

Abstract
There is widespread interest in defining factors and mechanisms that suppress the proliferation of cancer cells. Retinoic acid (RA) is a potent suppressor of mammary cancer and developmental embryonic cell proliferation. However, the molecular mechanisms by which 9-cis-RA signaling induces growth inhibition in RA-sensitive breast cancer and embryonic cells are not apparent. Here, we provide evidence that the inhibitory effect of 9-cis-RA on cell proliferation depends on 9-cis-RA-dependent interaction of retinoid X receptor α (RXRα) with replication factor C3 (RFC3), which is a subunit of the RFC heteropentamer that opens and closes the circular proliferating cell nuclear antigen (PCNA) clamp on DNA. An RFC3 ortholog in a sea urchin cDNA library was isolated by using the ligand-binding domain of RXRα as bait in a yeast two-hybrid screening. The interaction of RFC3 with RXRα depends on 9-cis-RA and bexarotene, but not on all-trans-RA or an RA receptor (RAR)-selective ligand. Truncation and mutagenesis experiments demonstrated that the C-terminal LXXLL motifs in both human and sea urchin RFC3 are critical for the interaction with RXRα. The transient interaction between 9-cis-RA-activated RXRα and RFC3 resulted in reconfiguration of the PCNA-RFC complex. Furthermore, we found that knockdown of RXRα or overexpression of RFC3 impairs the ability of 9-cis-RA to inhibit proliferation of MCF-7 breast cancer cells and sea urchin embryogenesis. Our results indicate that 9-cis-RA-activated RXRα suppresses the growth of RA-sensitive breast cancer and embryonic cells through RFC3.
The nuclear receptor (NR) superfamily of transcription factors controls a wide range of physiological processes. Within the NR superfamily, the class II NR, such as the retinoic acid (RA) receptor (RAR), thyroid hormone receptor, and peroxisome proliferator-activated receptor, heterodimerize with a common partner, the retinoid X receptor (RXR) (1, 2). The modular structure of NR reveals a distinct functional domain, the activation factor-2 core region, which is located at the extreme C terminus of the ligand-binding domain (LBD). This region has been shown to play a critical role in mediating transactivation by serving as a ligand-dependent interaction interface with coactivators. These coactivators contain NR-interacting domains that consist of LXXLL (where L and X denote leucine and any amino acid, respectively), which mediates ligand-dependent interactions with the LBD of NR (3).
Retinoic acid (RA), a natural derivative of vitamin A, regulates the expression of specific genes through two families of NR, RAR and RXR (4). Vitamin A deficiency in experimental animals has been associated with a higher incidence of cancer and an increased susceptibility to chemical carcinogens (5). Many cellular and preclinical models have demonstrated the efficacy of natural and synthetic RA as chemopreventive and chemotherapeutic agents in the treatment of cancer (6). The major and endogenous agonist for RAR, all-trans-RA (ATRA), influences the cell cycle, which is driven by complexes of cyclin-dependent kinases (CDK) and cyclins, as well as differentiation and apoptosis in various human cancer cells by RAR-dependent mechanisms (7, 8). This is also highlighted by the successful therapeutic application of ATRA to acute promyelocytic leukemia (9, 10). The ability of ATRA to induce differentiation and arrest proliferation has been associated not only with the modulation of cyclins, CDK, and cell-cycle inhibitors but also with direct interaction with RAR-binding proteins, such as tumor antigen preferentially expressed antigen in melanoma (PRAME) and antiapoptotic Bcl-2-associated athanogene 1 (BAG1) (11, 12). An RXR-specific ligand, 9-cis-RA, also inhibits the growth of cancer cells by inducing G1 cell-cycle blockage (13, 14), although whether this compound is a natural bioactive RA for RXR remains controversial (15). The molecular mechanisms by which 9-cis-RA signaling induces growth inhibition and apoptosis in RA-sensitive cancer cells remain poorly understood, with the exception of the report of a regulatory mechanism in which the activation of RXR-peroxisome proliferator-activated receptor-γ heterodimer by RXR agonists induces the nitric oxide-mediated intrinsic death pathway in leukemic and solid cancer cells (16).
In chordates and nonchordate deuterostomes, vitamin A and RA exert a wide variety of effects on vertebrate embryonic body shaping and organogenesis, cell proliferation, and differentiation (17–19). Altering the levels of endogenous RA signaling (especially ATRA) during the early developmental stages leads to severe malformations and embryonic death, mainly due to embryo mispatterning (18, 20). RAR isoforms are known to be closely related in the successive steps of early embryonic development and in later stages in various differentiating organ systems (21, 22). In contrast, few mechanistic studies have examined 9-cis-RA- and RXR-dependent developmental abnormalities in embryos, which hampers our understanding of the specific functions of 9-cis-RA during embryogenesis, although it is a definite morphogen in deuterostomes (23).
In this report, we describe a novel mechanism by which 9-cis-RA signaling inhibits the proliferation of RA-sensitive mammary cancer cells. By using the LBD of RXRα as bait in a yeast two-hybrid screening, we isolated an ortholog of human replication factor C subunit 3 (hRFC3) in a sea urchin library. The interaction of RFC3 with RXRα depends on 9-cis-RA through the C-terminal LXXLL motifs of RFC3 and regulates the ability of 9-cis-RA to inhibit proliferation of MCF-7 breast cancer cells and sea urchin embryogenesis. This study provides the first example of cooperation between RA-activated RXRα and RFC complex for processive DNA synthesis and replication and their implications in RA-sensitive mammary cancer cells and embryonic development.
Materials and Methods
Yeast two-hybrid screening
Gal4DBD/RXRα-LBD (24) was used as bait to screen the sea urchin Strongylocentrotus purpuratus oocyte cDNA library in the pACT2 vector (Clontech, Palo Alto, CA) for RXRα-interacting proteins in the presence of ATRA (10−6 m; Sigma-Aldrich, St. Louis, MO), as described previously (25). The library-origin plasmids rescued from positive clones expressing both HIS3 LacZ reporters were retransformed into Y190 yeast cells, together with bait plasmid, for testing of the specificity of protein-protein interaction in the presence of ATRA or 9-cis-RA.
Plasmids
Mammalian expression plasmids for hemagglutinin (HA) and nuclear localization signal (NLS) fusion proteins (HA-NLS-hRFC1—hRFC5) were constructed by inserting PCR-amplified cDNA into EcoRI/XhoI (for hRFC2–5) and MunI/XhoI (for hRFC1) sites, respectively, in the pcDNA3-HA-NLS vector (25). The PCR templates for each hRFC were kindly provided by Dr. Jerard Hurwitz (Memorial Sloan-Kettering Cancer Center, New York, NY). The hRXRα, hRXRβ, and hRXRγ PCR products were digested with EcoRI/XhoI and ligated into the pcDNA3 vector (Invitrogen, Carlsbad, CA). The templates for hRXRα, hRXRβ, hRXRγ, and hRARα were kind gifts from Dr. Seung-Whan Kim (University of Ulsan College of Medicine, Seoul, Korea) or purchased from a provider (Source BioScience, Nottingham, UK). pcDNA3-hERα was kindly provided by Dr. Hueng-Sik Choi (Chonnam National University, Gwangju, Korea). The expression vectors for steroid receptor coactivator-1 (SRC1)-C, SRC1-D, and silencing mediators of retinoid and thyroid hormone receptor (SMRT)-D are as described previously (26, 27). To construct the pcDNA3-RXR and pcDNA3 (HA-NLS)-RFC families, primer sets were used in conventional PCR amplification as follows: hRXRβ (5 and 6), hRXRγ (7 and 8), hRFC1 (9 and 10), hRFC2 (11 and 12), hRFC3 (13 and 14), hRFC4 (15 and 16), and hRFC5 (17 and 18; Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). To express the Gal4-DBD fusion SnRXRα and hRXR families, primer sets for SnRXRα (19 and 20), hRXRβ, and hRXRγ (5–8) were used in conventional PCR amplification, and the cDNA amplicons were cloned into the EcoRI/PstI (for SnRXRα) or EcoRI/XhoI (for hRXRβ and hRXRγ) sites of the pCMX vector (25). To construct the VP16-fused RFC3 expression vectors for LXXLL mutants and fragments, cDNA for hRFC3-m1 (primers 21 and 22), -m2 (23 and 24), -m3 (23 and 24 with -m1 template), -A (ATPase domain; 25 and 26), -B (intermediate region; 27 and 28), and -C (C-terminal region; 29 and 14) and for SnRFC3-mut (30 and 31), -A (32 and 33), -B (34 and 35), and -C (36 and 37; Supplemental Table 1) were amplified by conventional PCR and then inserted into the XbaI/XhoI sites of the pCMX-VP16-N vector (25). cDNA for hRFC1 (38 and 39), hRFC2 (40 and 12), hRFC4 (41 and 16), hRFC5 (42 and 18), and SnRFC4 (43 and 44; Supplemental Table 1) were also cloned into the XbaI/XhoI sites of the pCMX-VP16-N vector. All constructs were verified by DNA sequencing. The nucleotide and predicted amino acid sequences of the sea urchin Strongylocentrotus nudus are available from GenBank [accession nos. JF320947 (SnRFC4), JF320948 (SnRFC3), and JF320949 (SnRXRα)].
Cell culture and reporter assays
The 293T and Cos7 cell cultures were performed as previously described (25). 9-Cis-RA, ATRA, T3, 4-[E-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]-benzoic acid, estradiol-17β, and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich. Bexarotene (LGD1069) was purchased from LC Laboratories (Woburn, MA). Transfection and luciferase assay were done as described previously (25). The transfection indicator construct pRSV-β-galactosidase (pRSV-β-gal), the Gal4-responsive reporter construct Gal4-thymidine kinase-luciferase (Gal4-Tk-Luc), luciferase reporters RXR response element (DR1-Luc) and estrogen-responsive element (ERE-Luc), and Gal4-DBD fusion hERα construct (Gal4-hERα) were described previously (25) or kindly provided by Dr. Jae W. Lee, Oregon Health and Science University (Portland, OR). After 24 h of transfection, ligands were applied to the culture medium at the indicated concentrations. After an additional 18 h, the cells were harvested and luciferase activities were assayed using a microplate luminometer (Berthold, Wildbad, Germany) and normalized by the β-galactosidase values. All transfection experiments were performed in triplicate determinations at least twice in different cells, yielding similar results. Statistical differences were determined by two-tailed Student's t test. All analyses were performed using the SPSS software (version 14.0; Chicago, IL), and data were considered statistically significant when P < 0.05.
Coimmunoprecipitation and immunoblotting
The 293T cells were transfected with the HA-NLS-hRFC1–5 and pcDNA3-hRXR(α, β, γ)/-hRARα expression plasmids. The cells were treated for 24 h with 1 μm 9-cis-RA or the same volume of DMSO. Cell lysates were prepared using a lysis buffer [50 mm Tris-Cl (pH 8.0), 170 mm NaCl, 5 mm EDTA, 0.5% Nonidet P-40, 1 mm dithiothreitol] supplemented with protease inhibitors (Sigma-Aldrich) and precleared with protein-A/G Plus-agarose beads (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 4 h at 4 C. The precleared lysates were sequentially incubated with 1.0 μg anti-HA antibody (H9658; Sigma-Aldrich) and 50 μl protein-A/G beads for 4 h at 4 C, and the beads were finally washed three times with the lysis buffer without protease inhibitors. Immunoadsorbed proteins were analyzed by Western blotting with specific antibodies.
The protein content of cell lysates or homogenized tissues was determined with Bradford reagent using BSA as the standard. After heating for 5 min at 100 C in 1× Laemmli sample buffer, the samples were separated by 10.0–12.5% SDS-PAGE. The resulting gels were transferred to nitrocellulose membranes (Pall Co., Ann Arbor, MI). For Western blotting, the membrane was incubated with the following antibodies: anti-HA (Sigma-Aldrich); RXRα, RARα, ERα, proliferating cell nuclear antigen (PCNA), glyceraldehyde-3-phosphate dehydrogenase, and secondary antibodies (goat antirabbit or antimouse horseradish peroxidase; all from Santa Cruz Biotechnology); and RXRβ and RXRγ (Millipore, Billerica, MA). After washing, the membranes were incubated in a chemiluminescent detection reagent, and the bands were visualized by exposure to x-ray films (GE Healthcare, Piscataway, NJ).
Analysis of cell growth and flow cytometry
MCF-7 and HCT-116 cell lines were obtained from the American Type Culture Collection (Rockville, MD). HepG2 and MDA-MB-231 cells were kindly provided by Dr. Chu Won Nho (Korea Institute of Science and Technology, Gangneung Institute, Gnagneung, Korea). MCF-7 and MDA-MB-231 cell lines were maintained at 37 C with 5% CO2 in RPMI-1640 and L-15 media (Welgene, Daegu, Korea), respectively, containing 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen) and 1% antibiotic-antimycotic reagent. The 293T and HepG2 cells were grown in DMEM supplemented with 10% FBS and 1% antibiotic reagent. HCT-116 cells were maintained in MEM with 10% FBS and 1% antibiotics. MCF-7 and MDA-MB-231 cells were seeded at a density of 5 × 104 cells per 24-well plate and incubated for 24 h. The cells were transfected with 0.8 μg small interfering RNA (siRNA)-RXRα (5′-CUGGAAUGAGCUGCUCUCAUC-dTdT-3′), siRNA-RFC3 (5′-AAGUAACUACCACCUUGAAGUUA-dTdT-3′), or siRNA-control (5′-CCUACGCCACCAAUUUCGU-dTdT-3′), along with the indicated amount of hRXRα, HA-hRFC3, and/or hERα expression vector. At 1 and 3 d after transfection, 9-cis-RA was added to the culture medium at 10−6 m concentration. Suitable aliquots of the cell suspension were placed on a hemocytometer, and the number of cells able to exclude trypan blue was counted under a light microscopy at 1, 3, and 5 d after transfection. Cell-cycle analysis by flow cytometry was performed according to a standard procedure (28). Briefly, at 5 d after transfection, the cells were harvested, fixed with 70% ethanol, and incubated with a staining solution containing ribonuclease A (30 μg/ml; Sigma-Aldrich) and propidium iodide (50 μg/ml; Sigma-Aldrich) in PBS. Cellular DNA content was analyzed by flow cytometry using a laser-based flow cytometer (BD Biosciences, Franklin Lakes, NJ). At least 20,000 cells were used for each analysis, and the results are displayed as histograms. Multiple comparisons between groups were performed by one-way ANOVA followed by Tukey's post hoc analysis. Statistical significance was accepted with P < 0.05. All statistical analyses were performed by using SPSS version 14.0.
To evaluate the effect of siRNA on mRNA expression of RXRα and RFC3, total RNA were prepared from the transfected cells using an RNA extraction kit (Bioneer, Daejeon, Korea). One microgram of total RNA was reverse transcribed to first-strand cDNA using Moloney murine leukemia virus reverse transcriptase and oligo(deoxythymidine) primer. PCR was performed in a 20-μl final volume containing 2 μl 10× reaction buffer including 0.3 μg synthesized cDNA, 15 mm MgCl2, 2.5 mm deoxynucleotide triphosphate, 1.6 μm RXRα or RFC3 primer set (27, 28, 45, and 46; Supplemental Table 1), and 2.5 U Taq DNA polymerase (Bioneer). As a control, human β-actin cDNA was also amplified under the same conditions using specific primers (47 and 48). Protein levels of endogenous RXRα and ectopic HA-RFC3 expression were also examined by Western blot.
Isolation of sea urchin mRNA and cDNA cloning
Messenger RNA extraction from the S. nudus 64-cell-stage embryos and the synthesis of first-strand cDNA were performed as described previously (25). The full-length cDNA encoding RFC3 and RXRα were amplified by rapid amplification of cDNA ends (RACE)-PCR using the first-strand cDNA and gene-specific primers based on cDNA sequences of S. purpuratus RFC3 and RXRα (GenBank Accession nos. XM_796686 and XM_779153) according to a established protocol (Clontech). The gene-specific primer sequences used in the 5′- and 3′-RACE-PCR are shown in Supplemental Table 1. The amplified PCR products were cloned into a TOPO vector (Invitrogen), and independent clones were sequenced. Nucleotide sequences of independent 5′- and 3′-RACE clones were identified by the NCBI BLAST search program. Amino acid sequence alignment was performed using the CLUSTAL W multiple sequence alignment program.
Microinjection of mRNA and morpholinos into sea urchin embryos
S. nudus adults were purchased from a fishermen's cooperative society in the Gangneung area during their breeding season (July to September) and maintained at 16–18 C. Gametes were obtained by injecting 1.0 m KCl directly into the coelomic cavity. Eggs were fertilized with a suspension of sperm in Millipore-filtered seawater containing 50 μg/ml streptomycin and 50 μg/ml kanamycin and cultured at 16 C. The open reading frame of SnRFC3 was cloned into the pcDNA3-HA-NLS vector behind a T7 promoter and transcribed in vitro with a Message Machine transcription kit (Ambion, Austin, TX) (Supplemental Fig. 8A). The standard control mRNA provided by the manufacturer was prepared as a control. Antisense morpholinos (MO) (Gene Tools, Portland, OR) for SnRXRα (5′-AATGCAAAACATCGGCTGTGCTTCG-3′) and SnRFC3 (5′-TCCACCCATAAACTCATTTTGAGG-3′) targeting their start sites were used to suppress its translation. The standard control MO provided by the manufacturer was used as a control. Microinjection was carried out as described previously, with some modifications (29, 30). The mRNA and MO were diluted in distilled water and mixed with fast green just before injection. MO (10 pg), or mRNA (110 or 330 ng) was injected into eggs just after fertilization. Western blot analysis was applied to detect HA-SnRFC3 protein expression in 293T cells using HA antibody. To define the concentration range of 9-cis-RA to be used for the sea urchin embryos, different doses of 9-cis-RA between 2 × 10−5 and 2 × 10−6 m were added to the embryos. 9-Cis-RA at concentrations of 4–8 μm caused severe disturbance of gastrulation and further development, and higher concentrations added from the two-cell stage were lethal to early-stage embryos. Thus, we decided to treat embryos with 6 μm 9-cis-RA.
Whole-mount in situ hybridization
Whole-mount in situ hybridization was performed as previously described (31). Briefly, digoxigenin-labeled antisense or sense probes were transcribed from a TOPO cloning vector (Invitrogen) including partial SnRXRα (770 bp, 646-1417 nucleotides) that had been linearized by digestion with EcoRV or KpnI using SP6 or T7 RNA polymerase and a digoxigenin-labeling mix (Roche Diagnostics GmbH, Mannheim, Germany). In the case of SnRFC3, the pcDNA3-HA-NLS vector including SnRFC3 (1036 bp, 1–1036 nucleotides) was linearized by digestion with BamHI or XbaI. The sizes of probes were then reduced to approximately 200 nucleotides by limited alkaline hydrolysis.
Quantitative RT-PCR analysis
Total RNA were extracted from fertilized eggs and embryos of the sea urchin S. nudus (from four maternal individuals) at various developmental stages using the RNeasy kit (QIAGEN, Valencia, CA). RNA (1 μg) were reverse transcribed to the first-strand cDNA using Moloney murine leukemia virus reverse transcriptase (Bioneer). Quantitative PCR was performed in a volume of 20 μl containing synthesized cDNA (0.4 μg each), 2× SYBR Premix Ex Taq, 50× ROX Reference Dye II (TaKaRa Bio, Otsu, Shiga, Japan), and 10 μm primer set (49 and 50 for SnRXRα; 51 and 52 for SnRFC3; Supplemental Table 1) designed by the Primer Express software (version 3.0; Applied Biosystems, Foster City, CA) on a quantitative PCR system (7500; Applied Biosystems). Thermal cycling was performed with a two-step PCR protocol: 50 C for 2 min, 95 C for 10 min, and 40 cycles of 95 C for 15 sec and 60 C for 1 min. Relative quantitation values are expressed using the 2−ΔΔCt method as fold changes in the SnRXRα or SnRFC3 mRNA normalized to ubiquitin mRNA (53 and 54; Supplemental Table 1).
Results
RXRα interacts with RFC3 in a 9-cis-RA-dependent mode
To identify potential coregulators of RXRα, we screened a sea urchin (S. purpuratus) oocyte cDNA library using the LBD of hRXRα (24) as bait in the yeast two-hybrid screening. Among 36 isolates that showed ATRA- or 9-cis-RA-dependent interaction, several cDNA were found to encode SRC homologs (25). The isolate showing the strongest interaction in the presence of 9-cis-RA encoded an ortholog of the mammalian RFC3 C-terminal region (287–356 amino acid residues; Supplemental Fig. 1, A and B). Because the Oriental purple sea urchin (S. nudus) belongs to the same genus and is easily obtained along the eastern coast of Korea (32), we obtained full-length cDNA encoding S. nudus RFC3 (SnRFC3) and RXRα (SnRXRα) (Supplemental Figs. 1B and 2). The RFC3-RXRα interaction was also enhanced by the RXR ligands 9-cis-RA and bexarotene (LGD1069) (13), as confirmed by the mammalian two-hybrid assays (Fig. 1A). In contrast, the RAR-selective retinoids ATRA and 4-[E-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]-benzoic acid and T3 did not noticeably affect the interaction (Fig. 1A).
Fig. 1.

RXRα interacts with RFC3 in a ligand- and LXXLL motif-dependent manner. A, 9-Cis-RA- and rexinoid-activated human RXRα LBD interacts with hRFC3. The 293T cells were transfected with Gal4-thymidine kinase luciferase (Gal4-Tk-Luc) reporter vector (200 ng), Gal4-hRXRα-LBD (20 ng), VP16-hRFC3 (200 ng), VP16-hSRC1-C (200 ng), and LacZ expression vectors (pRSV-β-gal, 200 ng). After 24 h of transfection, the cells were incubated in the presence of the indicated ligands for 18 h. All cells were lysed, and the luciferase activities were measured and normalized against the LacZ expression as an internal control. Values are the average of three independent assays (mean ± sd), which were normalized to the control sample transfected with Gal4-hRXRα-LBD and VP16-hRFC3, and treated with DMSO instead of ligands. B, LXXLL motifs of RFC3 are critical for 9-cis-RA-dependent RXRα interaction. The hRFC3 fragments (hRFC-A, -B, and -C) and site-directed mutants (hRFC3–m1, -m2, and -m3) were fused to the VP16 gene and the constructs were transfected in 293T cells as described in A. Statistical changes were determined by the Student's t test (two-tailed) and are denoted as follows: *, P < 0.05; **, P < 0.01.
LXXLL motifs of RFC3 are essential for 9-cis-RA-dependent interaction with RXRα
Two LXXLL motifs in hRFC3 and one LXXLL motif in sea urchin RFC3 may serve as RA-dependent interaction interfaces with RXRα (3). To test this idea, we constructed mammalian expression vectors encoding chimeric proteins of the VP16 activation domain fused to several RFC3 fragments and site-directed mutants at the LXXLL motifs (Fig. 1B). As expected, hRFC3-A and hRFC3-m3 (the double-LXXAA mutant) did not show 9-cis-RA-dependent interactions with the hRXRα LBD. However, the hRFC3-B and -C fragments, which included the first and second LXXLL motifs, respectively, and hRFC3-m1 and -m2 (the single-LXXAA mutants) showed basal interactions with the holo-hRXRα LBD (Fig. 1B). Similarly, the SnRFC3-C fragment possessing the LXXLL motif, but not SnRFC3-A, -B, or the LXXAA mutant, interacted with human and sea urchin RXRα (Fig. 2, A and B). To directly test whether RFC3 competes with the LXXLL motifs in NR coactivators, a SRC1 fragment (SRC1-C, 568–779 residues containing three LXXLL motifs) (27) was subjected to the mammalian two-hybrid assays. Cotransfection with the SRC1-C expression vector but not SRC1-D (759–1141 residues), concentration-dependently abrogated 9-cis-RA-dependent interaction between hRFC3 and hRXRα (Fig. 3). These results clearly demonstrate that deuterostome RFC3 uses its LXXLL motifs to interact with RXRα in a 9-cis-RA-dependent manner.
Fig. 2.

Sea urchin RFC3 interacts with RXRα of human or sea urchin in a 9-cis-RA- and LXXLL-dependent manner. A, Full-length sea urchin RFC3 (SnRFC3), fragments (SnRFC3-A, -B, and -C), and a site-directed mutant at the LXXLL motif (SnRFC3-mut) were fused to the VP16 gene, as depicted in the schematic diagram, and the constructs (200 ng each) were transfected into 293T cells along with the Gal4-Tk-Luc reporter vector (200 ng), Gal4-hRXRα-LBD (20 ng), and LacZ expression vector (pRSV-β-gal, 200 ng). At 24 h after transfection, the cells were incubated in the presence of 9-cis-RA at three different dosages (10−9, 10−8, and 10−7 m) for 18 h. All cells were lysed and the luciferase activities were measured and normalized against LacZ expression as an internal control. Values are the average of three independent assays (mean ± SD), which were normalized to the same amount of DMSO-treated cells transfected with the Gal4-hRXRα-LBD and VP16-SnRFC3 series. (B) Sea urchin RXRα-LBD (SnRXRα-LBD) interacts with SnRFC3 in a 9-cis-RA-dependent manner. The SnRXRα-LBD region (229–463 amino acids) was inserted to the Gal4-DBD expression vector and this construct (20 ng) was used in the mammalian two-hybrid assays as described in (A). Statistical changes were determined by the Student's t test (two-tailed) and are denoted as (*) for P < 0.05.
Fig. 3.

SRC1-LXXLL motifs block hRXRα–RFC3 interaction. Gal4-DBD–fused hRXRα-LBD, VP16-hRFC3, pcDNA3-SRC1-C, and -SRC1-D expression vectors were transfected in 293T cells, along with reporter vectors (200 ng) as indicated. At 24 h after transfection, the cells were treated with 9-cis-RA and the luciferase activities were measured and normalized as described in Fig. 1.
Next, we examined whether RFC3 also interacts with other retinoid receptors, including RXRβ/γ and RARα, using coimmunoprecipitation. We were unable to find an antibody that could detect endogenous RFC3, so we constructed an expression vector for HA-tagged and ectopically expressed RFC3 in 293T cells. As expected, RFC3 strongly copurified with RXRα in the presence of 9-cis-RA (Fig. 4A), whereas RXRβ/γ and RARα showed no significant interaction with RFC3, even in the presence of 9-cis-RA (Supplemental Fig. 3). Mammalian two-hybrid assays also exhibited the specific interaction between 9-cis-RA-activated RXRα and RFC3 (Supplemental Fig. 4, A and B). Conversely, liganded RXRα interacted with RFC3 but not with other RFC components (RFC1, -2, -4, and -5; Fig. 4B). RFC4 interacted with RXRα, but these interactions were inhibited by 9-cis-RA in sharp contrast with the interactions of RXRα and RFC3. In the absence of ligands, many NR, including RXRα, bind to transcriptional corepressors, such as SMRT and NR corepressor (33, 34). In an immunoprecipitation experiment, we revealed that SMRT-D (SMRT residues 1060–1495 containing the NR-interacting motifs) (27) disrupts the RFC4-RXRα interaction (Fig. 4C), suggesting that this interaction is similar to that of NR and NR corepressors. Altogether, these results suggest that 9-cis-RA-activated RXRα interacts with RFC3 via its C-terminal LXXLL motifs and that these interactions are not conserved among other retinoid receptors or RFC proteins.
Fig. 4.
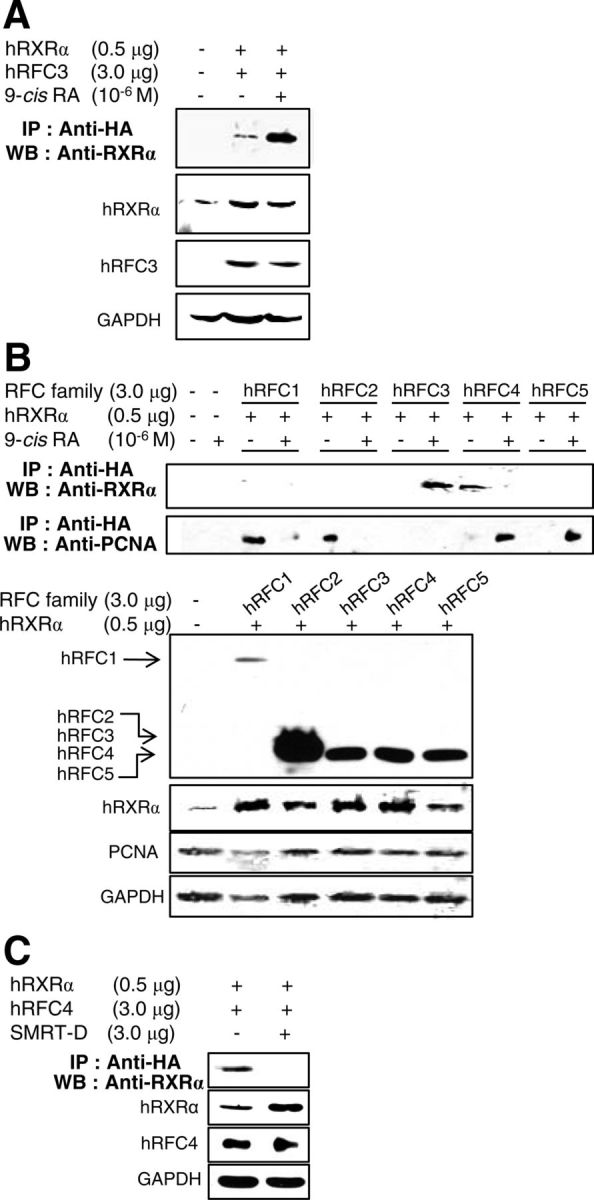
9-Cis-RA-activated RXRα specifically interacts with RFC3 and destabilizes the RFC-PCNA complex. A, hRFC3 immunoprecipitates human RXRα in the presence of 9-cis-RA. Because no antibody that could detect endogenous RFC3 was found, HA-tagged hRFC3 was coexpressed with hRXRα in 293T cells. Immune complexes were resolved by SDS-PAGE and blotted with the indicated antibodies. B, HA-tagged hRFC1-hRFC5 were coexpressed with hRXRα in 293T cells, with or without 9-cis-RA treatment. Immune complexes were resolved by SDS-PAGE and blotted with the indicated antibodies. Expression of HA-hRFC family members hRXRα, PCNA, and GAPDH was detected by Western blotting. C, Expression vectors for hRXRα and HA-tagged hRFC4 were transfected with an expression vector of the SMRT-D fragment or maternal vector in 293T cells. IP, Immunoprecipitation; WB, Western blot.
9-Cis-RA-activated RXRα results in reconfiguration of the RFC-PCNA complex
We further investigated the involvement of 9-cis-RA in the interaction between the five RFC subunits and the PCNA, because PCNA complexed with RFC plays an essential role in processive DNA synthesis (35, 36). Consistent with the previous reports of RFC-PCNA crystal structures (37, 38), HA-tagged hRFC1 and hRFC2 immunoprecipitated PCNA, whereas RFC4 and RFC5 copurified with PCNA in the presence of 9-cis-RA (Fig. 4B). We also found that RFC3 did not directly interact with PCNA, regardless of the presence of 9-cis-RA. An earlier report noted that RFC3 was essential for the interaction between a core RFC complex (RFC2/4/5) and the larger RFC1 subunit (39). This study showed that, at least in part, unliganded RXRα immunoprecipitates the core RFC-like complex through RFC4, whereas 9-cis-RA causes the dissociation of this complex (Fig. 5). 9-Cis-RA also moved RFC4 from the core complex to PCNA, and the liganded RXRα recruited RFC3 (Figs. 4B and 5). Considering the essential role of RFC-PCNA in processive DNA synthesis (35, 36, 40), 9-cis-RA-evoked conformational changes in RXRα may disrupt the stable RFC-PCNA complex through RFC3 inhibition of DNA replication and proliferation of RA-sensitive cells.
Fig. 5.
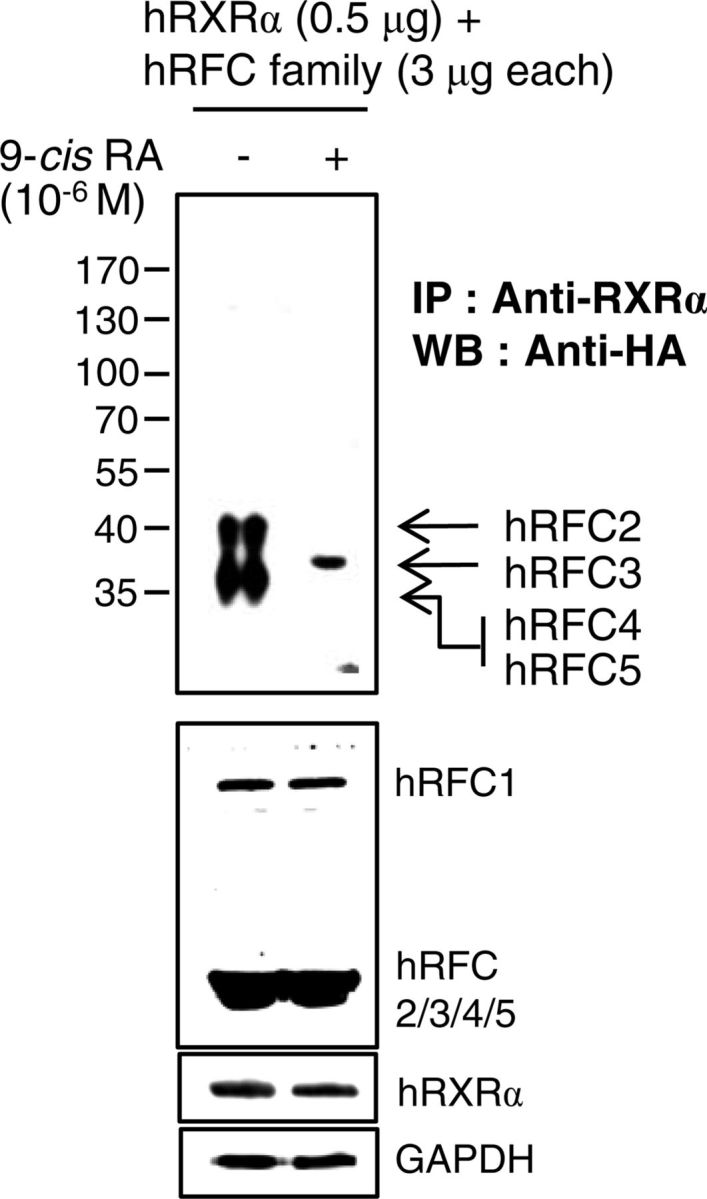
9-Cis-RA enforces a dissociation of the core RFC complex (hRFC2/4/5) and association of hRFC3 with hRXRα. Expression vectors for five HA-hRFC family members were cotransfected with hRXRα expression vector in 293T cells. The cells were incubated for 24 h with 1 μm 9-cis-RA (+) or the same volume of DMSO (−). Immune complexes were analyzed as described in Fig. 4. IP, Immunoprecipitation; WB, Western blot.
Knockdown of RXRα and overexpression of RFC3 impairs the ability of 9-cis-RA to block proliferation of MCF-7 breast cancer cells
Although several studies have demonstrated the ability of 9-cis-RA to inhibit the growth of estrogen receptor (ER)-positive breast cancer cells (41, 42), the signaling pathway through the cognate receptor RXR remains obscure. We observed reduced endogenous mRNA and/or protein levels in two siRNA targeting RXRα and RFC3 (Supplemental Fig. 5). We also confirmed that 9-cis-RA inhibits the growth of ER+ MCF-7 cells in a dose-dependent manner (Fig. 6A), as reported previously (41). Strikingly, the RXRα-knockdown MCF-7 cells strongly recovered the growth pattern despite the presence of 9-cis-RA, and this recovery was completely attenuated by RXRα overexpression (Fig. 6B). Consistent with these results, we observed a smaller percentage in the S-phase of the cell cycle than in normal cells after 5 d of 9-cis-RA treatment, similar to the previous report (41), whereas RXRα knockdown led to a normal cell-cycle pattern (Table 1 and Supplemental Fig. 6). Considering that liganded RXRα recruits RFC3, which is essential for the interaction between a core RFC complex (RFC2/4/5) and the larger RFC1 subunit (39), we hypothesized that RFC3 played a key role in the inhibition of MCF-7 cell growth by 9-cis-RA. This idea is clearly supported by RFC3-knockdown cells showing suppressed cell growth and S-phase entry and by full recoveries of the cell growth pattern and S-phase progression by RFC3 overexpression even in the presence of 9-cis-RA (Table 1 and Fig. 6C). These findings suggest that recruitment of RFC3 by 9-cis-RA-activated RXRα resulted in the dissociation of the RFC-PCNA complex and consequently suppressed the cell proliferation. We also found that among RFC components, only RFC3 restored MCF-7 cell growth (Fig. 6D). To further examine the cell specificity of this mechanism, we analyzed the correlation between the susceptibility of additional human cell lines (MDA-MB-231, 293T, HCT-116, and HepG2) to 9-cis-RA and protein expression levels of RXRα in the cells. All examined human cells except MCF-7 cells showed normal growth patterns regardless of 9-cis-RA, but a relatively high expression level of RXRα and the specific expression of ERα were detected in MCF-7 cells (Supplemental Fig. 7). ER− breast cancer cell lines, such as MDA-MB-231, are known to be relatively resistant to the growth-inhibitory effects of 9-cis-RA, and ectopic ER expression confers responsiveness to 9-cis-RA growth inhibition (41, 42). Consistent with the previous studies, we also demonstrated the involvement of ERα by recovery of 9-cis-RA sensitivity in ERα-transfected MDA-MB-231 cells (Supplemental Fig. 8A). Interestingly, the ERα-transfected MDA-MB-231 cells showed growth recovery by RXRα knockdown despite the presence of 9-cis-RA, as in MCF-7 cells, and this recovery was partially attenuated by RXRα overexpression in a concentration-dependent manner (Supplemental Fig. 8B). However, additional studies are clearly needed to reveal the involvement of ERα in the ligand-activated RXRα-RFC3 complex to gain better insights into cell-specific responsiveness to 9-cis-RA, although we observed that ERα does not interact with RFC3 but inhibits the interaction of liganded RXRα with RFC3 (Supplemental Fig. 8D). Collectively, our results suggest that 9-cis-RA-dependent inhibition of ER+ MCF-7 cell proliferation is due to the relative RXRα expression level and the specific interaction of RFC3 with liganded RXRα.
Fig. 6.

Knockdown of RXRα or overexpression of RFC3 restores proliferation of MCF-7 human breast cancer cells inhibited by 9-cis-RA. A, MCF-7 cells were inhibited by 9-cis-RA in a dose-dependent manner. The cells were plated at a density of 5 × 104 cells per 24-well plate culture and incubated for 5 d. At 1 and 3 elapsed days, 9-cis-RA was added to the culture medium. Aliquots of the cell suspension were placed on a hemocytometer, and the number of cells able to exclude trypan blue was counted under light microscopy at 1, 3, and 5 elapsed days. Cell numbers are expressed as the mean ± sd of three independent wells. B, Knockdown of human RXRα rescues MCF-7 cell proliferation suppressed by 9-cis-RA. MCF-7 cells were plated at a density of 5 × 104 cells as in A, and the cells were transfected with 0.8 μg siRNA-RXRα or siRNA-control, along with the indicated amount of the hRXRα expression vector. At 1 and 3 d after transfection, the cells were treated with 9-cis-RA (10−6 m). At 1, 3, and 5 d after transfection, the cell numbers were counted as in A. C, hRFC3 restores MCF-7 cell proliferation suppressed by 9-cis-RA. MCF-7 cells were transfected with 0.8 μg siRNA-RFC3 or siRNA-control, along with the indicated amount of hRFC3 expression vector. The cells were treated with 9-cis-RA (10−6 m), and the numbers of cells were counted as in A. D, hRFC3 specifically recovers proliferation of 9-cis-RA-treated MCF-7 cells. MCF-7 cells were transfected with RFC expression vector, as indicated, and then treated with 9-cis-RA and counted as in A. Different letters indicate significant differences (P < 0.05) among treatment groups at 5 d after transfection.
Table 1.
Percentage of MCF-7 cells in G0/G1, S, and G2/M phases
| Cell cycle | Treatment |
|||||||
|---|---|---|---|---|---|---|---|---|
| DMSO | DMSO si-RXRα | DMSO si-RFC3 | 9-cis-RA | 9-cis-RA si-RXRα | 9-cis-RA si-RXRαhRXRα | 9-cis-RA hRFC3 | 9-cis-RA si-RFC3 hRFC3 | |
| G0/G1 | 52.8 ± 0.3a | 53.2 ± 0.7a | 61.3 ± 2.9b | 74.8 ± 0.1c | 53.9 ± 2.9a | 74.7 ± 0.6c | 49.0 ± 1.9a | 62.3 ± 1.4b |
| S | 34.3 ± 0.3c,d | 36.2 ± 3.1c,d | 23.2 ± 2.0b | 16.9 ± 1.3a | 33.1 ± 0.9c | 15.9 ± 0.4a | 38.4 ± 3.1d | 27.9 ± 0.8b |
| G2/M | 12.9 ± 0.1a,b | 10.6 ± 2.4a,b | 15.5 ± 4.9b | 8.3 ± 0.4a | 13.0 ± 2.0a,b | 9.4 ± 1.0a | 12.6 ± 1.1a,b | 9.7 ± 0.7a,b |
See Materials and Methods and Supplemental Fig. 6 for details. Values (mean ± sd) within a row with the same superscript are not significantly different.
Sea urchin RXRα and RFC3 modulate early embryonic development
To define the global involvement of RXRα and RFC3 in cell proliferation, the spatiotemporal expression patterns of SnRXRα and SnRFC3 mRNA were first analyzed at various stages of sea urchin embryogenesis. The SnRXRα mRNA is gradually increased from the 32-cell to pluteus stage (Fig. 7A), indicating its zygotic expression. Whole-mount in situ hybridization revealed that SnRXRα was mainly expressed in mesomere derivatives (an1 and an2) at the 32- and 64-cell stages, in the archenteron tip and endoderm cells in the late gastrula stage, and in the anterolateral arm-tip ectoderm and gut at the pluteus stage (Fig. 7B). In contrast, SnRFC3 mRNA expression was detected from the one-cell stage and showed a widespread pattern during embryogenesis (Fig. 7, A and C). In the late gastrula and pluteus stages, SnRFC3 mRNA signals were colocalized with those of SnRFC3 in the archenteron tip and gut.
Fig. 7.
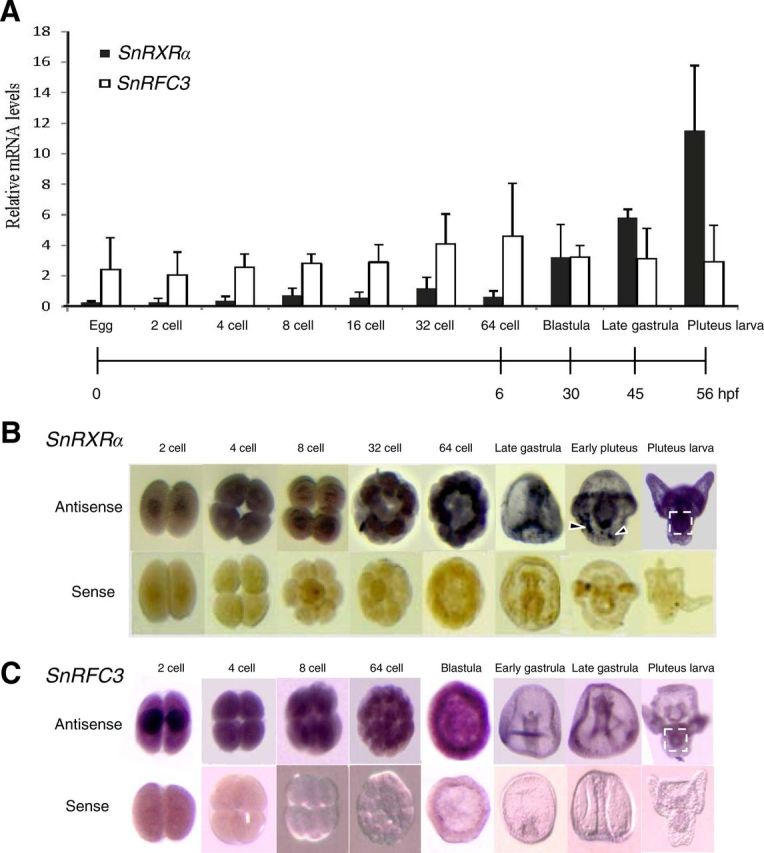
Temporal and spatial mRNA expression levels of RXRα and RFC3 in sea urchin embryonic development. A, Temporal expression levels of S. nudus RXRα (SnRXRα) and RFC3 (SnRFC3) mRNA using real-time PCR. The relative SnRXRα and RFC3 mRNA levels were normalized by ubiquitin values and expressed as mean ± sd (n = 4 maternally independent individuals). B and C, Spatial distribution of SnRXRα or SnRFC3 transcripts during normal embryonic development analyzed by whole-mount in situ hybridization. The embryos were hybridized with an antisense or sense probe as indicated. The 32-cell, 64-cell, and blastula stages are animal-pole views. The gastrula- and pluteus-stage embryos are in lateral view with the animal pole at the top. Dashed-line squares and arrowheads indicate the gut and nonspecific signals, respectively. hpf, Hours post-fertilization.
To further investigate the roles of SnRXRα and SnRFC3 in sea urchin embryonic development, we designed antisense MO oligonucleotides (SnRXRα and SnRFC3 MO) and microinjected them into fertilized eggs of S. nudus. SnRXRα MO-injected embryos (SnRXRα morphants) developed abnormally from the early blastula stage, showing a thick, monolayered epithelium (Fig. 8). Further developmental progress was stopped at the early gastrula stage without initiating primary invagination or subsequent archenteron formation. In contrast to the lethal SnRXRα morphants, SnRFC3 morphants survived beyond the pluteus larval stage (>118 h after fertilization); however, they showed a thick, monolayered epithelium at the earlier hatched-blastula stage (Fig. 8). Note that the SnRFC3 morphants displayed a morphological abnormality: the gut rudiment was short and disorganized, and the outer prism failed to form oral arms. Pigment cells eventually formed, however, indicating that some differentiation occurs. Taken together, these results suggest that SnRXRα and SnRFC3 play pivotal roles during early embryogenesis in the sea urchin.
Fig. 8.

Effects of microinjection of antisense MO oligonucleotides targeting SnRXRα or SnRFC3 on sea urchin S. nudus embryogenesis. Embryos are lateral views with the animal pole at the top. Uninjected embryos and embryos injected with a control MO developed to the complete pluteus larval stage. SnRXRα MO-injected embryos developed abnormally from the early blastula stage [21–30 h post-fertilization (hpf)], and further development was stopped at this stage. SnRFC3 MO-injected embryos showed failures of complete archenteron (arrowheads) and oral arm (asterisks) formation. #, Number of embryos/total number of injected embryos.
Overexpression of RFC3 recovers normal sea urchin embryogenesis despite the presence of 9-cis-RA
Finally, we tested the effects of RFC3 overexpression on 9-cis-RA-treated embryos by direct injection of SnRFC3 mRNA into fertilized eggs of sea urchins. As shown in previous studies (43, 44), excessive 9-cis-RA (6 μm in seawater) arrests development before the gastrula stage and causes eventual embryonic death (Fig. 9). We found that injection with a lower concentration (110 ng) of SnRFC3 mRNA was not able to fully recuperate the developmental process in the 9-cis-RA-treated embryos. Instead, it resulted in a significant delay in embryogenesis and severe abnormalities, with enlarged pigment cells and skeletal mispatterning at the gastrula and pluteus stages. However, injection of a higher concentration (330 ng) of SnRFC3 mRNA recovered a normal developmental pattern despite the presence of 9-cis-RA (Fig. 9).
Fig. 9.

SnRFC3 modulates early embryonic development in the sea urchin. mRNA were injected into eggs just after fertilization at the indicated doses. The injected eggs were maintained in filtered seawater containing 9-cis-RA (final concentration 6 μm) or the same amount of DMSO. The number of embryos/number of injected embryos at each stage is in parentheses. hpf, Hours post-fertilization.
Discussion
We were interested in defining the mechanisms by which RA inhibited the growth of RA-sensitive mammalian cells and the development of sea urchin embryos as a first step in the development of targets that may interact with RXRα, in which RA has been implicated. In this study, we isolated SnRFC3 as an interacting partner for RXRα in a yeast two-hybrid screening. Using several approaches, our studies demonstrated that 9-cis-RA-activated RXRα is able to functionally interact with RFC3 and consequently destabilizes the RFC-PCNA complex. Evidently, RXRα knockdown or RFC3 overexpression resulted in the attenuation of the ability of 9-cis-RA to inhibit not only the proliferation of RA-sensitive breast cancer cells but also normal development of sea urchin embryos. These findings may explain one of the mechanisms by which 9-cis-RA induces growth inhibition in RA-sensitive cells and developing embryos.
Here, we demonstrated that RFC3 contains functional LXXLL motifs to interact with 9-cis-RA-activated RXRα. Because RXRα was initially identified as a heterodimeric partner of NR (15), many coactivators have been characterized by biophysical analysis of NR-interacting regions containing LXXLL motifs (1). In fact, the coactivator SRC1-C domain containing multiple LXXLL motifs competed for RFC3 (Fig. 3), suggesting that RFC3 uses these motifs to interact with RXRα in a 9-cis-RA-dependent manner. In addition to NR coactivators, several classes of proteins, such as CLOCK, neuronal PAS domain protein 2 (NPAS2, also known as MOP4), and inhibitor of nuclear factor-κBβ (IκBβ), were also discovered to be RXRα-interacting proteins (45, 46). Similar to RFC3, the interaction of MOP4 and IκBβ with RXRα depended on their C-terminal LXXLL motifs in addition to 9-cis-RA. 9-Cis-RA-induced transcription via the activation of RXR-RXR but not RXR-RAR correlated with carcinoma cell growth suppression (47), suggesting that RXR homodimer activation leads to growth inhibition. However, the involvement of RXR heterodimers such as RAR and TR, which may interact with RFC3-RXRα, cannot be ruled out, although RAR- and thyroid hormone receptor-specific ligands did not influence the interaction. RFC4 appears to interact with RXRα only in the absence of 9-cis-RA, similar to the interactions of NR and NR corepressors, showing a sharp contrast to the interacting modes of RFC3 for RXRα. Notably, the corepressor SMRT competed with RFC4, and an NR-interacting motif-like sequence (326-IIXXXL-331) was observed in a putative helix region of hRFC4. These results indicate that RFC components are a novel class of RXRα-interacting proteins, potentially implicating RXRα in RA-controlled cellular proliferation.
An equally important issue raised by this study is the nature of the target genes and downstream proteins of the transient RFC3-RXRα complex. Interestingly, the largest subunit of the RFC heteropentamer RFC1, which exhibits greatly accelerated cell proliferation (48), and RFC2 showed a weakened interaction with PCNA by 9-cis-RA (Fig. 4B). PCNA plays an essential role in nucleic acid metabolism as a component of the replication and repair machinery and tethers the polymerase catalytic domain to the DNA template for processive DNA synthesis (35, 36, 40, 49, 50). In particular, PCNA has been shown to be clamped to DNA through the action of RFC (38, 51) and to interact with proteins involved in cell-cycle progression that are not part of the DNA polymerase apparatus (49). 9-Cis-RA appears to cause dissociation of the core RFC subunits (RFC2/4/5) and PCNA by the high affinity of liganded RXRα for RFC3 (Fig. 10), even though a weak interaction was observed between unliganded RXRα and RFC3 (Figs. 1A and 4A). The ligand-independent weak interaction may reflect either a low level of RXR ligand in serum or act as a bridge that functions to increase the local concentration of RXR in the vicinity of the RFC complex so that the ligand-dependent, strong interaction can kick in immediately after 9-cis-RA becomes available. Together, these results suggest that 9-cis-RA-evoked conformational change of RXRα disrupts the stable RFC-PCNA complex through RFC3 to inhibit the DNA replication and proliferation of RA-sensitive cells, as discussed below. It would be of interest to determine whether 9-cis-RA-bound RXRα is directly linked to the DNA replication machinery, particularly during the S-phase of a cell cycle, and whether RFC3 is recruited to the promoter/enhancer regions of RXRα target genes. For the latter question, we examined a conventional RXR response element (DR-1) in target genes. The transcriptional activity of DNA-bound RXRα was not affected by RFC3 overexpression (Supplemental Fig. 9), suggesting that at least the DR-1 is likely excluded from the target sequences of RXRα-RFC3 complex.
Fig. 10.

A putative model for how 9-cis-RA-activated RXRα may affect the steady-state RFC-PCNA complex to inhibit cell proliferation. In the absence of ligand (normal state), a multiprotein complex (five RFC subunits) binds and opens a ring-shaped sliding-clamp PCNA in the first stage of processive DNA synthesis for cell-cycle control, DNA replication and repair, and the apoptotic pathway (38, 40). RXRα appears to contact these complexes through RFC4 and partially RFC3, whose exact composition is still unknown. Upon ligand binding (9-cis-RA-activated state), RXRα may interact with RFC3, and this interaction appears to cause dissociation of the core RFC subunits (RFC2/4/5) (39) and PCNA to inhibit cell proliferation. Knockdown of RXRα or overexpression of RFC3 may drive backward partially to the normal state even in the presence of 9-cis-RA.
Data from cellular and animal models and clinical trials revealed the potent antitumor activity of RXR agonists (9, 52). The physiological relevance of the interaction of liganded RXRα and RFC3 has been elucidated by the novel finding that RFC3 overexpression overrides 9-cis-RA-induced MCF-7 cell growth inhibition. RXRα overexpression sensitizes several cancer cells, including breast cancers, to the antigrowth effects of RXR ligands for the induction of cellular differentiation and the control of aberrant cell growth (47, 53, 54). In line with these observations, we showed RXRα knockdown in MCF-7 cells recovered cellular growth and forced reentry into the S-phase without the effect of 9-cis-RA, indicating that RXRα may play a key role in RA responsiveness in breast cancer cells. Even in ER−/RA-resistant breast cancer cells, such as MDA-MB-231, ectopic expression of ERα conferred responsiveness to 9-cis-RA growth inhibition (Supplemental Fig. 8) and endogenous ERα expression was suppressed by 9-cis-RA treatment (42). These findings suggest that 9-cis-RA inhibits ER activity in ER+ breast cancer cells by actions downstream of ER. The predominant factor leading to growth arrest by 9-cis-RA was revealed to be G1 cell-cycle blockade, which results from the down-regulation of cyclin D1 and D3 (13). In contrast, Yasmin et al. (55) demonstrated that inhibition of MCF-7 cell growth by RXR ligands is due to the ligand-controlled switch of the RXRα oligomeric state and is independent of the receptor's direct transcriptional activity, although a classical and direct pathway was reported in which ligand-activated RXRα homodimers facilitated G1 arrest of MDA-MB-231 cells by induction of p21, a CDK inhibitor (53). Interestingly, p21 directly interacts with PCNA and inhibits PCNA-dependent DNA replication in the absence of a cyclin/CDK (56). Although the exact nature of the RXRα complex in 9-cis-RA-evoked breast cancer cells remains to be clarified, ligand-activated RXRα is assumed to be essential for the antiproliferation of breast cancer cells by direct binding to RFC3 as an RXR-permissive heterodimeric partner, and p21 is implicated in preventing its ability to drive coordination of the RFC-PCNA complex.
In vertebrates, mouse Rxra-null mutants died in utero and displayed myocardial and ocular malformations (57), and precocious expression of Xenopus RXRα led to drastic and distinct embryonic abnormalities (58). During embryogenesis, zygotic RXRα expression was observed from the gastrula stage, with somewhat specific localization in later stages, such as in animal pole derivatives, forebrain, and digestive system epithelia (59–61). In accordance with these observations, the invertebrate SnRXRα mRNA gradually increased from the 32-cell to pluteus stage and was mainly expressed in the archenteron tip, endoderm cells, and gut. In contrast, Rxrb expression is mostly ubiquitous in mouse and zebrafish development, whereas Rxrg is likely dispensable for embryonic development (62) and shows a highly restricted expression pattern in the hindbrain, spinal cord, retina, and pituitary gland (59–61). Together with a higher sequence homology between vertebrate RXRα and SnRXRα (>60%) compared with that between RXRβ and RXRγ (<55%), SnRXRα may be suggested to be a functional homolog of vertebrate RXRα, rather than RXRβ and RXRγ. Importantly, SnRXRα MO-injected sea urchin embryos showed abnormalities from the early blastula stage, and their development stopped at the early gastrula stage without initiating the primary invagination. Of the three RXR subtypes, RXRα is thought to have the greatest functional significance during mouse embryogenesis (62). The determination of 9-cis-RA along with RXRα activity in vertebrate and invertebrate embryos argues for their importance during development (63–65), although 9-cis-RA was undetectable in rodent embryos (66). These results indicate that the role of RXRα is critical and conserved for early embryonic development in deuterostomes, supported by the lethality of SnRXRα-knockdown sea urchin embryos.
The SnRFC3 mRNA expression profile showed a relatively constant and widespread pattern during sea urchin embryogenesis, ranging from the unfertilized egg to the larval stage. Similar to this observation, zebrafish rfc3 mRNA expression was not spatially restricted during the early embryonic and hatching stages (http://zfin.org). Mouse Rfc3 mRNA was also uniformly, although weakly, detected in the embryonic d-14.5 embryo (67). Unexpectedly, we observed that SnRFC3 MO-injected embryos were able to survive beyond the pluteus larval stage, despite some morphological abnormalities and developmental retardation. Although the effect of RFC3-MO in embryos is modest, this experiment may reflect the significant role of RFC3 during early embryogenesis in the sea urchin.
The roles of RA during embryonic development have been extensively investigated in vertebrates and invertebrates, particularly in axial and regional patterning, organogenesis, limb formation, and neurogenesis (18, 19, 68). Sea urchin embryos provide an excellent model for studying the signaling pathways of cell-cycle control and cell proliferation in vivo (69, 70). For example, cyclin E/CDK2 activity promotes DNA replication and S-phase progression, along with PCNA accumulation in the nucleus (71, 72), suggesting that sea urchin embryos conform to the pattern observed in mammalian somatic cells. Thus, we examined the implication of SnRFC3 in RA-induced delay of embryo development (43). Here we show that overexpression of SnRFC3 recovers almost normal developmental patterns in sea urchin embryos, despite treatment with a lethal dose of 9-cis-RA. In prokaryotic and eukaryotic cells, the RFC components form a stable ATP-dependent complex with PCNA to receive DNA polymerase for DNA synthesis (35, 36, 40, 50, 51). In line with our experiments using MCF-7 breast cancer cells, these findings suggest that RFC3 is a physiological partner of 9-cis-RA-activated RXRα and that the transient RXRα-RFC3 complex interferes with RFC-PCNA formation to eventually prevent S-phase progression, even in early developmental embryos.
In summary, we identified RFC3 as an interacting protein with 9-cis-RA-activated RXRα. RFC3 interacts with liganded RXRα through the LXXLL motifs, and the transient interaction results in reconfiguration of the RFC-PCNA complex and inhibits the proliferation of breast cancer cells (Fig. 10). In this respect, RFC3 recruited by 9-cis-RA signaling is distinct from the major cell-cycle regulators identified to date (13, 73). The present data represent a significant advance in understanding the molecular mechanisms of RXRα-selective RA to prohibit proliferation of breast cancer cells, which may aid future efforts to develop safe and effective RA for the prevention of breast cancer. In addition, we propose the sea urchin embryo as a pertinent alternative and complementary model for studies of RA-sensitive cell proliferation.
Acknowledgments
We are grateful to Dr. Jae W. Lee, Oregon Health and Science University (Portland, OR), and Dr. Yeon-Soo Seo, Korea Advanced Institute of Science and Technology, Korea, for their comments and critical reading of the manuscript and Dr. Tae Kyung Kim, Gangneung-Wonju National University, Korea, for help with illustration.
This work was supported by the Ministry of Land, Transport, and Maritime Affairs, Korea (20088033-1 and B10400207A290-100170), the National Fisheries Research and Development Institute of Korea (RP-2011-AQ-079), and the National Research Foundation of Korea funded by the Korean government (NRF-2009-0070344).
Present address for S.M.: Age-Related and Brain Diseases Research Center, Kyung Hee University, Seoul 130-170, Republic of Korea.
Present address for E.J.C.: Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California, San Diego, La Jolla, California 92093-0204.
Disclosure Summary: The authors have nothing to disclose.
NURSA Molecule Pages†:
Nuclear Receptors: RXR-α;
Ligands:9-cis-retinoic acid.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- ATRA
- all-trans-RA
- CDK
- cyclin-dependent kinase
- DMSO
- dimethylsulfoxide
- ER
- estrogen receptor
- FBS
- fetal bovine serum
- hRFC3
- human replication factor C subunit 3
- IκBβ
- inhibitor of nuclear factor-κBβ
- LBD
- ligand-binding domain
- MO
- morpholino
- NLS
- nuclear localization signal
- NR
- nuclear receptor
- PCNA
- proliferating cell nuclear antigen
- RA
- retinoic acid
- RACE
- rapid amplification of cDNA ends
- RAR
- RA receptor
- RXR
- retinoid X receptor
- siRNA
- small interfering RNA
- SMRT
- silencing mediators of retinoid and thyroid hormone receptor
- SRC1
- steroid receptor coactivator-1.
References
- 1. McKenna NJ , O'Malley BW. 2002. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108:465–474 [DOI] [PubMed] [Google Scholar]
- 2. Aranda A , Pascual A. 2001. Nuclear hormone receptors and gene expression. Physiol Rev 81:1269–1304 [DOI] [PubMed] [Google Scholar]
- 3. Torchia J , Rose DW , Inostroza J , Kamei Y , Westin S , Glass CK , Rosenfeld MG. 1997. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387:677–684 [DOI] [PubMed] [Google Scholar]
- 4. Lefebvre P , Martin PJ , Flajollet S , Dedieu S , Billaut X , Lefebvre B. 2005. Transcriptional activities of retinoic acid receptors. Vitam Horm 70:199–264 [DOI] [PubMed] [Google Scholar]
- 5. Sun SY , Lotan R. 2002. Retinoids and their receptors in cancer development and chemoprevention. Crit Rev Oncol Hematol 41:41–55 [DOI] [PubMed] [Google Scholar]
- 6. Fields AL , Soprano DR , Soprano KJ. 2007. Retinoids in biological control and cancer. J Cell Biochem 102:886–898 [DOI] [PubMed] [Google Scholar]
- 7. Tang XH , Gudas LJ. 2011. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol 6:345–364 [DOI] [PubMed] [Google Scholar]
- 8. Mongan NP , Gudas LJ. 2007. Diverse actions of retinoid receptors in cancer prevention and treatment. Differentiation 75:853–870 [DOI] [PubMed] [Google Scholar]
- 9. Altucci L , Leibowitz MD , Ogilvie KM , de Lera AR , Gronemeyer H. 2007. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov 6:793–810 [DOI] [PubMed] [Google Scholar]
- 10. Fenaux P , Wang ZZ , Degos L. 2007. Treatment of acute promyelocytic leukemia by retinoids. Curr Top Microbiol Immunol 313:101–128 [DOI] [PubMed] [Google Scholar]
- 11. Epping MT , Wang L , Edel MJ , Carlée L , Hernandez M , Bernards R. 2005. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 122:835–847 [DOI] [PubMed] [Google Scholar]
- 12. Liu R , Takayama S , Zheng Y , Froesch B , Chen GQ , Zhang X , Reed JC , Zhang XK. 1998. Interaction of BAG-1 with retinoic acid receptor and its inhibition of retinoic acid-induced apoptosis in cancer cells. J Biol Chem 273:16985–16992 [DOI] [PubMed] [Google Scholar]
- 13. Wu K , DuPré E , Kim H , Tin-U CK , Bissonnette RP , Lamph WW , Brown PH. 2006. Receptor-selective retinoids inhibit the growth of normal and malignant breast cells by inducing G1 cell cycle blockade. Breast Cancer Res Treat 96:147–157 [DOI] [PubMed] [Google Scholar]
- 14. Naka K , Yokozaki H , Domen T , Hayashi K , Kuniyasu H , Yasui W , Lotan R , Tahara E. 1997. Growth inhibition of cultured human gastric cancer cells by 9-cis-retinoic acid with induction of cdk inhibitor Waf1/Cip1/Sdi1/p21 protein. Differentiation 61:313–320 [DOI] [PubMed] [Google Scholar]
- 15. Germain P , Chambon P , Eichele G , Evans RM , Lazar MA , Leid M , De Lera AR , Lotan R , Mangelsdorf DJ , Gronemeyer H. 2006. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol Rev 58:760–772 [DOI] [PubMed] [Google Scholar]
- 16. Shankaranarayanan P , Rossin A , Khanwalkar H , Alvarez S , Alvarez R , Jacobson A , Nebbioso A , de Lera AR , Altucci L , Gronemeyer H. 2009. Growth factor-antagonized rexinoid apoptosis involves permissive PPARγ/RXR heterodimers to activate the intrinsic death pathway by NO. Cancer Cell 16:220–231 [DOI] [PubMed] [Google Scholar]
- 17. Mark M , Ghyselinck NB , Chambon P. 2006. Function of retinoid nuclear receptors: lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol 46:451–480 [DOI] [PubMed] [Google Scholar]
- 18. Marlétaz F , Holland LZ , Laudet V , Schubert M. 2006. Retinoic acid signaling and the evolution of chordates. Int J Biol Sci 2:38–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Albalat R. 2009. The retinoic acid machinery in invertebrates: ancestral elements and vertebrate innovations. Mol Cell Endocrinol 313:23–35 [DOI] [PubMed] [Google Scholar]
- 20. Niederreither K , Subbarayan V , Dollé P , Chambon P. 1999. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet 21:444–448 [DOI] [PubMed] [Google Scholar]
- 21. Dollé P. 2009. Developmental expression of retinoic acid receptors (RAR). Nucl Recept Signal 7:e006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mark M , Ghyselinck NB , Chambon P. 2009. Function of retinoic acid receptors during embryonic development. Nucl Recept Signal 7:e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campo-Paysaa F , Marlétaz F , Laudet V , Schubert M. 2008. Retinoic acid signaling in development: tissue-specific functions and evolutionary origins. Genesis 46:640–656 [DOI] [PubMed] [Google Scholar]
- 24. Lee SK , Anzick SL , Choi JE , Bubendorf L , Guan XY , Jung YK , Kallioniemi OP , Kononen J , Trent JM , Azorsa D , Jhun BH , Cheong JH , Lee YC , Meltzer PS , Lee JW. 1999. A nuclear factor, ASC-2, as a cancer-amplified transcriptional coactivator essential for ligand-dependent transactivation by nuclear receptors in vivo. J Biol Chem 274:34283–34293 [DOI] [PubMed] [Google Scholar]
- 25. Kim MA , Kim GJ , Maeng S , Jin DH , Sohn YC. 2011. Characterization of steroid receptor coactivator in sea urchin, Strongylocentrotus nudus, and its involvement in embryonic development. Mol Cell Endocrinol 331:89–101 [DOI] [PubMed] [Google Scholar]
- 26. Lee SK , Kim HJ , Na SY , Kim TS , Choi HS , Im SY , Lee JW. 1998. Steroid receptor coactivator-1 coactivates activating protein-1-mediated transactivations through interaction with the c-Jun and c-Fos subunits. J Biol Chem 273:16651–16654 [DOI] [PubMed] [Google Scholar]
- 27. Sohn YC , Kim SW , Lee S , Kong YY , Na DS , Lee SK , Lee JW. 2003. Dynamic inhibition of nuclear receptor activation by corepressor binding. Mol Endocrinol 17:366–372 [DOI] [PubMed] [Google Scholar]
- 28. Kang K , Lee HJ , Kim CY , Lee SB , Tunsag J , Batsuren D , Nho CW. 2007. The chemopreventive effects of Saussurea salicifolia through induction of apoptosis and phase II detoxification enzyme. Biol Pharm Bull 30:2352–2359 [DOI] [PubMed] [Google Scholar]
- 29. Cheers MS , Ettensohn CA. 2004. Rapid microinjection of fertilized eggs. Methods Cell Biol 74:287–310 [DOI] [PubMed] [Google Scholar]
- 30. Kim GJ , Kumano G , Nishida H. 2007. Cell fate polarization in ascidian mesenchyme/muscle precursors by directed FGF signaling and role for an additional ectodermal FGF antagonizing signal in notochord/nerve cord precursors. Development 134:1509–1518 [DOI] [PubMed] [Google Scholar]
- 31. Arenas-Mena C , Cameron AR , Davidson EH. 2000. Spatial expression of Hox cluster genes in the ontogeny of a sea urchin. Development 127:4631–4643 [DOI] [PubMed] [Google Scholar]
- 32. Lee YH. 2003. Molecular phylogenies and divergence times of sea urchin species of Strongylocentrotidae, Echinoida. Mol Biol Evol 20:1211–1221 [DOI] [PubMed] [Google Scholar]
- 33. Perissi V , Staszewski LM , McInerney EM , Kurokawa R , Krones A , Rose DW , Lambert MH , Milburn MV , Glass CK , Rosenfeld MG. 1999. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev 13:3198–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu X , Lazar MA. 1999. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 402:93–96 [DOI] [PubMed] [Google Scholar]
- 35. Lee SH , Kwong AD , Pan ZQ , Hurwitz J. 1991. Studies on the activator 1 protein complex, an accessory factor for proliferating cell nuclear antigen-dependent DNA polymerase δ. J Biol Chem 266:594–602 [PubMed] [Google Scholar]
- 36. Tsurimoto T , Stillman B. 1990. Functions of replication factor C and proliferating-cell nuclear antigen: functional similarity of DNA polymerase accessory proteins from human cells and bacteriophage T4. Proc Natl Acad Sci USA 87:1023–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yao NY , Johnson A , Bowman GD , Kuriyan J , O'Donnell M. 2006. Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C. J Biol Chem 281:17528–17539 [DOI] [PubMed] [Google Scholar]
- 38. Bowman GD , O'Donnell M , Kuriyan J. 2004. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 429:724–730 [DOI] [PubMed] [Google Scholar]
- 39. Uhlmann F , Cai J , Flores-Rozas H , Dean FB , Finkelstein J , O'Donnell M , Hurwitz J. 1996. In vitro reconstitution of human replication factor C from its five subunits. Proc Natl Acad Sci USA 93:6521–6526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Indiani C , O'Donnell M. 2006. The replication clamp-loading machine at work in the three domains of life. Nat Rev Mol Cell Biol 7:751–761 [DOI] [PubMed] [Google Scholar]
- 41. Zhao Z , Zhang ZP , Soprano DR , Soprano KJ. 1995. Effect of 9-cis-retinoic acid on growth and RXR expression in human breast cancer cells. Exp Cell Res 219:555–561 [DOI] [PubMed] [Google Scholar]
- 42. Rubin M , Fenig E , Rosenauer A , Menendez-Botet C , Achkar C , Bentel JM , Yahalom J , Mendelsohn J , Miller WH. 1994. 9-Cis retinoic acid inhibits growth of breast cancer cells and down-regulates estrogen receptor RNA and protein. Cancer Res 54:6549–6556 [PubMed] [Google Scholar]
- 43. Sciarrino S , Matranga V. 1995. Effects of retinoic acid and dimethylsulfoxide on the morphogenesis of the sea urchin embryo. Cell Biol Int 19:675–680 [DOI] [PubMed] [Google Scholar]
- 44. Sconzo G , Fasulo G , Romancino D , Cascino D , Giudice G. 1996. Effect of retinoic acid and valproate on sea urchin development. Pharmazie 51:175–180 [PubMed] [Google Scholar]
- 45. Na SY , Kim HJ , Lee SK , Choi HS , Na DS , Lee MO , Chung M , Moore DD , Lee JW. 1998. IκBβ interacts with the retinoid X receptor and inhibits retinoid-dependent transactivation in lipopolysaccharide-treated cells. J Biol Chem 273:3212–3215 [DOI] [PubMed] [Google Scholar]
- 46. McNamara P , Seo SB , Rudic RD , Sehgal A , Chakravarti D , FitzGerald GA. 2001. Regulation of CLOCK and MOP4 by nuclear hormone receptors in the vasculature: a humoral mechanism to reset a peripheral clock. Cell 105:877–889 [DOI] [PubMed] [Google Scholar]
- 47. Wan H , Dawson MI , Hong WK , Lotan R. 1998. Overexpressed activated retinoid X receptors can mediate growth inhibitory effects of retinoids in human carcinoma cells. J Biol Chem 273:26915–26922 [DOI] [PubMed] [Google Scholar]
- 48. Jaharul Haque S , van der Kuip H , Kumar A , Aulitzky WE , Rutherford MN , Huber C , Fischer T , Williams BR. 1996. Overexpression of mouse p140 subunit of replication factor C accelerates cellular proliferation. Cell Growth Differ 7:319–326 [PubMed] [Google Scholar]
- 49. Kelman Z. 1997. PCNA: structure, functions and interactions. Oncogene 14:629–640 [DOI] [PubMed] [Google Scholar]
- 50. Prelich G , Tan CK , Kostura M , Mathews MB , So AG , Downey KM , Stillman B. 1987. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-δ auxiliary protein. Nature 326:517–520 [DOI] [PubMed] [Google Scholar]
- 51. Zhang G , Gibbs E , Kelman Z , O'Donnell M , Hurwitz J. 1999. Studies on the interactions between human replication factor C and human proliferating cell nuclear antigen. Proc Natl Acad Sci USA 96:1869–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Conzen SD. 2008. Minireview: nuclear receptors and breast cancer. Mol Endocrinol 22:2215–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tanaka T , Suh KS , Lo AM , De Luca LM. 2007. p21WAF1/CIP1 is a common transcriptional target of retinoid receptors: pleiotropic regulatory mechanism through retinoic acid receptor (RAR)/retinoid X receptor (RXR) heterodimer and RXR/RXR homodimer. J Biol Chem 282:29987–29997 [DOI] [PubMed] [Google Scholar]
- 54. Crowe DL , Chandraratna RA. 2004. A retinoid X receptor (RXR)-selective retinoid reveals that RXR-α is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Res 6:R546–R555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yasmin R , Kannan-Thulasiraman P , Kagechika H , Dawson MI , Noy N. 2010. Inhibition of mammary carcinoma cell growth by RXR is mediated by the receptor's oligomeric switch. J Mol Biol 397:1121–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Waga S , Hannon GJ , Beach D , Stillman B. 1994. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 369:574–578 [DOI] [PubMed] [Google Scholar]
- 57. Kastner P , Grondona JM , Mark M , Gansmuller A , LeMeur M , Decimo D , Vonesch JL , Dollé P , Chambon P. 1994. Genetic analysis of RXR alpha developmental function: convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 78:987–1003 [DOI] [PubMed] [Google Scholar]
- 58. Puzianowska-Kuznicka M , Damjanovski S , Shi YB. 1997. Both thyroid hormone and 9-cis retinoic acid receptors are required to efficiently mediate the effects of thyroid hormone on embryonic development and specific gene regulation in Xenopus laevis. Mol Cell Biol 17:4738–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dollé P , Fraulob V , Kastner P , Chambon P. 1994. Developmental expression of murine retinoid X receptor (RXR) genes. Mech Dev 45:91–104 [DOI] [PubMed] [Google Scholar]
- 60. Mangelsdorf DJ , Borgmeyer U , Heyman RA , Zhou JY , Ong ES , Oro AE , Kakizuka A , Evans RM. 1992. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev 6:329–344 [DOI] [PubMed] [Google Scholar]
- 61. Tallafuss A , Hale LA , Yan YL , Dudley L , Eisen JS , Postlethwait JH. 2006. Characterization of retinoid-X receptor genes rxra, rxrba, rxrbb, and rxrg during zebrafish development. Gene Expr Patterns 6:556–565 [DOI] [PubMed] [Google Scholar]
- 62. Krezel W , Dupe V , Mark M , Dierich A , Kastner P , Chambon P. 1996. RXR gamma null mice are apparently normal and compound RXRα+/−/RXRβ−/−/RXRγ−/− mutant mice are viable. Proc Natl Acad Sci USA 93:9010–9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nowickyj SM , Chithalen JV , Cameron D , Tyshenko MG , Petkovich M , Wyatt GR , Jones G , Walker VK. 2008. Locust retinoid X receptors: 9-cis-retinoic acid in embryos from a primitive insect. Proc Natl Acad Sci USA 105:9540–9545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kraft JC , Schuh T , Juchau M , Kimelman D. 1994. The retinoid X receptor ligand, 9-cis-retinoic acid, is a potential regulator of early Xenopus development. Proc Natl Acad Sci USA 91:3067–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thaller C , Hofmann C , Eichele G. 1993. 9-Cis-retinoic acid, a potent inducer of digit pattern duplications in the chick wing bud. Development 118:957–965 [DOI] [PubMed] [Google Scholar]
- 66. Horton C , Maden M. 1995. Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo. Dev Dyn 202:312–323 [DOI] [PubMed] [Google Scholar]
- 67. Diez-Roux G , Banfi S , Sultan M , Geffers L , Anand S , Rozado D , Magen A , Canidio E , Pagani M , Peluso I , Lin-Marq N , Koch M , Bilio M , Cantiello I , Verde R , De Masi C , Bianchi SA , Cicchini J , Perroud E , Mehmeti S , Dagand E , Schrinner S , Nürnberger A , Schmidt K , Metz K , et al. . 2011. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 9:e1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Conlon RA. 1995. Retinoic acid and pattern formation in vertebrates. Trends Genet 11:314–319 [DOI] [PubMed] [Google Scholar]
- 69. Le Bouffant R , Cormier P , Cueff A , Bellé R , Mulner-Lorillon O. 2007. Sea urchin embryo as a model for analysis of the signaling pathways linking DNA damage checkpoint, DNA repair and apoptosis. Cell Mol Life Sci 64:1723–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fernandez-Guerra A , Aze A , Morales J , Mulner-Lorillon O , Cosson B , Cormier P , Bradham C , Adams N , Robertson AJ , Marzluff WF , Coffman JA , Genevière AM. 2006. The genomic repertoire for cell cycle control and DNA metabolism in S. purpuratus. Dev Biol 300:238–251 [DOI] [PubMed] [Google Scholar]
- 71. Philipova R , Kisielewska J , Lu P , Larman M , Huang JY , Whitaker M. 2005. ERK1 activation is required for S-phase onset and cell cycle progression after fertilization in sea urchin embryos. Development 132:579–589 [DOI] [PubMed] [Google Scholar]
- 72. Kisielewska J , Philipova R , Huang JY , Whitaker M. 2009. MAP kinase-dependent cyclinE/cdk2 activity promotes DNA replication in early sea urchin embryos. Dev Biol 334:383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhou Q , Stetler-Stevenson M , Steeg PS. 1997. Inhibition of cyclin D expression in human breast carcinoma cells by retinoids in vitro. Oncogene 15:107–115 [DOI] [PubMed] [Google Scholar]