

Abstract
Prostate tumor growth initially depends on androgens, which act via the androgen receptor (AR). Despite androgen ablation therapy, tumors eventually progress to a castrate-resistant stage in which the AR remains active. The mechanisms are poorly understood but it may be that changes in levels or activity of AR coregulators affect trafficking and activation of the receptor. A key stage in AR signaling occurs in the cytoplasm, where unliganded receptor is associated with the heat shock protein (HSP)90 foldosome complex. p23, a key component of this complex, is best characterized as a cochaperone for HSP90 but also has HSP90-independent activity and has been reported as having differential effects on the activity of different steroid receptors. Here we report that p23 increases activity of the AR, and this appears to involve steps both in the cytoplasm (increasing ligand-binding capacity, possibly via direct interaction with AR) and the nucleus (enhancing AR occupancy at target promoters). We show, for the first time, that AR and p23 can interact, perhaps directly, when HSP90 is not present in the same complex. The effects of p23 on AR activity are at least partly HSP90 independent because a mutant form of p23, unable to bind HSP90, nevertheless increases AR activity. In human prostate tumors, nuclear p23 was higher in malignant prostate cells compared with benign/normal cells, supporting the utility of p23 as a therapeutic target in prostate cancer.
A variety of transcription factors involved in signal transduction are associated with heat shock protein 90 (HSP90), which is in many instances essential for their proper functioning. Signaling pathways that require HSP90 include the steroid classes of the nuclear receptor superfamily (1). Steroid receptors, including the androgen receptor (AR), glucocorticoid receptor (GR), progesterone receptor (PR), and estrogen receptor (ER), are associated with large multiprotein complexes that include HSP90 (1–3). The steroid receptor-HSP90 heterocomplex assembly system involves the transient association of various chaperones including the constitutive and stress-inducible members of the HSP70 family, HSP40, HSP90, and HSP70-organizing protein (HOP), p23, BAG1L, and immunophilins (FKBP51 and FKBP52) (4–9). From these early studies the concept of a foldosome complex emerged: a complex that mediates the assembly of a biologically active protein by regulating its correct folding (10). It was previously believed that HSP were exclusively cytoplasmic and dissociated from steroid receptors before nuclear localization. However, more recent studies have shown nuclear localization and function of several HSP and chaperones, including p23, in certain cell lines (11, 12). The functional properties of p23 have been previously investigated in relation to the ER, GR, and PR (12–14) whereas its role in AR signaling in mammalian cells is largely uncharacterized. Further, the effect of p23 on activity of different steroid receptors is a matter of debate: for instance, it has been reported as both repressing and increasing activity of GR and ER in different studies (12–15). Here we show that p23 interacts with AR and potentiates AR activity and, unlike the situation for the other steroid receptors, this appears to occur, at least in part, independently of HSP90.
The AR shares a common domain structure with other steroid receptors: an N-terminal activation domain containing activation function 1, a central DNA-binding domain (DBD), and a C-terminal ligand binding domain (LBD) that also contains activation function 2. Upon binding ligand, steroid receptors adopt an active conformation, translocate to the nucleus, and bind to sequence-specific response elements in the regulatory regions of target genes. At these target sites, ligand-activated steroid receptors recruit accessory proteins (cofactors) to regulate transcription (16). The conformational changes that generate a high-ligand-binding affinity steroid receptor molecule require an orchestrated signaling cascade initiated by the foldosome complex; p23 and HSP90 are the final components that stabilize the unliganded steroid receptor complex (2, 9, 17, 18). p23 is a 23-kDa ubiquitously expressed acidic protein that binds directly to HSP90 via its highly conserved and structured amino-terminal domain (10, 19). Its carboxyl-terminal domain (CTD) is largely unstructured and flexible and contains chaperone activity capable of preventing aggregation of denatured proteins (20, 21). p23 has been found to complex with a variety of HSP90 substrates and to have cytoplasmic prostaglandin synthase activity (22) as well as a role in regulating endoplasmic reticulum stress-induced cell death (23). What remains unclear is to what extent p23 can exert effects independently of HSP90. Biochemical studies have demonstrated that in the absence of HSP90, p23 can still suppress the aggregation of denatured proteins (20, 24, 25).
Androgen signaling is a key pathway in the development and treatment of prostate cancer: androgens drive prostate tumor growth; hence many therapies are aimed at reducing synthesis of circulating androgens and/or inhibiting the AR itself (26). HSP90 inhibitors, which induce degradation of client proteins, have been shown to demonstrate antitumor effects, notably in hormone-dependent tumors such as breast and prostate cancer (27, 28). A derivative of the HSP90-specific inhibitor geldanamycin (GA), 17-allylaminogeldanamycin (17-AAG or Tanespimycin), is currently undergoing clinical trials for solid tumors and advanced malignancies (29). Because p23 regulates the activity of HSP90 (30), it could also be a target to reduce HSP90 activity within the tumor microenvironment. In support of this, deletion of the p23 gene in yeast confers hypersensitivity to GA (31), whereas p23 itself is up-regulated in primary tumors and metastases (32–35). Here, we show that nuclear p23 levels are increased in prostate tumors and that p23 interacts with the AR and increases AR activity by increasing both ligand binding capacity and DNA binding, with functional consequences on expression of androgen-regulated genes. The effect of p23 on AR signaling appears to be largely, but not wholly, independent of its interaction with HSP90. This supports the hypothesis that p23 may be a therapeutic target in prostate cancer, in its own right, and/or an effective secondary therapeutic target for HSP90 inhibition.
Materials and Methods
Plasmid construction
All constructs were created by PCR amplification and verified by sequencing. The following plasmids have been described previously: pSG5-SRC-1e (36), pSVARΔLBD (amino acids 1–653) (37), pGAD424-ARNTD, and pGAD424-ARLBD (38). The following were kind gifts: pSVAR from A. Brinkmann (Rotterdam, The Netherlands), TAT-GRE-EIB-Luc from G. Jenster (Rotterdam, The Netherlands), mouse mammary tumor virus (MMTV)-Luc from P. Chambon (Illkirch, France) and pRK5-p23W106A from T. Rein (Munich, Germany) (39). p23 cDNA was cloned into the pSG5 expression vector from an LNCaP cDNA library and then subcloned in frame with a V5 tag using primers 5′-CCCGGATCCGACATGGACGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGATGCAGCCTGCTTCTGCAAAGTGG-3′ and 5′-CCCCTCGAGTTACTCCAGATCTGG-3′, also in frame with the LexA-DBD in the yeast expression vector pBTM116 (CLONTECH Laboratories, Palo Alto, CA), in frame with glutathione-S-transferase (GST) in pGEX-6P-1 (GE Healthcare, Piscataway, NJ) and in frame with the Gal4 DBD in pMGal4 (CLONTECH). Full-length wild-type p23 and p23W106A were each subcloned into the expression vectors pcDNA4TO and pSG5 in frame with V5 and 2x FLAG tag, respectively. The NH3 domain of p23 (a.a 1–86) was subcloned in frame with GST in pGEX-6P-1 (GE Healthcare). The AR N-terminal domain (NTD) and C-terminal domain (CTD) were cloned in frame with the Gal4-AD in the yeast vector pACT2 (CLONTECH).
Cell culture
MCF-7 and COS-1 cells were cultured in DMEM (Sigma Chemical Co., St. Louis, MO) supplemented with 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mmol/liter glutamine (Sigma) and 10% fetal bovine serum (Labtech International, Ringmer, East Sussex, UK). PC3 and LNCaP cells were cultured in RPMI-1640 medium (Sigma), supplemented as above. PC3wtAR cells (40) and LNCaP-TR2-MAR4 cells (41) were additionally supplemented with 4 μg/ml of Geneticin (Life Technologies, Gaithersburg, MD), and the LNCaP-TR2-MAR4 cells were grown in certified tetracycline-free medium (Firstlink UK, Sheffield, UK). For androgen regulation experiments, cells were cultured in starvation media, comprising phenol red-free DMEM or RPMI (Sigma) supplemented as above but with 5% charcoal-stripped fetal bovine serum (Labtech International).
Reporter assay
Cells grown to 70% confluency in 24-well plates were cultured in the relevant starvation media for 24 h and then transfected using FuGENE6 (Roche Diagnostics, Indianapolis, IN), following the manufacturers' instructions. The transfected DNA per well included TAT-GRE-E1B-Luc or MMTV-Luc (500 ng), pdmLacZ-β-Gal (250 ng), pSVAR or pSVAR-ΔLBD (50 ng, only in the AR-negative cell lines), pSG5-SRC1e (200 ng), pSG5-p23 (50–200 ng) and pSG5 control plasmid to standardize the amounts of DNA. After incubation for 16 h, cells were washed, placed in the corresponding starvation media, and treated with hormone [mibolerone (MB) (PerkinElmer Life Sciences, Wellesley, MA) or dexamethasone (Sigma)] or equivalent volume of vehicle (ethanol) for 24 h. Luciferase data were normalized to β-galactosidase activity, as previously described (42).
Yeast culture, transformation and two-hybrid assay
The Saccharomyces cerevisiae L40 yeast strain [MAT, Trp1, His3, Leu2, Ade2, LYS::(LexAop) 4-HIS3, URA3:(LexAop) 8-LacZ] containing LexA-responsive LacZ reporter (Stratagene, La Jolla, CA) was maintained on rich media plates (yeast extract peptone dextrose) (Anachem Ltd., Luton, UK) excluding selection. A single yeast colony was inoculated into 100 ml of minimal yeast media with the appropriate selection (−Leu, −Trp, or −Leu/−Trp drop-out medium) and grown overnight to mid-log phase at 30 C. After transformation using the Alkali-Cation Yeast Transformation Kit (Anachem), transformed yeast cells were grown to late log phase (overnight at 30 C) in 10 ml of Minimal SD Base Selective media (CLONTECH; BD Bioscience, Palo Alto, CA) with the appropriate selection and MB or vehicle where appropriate. The β-galactosidase assay was carried out as previously described (43). β-Galactosidase reading was measured at OD400, and cell growth was measured at OD600. β-Galactosidase was measured at OD400, cell growth was measured at OD600, and activity was calculated as Miller units: (1000 × OD400)/ (OD600 × reaction time).
Sulforhodamine B assay
MCF7 cells were grown to 70% confluency in 24-well plates, medium was changed to starvation media for 48 h and treated with 10 nm MB and/or 1 nm, 10 nm, and 100 nm of GA added for 16 h. The sulforhodamine assay was performed as described elsewhere (44).
Scatchard analysis
COS-1 cells were plated in 24-well plates, transfected using FuGENE 6 (Roche Diagnostics) with 50 ng pSVAR and 0, 100, or 200 ng pSG5-p23 per well and left for 24 h before addition of 10 nm GA for 2 h. Media were changed and 0.125, 0.25, 0.5, 1, 2 or 4 nm [3H]MB with or without a 200-fold excess of unlabeled MB was added. After 1 h incubation at 37 C, cells were washed three times in ice-cold PBS and lysed in 100% ethanol. Lysates were mixed with scintillation fluid (Hyonic Fluor) and read on a scintillation counter (Beckman Coulter Ltd, Buckinghamshire, UK). Specific binding was calculated and maximum binding capacities (Bmax) were calculated using Prism software (GraphPad Software, San Diego, CA).
Immunohistochemistry
A prostate tissue microarray (A302II, AccuMax, ISU ABXIS Co. Ltd, Seoul, Korea) containing 32 samples of prostate cancer tissue with corresponding normal tissues was stained for p23. Slides were rehydrated in decreasing concentrations of ethanol, and endogenous peroxidase activity was blocked using 3% hydrogen peroxide. Antigen retrieval was carried out by microwaving at 750 W in 0.01m trisodium citrate, pH 6, three times for 5 min. Sections were probed overnight at 4 C with anti-p23 (1:50), washed, incubated with biotinylated rabbit-antimouse IgG (1:200, 45 min; DAKO Corp., Carpenteria, CA), followed by incubation with peroxide conjugated with streptavidin (1:100, 30 min; DAKO). Staining was visualized using 3,3′diaminobenzidine tetrahydrochloride (DAKO) and nuclei were counterstained with hematoxylin (Vector Laboratories, Inc., Burlingame, CA) as previously described (45). Tissue scoring was performed by a consultant histopathologist (M.W.). p23 nuclear staining was assessed as negative, weak, or strong in the different Gleason prostate cancer grades and compared with benign prostate tissue using a Wilcoxon test to show significance.
Transient knockdowns
LNCaP cells were grown to 70% confluence in six-well plates, transferred to starvation medium for 24 h before transfection with 100 nm/well p23-targeting small interfering RNA (siRNA) pool (Dharmafect L-004496–00; Dharmacon, Lafayette, CO) or scrambled negative control using 2 μl of DharmaFECT-2 transfection reagent (Dharmacon). After 72 h, 10 nm MB was added and cells were cultured for a further 24 or 48 h before being harvested for RNA or protein extraction. respectively. Protein expression was assessed by Western blotting using 10 μg of protein lysate.
Coimmunoprecipitation
Cells were grown to 70% confluency in 10-cm2 dishes, starved for 24 h, and treated with 10 nm MB for 3 h before lysis on ice in immunoprecipitation (IP) buffer [150 mm NaCl, 1% (vol/vol) Nonidet P40, 50 mm Tris (pH 7.9), 0.2 mm phenylmethylsulfonyl fluoride (PMSF), 5 μl/ml of protease inhibitors (Sigma)]. After preclearing with protein-G-sepharose beads (Sigma), 2 μl of anti-p23 (ab2814; Abcam, Cambridge, MA) or 2 μg of anti-AR (N20, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were added to the supernatant and incubated overnight at 4 C with gentle rotation before incubation with 100 μl of protein G beads for 1 h at 4 C. Beads were washed with IP buffer, resuspended in Laemmli sodium dodecyl sulfate sample buffer, and boiled for 5 min before separation by SDS-PAGE.
Immunoblotting
Equal amounts of total protein were separated by SDS-PAGE transferred onto nitrocellulose membrane (Merck Millipore, Darmstadt, Germany) and probed using primary antibodies: mouse anti-p23 (1:2000, Abcam), mouse anti-β-actin (ab8226, 1:10000; Abcam), rabbit anti-AR (sc816, 1:1000; SantaCruz), rabbit anti-HSP90 (sc7977, 1:500; SantaCruz), mouse anti- poly(ADP-ribose) polymerase 1 (sc8007, 1:1000; SantaCruz), mouse anti-AR (AR441, 1:1000; DAKO) and mouse anti-HSP70 (SPA806, 1:2000; Stressgen, Ann Arbor, MI). Proteins were detected using horseradish peroxidase-conjugated antimouse or antirabbit secondary antibodies (1:2000, DAKO) and visualized using the ECL system (GE Healthcare).
Cell fractionation
COS-1 cells were seeded in 15-cm dishes at 70% confluence and transfected as described previously with 6 μg of pSG5-GFP-p23 expression vector and 6 μg of pSVAR. Twenty four hours after transfection, cells were treated ± 10 nm MB for 2 h, harvested, and pelleted by centrifugation for 3 min at 1500 × g. Cells (3 × 106) were resuspended in buffer A (10 mm HEPES, 10 mm KCl, 1.5 mm MgCl2, 0.34 m sucrose, 10% glycerol, 1 mm dithiothreitol, 0.2 mm PMSF, and 5 μl/ml proteinase inhibitor cocktail). After addition of 20 μl 0.1% Triton, cell suspensions were incubated on ice for 5 min and then centrifuged at 1300 × g for 4 min at 4 C. Supernatant containing the cytoplasmic fraction was removed and retained, and the cell pellets were washed once with buffer A. Pellets were then resuspended in 200 μl buffer B (3 mm EDTA, 0.2 mm EGTA, 1 mm dithiothreitol, 0.2 mm PMSF and 5 μl/ml proteinase inhibitor cocktail) and incubated on ice for 30 min. Pellets were centrifuged at 1700 × g for 4 min at 4 C, and the supernatant containing the soluble nuclear fraction was removed. The remaining pellets, containing insoluble chromatin, were washed in buffer B and then resuspended in 200 μl of ddH2O.
GST pull-down assay
Vectors expressing GST fused to full-length p23 (GST-p23); its N terminus (GST-NTD) or empty vector (pGEX-6P-1) was expressed in BL21-codon plus Escherichia coli. The GST-fused protein was purified from 5-mg aliquots of supernatant using 200 μl of glutathione sepharose beads (GE Healthcare). Equal amounts of beads were incubated with 500 μg of LNCaP or AR-transfected COS-1 cell extract in GST buffer [150 mm NaCl, 20 mm Tris (pH 8), 1 mm EDTA and 0.5% (vol/vol) NP-40] overnight at 4 C before being washed five times in the same buffer before resuspension in sodium dodecyl sulfate loading sample buffer, SDS-PAGE, and immunoblotting.
Generation of stable cell lines
LNCaP-TR2-MAR4 cells (41) were used to establish cell lines expressing exogenous p23 under doxycycline control. Cells were seeded in six-well plates and transfected with 2 μg pCDNA4TO-p23 expression vector using FuGene-6 transfection reagent according to manufacturer's protocol. Cells were selected with Zeocin (Invitrogen; final concentration 260 μm), Blastocidin (Melford Labs; final concentration 26 μm), and G418 (Sigma; final concentration 720 μm). p23 levels in individual colonies after doxycycline induction were characterized at the mRNA and protein levels. Resultant cells were named LNCaP-TR2-p23.
RNA preparation and quantitative real-time PCR
Total RNA was extracted using the RNeasy extraction kit (QIAGEN, Chatsworth, CA) and 500 ng RNA reverse transcribed using the transcriptor reverse transcriptase kit (Roche). PSA and KLK2 expression was assessed by quantitative real-time PCR using Taqman Fast universal PCR Master Mix (Applied Biosystems, Foster City, CA), FAM-labeled oligo sets for p23, PSA, and KLK2, and Fast Reaction SYBR-Green mixture (Applied Biosystems). Data were normalized to L19 or GAPDH expression. Primer sequences are shown in Supplemental Table 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org.
Chromatin immunoprecipitation (ChIP)
LNCaP cells were grown to 80% confluence, starved for 72 h, and treated with 10 nm MB or vehicle for 2 h and then cross-linked with formaldehyde (Sigma) for 10 min at room temperature. ChIP was performed using the Magna-ChIP kit (Merck Millipore) according to the manufacturer's instructions but with sonication (Biorupter; Diagenode, Liège, Belgium) for 10 min at 4 C, using 30-sec high-power pulses. Immunoprecipitation was performed using 2 μg of p23- (JJ3, Abcam), HSP90- (Sc-69703, Santa Cruz), or AR-specific (N20, Santa Cruz) antibodies. DNA was recovered using the PCR purification kit (QIAGEN) or phenol/chloroform extraction and analyzed using RT-quantitative PCR (RT-qPCR): primers (see Supplemental Table 1) were designed at either side of ARE or negative control regions upstream of KLK2, PSA, and TMPRSS2. For LNCaP-TR2-p23 cells, ChIP was performed as above with the addition of an incubation step with 100 nm doxycycline for 48 h in full media before cells were incubated in a serum-free media also containing 100 nm doxycycline for 72 h before the addition of MB or vehicle.
Results
p23 interacts with the AR in a ligand-independent manner
We previously identified p23 in an AR-interacting immune complex purified from the PC3wtAR prostate cancer cell line (Supplemental Fig. 1A). These AR-negative prostate epithelial cells are stably transfected with wild-type AR expression vector (40, 46). To confirm this interaction for endogenous AR, anti-p23 antibody was used to immunoprecipitate the AR from the AR-positive breast cancer MCF7 cell line, and immunoblotting revealed that the AR associates with p23 in the presence and absence of ligand (Fig. 1A). We then performed an in vitro GST pull-down interaction assay using full-length p23 fused to GST as bait protein and incubated with whole-cell extracts from COS-1 cells transfected with either wild-type AR (wt) or a mutant AR previously associated with PCa and present in the LNCaP PCa cell line (T877A). Separation of the immobilized GST-p23 complex by SDS-PAGE and immunoblotting with anti-AR antibody demonstrated a positive interaction between p23 and either form of AR (Fig. 1B and Supplemental Fig. 1B). To establish the region of AR that interacts with p23, a yeast two-hybrid interaction assay was performed. p23 fused in frame with the LexA DNA-binding domain was assayed for interaction with the AR N-terminus (NTD, residues 1–556) or C-terminal LBD (CTD, residues 625–919) fused with the Gal4 activation domain. p23 showed positive interaction with the CTD of the AR, and this was irrespective of ligand treatment (Fig. 1C). There was also a slight but significant increase in β-galactosidase activity in the presence of the AR NTD, suggesting that p23 may also interact weakly with the N terminus of AR.
Fig. 1.

p23 interacts with AR. A, MCF7 cells were treated with 10 nm MB or vehicle (ethanol) for 3 h before harvesting and whole-cell extract preparation for incubation with anti-p23 antibody. Immunoprecipitated complexes were resolved and probed with p23 and AR specific antibodies. B, GST alone or GST-fused to full-length p23 was expressed in Escherichia coli, purified using Glutathione sepharose beads, and incubated with whole-cell extract COS-1 cells expressing exogenous AR. Bound proteins were eluted and resolved by SDS-PAGE for Western blotting with a specific antibody against AR. C, Yeast two-hybrid analysis of the interaction between LexA DBD alone or fused to full-length p23 with the Gal4 activation domain alone, or fused to the AR NTD or hinge/ligand-binding domain (CTD). Interaction was assayed by ß-galactosidase readout, normalized for protein. β-Galactosidase expression was measured using a colorimetric assay. Results shown are mean ± sd for one clone assayed in triplicate and representative of data for three independent clones. *, P < 0.05; **, P < 0.01. MB was used at 500 nm. Wt, Wild type.
p23 increases transcriptional activity of the AR
To determine whether overexpression of p23 altered AR transcriptional activity, a reporter assay was carried out in PC3wtAR cells transiently cotransfected with an androgen-dependent luciferase reporter (TAT-GRE-EIB-Luc) (47) and increasing concentrations of p23 expression plasmid (pSG5-p23). Immunoblotting confirmed that increased p23 plasmid resulted in increased p23 protein levels whereas AR levels remained unchanged (Supplemental Fig. 2, A and B). We observed a dose-dependent increase in AR transcriptional activity in response to increasing p23 (Fig. 2A). To determine whether this increase in AR activity was cell- or promoter specific, we examined the effect of p23 on endogenously expressed AR in MCF7 cells using the androgen-responsive MMTV-ARE2-Luc reporter vector (48) and demonstrated that AR activity was significantly increased by up to 3.5-fold (Fig. 2B).
Fig. 2.

p23 increases AR transcriptional activity. A, PC3wtAR cells (panel A) or MCF7 cells (panel B) were transiently transfected with the luciferase reporter TAT-GRE-Luc, a β-galactosidase expression vector (pdm-LacZ-β-gal) and increasing amounts of p23 expression vector (pSG5–p23). Luciferase activity was normalized for β-galactosidase activity and expressed as a percentage of AR activity in the presence of 10 nm MB and absence of exogenous p23. C, COS-1 cells expressing transiently expressed GR and treated with 10 nm Dexamethasone (DEX), or AR treated with 10 nm MB (panel D), were cotransfected with the luciferase reporter MMTV-Luc, a β-galactosidase expression vector (pdm-LacZ-β-gal), and increasing amounts of p23 expression vector (pSG5–p23). Luciferase activity was normalized and expressed as for panel A. All results are mean ± se of four independent experiments performed in duplicate. *, P < 0.05; **, P < 0.005, ***, P < 0.001 (Student's two-tailed t test).
p23 has previously been demonstrated to repress GR activity (12, 39). To rule out the possibility that the differences observed between the AR and the GR are due to promoter-specific effects, each receptor was co-transfected into COS-1 cells with the same MMTV reporter construct and increasing amounts of p23. As reported, increasing levels of p23 decreased ligand-dependent activity of GR by up to 70% (Fig. 2C). However, again AR transcriptional activity was enhanced, by almost 80% (Fig. 2D).
We next investigated the effect of p23 on endogenous androgen-responsive parameters by using a cell line with endogenous AR (LNCaP) and stably transfected with doxycycline-inducible V5-tagged p23 (Supplemental Fig. 3A). After induction, we measured the effect of increased p23 levels on several endogenous androgen target genes: PSA, KLK2, DRG1, and TMPRSS2 (Fig. 3A) and saw significant enhancement of ligand-dependent activation at each of the target genes. Although the effects were of different magnitude, the overriding trend is that increased expression of p23 enhanced ligand-induced activity of the AR. When the levels of endogenous p23 in LNCaP cells were reduced by siRNA (Supplemental Fig. 3B), a reciprocal decrease in androgen-dependent expression of PSA and KLK2 was seen (Fig. 3B). These observations suggest that changes in the expression level of p23 can have downstream functional consequences in prostate epithelial cells.
Fig. 3.

p23 enhances activation of endogenous AR target genes. A, LNCaP-TR2–p23 cells (stably transfected with a doxycycline-inducible plasmid expressing V5-tagged p23) were cultured in media containing 100 nm doxycycline (DOX) for 48 h before starvation for 72 h in the presence of 100 mm DOX. Cells were then treated ± 10 nm MB for 6, 10, or 16 h, harvested, and RNA extracted for reverse transcription. The resulting cDNA was then analyzed by RT-qPCR with specific primers for the AR target genes PSA, KLK2, DRG1, and TMPRSS2. L19 was used as the reference gene. All primer sequences are listed in Supplemental Table 1. Results shown are a mean ± se of three independent experiments performed in triplicate. B, LNCaP cells were transiently transfected with siRNA pool against p23 or scrambled negative control. Forty eight hours later, cells were treated ± 10 nm MB for 24 h before harvesting for total RNA extraction. The resulting cDNA was then analyzed for AR target genes, PSA and KLK2 by RT-qPCR. GAPDH was used as the reference gene. Primer sequences used are listed in Supplemental Table 1. Results shown are the mean of three independent experiments performed in triplicate ± 1 se. *, P < 0.05; **, P < 0.005.
p23 affects AR activity within the nuclear compartment
Previous studies have demonstrated a nuclear role for p23 in disassembly of transcriptional regulatory complexes (12, 15). To determine whether endogenous p23 is present in the nucleus of prostate cells, we initially performed immunofluorescence staining in prostate cancer cells (Supplemental Fig. 4A), which demonstrated that p23 was present in both the cytoplasm and nucleus. Nuclear p23 was in pronounced nuclear speckles, which became larger and more diffuse upon ligand treatment. To confirm the nuclear p23, cellular fractionation of the PC3wtAR cell line was performed, and efficient fractionation was confirmed by immunoblotting for the mitochondrial marker MnSOD (cytoplasmic fraction) and histone H3 (nuclear fraction). Although the majority of p23 expression was cytoplasmic, a nuclear pool (both soluble nuclear and chromatin associated) was evident (Fig. 4A). As expected, there was more AR associated with the chromatin in the presence than in the absence of androgen, and, interestingly, this was also the case for p23.
Fig. 4.
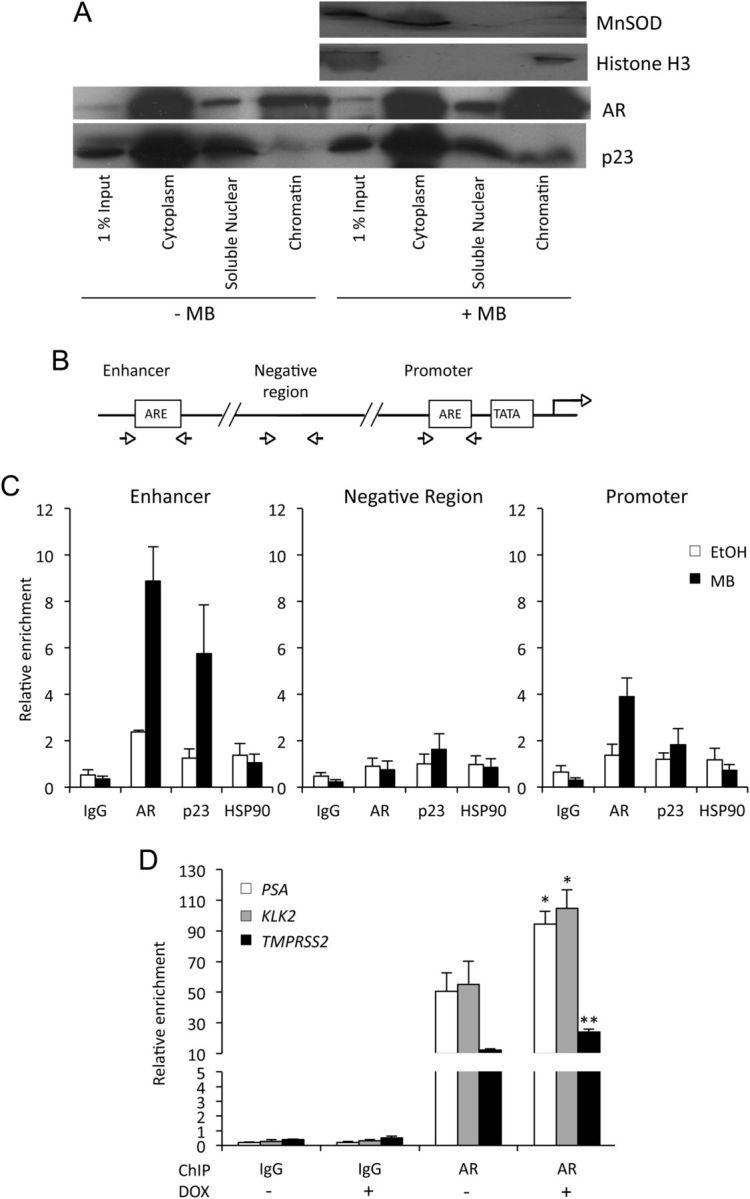
p23 is associated with chromatin and affects recruitment of the AR to its target genes. A, COS-1 cells were transfected with expression vectors for AR and green fluorescent protein-tagged p23. After 24 h, the cells were treated for 2 h ± 10 nm MB before fractionation. Equal amounts of protein fraction from the ± MB cells were resolved and probed using specific antibodies against green fluorescent protein and AR. MnSOD and Histone H3 were used as cytoplasmic and nuclear fractionation controls, respectively. B, Representative schematic depicting the enhancer and promoter regions of the AR-regulated kallikrein genes used for ChIP. C, LNCaP cells were treated ± 10 nm MB for 2 h and fixed. IP was then performed using an antibody specific for p23, AR, HSP90, or control IgG, and RT-qPCR was performed on the immunoprecipitated DNA to detect the presence of one non-ARE (negative region) and two specific ARE sequences (enhancer and promoter) of the AR target gene, PSA. Relative enrichment for each of the studied regions was calculated as a percentage of the corresponding input. Data are representative of three independent experiments ± se. D, Exogenous expression of p23 was induced in LNCaP-TR2–p23 cells as for Fig. 3A. Cells were then treated ± 10 nm MB for 2 h, and ChIP was performed using anti-AR antibody or control IgG. RT-qPCR was performed as above for the relevant regions of the AR target genes, PSA, KLK2, and TMPRSS2 and is presented as for panel C. DOX, Doxycycline; *, P < 0.05; **, P < 0.005.
Previously, Freeman and Yamamoto (12) demonstrated that the repressive effect of p23 on GR activity was due to destabilization of GR-DNA binding. We investigated AR and p23 binding to regions with and without androgen response elements (ARE) in the regulatory region of androgen-responsive genes (Fig. 4B) by chromatin immunoprecipitation (ChIP) in LNCaP cells. We found that p23 is associated with endogenous ARE in the enhancers of the androgen-responsive genes such as PSA, but not non-AR-binding regions, in a ligand-dependent manner (Fig. 4C and Supplemental Fig. 4B). We did not see HSP90 corecruitment to the regulatory regions of PSA, in the presence or absence of androgen. Furthermore, when p23 levels were increased in the inducible LNCaP-TR2-p23 cell line, the relative amount of AR associated with ARE in the enhancer region of several androgen-responsive genes increased by approximately 2-fold in each case (Fig. 4D), suggesting that p23 may enhance or stabilize AR binding to ARE.
p23 interacts with AR independently of HSP90
A GST pull-down assay showed that the isolated N-terminal domain (NTD, residues 1–86) retains the ability to interact with p23 (Fig. 5A and Supplemental Fig. 1C). Because this construct does not contain the region (residues 86–108) known to mediate HSP90 interaction, we surmised that p23 may be interacting with the AR independently of HSP90. To determine the contribution of HSP90 to the effect of p23 on AR activity, we used the HSP90 inhibitors GA and its analog 17-AAG to disrupt the p23-HSP90 interaction (49–51). By immunoprecipitating p23 from LNCaP cells we confirmed that treatment with 17-AAG disrupted the HSP90-p23 complex entirely (while not affecting cell viability; Supplemental Fig. 5A), whereas AR interaction was retained (Fig. 5B), suggesting that there is AR-p23 interaction even when HSP90 is stripped from the complex, i.e. HSP90-independent interaction between AR and p23. In support of this, immunoprecipitation assays performed using Flag-tagged wild-type and W106A p23 constructs confirmed that the latter did not pull down endogenous HSP90, but was unimpaired in its interaction with AR (Fig. 5C). The W106A mutant p23 was also able to increase ligand-dependent activity of AR to a similar extent as wild-type despite not interacting with HSP90 (Fig. 5D), whereas it is reported as not retaining the repressive effect on GR ligand-dependent activity (39).
Fig. 5.

Physical and functional interaction of AR, p23, and HSP90. A, GST alone or GST fused to either full-length p23 or p23 aa1–85 were expressed in E. coli, purified using Glutathione sepharose beads, and incubated with whole-cell extract COS-1 cells. Bound proteins were eluted and resolved by SDS-PAGE for Western blotting with a specific antibody against AR. B, LNCaP cells were treated with vehicle (ethanol), and 10 nm MB alone or in combination with 5 μm 17-AAG for 2 h before being harvested, and whole-cell extract was prepared for incubation with anti-p23 antibody. Immunoprecipitated complexes were resolved and probed with p23-, HSP90-, and AR-specific antibodies. C, COS-1 cells were transfected with 5 μg of either FLAG-wtp23 or FLAG-W106Ap23 and 5 μg of the AR expression vector pSVAR. After 24 h, cells were treated with 10 nm MB or vehicle (ethanol) for 2 h and harvested after which whole-cell extract was prepared for incubation with anti-FLAG antibody. Immunoprecipitated complexes were resolved and probed with FLAG-, HSP90-, and AR-specific antibodies. D, COS-1 cells were transiently transfected with the luciferase reporter TAT-GRE-Luc, a β-galactosidase expression vector (pdm-LacZ-β-gal), and increasing amounts of either p23 expression vector (pCDNA4TO-p23) or W106Ap23 expression vector (pCDNA4TO-W106Ap23). Luciferase activity was normalized for β-galactosidase activity and expressed as a percentage of AR activity in the presence of 10 nm MB and absence of exogenous p23. All results are mean ± se of four independent experiments performed in duplicate. *, P < 0.05; **, P < 0.005 (Student's two-tailed t test). WT, Wild type.
p23 increases binding capacity of the AR
Previously, it has been proposed that p23 may function by increasing the pool of ligand binding-competent steroid receptor rather than by increasing the ligand binding affinity (52). To investigate whether p23 exerts a similar effect on the AR, we carried out a ligand-binding assay using COS-1 cells exogenously expressing AR and increasing concentrations of p23, with and without 10 nm GA, which disrupts the p23-HSP90 interaction without compromising cell viability (Supplemental Fig. 5, B and C). Scatchard analysis demonstrated that the binding capacity (BMAX) of the AR increased significantly at the higher concentration (200 ng) of transiently transfected p23 regardless of whether cells were treated with GA (Fig. 6). The lower amount (100 ng) also resulted in a significant increase in the BMAX value of the AR in cells treated with GA but had no effect in untreated cells. Hence it appears that lower amounts of p23 are able to affect AR ligand binding when the HSP90-p23 interaction is disrupted, albeit perhaps not completely. This may be due to GA increasing the pool of bioavailable p23 able to interact with the AR and hence enhance ligand-binding competency.
Fig. 6.
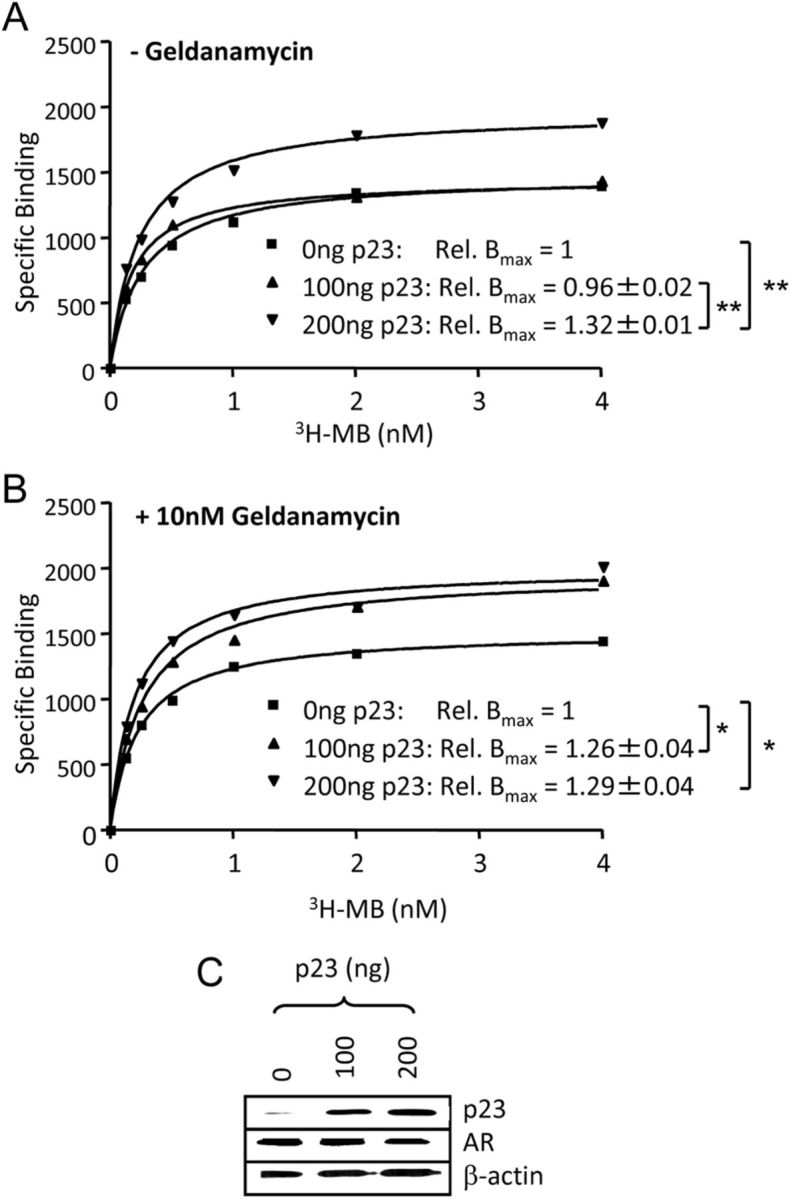
Increased expression of p23 increases AR binding capacity. COS-1 cells were transfected with AR expression vector and 100 or 200 ng of pSG5–p23 using FuGENE6 and 24 h later treated for 2 h without (A) or with (B) 10 nm GA, before addition of [3H]MB at indicated concentrations in the presence or absence of 200-fold excess unlabeled MB. Scatchard analysis was performed and relative Bmax (as compared with Bmax in the absence of exogenous p23) calculated. Values given are mean ± sd of three independent experiments and graphs are representative experiments. *, P < 0.05; **, P < 0.005. C, COS cells were transfected as above. C, After 24 h, whole-cell extracts were prepared and immunoblotted using antibodies against AR, p23, and β-actin. Rel., Relative.
p23 expression in human prostate
p23 is generally described as a ubiquitously expressed protein and is highly conserved across many species ranging from yeast to humans (15, 53). We investigated its expression in a panel of human prostate and prostate cancer cell lines: the AR-negative PC3, DU145 (cancer), BPH1 and RWPE1 (non-cancer) lines, and the AR-positive VCaP, DuCaP, LNCaP and its derivative C4–2 (all cancer). p23 was expressed in all of these cell lines (Fig. 7A), and, when grouped, levels in the AR-positive lines were significantly higher than the AR-negative lines (P = 0.0006). Using immunohistochemistry, we investigated p23 expression in human prostate tissue samples from patients with prostate cancer compared with adjacent benign tissue. Both the epithelial and stromal layers stained positive for p23 (Fig. 7B). Staining in the luminal epithelium was absent or weak in the normal/benign tissue; however there was a significant increase in staining (both nuclear and cytoplasmic) in tumors (Fig. 7, B and C), indicating that increased p23 protein expression may be linked with the development of prostate cancer. Similar to previous studies of prostate cancer cohorts (34, 35), we did not see a correlation with Gleason grade.
Fig. 7.
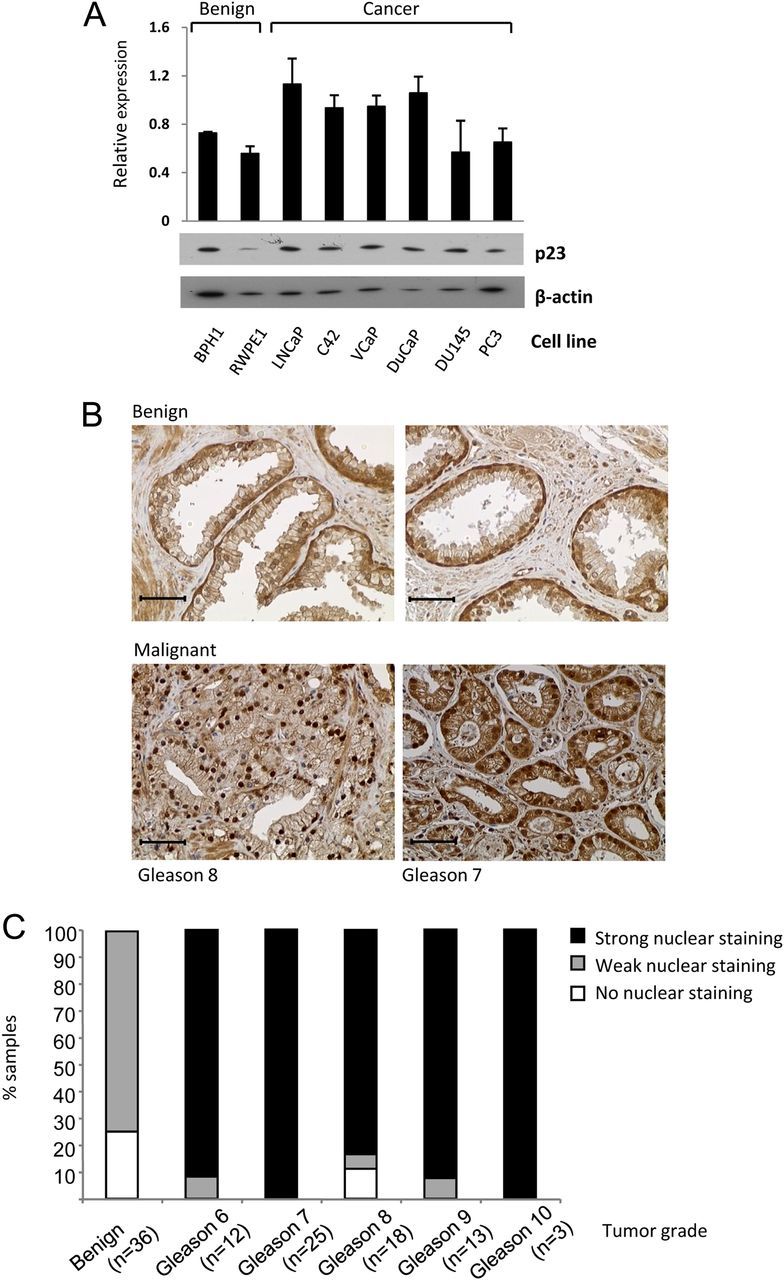
Expression of p23 in human prostate cell lines and tissue. A, Total protein (10 μg) was separated by SDS-PAGE and immunoblotted using antibodies against p23 and β-actin. Band intensities were quantified using ImageJ software, normalized for ß-actin, and graph shows mean ± sd of three independent repeats (image is representative). B, Representative sections of human prostate stained with anti-p23 antibody (brown) and nuclei counterstained with hemotoxylin (blue). C, Tissue scoring depicting the intensity of p23 nuclear staining. The bars indicate the percentage of samples with strong, weak, or no nuclear p23 staining. Tumors of all Gleason grades express significantly more nuclear p23 relative to benign tissue (Wilcoxon test: n = 88; Z = 2.10; P < 0.0018).
Discussion
The best-characterized role of p23 is as a cochaperone of HSP90 in the foldosome complex, where it is involved in correct folding of client proteins including steroid receptors (10). However, an ever-growing number of studies suggest that p23 has numerous and complex biological functions including regulation of telomerase, regulation of endoplasmic reticulum-mediated apoptosis and prostaglandin synthesis, as well as regulation of steroid receptor signaling (1, 22, 23, 54). Several of these activities may be partly or wholly independent of HSP90, including chaperone function: the C-terminal tail of p23 has intrinsic chaperone activity and even p23 variants lacking the HSP90-interacting domain retain chaperone function (20, 31). This dichotomy even within its role in protein folding may explain why it can have opposing effects on steroid receptor function, even to the extent that it has been reported as variously increasing and repressing activity of both GR and ER. It was shown by Garabedian and co-workers (13, 14) that p23 overexpression enhanced the ligand binding efficiency and the rate of transcriptional activity of the ER in a yeast system, and that this required HSP90 interaction. However, Freeman et al. (15) showed that p23 repressed ER activity, while enhancing activity of GR and PR. In an important subsequent study they demonstrated a nuclear role of p23, whereby p23 overexpression promotes disassembly of the transcriptional machinery at hormone response elements, resulting, in this case, in a reduction in GR activity (12). GR activity was later reported as being inhibited by p23 in an HSP90-dependent manner by the Rein laboratory (39). The effect of p23 on AR activity has to date been little studied, but Freeman et al. (15) reported it as inhibiting AR activity, whereas Fang et al. (56) saw little effect on AR activity of depleting the p23 analog in S. cerevisiae at the shorter time point of 2 h. Conversely, within similar time frames as Freeman and Yamamoto, we saw a robust enhancement of AR activity in several cell lines, including an effect on endogenous target genes.
These apparent discrepancies in p23 effects on steroid receptors could be due to differential effects on target genes perhaps attributable to the mode of regulation, as suggested by Oxelmark et al. (57), or to differences in yeast vs. mammalian cells. However, we suggest that the apparently contradictory effects of p23 on steroid receptors could reflect a true dichotomy in regulation caused by different contributions of the HSP90-dependent vs. HSP90-independent functions of p23 and/or different extent of its role in the cytoplasm vs. the nucleus. The relative contributions of these could be affected by a host of cellular factors.
We show here, for the first time, that modulating p23 levels affects the expression of endogenous AR target genes consistent with it acting as an activator of AR, and also that p23 is recruited to the regulatory regions of such genes. Further, increasing p23 levels resulted in increased AR association with ARE; thus, unlike the situation for the GR (12), p23 does not induce disassembly of the AR from the promoter region or target genes. Steroid receptors are highly dynamic proteins that shuttle between the cytoplasm and nucleus and which, when activated, undergo cycles of association and dissociation with their cognate response elements (58–60). There are various mechanisms that regulate the rate of receptor recycling; however, the main limiting factor is thought to be the levels of hormone present coupled with the available pool of ligand binding-competent receptor; this latter is dependent on the activity of the foldosome. Our data suggest that increasing expression levels of p23 enhanced the population of ligand-bound holoreceptor complex, which correlates with published reports that p23 alters the ligand efficacy of steroid receptors (15). Interestingly, we were unable to demonstrate association of HSP90 at regulatory regions of target genes that recruit AR and p23. This may support the theory that HSP90 is not required for the nuclear function of p23 in AR signaling; however we cannot rule out HSP90 recruitment at different regions of the promoter/enhancer that may be associated with the ARE by other means, e.g. chromatin looping.
To determine whether the effects of p23 on AR activity were, as for ER and GR, dependent on HSP90, we used a mutant form of p23 unable to bind HSP (39) and also the HSP90-specific inhibitors GA and the clinically used analog 17-AAG. GA is a benzoquinone ansamycin antibiotic that has been identified as having potent antitumor activity (61). It specifically targets the ATP-binding site of HSP90, which as a result disrupts p23 binding to that same site (62). We found that treatment with these inhibitors resulted in the loss of HSP90 from the p23-interacting complex, but that AR remained associated. This result suggests that a three-way interaction between the three proteins is occurring and that p23 may be integral, interacting with both components rather than simply recruited via HSP90. Until now, direct interaction of p23 with HSP90 client proteins has not been demonstrated, but using the W106A p23 mutant we showed that AR interaction is retained when HSP90 is absent from the complex, and also that p23 retains its positive effect on AR activity. Thus we have demonstrated an HSP90-independent function of p23 in AR signaling as well as a direct interaction of p23 with AR.
Ligand binding assays demonstrated that, even in the presence of an HSP90 inhibitor, p23 was able to promote or stabilize the population of ligand-bound AR. This effect may be a result of the interaction we observed with both termini of the AR, because it is well established that the AR N- and C termini interact, and this slows ligand dissociation, which then increases the pool of ligand-bound receptor (63). Altering p23 levels had more of an effect when HSP90 function was inhibited: an effect was seen at lower concentrations of exogenous p23 in the presence of GA. These results are in line with studies showing that overexpression of p23 will confer a protective role against the effects of HSP90 inhibition whereas p23 depletion renders cells hypersensitive to HSP90 inhibitors (31). Thus, targeting both p23 and HSP90 simultaneously is likely to have a greater effect on downstream effects than either alone.
To understand whether the observed effects of p23 on androgen signaling may be altered in prostate cancer, we performed immunohistochemistry staining on a panel of human prostate cancer tissue biopsies together with corresponding normal tissue. Nuclear p23 expression levels were significantly higher in the tumor samples at all stages. No correlation was seen with Gleason grade, and studies of further patient material are required to ascertain whether p23 levels correlate with clinical parameters. Others have reported increased p23 levels in various cancers including prostate cancer, although in some cases this was originally erroneously attributed to apoptosis-linked gene 2 (32–35). In one report, increased cytoplasmic p23 correlated with shorter disease-free survival in breast cancer, but it was notable that high cytoplasmic p23 was significantly associated with nuclear p23 expression (55). Our results support p23 as a therapeutic target in prostate cancer. It has been suggested that cochaperones of HSP90 delineate subgroups of HSP90 client proteins, and we suggest that targeting p23 may provide a measure of selectivity not only for steroid receptors but for the AR in particular, given its coactivation potential. Targeting HSP90 function via p23 in this way could thus avoid the toxicity of HSP90 inhibitors. Further, due to the HSP90-dependent and -independent effects of p23, and the observed increase in nuclear p23 in prostate cancer coupled with the reports that increased p23 can protect against HSP90 inhibition, p23 inhibitors may demonstrate additive, independent, or synergistic effects in combination with HSP90 inhibitors.
Acknowledgments
We thank Simak Ali (Imperial College London, UK) and members of the Androgen Signaling Laboratory for extensive discussion and critical reading of the manuscript; Didier Picard (University of Geneva, Switzerland) for helpful discussion; and Theo Rein (Max Planck Institute of Psychiatry, Germany) for the kind gift of mutant p23 construct.
This work was supported by grants from the Cancer Research UK, the Association for Cancer Research Prostate Action and the Imperial College Experimental Cancer Medicine Centre. G.B. is the recipient of the Martin Harris Research Fellowship.
Disclosure Summary: The authors declare no conflict of interest.
NURSA Molecule Pages†:
Nuclear Receptors: AR.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- 17-AAG
- 17-Allylaminogeldanamycin
- AR
- androgen receptor
- ARE
- androgen response element
- ChIP
- chromatin immunoprecipitation
- CTD
- C-terminal domain
- DBD
- DNA-binding domain
- ER
- estrogen receptor
- GA
- geldanamycin
- GR
- glucocorticoid receptor
- GST
- glutathione-S-transferase
- HSP
- heat shock protein
- IP
- immunoprecipitation
- LBD
- ligand-binding domain
- MB
- mibolerone
- MMTV
- mouse mammary tumor virus
- NTD
- N-terminal domain
- PMSF
- phenylmethylsulfonyl fluoride
- PR
- progesterone receptor
- RT-qPCR
- RT-quantitative PCR
- siRNA
- small interfering RNA.
References
- 1. Smith DF , Toft DO. 2008. Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol 22:2229–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith DF , Faber LE , Toft DO. 1990. Purification of unactivated progesterone receptor and identification of novel receptor-associated proteins. J Biol Chem 265:3996–4003 [PubMed] [Google Scholar]
- 3. Pratt WB. 1993. The role of heat shock proteins in regulating the function, folding, and trafficking of the glucocorticoid receptor. J Biol Chem 268:21455–21458 [PubMed] [Google Scholar]
- 4. Smith DF , Toft DO. 1992. Composition, assembly and activation of the avian progesterone receptor. J Steroid Biochem Mol Biol 41:201–207 [DOI] [PubMed] [Google Scholar]
- 5. Hendrick JP , Hartl FU. 1993. Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem 62:349–384 [DOI] [PubMed] [Google Scholar]
- 6. Hutchison KA , Dittmar KD , Czar MJ , Pratt WB. 1994. Proof that hsp70 is required for assembly of the glucocorticoid receptor into a heterocomplex with hsp90. J Biol Chem 269:5043–5049 [PubMed] [Google Scholar]
- 7. Dittmar KD , Banach M , Galigniana MD , Pratt WB. 1998. The role of DnaJ-like proteins in glucocorticoid receptor.hsp90 heterocomplex assembly by the reconstituted hsp90.p60.hsp70 foldosome complex. J Biol Chem 273:7358–7366 [DOI] [PubMed] [Google Scholar]
- 8. Froesch BA , Takayama S , Reed JC. 1998. BAG-1L protein enhances androgen receptor function. J Biol Chem 273:11660–11666 [DOI] [PubMed] [Google Scholar]
- 9. Johnson JL , Toft DO. 1994. A novel chaperone complex for steroid receptors involving heat shock proteins, immunophilins, and p23. J Biol Chem 269:24989–24993 [PubMed] [Google Scholar]
- 10. Hutchison KA , Stancato LF , Owens-Grillo JK , Johnson JL , Krishna P , Toft DO , Pratt WB. 1995. The 23-kDa acidic protein in reticulocyte lysate is the weakly bound component of the hsp foldosome that is required for assembly of the glucocorticoid receptor into a functional heterocomplex with hsp90. J Biol Chem 270:18841–18847 [DOI] [PubMed] [Google Scholar]
- 11. Elbi C , Walker DA , Romero G , Sullivan WP , Toft DO , Hager GL , DeFranco DB. 2004. Molecular chaperones function as steroid receptor nuclear mobility factors. Proc Natl Acad Sci USA 101:2876–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Freeman BC , Yamamoto KR. 2002. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296:2232–2235 [DOI] [PubMed] [Google Scholar]
- 13. Knoblauch R , Garabedian MJ. 1999. Role for Hsp90-associated cochaperone p23 in estrogen receptor signal transduction. Mol Cell Biol 19:3748–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oxelmark E , Knoblauch R , Arnal S , Su LF , Schapira M , Garabedian MJ. 2003. Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J Biol Chem 278:36547–36555 [DOI] [PubMed] [Google Scholar]
- 15. Freeman BC , Felts SJ , Toft DO , Yamamoto KR. 2000. The p23 molecular chaperones act at a late step in intracellular receptor action to differentially affect ligand efficacies. Genes Dev 14:422–434 [PMC free article] [PubMed] [Google Scholar]
- 16. McKenna NJ , Lanz RB , O'Malley BW. 1999. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 20:321–344 [DOI] [PubMed] [Google Scholar]
- 17. Dittmar KD , Demady DR , Stancato LF , Krishna P , Pratt WB. 1997. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery. The role of p23 is to stabilize receptor.hsp90 heterocomplexes formed by hsp90.p60.hsp70. J Biol Chem 272:21213–21220 [DOI] [PubMed] [Google Scholar]
- 18. Sullivan W , Stensgard B , Caucutt G , Bartha B , McMahon N , Alnemri ES , Litwack G , Toft D. 1997. Nucleotides and two functional states of hsp90. J Biol Chem 272:8007–8012 [DOI] [PubMed] [Google Scholar]
- 19. Martinez-Yamout MA , Venkitakrishnan RP , Preece NE , Kroon G , Wright PE , Dyson HJ. 2006. Localization of sites of interaction between p23 and Hsp90 in solution. J Biol Chem 281:14457–14464 [DOI] [PubMed] [Google Scholar]
- 20. Weikl T , Abelmann K , Buchner J. 1999. An unstructured C-terminal region of the Hsp90 co-chaperone p23 is important for its chaperone function. J Mol Biol 293:685–691 [DOI] [PubMed] [Google Scholar]
- 21. Weaver AJ , Sullivan WP , Felts SJ , Owen BA , Toft DO. 2000. Crystal structure and activity of human p23, a heat shock protein 90 co-chaperone. J Biol Chem 275:23045–23052 [DOI] [PubMed] [Google Scholar]
- 22. Tanioka T , Nakatani Y , Semmyo N , Murakami M , Kudo I. 2000. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem 275:32775–32782 [DOI] [PubMed] [Google Scholar]
- 23. Rao RV , Niazi K , Mollahan P , Mao X , Crippen D , Poksay KS , Chen S , Bredesen DE. 2006. Coupling endoplasmic reticulum stress to the cell-death program: a novel HSP90-independent role for the small chaperone protein p23. Cell Death Differ 13:415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bose S , Weikl T , Bügl H , Buchner J. 1996. Chaperone function of Hsp90-associated proteins. Science 274:1715–1717 [DOI] [PubMed] [Google Scholar]
- 25. Freeman BC , Toft DO , Morimoto RI. 1996. Molecular chaperone machines: chaperone activities of the cyclophilin Cyp-40 and the steroid aporeceptor-associated protein p23. Science 274:1718–1720 [DOI] [PubMed] [Google Scholar]
- 26. Jenster G. 1999. The role of the androgen receptor in the development and progression of prostate cancer. Semin Oncol 26:407–421 [PubMed] [Google Scholar]
- 27. Trepel J , Mollapour M , Giaccone G , Neckers L. 2010. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10:537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bagatell R , Khan O , Paine-Murrieta G , Taylor CW , Akinaga S , Whitesell L. 2001. Destabilization of steroid receptors by heat shock protein 90-binding drugs: a ligand-independent approach to hormonal therapy of breast cancer. Clin Cancer Res 7:2076–2084 [PubMed] [Google Scholar]
- 29. Powers MV , Workman P. 2006. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer 13(Suppl 1):S125–S135 [DOI] [PubMed] [Google Scholar]
- 30. McLaughlin SH , Sobott F , Yao ZP , Zhang W , Nielsen PR , Grossmann JG , Laue ED , Robinson CV , Jackson SE. 2006. The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J Mol Biol 356:746–758 [DOI] [PubMed] [Google Scholar]
- 31. Forafonov F , Toogun OA , Grad I , Suslova E , Freeman BC , Picard D. 2008. p23/Sba1p protects against Hsp90 inhibitors independently of its intrinsic chaperone activity. Mol Cell Biol 28:3446–3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krebs J , Saremaslani P , Caduff R. 2002. ALG-2: a Ca2+ -binding modulator protein involved in cell proliferation and in cell death. Biochim Biophys Acta 1600:68–73 [DOI] [PubMed] [Google Scholar]
- 33. Mollerup J , Krogh TN , Nielsen PF , Berchtold MW. 2003. Properties of the co-chaperone protein p23 erroneously attributed to ALG-2 (apoptosis-linked gene 2). FEBS Lett 555:478–482 [DOI] [PubMed] [Google Scholar]
- 34. Elmore LW , Forsythe R , Forsythe H , Bright AT , Nasim S , Endo K , Holt SE. 2008. Overexpression of telomerase-associated chaperone proteins in prostatic intraepithelial neoplasia and carcinomas. Oncol Rep 20:613–617 [PubMed] [Google Scholar]
- 35. Akalin A , Elmore LW , Forsythe HL , Amaker BA , McCollum ED , Nelson PS , Ware JL , Holt SE. 2001. A novel mechanism for chaperone-mediated telomerase regulation during prostate cancer progression. Cancer Res 61:4791–4796 [PubMed] [Google Scholar]
- 36. Kalkhoven E , Valentine JE , Heery DM , Parker MG. 1998. Isoforms of steroid receptor co-activator 1 differ in their ability to potentiate transcription by the oestrogen receptor. EMBO J 17:232–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jenster G , van der Korput HA , van Vroonhoven C , van der Kwast TH , Trapman J , Brinkmann AO. 1991. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol 5:1396–1404 [DOI] [PubMed] [Google Scholar]
- 38. Bevan CL , Hoare S , Claessens F , Heery DM , Parker MG. 1999. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol Cell Biol 19:8383–8392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wochnik GM , Young JC , Schmidt U , Holsboer F , Hartl FU , Rein T. 2004. Inhibition of GR-mediated transcription by p23 requires interaction with Hsp90. FEBS Lett 560:35–38 [DOI] [PubMed] [Google Scholar]
- 40. Peterziel H , Mink S , Schonert A , Becker M , Klocker H , Cato AC. 1999. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene 18:6322–6329 [DOI] [PubMed] [Google Scholar]
- 41. Dart DA , Spencer-Dene B , Gamble SC , Waxman J , Bevan CL. 2009. Manipulating prohibitin levels provides evidence for an in vivo role in androgen regulation of prostate tumours. Endocr Relat Cancer 16:1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brooke GN , Parker MG , Bevan CL. 2008. Mechanisms of androgen receptor activation in advanced prostate cancer: differential co-activator recruitment and gene expression. Oncogene 27:2941–2950 [DOI] [PubMed] [Google Scholar]
- 43. Dodou E , Treisman R. 1997. The Saccharomyces cerevisiae MADS-box transcription factor Rlm1 is a target for the Mpk1 mitogen-activated protein kinase pathway. Mol Cell Biol 17:1848–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skehan P , Storeng R , Scudiero D , Monks A , McMahon J , Vistica D , Warren JT , Bokesch H , Kenney S , Boyd MR. 1990. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82:1107–1112 [DOI] [PubMed] [Google Scholar]
- 45. Belandia B , Powell SM , García-Pedrero JM , Walker MM , Bevan CL , Parker MG. 2005. Hey1, a mediator of notch signaling, is an androgen receptor corepressor. Mol Cell Biol 25:1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whitaker HC , Hanrahan S , Totty N , Gamble SC , Waxman J , Cato AC , Hurst HC , Bevan CL. 2004. Androgen receptor is targeted to distinct subcellular compartments in response to different therapeutic antiandrogens. Clin Cancer Res 10:7392–7401 [DOI] [PubMed] [Google Scholar]
- 47. Verrijdt G , Schoenmakers E , Haelens A , Peeters B , Verhoeven G , Rombauts W , Claessens F. 2000. Change of specificity mutations in androgen-selective enhancers. Evidence for a role of differential DNA binding by the androgen receptor. J Biol Chem 275:12298–12305 [DOI] [PubMed] [Google Scholar]
- 48. Christiaens V , Bevan CL , Callewaert L , Haelens A , Verrijdt G , Rombauts W , Claessens F. 2002. Characterization of the two coactivator-interacting surfaces of the androgen receptor and their relative role in transcriptional control. J Biol Chem 277:49230–49237 [DOI] [PubMed] [Google Scholar]
- 49. Johnson JL , Toft DO. 1995. Binding of p23 and hsp90 during assembly with the progesterone receptor. Mol Endocrinol 9:670–678 [DOI] [PubMed] [Google Scholar]
- 50. Segnitz B , Gehring U. 1997. The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J Biol Chem 272:18694–18701 [DOI] [PubMed] [Google Scholar]
- 51. Marcu MG , Schulte TW , Neckers L. 2000. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst 92:242–248 [DOI] [PubMed] [Google Scholar]
- 52. Felts SJ , Toft DO. 2003. p23, a simple protein with complex activities. Cell Stress Chaperones 8:108–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garcia-Ranea JA , Mirey G , Camonis J , Valencia A. 2002. p23 and HSP20/α-crystallin proteins define a conserved sequence domain present in other eukaryotic protein families. FEBS Lett 529:162–167 [DOI] [PubMed] [Google Scholar]
- 54. Toogun OA , Zeiger W , Freeman BC. 2007. The p23 molecular chaperone promotes functional telomerase complexes through DNA dissociation. Proc Natl Acad Sci USA 104:5765–5770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Simpson NE , Lambert WM , Watkins R , Giashuddin S , Huang SJ , Oxelmark E , Arju R , Hochman T , Goldberg JD , Schneider RJ , Reiz LF , Soares FA , Logan SK , Garabedian MJ. 2010. High levels of Hsp90 cochaperone p23 promote tumor progression and poor prognosis in breast cancer by increasing lymph node metastases and drug resistance. Cancer Res 70:8446–8456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fang Y , Fliss AE , Rao J , Caplan AJ. 1998. SBA1 encodes a yeast hsp90 cochaperone that is homologous to vertebrate p23 proteins. Mol Cell Biol 18:3727–3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oxelmark E , Roth JM , Brooks PC , Braunstein SE , Schneider RJ , Garabedian MJ. 2006. The cochaperone p23 differentially regulates estrogen receptor target genes and promotes tumor cell adhesion and invasion. Mol Cell Biol 26:5205–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McNally JG , Müller WG , Walker D , Wolford R , Hager GL. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287:1262–1265 [DOI] [PubMed] [Google Scholar]
- 59. Freeman BC , Yamamoto KR. 2001. Continuous recycling: a mechanism for modulatory signal transduction. Trends Biochem Sci 26:285–290 [DOI] [PubMed] [Google Scholar]
- 60. Métivier R , Penot G , Hübner MR , Reid G , Brand H , Kos M , Gannon F. 2003. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115:751–763 [DOI] [PubMed] [Google Scholar]
- 61. Scheibel T , Buchner J. 1998. The Hsp90 complex–a super-chaperone machine as a novel drug target. Biochem Pharmacol 56:675–682 [DOI] [PubMed] [Google Scholar]
- 62. Neckers L , Mimnaugh E , Schulte TW. 1999. Hsp90 as an anti-cancer target. Drug Resist Updat 2:165–172 [DOI] [PubMed] [Google Scholar]
- 63. Zhou ZX , Lane MV , Kemppainen JA , French FS , Wilson EM. 1995. Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol Endocrinol 9:208–218 [DOI] [PubMed] [Google Scholar]