

Abstract
Patients suffering from low-grade chronic inflammatory diseases, such as rheumatoid arthritis, osteoarthritis, diabetes, and obesity, have low plasma sex hormone-binding globulin (SHBG) levels. These diseases are characterized among other features by high plasma IL1β levels. The aim of the present study is to explore whether IL1β could regulate hepatic SHBG production to account for low SHBG levels in these diseases. We provide evidence that daily IL1β treatment reduces SHBG production in HepG2 cells by the down-regulation of HNF-4A via the MAPK kinase (MEK)-1/2 and c-Jun N-terminal kinase (JNK) MAPK signaling pathways through the activation c-Jun transcription factors. The human SHBG promoter sequence contains two putative activator protein 1 (AP1) binding sites recognized by c-Jun transcription factors, but they are not necessary for the IL1β-induced down-regulation of SHBG promoter activity in luciferase reporter gene assays. Daily treatment with IL1β reduces hepatic nuclear factor (HNF)-4α mRNA and protein levels via the MEK-1/2 and JNK MAPK signaling pathways. Moreover, IL1β rapidly decreased HNF-4α mRNA and protein levels while increased phospho-c-Jun protein levels after the treatment. Finally, daily IL1β treatment of human SHBG transgenic mice reduced plasma SHBG and SHBG mRNA levels. Moreover, IL1β treatment also reduced HNF-4α mRNA and protein levels while increased hepatic phospho-c-Jun protein levels. Our results show that IL1β reduces hepatic SHBG production by decreasing HNF-4α via MEK-1/2 and JNK MAPK pathways. In addition, our findings suggest that IL1β could be involved the low plasma SHBG levels reported in chronic low-grade inflammatory diseases.
Sex hormone-binding globulin (SHBG) is produced and secreted by the human liver, and it binds androgens and estrogens with high affinity. In blood, SHBG acts as a carrier of these sex steroids and regulates their bioavailability (1). Apart from metabolic syndrome, obesity, and type 2 diabetes (2–4), there are other chronic inflammatory diseases, such as rheumatoid arthritis and osteoarthritis, where low SHBG levels have been reported (5, 6).
Chronic inflammatory diseases are characterized among other features by elevated levels of IL1β. IL1β is a pivotal inflammatory cytokine mainly produced by macrophage cells, which has pleiotropic effects (7). Plasmatic IL1β binds to the type 1 IL1 receptor together with the IL1 receptor accessory protein. IL1β binding causes the activation of two receptor-associated kinases, interleukin 1 receptor-associated kinase 1 and 2, which activate and recruit tumor necrosis factor receptor associated factor 6 (TRAF6) to the IL1 receptor complex. TRAF6 activates two different pathways, one leading to nuclear factor kappa B (NF-κB) activation and another leading to c-Jun activation through the MAPK/c-Jun N-terminal kinase (JNK) signaling system (8).
The human SHBG proximal promoter has been analyzed by deoxyribonuclease I footprinting (FP), and several FP regions have been defined and studied, to identify which transcription factors (TF) regulate SHBG expression (9). Several reports have identified hepatic nuclear factor (HNF)-4α, chicken ovalbumin upstream promoter-TF, upstream stimulatory factor, and peroxisome-proliferator receptor γ as a TF that regulates SHBG expression (9–11). Among them, the HNF-4α has been identified as the most important TF that regulates SHBG expression (9). Our recent work has also shown that SHBG gene expression can be regulated indirectly by affecting HNF4A gene expression. In this regard, we have identified that an increase in de novo lipogenesis induced by high carbohydrate diets (glucose or fructose) was able to reduce hepatic HNF-4α protein levels, which in turn decreased SHBG production (12). We have recently identified the TNFα as another factor that reduces hepatic SHBG production by decreasing HNF-4α protein levels via NF-κB (13, 14). Finally, we have also elucidated the molecular mechanism by which thyroid hormones increase SHBG gene expression indirectly by affecting hepatic HNF-4α levels (15).
In the present work, we explore the possibility that IL1β down-regulates SHBG expression by reducing hepatic HNF-4α levels, which would explain why patients suffering from chronic inflammatory diseases present low plasma SHBG levels. We have performed in vitro and in vivo studies using human HepG2 hepatoblastoma cells and human SHBG transgenic mice, which is the only available mouse model to study human SHBG regulation, because normal mice do not express SHBG gene in their livers (9). Our results provide first evidence that IL1β is able to reduce hepatic production of SHBG. In addition, the molecular mechanisms involved in this effect have been elucidated.
Results
IL1β treatment decreases SHBG production via MAPK kinase (MEK)-1/2 and JNK MAPK pathways in HepG2 cells
We first examined the effects of daily supplementation with IL1β on SHBG production by HepG2 cells over the course of 5 d. The dosages of IL1β were similar to previously reported in the literature using this cell line (16–18). The studies were performed by comparing medium concentrations of SHBG on d 1 and 5 of vehicle and IL1β (50, 100, and 200 ng/ml) treatments. These treatments showed that cells treated with IL1β at all three concentrations had significantly reduced SHBG production when compared with vehicle-treated cells (Fig. 1A). In addition, the amount of SHBG mRNA in HepG2 cells after 5 d of treatment with IL1β (50, 100, and 200 ng/ml) was significantly decreased in relation to the 18S mRNA control when compared with vehicle-treated cells (Fig. 1B).
Fig. 1.
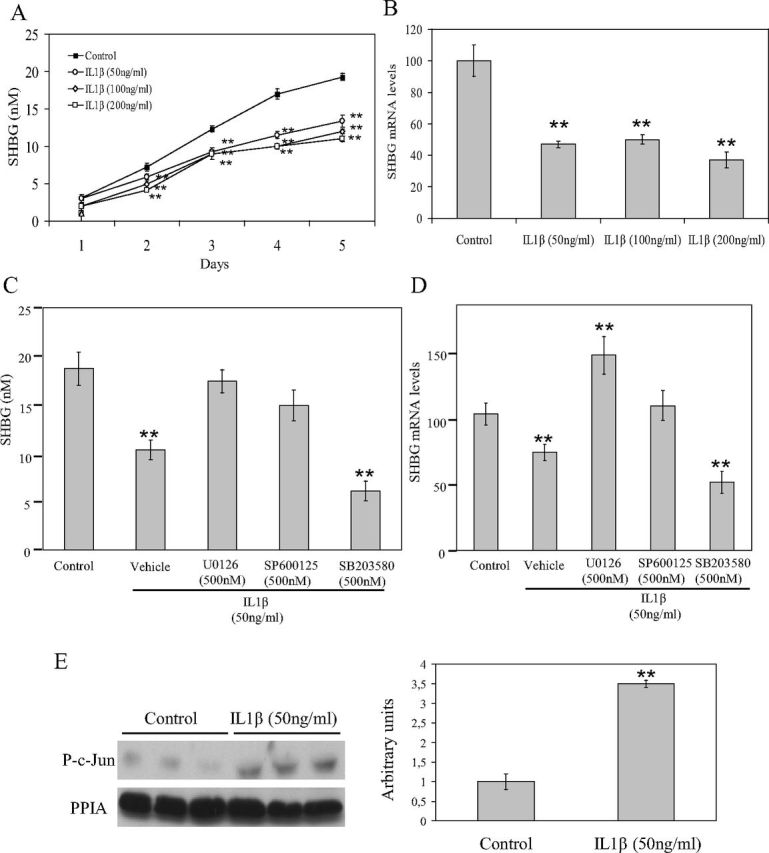
Daily treatment with IL1β decreases SHBG production over 5 d via MEK-1/2 and JNK MAPK by c-Jun activation in HepG2 cells. A, HepG2 cells were treated daily with vehicle or IL1β (50 ng/ml) for 5 d. SHBG accumulation in the medium was measured using an ELISA. B, Analysis of SHBG mRNA levels in HepG2 cells treated as in A. Human 18S (h18S) mRNA was amplified as a control. Data points are shown as mean ± sd of triplicates. **, P < 0.01 compared with the control. C, HepG2 cells were treated daily with vehicle, IL1β (50 ng/ml) alone or with U0126 (500 nm), and SP600125 (500 nm) or SB203580 (500 nm). SHBG accumulation in the medium was measured using an ELISA. D, Analysis of SHBG mRNA levels in HepG2 cells treated as in C. Human 18S mRNA was amplified as a control. Data points are shown as mean ± sd of triplicates. **, P < 0.01 compared with the control. E, Western blotting of phospho-c-Jun and PPIA in total cell protein extracts of HepG2 cells treated as in A. **, P < 0.01 compared with the control.
To explore the signaling pathway by which IL1β treatment was down-regulating SHBG production, we next treated HepG2 cells over the course of 5 d with vehicle, IL1β alone, or IL1β in the presence of U0126 (500 nm), SP600125 (500 nm), SB203580 (500 nm), or QNZ (500 nm) inhibitors of MEK-1/2, JNK, p38, or NF-κB, respectively. IL1β-induced reduction of SHBG production by HepG2 cells was blocked by cotreatment with U0126 or SP600125 inhibitors, whereas cotreatment with the SB203580 inhibitor did not have any effect (Fig. 1C). The HepG2 cells treated with QNZ inhibitor for 5 d died before the end of the treatment (data not shown). Moreover, IL1β-induced reduction of SHBG mRNA levels was also blocked by cotreatment with U0126 or SP600125 inhibitors (Fig. 1D).
It has been described in the literature that one of the common TF activated by MEK-1/2 and JNK MAPK in the signaling cascade of IL1β binding to its receptor is c-Jun (8). To explore whether this is our case, in our HepG2 cell treatments with IL1β, we performed a Western blotting of protein extracts from HepG2 cells treated with vehicle or IL1β using an antibody against phospho-c-Jun. The results showed a clear increase in phospho-c-Jun protein levels in the HepG2 cells treated daily with IL1β when compared with vehicle-treated cells (Fig. 1E).
IL1β treatment reduces human SHBG promoter activity in luciferase reporter gene assays in HepG2 cells
Because we found, after daily IL1β treatment of HepG2 cells, a reduction in SHBG mRNA expression and we detected an increase of c-Jun phosphorylation, we next scanned the human SHBG promoter sequence for putative binding sites for activator protein 1 (AP1) TF (http://tfbind.hgc.jp/), which are recognized by c-Jun. We found two putative AP1 binding sites located from −112 to −103 bp and from −78 to −68 bp upstream of the transcription start site, and importantly, they overlap with hepatic nuclear protein-binding sites previously identified by deoxyribonuclease I FP (9) (Fig. 2A). These two AP1 binding sites were located within the FP 4 and FP 3 regions of the human SHBG promoter. It has been previously described that FP 3 and FP 4 regions are bound by upstream stimulatory factor and HNF-4α, respectively (Fig. 2A) (9, 10).
Fig. 2.
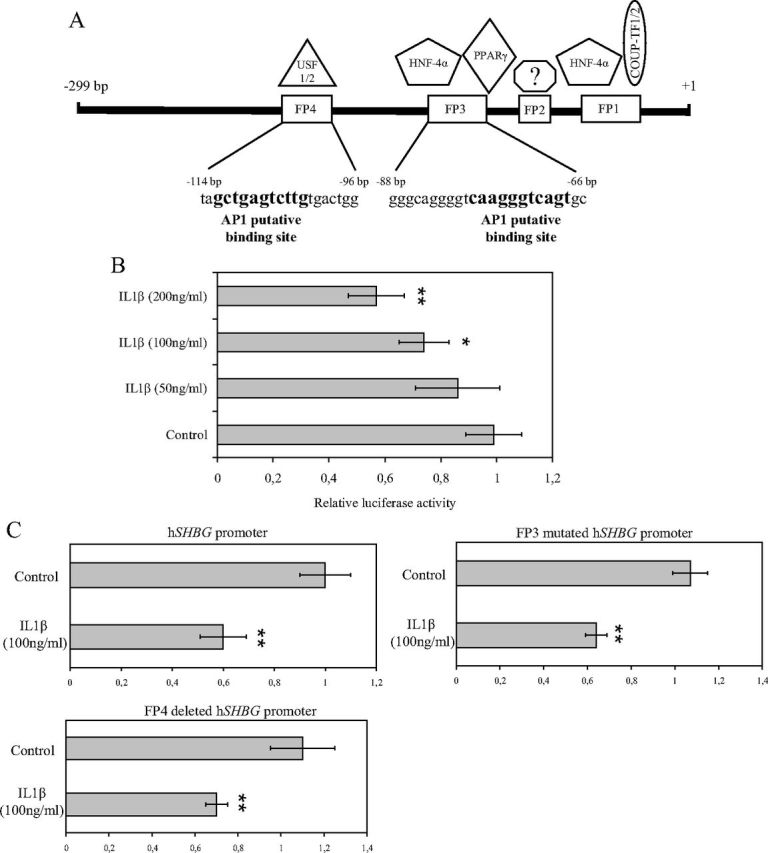
The SHBG promoter contains two AP1 binding sites, and it responds to IL1β in luciferase reporter gene assays. A, SHBG promoter scheme of the previously described FP regions and the TF that binds them. The putative AP1 binding sites are located within the FP 3 and FP 4 regions. B, The SHBG promoter activity was analyzed in HepG2 cells treated with vehicle or IL1β (50, 100, or 200 ng/ml) in luciferase reporter gene assays. Data points are mean ± sd of triplicate measurements. **, P < 0.01 compared with the control. C, The SHBG promoter activity of the wild type, FP 3 mutated, or FP 4 deleted was analyzed in HepG2 cells using treated with vehicle or IL1β (100 ng/ml) in luciferase reporter gene assays. Data points are mean ± sd of triplicate measurements. **, P < 0.01 compared with the control. USF, Upstream stimulatory factor; COUP-TF, chicken ovalbumin upstream promoter-TF.
To study whether IL1β was regulating SHBG gene expression, we performed luciferase reporter gene assays with SHBG promoter treated with vehicle or gradually increased concentrations of IL1β (50, 100, and 200 ng/ml). The SHBG promoter activity was reduced by IL1β treatment at 100 and 200 ng/ml when compared with the vehicle or IL1β-treated (50 ng/ml) HepG2 cells in luciferase reporter gene assays (Fig. 2B).
We next wanted to study whether IL1β down-regulation of the human SHBG promoter activity was mediated through the AP1 putative binding sites found in the human SHBG promoter sequence. For this purpose, we examined the effect of IL1β treatment in three different constructs: the wild-type, the FP 3-mutated, and the FP 4-deleted human SHBG promoter in luciferase reporter gene assays. The results showed that IL1β treatment was able to reduce the promoter activity of the three different constructs (Fig. 2C), thus suggesting that IL1β-induced down-regulation of SHBG gene expression is not mediated by the binding of c-Jun to any of the two AP1 putative binding sites present in the human SHBG promoter.
IL1β reduction of SHBG production is mediated indirectly by decreasing HNF-4α levels through activation of c-Jun
Because the human SHBG promoter did not respond to IL1β through the AP1 biding sites and our recent works described a new molecular mechanism by which affecting the HNF-4α levels it is possible to regulate SHBG expression, we examined the HNF-4α mRNA and protein levels in HepG2 cells after daily treatment with vehicle or IL1β (50, 100, and 200 ng/ml). The results showed that HNF-4α mRNA levels were significantly decreased (P < 0.01) after IL1β treatment at all concentrations when compared with vehicle-treated cells (Fig. 3A), and this resulted in a significant decrease (P < 0.01) in HNF-4α protein levels (Fig. 3B). Moreover, IL1β-induced reduction of HNF-4α mRNA and protein levels was blocked by cotreatment with U0126 (500 nm) and SP600125 (500 nm) when compared with the vehicle-treated or SB203580-treated (500 nm) HepG2 cells (Fig. 3C).
Fig. 3.
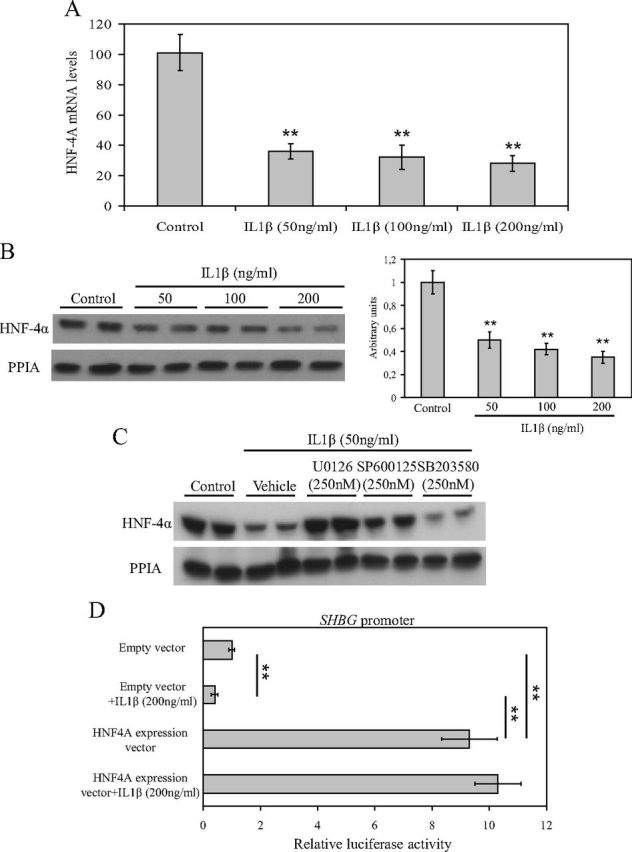
IL1β treatment decreases HNF-4α mRNA and protein levels via MEK-1/2 and JNK MAPK in HepG2 cells. A, Analysis of HNF-4α mRNA levels in HepG2 cells treated with vehicle or IL1β (50 ng/ml) over the course of 5 d. Human 18S (h18S) mRNA was amplified as a control. Data points are shown as mean ± sd of triplicates. **, P < 0.01 compared with the control. B, Western blotting of HNF-4α and PPIA in total cell protein extracts of HepG2 cells treated as in A. **, P < 0.01 compared with the control. C, Western blotting of HNF-4α and PPIA in total cell protein extracts of HepG2 cells treated with vehicle, IL1β (50 ng/ml) alone, or IL1β (50 ng/ml) in the presence of U0126 (500 nm), SP600125 (500 nm), or SB203580 (500 nm). **, P < 0.01 compared with the control. D, The SHBG promoter activity was analyzed in HepG2 cells cotransfected with an empty vector or a HNF-4α expression vector in the absence or presence of IL1β (200 ng/ml) in luciferase reporter gene assays. Data points are mean ± sd of triplicate measurements. **, P < 0.01 compared with the control.
We next used an HNF4A expression vector to study whether we could block the IL1β-induced reduction of SHBG promoter activity in luciferase reporter gene assays in HepG2 cells. The results showed that although IL1β treatment reduced SHBG promoter activity by about 50% in the empty vector transfected HepG2 cells, cotransfection of an HNF4A expression vector was able to block the IL1β-induced reduction of the SHBG promoter activity (Fig. 3D).
Finally, given that HNF-4α is the key TF identified in the regulation of liver SHBG, we examined the expression of other HNF-4α dependent genes (Apolipoprotein B, Apolipoprotein E, Cytochrome P450 2C9, and Cytochrome P450 7A1) that could be repressed by IL1β treatment. The results showed that all genes analyzed were reduced by IL1β treatment when compared with vehicle-treated cells (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org).
IL1β treatment rapidly decreases HNF-4α levels via c-Jun activation in HepG2 cells
Our results showed that IL1β treatment was able to reduce the human SHBG promoter activity after a 24-h treatment and that this effect seems to be mediated through the down-regulation of HNF-4α. To examine this issue, we performed IL1β (100 ng/ml) treatments in HepG2 cells at different time points, 0, 2, and 4 h. The results showed that HNF-4α mRNA and protein levels were reduced at 2 and 4 h after the IL1β treatment when compared with the initial levels before the treatment (Fig. 4, A and B). More importantly, SHBG mRNA levels were also reduced at 2 and 4 h after the IL1β treatment when compared with the initial levels before the treatment (Fig. 4A). Moreover, the IL1β treatment also activated c-Jun by increasing the phospho-c-Jun protein levels at 2 and 4 h when compared with the initial levels before the treatment (Fig. 4B). This rapid IL1β reduction of HNF-4α protein levels was also mediated by inducing its proteasomal degradation, because cotreatment with MG132 (10 μm) was able to block the HNF-4α degradation at 4 h after IL1β treatment when compared with the IL1β-treated HepG2 cells (Fig. 4C).
Fig. 4.
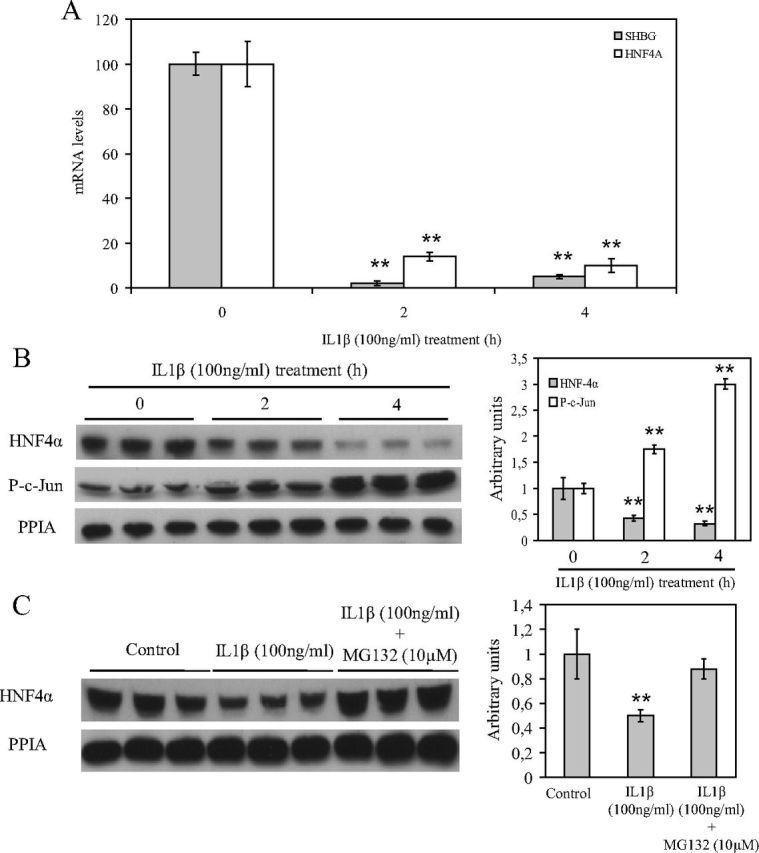
IL1β treatment reduces HNF-4α mRNA and protein levels after 2–4 h via c-Jun activation. A, Analysis of SHBG and HNF-4α mRNA levels in HepG2 cells treated with vehicle or IL1β (100 ng/ml) at 0, 2, and 4 h. Human 18S (h18S) mRNA was amplified as a control. Data points are shown as mean ± sd of triplicates. **, P < 0.01 compared with the control. B, Western blotting of HNF-4α, phospho-c-Jun, and PPIA in total cell protein extracts of HepG2 cells treated as in A. C, Western blotting of HNF-4α and PPIA in total cell protein extracts of HepG2 cells treated with vehicle, IL1β (100 ng/ml) alone, or IL1β (50 ng/ml) with MG132 (10 μm).
The strong repression of SHBG transcription observed at 2–4 h after IL1β treatment was not in accordance with our luciferase reporter gene assays, where SHBG promoter activity was only reduced by about 30–40% when compared with the vehicle-treated cells. To explore this issue, we decided to perform a longer time-course treatment with IL1β. We treated HepG2 cells with IL1β (50, 100, and 200 ng/ml) to determine SHBG and HNF-4α levels at 6 and 12 h. The results showed that SHBG and HNF-4α mRNA levels remained reduced when compared with the control 6 h after treatment, whereas they returned to the control levels at 12 h (Fig. 5A). Moreover, HNF-4α protein levels remained reduced when compared with the control 6 h after the treatment, whereas they also returned to the control levels at 12 h (Fig. 5B).
Fig. 5.
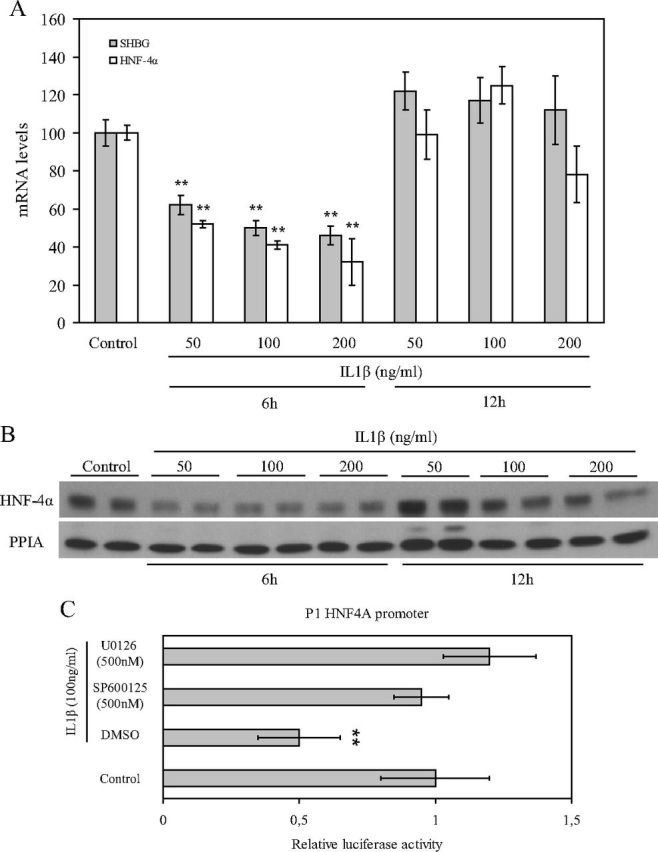
IL1β reduces HNF-4α mRNA and protein levels up to 12 h after treatment. A, Analysis of SHBG and HNF-4α mRNA levels in HepG2 cells treated with vehicle or IL1β (50, 100, and 200 ng/ml) at 6 and 12 h. Human 18S (h18S) mRNA was amplified as a control. Data points are shown as mean ± sd of triplicates. **, P < 0.01 compared with the control. B, Western blotting of HNF-4α and PPIA in total cell protein extracts of HepG2 cells treated as in A. C, The HNF4A promoter activity was analyzed in HepG2 cells treated with vehicle, IL1β (50 ng/ml) alone, or IL1β (50 ng/ml) with SP600125 (500 nm) or U0126 (500 nm) in luciferase reporter gene assays. Data points are mean ± sd of triplicate measurements. **, P < 0.01 compared with the control. DMSO, Dimethylsulfoxide.
We next scanned the human HNF4A promoter looking for putative AP1 binding sites (http://tfbind.hgc.jp/) and found 10 putative binding sites within the sequence present in our promoter (data not shown). We therefore analyzed the effect of IL1β treatment in the absence or presence of U0126 (500 nm) or SP600125 (500 nm) using a promoter 1 (P1) HNF4A promoter in luciferase reporter gene assays. The results indicated that IL1β treatment was able to reduce luciferase activity of the P1 HNF4A promoter, and this IL1β-induced reduction was blocked completely by cotreatment with U0126 (500 nm) or SP600125 (500 nm) in luciferase reporter gene assays (Fig. 5C).
IL1β treatment decreases SHBG production by reducing HNF-4α levels via c-Jun activation in human SHBG transgenic mice
To assess whether IL1β decreases hepatic production of plasma SHBG in vivo, we treated mice (n = 3) with daily ip injections of vehicle or 1.5 μg of IL1β. Blood was collected immediately before treatment and every day before the mice were killed. The results indicated that plasma SHBG levels were reduced after 24 h from the first IL1β ip injection and remain reduced at d 2 and 3, whereas no changes in SHBG levels were observed in the vehicle-treated mice during the 3-d treatment (Fig. 6A). In addition, the SHBG and HNF-4α mRNA levels were reduced in relation to 18S mRNA levels when compared with the vehicle-treated mice (Fig. 6B). Furthermore, the HNF-4α protein levels were significantly reduced, and the phospho-c-Jun protein levels were significantly increased in the livers of IL1β-treated mice (Fig. 6C).
Fig. 6.
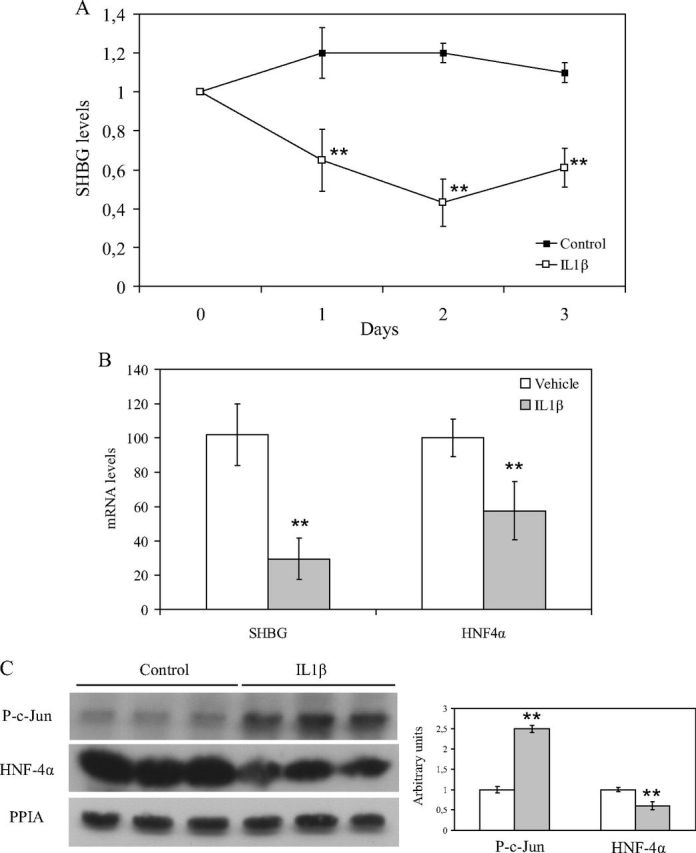
Daily treatment with IL1β reduces plasma SHBG levels in human SHBG transgenic mice. A, Human SHBG transgenic mice (n = 3) were treated over 3 d with ip injections of PBS or IL1β (1.5 μg), and blood SHBG levels were measured by ELISA. The SHBG levels are expressed as mean ± sd relative to pretreatment levels to compensate for between-animal variability. B, SHBG and HNF-4α mRNA abundance was determined in relation to 18S RNA (mean ± sd) in liver of vehicle-treated (n = 3) and IL1β-treated (n = 3) mice. C, Liver HNF-4α and phospho-c-Jun protein levels were measured by Western blotting using PPIA as a housekeeping reference protein in liver of vehicle-treated (n = 3) and IL1β-treated (n = 3) mice. Data points are mean ± sd of triplicate measurements. **, P < 0.01 compared with the control.
IL1β treatment decreases total testosterone plasma levels in human SHBG transgenic mice
To study whether IL1β treatment reduces total testosterone plasma levels in vivo by reducing the SHBG protein levels, we measured the total testosterone plasma levels in blood from human SHBG transgenic mice before (d 0) and after 3-d treatment with IL1 β. Total testosterone plasma levels were significantly reduced in human SHBG transgenic mice treated with IL1β at d 3 when compared with their initial pretreatment levels (0.089 ± 0.01 vs. 0.11 ± 0.005 nmol/ml; P < 0.01).
Discussion
Low plasma SHBG levels are found in chronic inflammatory diseases, such as obesity, type 2 diabetes, rheumatoid arthritis, and osteoarthritis (3–6). These diseases are characterized among other features by high plasma levels of proinflammatory cytokines, such as IL1β (19, 20). In this work we have tested the hypothesis that IL1β down-regulates SHBG production, which permits us to explain, at least in part, why patients with chronic inflammatory diseases have low plasma SHBG levels. We have shown that when the human HepG2 hepatoblastoma cell line and human SHBG transgenic mice were treated daily with IL1β, the SHBG production was reduced when compared with vehicle-treated cells or mice.
IL1β is an important proinflammatory cytokine that, on the one hand, activates monocytes, macrophages, and neutrophils and, on the other hand, induces T helper 1 and T helper 17 adaptive responses. IL1β is secreted as an inactive precursor and the processing of pro-IL1β depends on cleavage by proteases (21, 22). One of the most important enzymes is caspase-1, which in turn is activated by several protein platforms called inflammasomes, which contribute to obesity-induced inflammation and insulin resistance (23, 24). Therefore, a strong link between inflammation and metabolism is becoming increasingly evident (25). In fact, it has recently been reported that lack of the IL1 receptor protects against high-fat diet-induced insulin resistance coincident with reduced local adipose tissue inflammation (26). IL1β actions in the liver occur through the IL1β receptor, and they are mediated by two different signaling pathways, the NF-κB and JNK and MEK-1/2 MAPK, which result in the activation by phosphorylation of NF-κB and c-Jun TF (8). In the present study, we have shown that IL1β reduces SHBG production in vitro via MEK-1/2 and JNK MAPK by activating c-Jun TF in HepG2 cells.
Because IL1β treatment reduces SHBG mRNA expression, we analyzed the human SHBG promoter sequence looking for AP1 binding sites that are bound by c-Jun TF (27). We found two putative AP1 binding sites in the human SHBG proximal promoter within the FP 3 and the FP 4 regions of the promoter previously described (9). Although the human SHBG promoter responded to IL1β treatment in luciferase reporter gene assays, the effect seemed not to be mediated through the AP1 binding sites, because the promoter activity was decreased in the same manner when we used two different promoters in which either the FP 4 was deleted or the FP 3 was mutated. These findings suggest that the IL1β regulation of the SHBG expression is mediated by a different mechanism.
Our previous work in vitro and in vivo using HepG2 cells and human SHBG transgenic mice have revealed how SHBG expression can be regulated by alterations in cellular HNF-4α levels, which is a key regulator of SHBG transcription (9). More recently, we have identified thyroid hormones, increased de novo lipogenesis, and TNFα as factors that regulate SHBG production by changing hepatic HNF4α levels (12–15). In the present study, we provide evidence that IL1β down-regulates the SHBG production by the same mechanism. In addition, it is worth mentioning that IL1β-induced reduction of human SHBG promoter activity was completely blocked by the cotransfection of an HNF-4α expression vector.
Given that IL1β treatment decreases human SHBG promoter activity by reducing hepatic HNF-4α levels, we assumed that this effect had to occur very rapidly at the transcriptional and protein level. In this regard, it has been shown in the literature that IL1β induces a reduction in the mRNA HNF-4α levels (28) and in protein HNF-4α levels by inducing its proteasomal degradation (29). Our results confirm this and showed a fast reduction in mRNA and protein HNF-4α levels after 2–4 h treatment with IL1β. This effect was blocked by the cotreatment with MG132, a proteasomal inhibitor. Importantly, these results were confirmed in vivo using human SHBG transgenic mice after daily treatment with IL1β for 3 d. IL1β-treated mice showed reduced plasma SHBG and hepatic SHBG mRNA levels. Furthermore, HNF-4α mRNA and protein levels were also reduced when compared with vehicle-treated mice, whereas hepatic phospho-c-Jun protein levels were increased. Overall our results point to IL1β as a putative factor in accounting for the low SHBG levels observed in chronic inflammatory diseases (3–6). This IL1β-induced reduction in HNF4α protein levels was accompanied by an increase in c-Jun phosphorylation in HepG2 cells. Finally, when analyzed in the context of a luciferase reporter gene assay in HepG2 cells, human HNF4A P1 promoter activity was decreased by IL1β treatment, and we provided evidence that this effect was mediated via MEK-1/2 and JNK MAPK.
Low sex steroid levels have been documented consistently in acute or chronic inflammation and in critical illness, but the mechanism involved has not been completely understood (30–33). Several observational studies have reported an inverse relationship between serum levels of IL1β and testosterone, which might be explained by an impairment of the hypothalamic-pituitary-gonadal axis induced by IL1β (34, 35). Our findings showing that IL1β is able to down-regulate SHBG suggest a new mechanism by which IL1β could participate in the low testosterone levels detected in both chronic and acute inflammatory processes. To confirm this mechanism in the clinical setting, a prospective study to test the hypothesis that a significant increase in SHBG and testosterone will occur after treatment with IL1β receptor antagonist (i.e. anakinra) seems warranted.
In addition, our results introduce a new pathway in the complex relationship between innate immunity and sex steroid levels. It is widely recognized that testosterone has an immune-modulating action. In fact, the greater incidence of immune-mediated disease in women and androgen-deficient men has been attributed to the immunosuppressive effect of androgen compared with estrogens (36). Laboratory studies using animals with experimentally induced inflammatory disease have reported a beneficial response with androgens (37). Our findings support recent evidence that cytokines are important partners in the bidirectional communication that exists between sex steroid levels and the innate immune system (32, 38, 39) and add a new IL1β-mediated pathway to this cross talk. Thus, although testosterone administration has been shown to be useful in abrogating the inflammatory response (37), our results suggest that the inflammatory response, and specifically IL1β, could also down-regulate circulating testosterone levels by decreasing SHBG production.
In conclusion, the administration of IL1β reduces hepatic production of SHBG by decreasing HNF-4α via the MEK-1/2 and JNK pathways, thus resulting in low circulating levels of testosterone. These results suggest that IL1β could play an essential role in accounting for the low levels of testosterone detected in acute and chronic inflammatory diseases. Because several factors, such as glucose, TNFα, and now IL1β, have been identified as down-regulating hepatic SHBG production, further research addressed to investigating their respective contribution to this effect appears to be warranted.
Materials and Methods
Animals
Mice C57BL6 expressing human SHBG transgenes have been characterized previously (12, 13). Mice were maintained under standard conditions with food (Global Diet 2018; Harlan Interfauna Iberica, Barcelona, Spain) and water provided ad libitum and a 12-h light, 12-h dark cycle. Experimental procedures were approved by the Institutional Animal Use Subcommittees of Hospital Vall d'Hebron Research Institute and the Universitat Autònoma de Barcelona.
In vivo experiments
Eight-week male mice (n = 3) were treated with daily ip injections of PBS or 1.5 μg of human IL1β (Miltenyi Biotech S.L, Madrid, Spain). Blood samples were taken by saphenous vein sampling for measurements of plasma SHBG levels immediately before the treatment and daily. At d 3, the animals were killed, and livers were taken for RNA and protein extraction.
Cell culture experiments
Cell culture reagents were from Life Technologies, Inc. (Invitrogen SA, Barcelona, Spain). HepG2 hepatoblastoma cells (catalog no. HB-8065; American Type Culture Collection, Manassas, VA) were maintained in DMEM supplemented with 10% fetal bovine serum and antibiotics (100 U of penicillin/ml and 100 μg of streptomycin/ml). For experiments, HepG2 cells were cultured to 50–70% confluence before the addition of vehicle (PBS or dimethylsulfoxide), IL1β (Miltenyi Biotech S.L), U0126 (Enzo Life Sciences, Exeter, UK), SP600125 (Sigma-Aldrich. St. Louis, MO), SB203580 (Sigma-Aldrich), QNZ (Enzo Life Sciences), and MG132 (Sigma-Aldrich). All experiments have been performed in triplicates at least two times.
Wild-type, FP 3-mutated, and FP 4-deleted human SHBG promoter plasmids were kindly provided by Geoffrey Hammond (Department of Cellular and Physiological Sciences, Life Sciences Center, The University of British Columbia, Vancouver, British Columbia, Canada). Transient transfections of human SHBG or HNF-4A P1 promoter-driven luciferase reporter plasmids together with a pCMRenilla control plasmid were performed using Lipofectamine 2000 (Invitrogen SA). Two days after transfection, luciferase and renilla activity were measured using the Dual-Luciferase Reporter Assay System (Promega, Barcelona, Spain).
SHBG and testosterone measurements
Human SHBG levels in culture medium taken from HepG2 cells or from mice plasma were measured using an ELISA (Demeditec Diagnostics GmbH, Kiel-Wellsee, Germany). Testosterone plasma levels from mice were measured using an ELISA (Demeditec Diagnostics GmbH).
RNA analysis
Total RNA was extracted from HepG2 cells and mice livers using TRIzol reagent (Invitrogen SA). RT was performed at 42 C for 50 min using 3 μg of total RNA and 200 U of Superscript II together with an oligo-dT primer and reagents provided by Invitrogen SA. An aliquot of the RT product was amplified in a 25-μl reaction using SYBR Green (Invitrogen SA) with appropriate oligonucleotide primer pairs corresponding to human HNF-4α, mouse HNF-4α, human SHBG, human 18S, and mouse 18S (Supplemental Table 1). Results were analyzed using the 7000 SDS program.
Western blot analysis
After treatments, mice livers or HepG2 cells were harvested and homogenized in radioimmunoprecipitation assay buffer supplemented with Complete Protease Inhibitor Cocktail (Roche Diagnostics, Barcelona, Spain) at 4 C, followed by centrifugation (12,000 rpm at 4 C) for 10 min to obtain total protein extracts. Total cell protein extracts were used for Western blotting with antibodies against human HNF-4α (C-19; catalog sc-6556; Santa Cruz Biotechnology, Inc., Heidelberg, Germany), human c-Jun (sc-114; Santa Cruz Biotechnology, Inc.), human phospho-c-Jun (Calbiochem, San Diego, CA), and human Cyclophilin A (PPIA) (SA-296; BIOMOL Int., Madrid, Spain). Specific antibody-antigen complexes were identified using an horseradish peroxidase-labeled goat antirabbit IgG or rabbit antigoat IgG and chemiluminescent substrates (Pierce Biotechnology, Inc., Barcelona, Spain) by exposure to x-ray film.
Statistical analyses
Data were analyzed using the unpaired Student's t test. Levels of statistical significance were set at P < 0.05 and P < 0.01.
Acknowledgments
We thank Dr. Geoffrey L. Hammond (Child and Family Research Institute, University of British Columbia, Vancouver, British Columbia, Canada) for letting us use the human SHBG transgenic mice in this work. We also thank Lorena Ramos (Research Institute Hospital Vall d'Hebron) for her technical assistance.
This work was supported by a grant from the Instituto de Salud Carlos III (D.M.S.) and by Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas, an initiative of Instituto de Salud Carlos III (R.S., A.B.-D., C.H., and D.M.S.). D.M.S. is the recipient of a Miguel Servet contract.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AP1
- Activator protein 1
- FP
- footprinting
- HNF
- hepatic nuclear factor
- JNK
- c-Jun N-terminal kinase
- MEK
- MAPK kinase
- NF-κB
- nuclear factor kappa B
- P1
- promoter 1
- PPIA
- cyclophilin A
- SHBG
- sex hormone-binding globulin
- TF
- transcription factor.
References
- 1. Siiteri PK , Murai JT , Hammond GL , Nisker JA , Raymoure WJ , Kuhn RW. 1982. The serum transport of steroid hormones. Recent Prog Horm Res 38:457–510 [DOI] [PubMed] [Google Scholar]
- 2. Peter A , Kantartzis K , Machann J , Schick F , Staiger H , Machicao F , Schleicher E , Fritsche A , Häring HU , Stefan N. 2010. Relationships of circulating sex hormone-binding globulin with metabolic traits in humans. Diabetes 59:3167–3173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li C , Ford ES , Li B , Giles WH , Liu S. 2010. Association of testosterone and sex hormone-binding globulin with metabolic syndrome and insulin resistance in men. Diabetes Care 33:1618–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ding EL , Song Y , Manson JE , Hunter DJ , Lee CC , Rifai N , Buring JE , Gaziano JM , Liu S. 2009. Sex hormone-binding globulin and risk of type 2 diabetes in women and men. N Engl J Med 361:1152–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mirone L , Altomonte L , D'Agostino P , Zoli A , Barini A , Magaro M. 1996. A study of serum androgen and cortisol levels in female patients with rheumatoid arthritis. Correlation with disease activity. Clin Rheumatol 15:15–19 [DOI] [PubMed] [Google Scholar]
- 6. Spector TD , Perry LA , Jubb RW. 1991. Endogenous sex steroid levels in women with generalised osteoarthritis. Clin Rheumatol 10:316–319 [DOI] [PubMed] [Google Scholar]
- 7. Guarda G , So A. 2010. Regulation of inflammasome activity. Immunology 130:329–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chung JY , Park YC , Ye H , Wu H. 2002. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci 115:679–688 [DOI] [PubMed] [Google Scholar]
- 9. Jänne M , Hammond GL. 1998. Hepatocyte nuclear factor-4 controls transcription from a TATA-less human sex hormone-binding globulin gene promoter. J Biol Chem 273:34105–34114 [DOI] [PubMed] [Google Scholar]
- 10. Selva DM , Hogeveen KN , Hammond GL. 2005. Repression of the human sex hormone-binding globulin gene in Sertoli cells by upstream stimulatory transcription factors. J Biol Chem 280:4462–4468 [DOI] [PubMed] [Google Scholar]
- 11. Selva DM , Hammond GL. 2009. Peroxisome-proliferator receptor γ represses hepatic sex hormone-binding globulin gene. Endocrinology 150:2183–2189 [DOI] [PubMed] [Google Scholar]
- 12. Selva DM , Hogeveen KN , Innis SM , Hammond GL. 2007. Monosaccharide-induced lipogenesis regulates the human hepatic sex hormone-binding globulin gene. J Clin Invest 117:3979–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simó R , Barbosa-Desongles A , Lecube A , Hernandez C , Selva DM. 2012. Potential role of tumor necrosis factor-α in downregulating sex hormone-binding globulin. Diabetes 61:372–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simó R , Barbosa-Desongles A , Sáez-Lopez C , Lecube A , Hernandez C , Selva DM. 2012. Molecular mechanism of TNFα-induced down-regulation of SHBG expression. Mol Endocrinol 26:438–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Selva DM , Hammond GL. 2009. Thyroid hormones act indirectly to increase sex hormone-binding globulin production by liver via hepatocyte nuclear factor-4α. J Mol Endocrinol 43:19–27 [DOI] [PubMed] [Google Scholar]
- 16. Dreher I , Jakobs TC , Köhrle J. 1997. Cloning and characterization of the human selenoprotein P promoter. Response of selenoprotein P expression to cytokines in liver cells. J Biol Chem 272:29364–29371 [DOI] [PubMed] [Google Scholar]
- 17. Frost RA , Nystrom GJ , Lang CH. 2000. Stimulation of insulin-like growth factor binding protein-1 synthesis by interleukin-1β: requirement of the mitogen-activated protein kinase pathway. Endocrinology 141:3156–3164 [DOI] [PubMed] [Google Scholar]
- 18. Haas MJ , Horani M , Mreyoud A , Plummer B , Wong NC , Mooradian AD. 2003. Suppression of apolipoprotein AI gene expression in HepG2 cells by TNFα and IL-1β. Biochim Biophys Acta 1623:120–128 [DOI] [PubMed] [Google Scholar]
- 19. Eastgate JA , Symons JA , Wood NC , Grinlinton FM , Di Giovine FS , Duff GW. 1988. Correlation of plasma interleukin I levels with disease activity in rheumatoid arthritis. Lancet 2:706–709 [DOI] [PubMed] [Google Scholar]
- 20. Maury CP , Andersson LC , Teppo AM , Partanen S , Juvonen E. 1988. Mechanism of anaemia in rheumatoid arthritis: demonstration of raised interleukin 1β concentrations in anaemic patients and of interleukin 1 mediated suppression of normal erythropoiesis and proliferation of human erythroleukaemia (HEL) cells in vitro. Ann Rheum Dis 47:972–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Netea MG , Simon A , van de Veerdonk F , Kullberg BJ , Van der Meer JW , Joosten LA. 2010. IL-1β processing in host defense: beyong the inflammasomes. PLoS Pathog 6:e1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bauernfeind F , Ablasser A , Bartok E , Kim S , Schmid-Burgk J , Cavlar T , Hornung V. 2011. Inflammasomes: current understanding and open questions. Cell Mol Life Sci 68:765–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stienstra R , Joosten LA , Koenen T , van Tits B , van Diepen JA , van den Berg SA , Rensen PC , Voshol PJ , Fantuzzi G , Hijmans A , Kersten S , Müller M , van den Berg WB , van Rooijen N , Wabitsch M , Kullberg BJ , van der Meer JW , Kanneganti T , Tack CJ , Netea MG. 2010. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab 12:593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vandanmagsar B , Youm YH , Ravussin A , Galgani JE , Stadler K , Mynatt RL , Ravussin E , Stephens JM , Dixit VD. 2011. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17:179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Nardo D , Latz E. 2011. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol 32:373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McGillicuddy FC , Harford KA , Reynolds CM , Oliver E , Claessens M , Mills KH , Roche HM. 2011. Lack of interleukin-1 receptor I (IL-1RI) protects mice from high-fat diet-induced adipose tissue inflammation coincident with improved glucose homeostasis. Diabetes 60:1688–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Curran T , Franza BR. 1988. Fos and Jun: the AP-1 connection. Cell 55:395–397 [DOI] [PubMed] [Google Scholar]
- 28. Jahan A , Chiang JY. 2005. Cytokine regulation of human sterol 12α-hydroxylase (CYP8B1) gene. Am J Physiol Gastrointest Liver Physiol 288:685–695 [DOI] [PubMed] [Google Scholar]
- 29. Krajewski J , Batmunkh C , Jelkmann W , Hellwig-Bürgel T. 2007. Interleukin-1β inhibits the hypoxic inducibility of the erythropoietin enhancer by suppressing hepatocyte nuclear factor-4α. Cell Mol Life Sci 64:989–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van den Berghe G , Weekers F , Baxter RC , Wouters P , Iranmanesh A , Bouillon R , Veldhuis JD. 2001. Five-day pulsatile gonadotropin-releasing hormone administration unveils combined hypothalamic-pituitary-gonadal defects underlying profound hypoandrogenism in men with prolonged critical illness. J Clin Endocrinol Metab 86:3217–3226 [DOI] [PubMed] [Google Scholar]
- 31. Chesnokova V , Melmed S. 2002. Minireview: neuro-immuno-endocrine modulation of the hypothalamic-pituitary-adrenal (HPA) axis by gp130 signaling molecules. Endocrinology 143:1571–1574 [DOI] [PubMed] [Google Scholar]
- 32. Kalyani RR , Gavini S , Dobs AS. 2007. Male hypogonadism in system disease. Endocrinol Metab Clin North Am 36:333–348 [DOI] [PubMed] [Google Scholar]
- 33. Tengstrand B , Carlström K , Hafström I. 2009. Gonadal hormones in men with rheumatoid arthritis-from onset through 2 years. J Rheumatol 36:887–892 [DOI] [PubMed] [Google Scholar]
- 34. Oktenli C , Doganci L , Ozgurtas T , Araz RE , Tanyuksel M , Musabak U , Sanisoglu SY , Yesilova Z , Erbil MK , Inal A. 2004. Transient hypogonadotrophic hypogonadism in males with acute toxoplasmosis: suppressive effect of interleukin-1β on the secretion of GnRH. Hum Reprod 19:859–866 [DOI] [PubMed] [Google Scholar]
- 35. Nettleship JE , Pugh PJ , Channer KS , Jones T , Jones RD. 2007. Inverse relationship between serum levels of interleukin-1β and testosterone in men with stable coronary artery disease. Horm Metab Res 39:366–371 [DOI] [PubMed] [Google Scholar]
- 36. Bouman A , Heineman MJ , Faas MM. 2005. Sex hormones and the immune response in humans. Hum Reprod Update 11:411–423 [DOI] [PubMed] [Google Scholar]
- 37. Malkin CJ , Pugh PJ , Jones RD , Kapoor D , Channer KS , Jones TH. 2004. The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J Clin Endocrinol Metab 89:3313–3318 [DOI] [PubMed] [Google Scholar]
- 38. Whitacre CC. 2001. Sex differences in autoimmune disease. Nature Immunol 2:777–780 [DOI] [PubMed] [Google Scholar]
- 39. Gaillard RC. 2003. Interactions between the immune and neuroendocrine systems: clinical implications. J Soc Biol 197:89–95 [PubMed] [Google Scholar]