Weisenhorn DM , Wurst W. Differential mRNA distribution of components of the ERK/MAPK signalling cascade in the adult mouse brain. J Comp Neurol. 2007;500:542–556. [DOI] [PubMed] [Google Scholar]
- 32. Thomas GM , Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. [DOI] [PubMed] [Google Scholar]
- 33. Samuels IS , Saitta SC , Landreth GE. MAP'ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davis S , Laroche S. Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: a review. Genes Brain Behav. 2006;5(Suppl 2):61–72. [DOI] [PubMed] [Google Scholar]
- 35. Meller E , Shen C , Nikolao TA , et al. . Region-specific effects of acute and repeated restraint stress on the phosphorylation of mitogen-activated protein kinases. Brain Res. 2003;979:57–64. [DOI] [PubMed] [Google Scholar]
- 36. Qi X , Lin W , Li J , Pan Y , Wang W. The depressive-like behaviors are correlated with decreased phosphorylation of mitogen-activated protein kinases in rat brain following chronic forced swim stress. Behav Brain Res. 2006;175:233–240. [DOI] [PubMed] [Google Scholar]
- 37. Einat H , Yuan P , Gould TD , et al. . The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23:7311–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ailing F , Fan L , Li S , Manji S. Role of extracellular signal-regulated kinase signal transduction pathway in anxiety. J Psychiatr Res. 2008;43:55–63. [DOI] [PubMed] [Google Scholar]
- 39. Todorovic C , Sherrin T , Pitts M , Hippel C , Rayner M , Spiess J. Suppression of the MEK/ERK signaling pathway reverses depression-like behaviors of CRF2-deficient mice. Neuropsychopharmacology. 2009;34:1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu J , Li L , Suo WZ. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009;84:267–271. [DOI] [PubMed] [Google Scholar]
- 41. Aminova LR , Chavez JC , Lee J , et al. . Prosurvival and prodeath effects of hypoxia-inducible factor-1α stabilization in a murine hippocampal cell line. J Biol Chem. 2005;280:3996–4003. [DOI] [PubMed] [Google Scholar]
- 42. Stanciu M , Wang Y , Kentor R , et al. . Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. [DOI] [PubMed] [Google Scholar]
- 43. Davis JB , Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–173. [DOI] [PubMed] [Google Scholar]
- 44. Caldwell JD , Shapiro RA , Jirikowski GF , Suleman F. Internalization of sex hormone-binding globulin into neurons and brain cells in vitro and in vivo. Neuroendocrinology. 2007;86:84–93. [DOI] [PubMed] [Google Scholar]
- 45. Li Y , Maher P , Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. [DOI] [PubMed] [Google Scholar]
- 46. Luo Y , DeFranco DB. Opposing roles for ERK1/2 in neuronal oxidative toxicity: distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J Biol Chem. 2006;281:16436–16442. [DOI] [PubMed] [Google Scholar]
- 47. van Leyen K , Siddiq A , Ratan RR , Lo EH. Proteasome inhibition protects HT22 neuronal cells from oxidative glutamate toxicity. J Neurochem. 2005;92:824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tobaben S , Grohm J , Seiler A , Conrad M , Plesnila N , Culmsee C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 2011;18:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bonfiglio JJ , Maccarrone G , Rewerts C , et al. . Characterization of the B-Raf interactome in mouse hippocampal neuronal cells. J Proteomics. 2011;74:186–198. [DOI] [PubMed] [Google Scholar]
- 50. Perkins DN , Pappin DJ , Creasy DM , Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. [DOI] [PubMed] [Google Scholar]
- 51. Davio CA , Cricco GP , Bergoc RM , Rivera ES. H1 and H2 histamine receptors in N-nitroso-N-methylurea (NMU)-induced carcinomas with atypical coupling to signal transducers. Biochem Pharmacol. 1995;50:91–96. [DOI] [PubMed] [Google Scholar]
- 52. Holmes KD , Babwah AV , Dale LB , Poulter MO , Ferguson SS. Differential regulation of corticotropin releasing factor 1α receptor endocytosis and trafficking by β-arrestins and Rab GTPases. J Neurochem. 2006;96:934–949. [DOI] [PubMed] [Google Scholar]
- 53. Vossler MR , Yao H , York RD , Pan MG , Rim CS , Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. [DOI] [PubMed] [Google Scholar]
- 54. Ritt DA , Zhou M , Conrads TP , Veenstra TD , Copeland TD , Morrison DK. CK2 Is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol. 2007;17:179–184. [DOI] [PubMed] [Google Scholar]
- 55. Brummer T , Martin P , Herzog S , Misawa Y , Daly RJ , Reth M. Functional analysis of the regulatory requirements of B-Raf and the B-Raf(V600E) oncoprotein. Oncogene. 2006;25:6262–6276. [DOI] [PubMed] [Google Scholar]
- 56. Tzivion G , Luo Z , Avruch J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature. 1998;394:88–92. [DOI] [PubMed] [Google Scholar]
- 57. Fischer A , Baljuls A , Reinders J , et al. . Regulation of RAF activity by 14-3-3 proteins: RAF kinases associate functionally with both homo- and heterodimeric forms of 14-3-3 proteins. J Biol Chem. 2009;284:3183–3194. [DOI] [PubMed] [Google Scholar]
- 58. Thorson JA , Yu LW , Hsu AL , et al. . 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol Cell Biol. 1998;18:5229–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grewal SS , Horgan AM , York RD , Withers GS , Banker GA , Stork PJ. Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase. J Biol Chem. 2000;275:3722–3728. [DOI] [PubMed] [Google Scholar]
- 60. Stork PJ. Does Rap1 deserve a bad Rap? Trends Biochem Sci. 2003;28:267–275. [DOI] [PubMed] [Google Scholar]
- 61. McKay MM , Ritt DA , Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci U S A. 2009;106:11022–11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pierce KL , Maudsley S , Daaka Y , Luttrell LM , Lefkowitz RJ. Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc Natl Acad Sci U S A. 2000;97:1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lefkowitz RJ , Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. [DOI] [PubMed] [Google Scholar]
- 64. Luini A , Lewis D , Guild S , Corda D , Axelrod J. Hormone secretagogues increase cytosolic calcium by increasing cAMP in corticotropin-secreting cells. Proc Natl Acad Sci U S A. 1985;82:8034–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee AK , Tse A. Mechanism underlying corticotropin-releasing hormone (CRH) triggered cytosolic Ca2+ rise in identified rat corticotrophs. J Physiol. 1997;504(Pt 2):367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lancaster B , Hu H , Gibb B , Storm JF. Kinetics of ion channel modulation by cAMP in rat hippocampal neurones. J Physiol. 2006;576:403–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Means AR. Regulatory cascades involving calmodulin-dependent protein kinases. Mol Endocrinol. 2000;14:4–13. [DOI] [PubMed] [Google Scholar]
- 68. Punn A , Chen J , Delidaki M , et al. . Mapping structural determinants within third intracellular loop that direct signaling specificity of type 1 corticotropin-releasing hormone receptor. J Biol Chem. 2012;287:8974–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Murphy LO , Smith S , Chen RH , Fingar DC , Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. [DOI] [PubMed] [Google Scholar]
- 70. Asimaki O , Mangoura D. Cannabinoid receptor 1 induces a biphasic ERK activation via multiprotein signaling complex formation of proximal kinases PKCϵ, Src, and Fyn in primary neurons. Neurochem Int. 2011;58:135–144. [DOI] [PubMed] [Google Scholar]
- 71. Ahn S , Shenoy SK , Wei H , Lefkowitz RJ. Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. [DOI] [PubMed] [Google Scholar]
- 72. Silberstein S , Vogl AM , Refojo D , et al. . Amygdaloid pERK1/2 in corticotropin-releasing hormone overexpressing mice under basal and acute stress conditions. Neuroscience. 2009;159:610–617. [DOI] [PubMed] [Google Scholar]
- 73. Bangasser DA , Curtis A , Reyes BA , et al. . Sex differences in corticotropin-releasing factor receptor signaling and trafficking: potential role in female vulnerability to stress-related psychopathology. Mol Psychiatry. 2010;15:877, 896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Waselus M , Nazzaro C , Valentino RJ , Van Bockstaele EJ. Stress-induced redistribution of corticotropin-releasing factor receptor subtypes in the dorsal raphe nucleus. Biol Psychiatry. 2009;66:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Song X , Coffa S , Fu H , Gurevich VV. How does arrestin assemble MAPKs into a signaling complex? J Biol Chem. 2009;284:685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Luttrell LM , Roudabush FL , Choy EW , et al. . Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci U S A. 2001;98:2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wimmer R , Baccarini M. Partner exchange: protein-protein interactions in the Raf pathway. Trends Biochem Sci. 2010;35:660–668. [DOI] [PubMed] [Google Scholar]
- 78. Rushworth LK , Hindley AD , O'Neill E , Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26:2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Galabova-Kovacs G , Catalanotti F , Matzen D , et al. . Essential role of B-Raf in oligodendrocyte maturation and myelination during postnatal central nervous system development. J Cell Biol. 2008;180:947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Valluet A , Druillennec S , Barbotin C , et al. . B-raf and C-raf are required for melanocyte stem cell self-maintenance. Cell Rep. 2012;2:774–780. [DOI] [PubMed] [Google Scholar]
- 81. MacNicol MC , Muslin AJ , MacNicol AM. Disruption of the 14-3-3 binding site within the B-Raf kinase domain uncouples catalytic activity from PC12 cell differentiation. J Biol Chem. 2000;275:3803–3809. [DOI] [PubMed] [Google Scholar]
- 82. Stork PJ , Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. [DOI] [PubMed] [Google Scholar]
- 83. Obara Y , Horgan AM , Stork PJ. The requirement of Ras and Rap1 for the activation of ERKs by cAMP, PACAP, and KCl in cerebellar granule cells. J Neurochem. 2007;101:470–482. [DOI] [PubMed] [Google Scholar]
- 84. Morozov A , Muzzio IA , Bourtchouladze R , et al. . Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron. 2003;39:309–325. [DOI] [PubMed] [Google Scholar]
- 85. Perlson E , Michaelevski I , Kowalsman N , et al. . Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J Mol Biol. 2006;364:938–944. [DOI] [PubMed] [Google Scholar]
- 86. Janosch P , Kieser A , Eulitz M , et al. . The Raf-1 kinase associates with vimentin kinases and regulates the structure of vimentin filaments. FASEB J. 2000;14:2008–2021. [DOI] [PubMed] [Google Scholar]
- 87. Vallee RB , Okamoto PM. The regulation of endocytosis: identifying dynamin's binding partners. Trends Cell Biol. 1995;5:43–47. [DOI] [PubMed] [Google Scholar]
- 88. Xiao K , McClatchy DB , Shukla AK , et al. . Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A. 2007;104:12011–12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. DeWire SM , Ahn S , Lefkowitz RJ , Shenoy SK. β-Arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. [DOI] [PubMed] [Google Scholar]
- 90. Illario M , Cavallo AL , Bayer KU , et al. . Calcium/calmodulin-dependent protein kinase II binds to Raf-1 and modulates integrin-stimulated ERK activation. J Biol Chem. 2003;278:45101–45108. [DOI] [PubMed] [Google Scholar]
- 91. Cipolletta E , Monaco S , Maione AS , Vitiello L , Campiglia P , Pastore L , Franchini C , Novellino E , Limongelli V , Bayer KU , Means AR , Rossi G , Trimarco B , Iaccarino G , Illario M. Calmodulin-dependent kinase II mediates vascular smooth muscle cell proliferation and is potentiated by extracellular signal regulated kinase. Endocrinology. 2010;151:2747–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mangmool S , Shukla AK , Rockman HA. β-Arrestin-dependent activation of Ca2+/calmodulin kinase II after β1-adrenergic receptor stimulation. J Cell Biol. 2010;189:573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Labasque M , Reiter E , Becamel C , Bockaert J , Marin P. Physical interaction of calmodulin with the 5-hydroxytryptamine2C receptor C terminus is essential for G protein-independent, arrestin-dependent receptor signaling. Mol Biol Cell. 2008;19:4640–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]