

Abstract
Placental CRH may be part of a clock that governs the length of human gestation. The mechanism underlying differential regulation of CRH in the human placenta is poorly understood. We report here that constitutively activated RelB/nuclear factor-κB2 (NF-κB)-2 (p100/p52) acts as an endogenous stimulatory signal to regulate CRH by binding to an NF-κB enhancer of CRH gene promoter in the human placenta. Nuclear staining of NF-κB2 and RelB in villous syncytiotrophoblasts and cytotrophoblasts was coupled with cytoplasmic CRH in syncytial knots of cytotrophoblasts. Chromatin immunoprecipitation identified that CRH gene associated with both RelB and NF-κB2 (p52). Dexamethasone increased synthesis and nuclear translocation of RelB and NF-κB2 (p52) and their association with the CRH gene. In contrast, progesterone, a down-regulator of placental CRH, repressed NF-κB2 (p100) processing, nuclear translocation of RelB and NF-κB2 (p52), and their association with the CRH gene. Luciferase reporter assay determined that the NF-κB enhancer of CRH was sufficient to regulate transcriptional activity of a heterologous promoter in primary cytotrophoblasts. RNA interference-mediated repression of RelB or NF-κB2 resulted in significant inhibition of CRH at both transcriptional and translational levels and prevented the dexamethasone-mediated up-regulation of CRH transcription and translation. These results suggest that the noncanonical NF-κB pathway regulates CRH production in the human placenta and is responsible for the positive regulation of CRH by glucocorticoids.
The complex mechanisms timing human birth involve the mother, fetus, and placenta; and a major role has been proposed for the euroendocrine mechanisms (1). CRH/urocortin (Ucn), including four ligands, CRH, Ucn, Ucn2, and Ucn3, represent important neuroendocrine mediators in physiology of human pregnancy and labor. CRH/Ucn, produced in the placenta, possess multiple functions that are associated with human gestation and parturition. On the one hand, CRH/Ucn may influence embryo implantation in early pregnancy (1) and protect the fetus from environmental stress by modulating hormone secretion of the placenta and fetal membranes (2–5), regulating fetoplacental circulation by vasodilatation (6, 7), and inhibiting premature myometrial contractions during most of pregnancy (8). On the other hand, CRH/Ucn may play an essential role in coordinating a switch from the state of quiescence to that of enhanced myometrial contractility at term (1). Indeed, this dual role for CRH/Ucn in regulation of human pregnancy suggests existence of a placenta clock that may determine the length of human gestation.
Consistent with the hypothesis that CRH is part of the placental clock timing human birth (9), several studies have shown that maternal plasma CRH, which is of placental origin, increased exponentially as pregnancy advanced, peaking at the time of delivery (10–12). Importantly, women who delivered preterm showed this exponential increase was rapid. Coupled with this exponential increase of CRH, the fall of maternal plasma CRH binding protein, which inactivates CRH and is constitutionally produced in the third trimester of pregnancy, was noted in the final weeks of term, still further with onset of labor (13, 14). In addition, CRH binding protein had the lowest measurable levels in both maternal and fetal circulation in preterm labor (15, 16). At term, CRH may promote myometrial contractions by increasing the production of prostaglandins in the fetal membrane (17) as well as cortisol and dehydroepiandrosterone by the fetal adrenal gland (18). In the placenta, dehydroepiandrosterone is converted to estrogen that in turn increases the number of oxytocin receptors on uterine muscle fibers for promoted uterine contractions (19).
In both the hypothalamus and placenta, CRH is a product of the same gene located on the long arm of chromosome 8 (20). In contrast with the fact that CRH is negatively regulated by glucocorticoid in the hypothalamus, CRH of placental origin is positively regulated by glucocorticoid, establishing a feed-forward loop between the fetus and placenta, which drives production of CRH during pregnancy (21). Although the mechanism underlying the negative regulation of hypothalamic CRH has been extensively investigated, the molecular mechanism of positive regulation of placental CRH remains elusive. In most mammals, labor is heralded by withdrawal of progesterone that is capable of inhibiting uterine contractions. In primary cultures from the term human placentas, cortisol competed with the action of progesterone in the regulation of CRH (22), which provided one clue to CRH regulation in the placenta.
Increasing evidence suggests that the activation of inflammatory transcription factors, such as inducible nuclear transcription factor κB (NF-κB), may play an important role in human labor (23, 24). NF-κB is essential to functioning of the immune system in mammalian and other species and is now known to regulate gene expression in many tissues. NF-κB has roles in multiple pathways that affect cell survival, differentiation, and proliferation (25–27). Mammalian NF-κB family consists of five members: RelA or p65, RelB, C-Rel, NF-κB1 p50 and its precursor p105, and NF-κB2 p52 and its precursor p100. All of them contain an N-terminal Rel homology domain responsible for homo- and heterodimerization that are capable of transactivating numerous target genes via translocation into the nucleus and binding to the NF-κB enhancer. Whereas RelB preferentially heterodimerizes with p100 to constitute the noncanonical pathway, p65 usually heterodimerizes with p50 that is constitutively processed from p105 to represent the canonical pathway. The canonical NF-κB, RelA/p50, although in an inactivated state, is located in the cytoplasma and complexed with the inhibitory protein, inhibitory-κB (IκBα). Numerous signals trigger activation or nuclear translocation of RelA/p50 by causing phosphorylation and ubiquitination of IκBα, dissociation of IκBα from RelA/p50, and eventual degradation of IκBα by the proteasomes. The noncanonical NF-κB pathway, triggered by a subset of TNF receptor (TNFR) family members, is mediated by activation of NF-κB-inducing kinase (NIK). Activated NIK phosphorylates inhibitory-κB kinase-α (IKKα) complex that in turn phosphorylates the IKB (inhibitory κB) domain of NF-κB2 (p100) to liberate RelB/NF-κB2 (p52) that subsequently translocates into the nucleus. The eventual degradation of phosphorylated/ubiquitinated IKB domain of p100 is also mediated by the proteasomes. NF-κB target genes are widely expressed in multiple cell types. These genes can be activated either in response to diverse stimuli or could be due to the constitutively nuclear localization of NF-κB heterodimers in some normal cells like B lymphocytes (28) and cancer cells (29, 30).
Provided that NF-κB activity has been identified in the nuclei of human term cytrotrophoblasts (31), we hypothesize that NF-κB may regulate CRH expression in the human placenta. To test this hypothesis, we performed a variety of approaches in human placental tissues and primary cytotrophoblasts from human term placentas to determine whether NF-κB pathway is activated in the human placenta and whether this activated NF-κB pathway is both sufficient and necessary for CRH expression. We have shown that the noncanonical NF-κB signaling, or RelB/NF-κB2, is constitutively activated in the human placenta, which binds to a previously undescribed NF-κB enhancer of CRH gene promoter to regulate CRH expression.
Results
The proteasomes regulate CRH transcription in cytotrophoblasts
Proteasomes are large protein complexes in both the cytoplasma and nucleus of the eukaryotes, the major function of which is to degrade unneeded or damaged proteins by proteolysis (32). The activation of NF-κB is unanimously coupled with proteasomal degradation of NF-κB inhibitory proteins, IκBα or IKB domain of NF-κB2 (p100) (27). To resolve the molecular mechanism of CRH regulation in the human placenta, we turned to our previous finding that nuclear extracts of human term cytotrophoblasts might contain NF-κB activity (31). Herein we first tested the effects of the proteasome inhibitors on CRH transcription in cytotrophoblasts. Activation of NF-κB signaling results from phosphorylation, ubiquitination, and eventually proteasomal degradation of IκBα or IKB domain of p100. As a result, inhibition of the proteasomes results in the loss of function of NF-κB. Human term cytotrophoblasts were purified and cultured for 24 h before being incubated with proteasome inhibitors, MG132, lactacyctin, or PSI [N-benzyloxycarbonyl-Ile-Glu(O-t-butyl)-Ala-leucinal]. A nonproteasomal inhibitor (cysteine protease inhibitor), 1,2-di-(N-carboxybenzoyl-l-leucyl)amino acetone (ZLL) was used as a negative control. A dose-dependent effect on CRH transcription was observed with MG132 (Fig. 1, lanes 2–5) and lactacystin (Fig. 1, lanes 6–9). In addition, pyrrolidine dithiocarbamate, a thiol compound used as an NF-κB mobilization inhibitor, repressed CRH transcription (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). CRH was not inhibited with the use of the nonproteasomal inhibitor ZLL, even at 3.0 μm (Fig. 1, lanes 14–17). cAMP positively regulates CRH transcription by binding to cAMP-response element (CRE) binding protein at the CRE of the CRH promoter (33). To investigate whether proteasome inhibitors modulate the levels of CRH transcription through cAMP in the human placenta, we measured the intracellular cAMP levels in cytotrohophoblasts that were subjected to the treatment of proteasome inhibitors. In three independent experiments, we did not find that any of the proteasome inhibitors displayed the inhibitory effects on cAMP level (Supplemental Fig. 2). These results suggest a mechanism that is dependent on NF-κB but independent of cAMP in regulation of CRH expression in the human placenta.
Fig. 1.

The proteasomes regulate CRH transcription in cytotrophoblasts. Human cytotrophoblasts were incubated with vehicle (lane 1) or proteasomal inhibitors MG132 (lanes 2–5), lactacystin (lanes 6–9), PSI (lanes 10–13) or nonproteasomal inhibitor, or ZLL (lanes 14–17) at a concentration ranging from 0.1 to 3.0 μm as indicated for 24 h. The total RNA were extracted and subjected to Northern blotting analysis with the use of GAPDH as the reference.
Typically, RelA (p65) heterodimerizes with NF-κB1 (p50) to constitute the canonical pathway, whereas RelB and NF-κB2 (p100/p52) form the noncanonical pathway (26). To distinguish the activities of NF-κB in the placenta, we incubated cytotrophoblasts with a proteasome inhibitor, PSI, to determine the NF-κB effect on CRH expression. PSI has been reported to reduce NF-κB2 activity by more than 70% but barely affect processing of NF-κB1 (p50) at 1.0 μm in human embryonic kidney 293 cells (34), suggesting that NF-κB1 is more resistant to inhibition by this proteasome inhibitor. Indeed, we found here that PSI was capable of completely repressing CRH transcription in cytotrophoblasts at concentrations from 0.1 to 3.0 μm (Fig. 1, lanes 10–13). Taken together, these results suggest that NF-κB, more likely RelB/NF-κB2, may be activated and participate in the regulation of CRH transcription in the human placenta.
The noncanonical NF-κB pathway is constitutively activated in the human placenta
Constitutive activation of the noncanonical NF-κB pathway regulates important biological functions in lymphocytes such as lymphoid organogenesis and B cell survival and maturation (28). A hallmark for activation of the noncanonical NF-κB pathway is that the precursor form of NF-κB2, p100, is proteolytically processed by proteasome into active form; p52 that heterodimerizes with RelB, in turn, translocates into the nucleus to transactivate target genes. To further determine which NF-κB pathway is activated in the placenta, the human term placentas after delivery were immediately formalin fixed, paraffin embedded, and sectioned for immunohistochemical analysis for direct visualization of subcellular localization of NF-κB components. The titration of RelB or NF-κB2 antibody was optimized with the recommendation of the manufacturers as well as the serial dilution of antibodies tested in the positive control tissue (Supplemental Fig. 3). As shown in Fig. 2, there was a differentially subcellular localization of NF-κB components, RelB, NF-κB2 (p100/p52), and p65. NF-κB2 and RelB demonstrated both cytoplasmic and nuclear staining in syncytiotrohoblasts in the syncytium and syncytial knots and the cells resembling cytotrophoblasts in morphology in the layer beneath the syncytium (Fig. 2, A and B). RelA (p65) was exclusively localized in the cytoplasma of trophoblasts (Fig. 2D), suggesting the specificity of nuclear localization of RelB and NF-κB2. Cytoplasmic CRH staining was primarily found in the syncytium and syncytial knots (Fig. 2C). Consistent with the fact that CRH enters into the fetal circulation and CRH receptor is expressed in the endothelium of fetal blood vessels (35), the endothelium of fetal blood vessels here was highlighted with CRH staining. In addition, strong positive CRH staining was observed in extravillous tissues, the decidua, as previously described (36). Quantitative analysis revealed that positive nuclear staining in trophoblsts was observed in approximately 27% of cross-sections of placental villi (Fig. 2, A and B, insets) for both RelB and NF-κB2. Collectively these results support that the noncanonical NF-κB pathway is constitutively activated in the human term placenta.
Fig. 2.
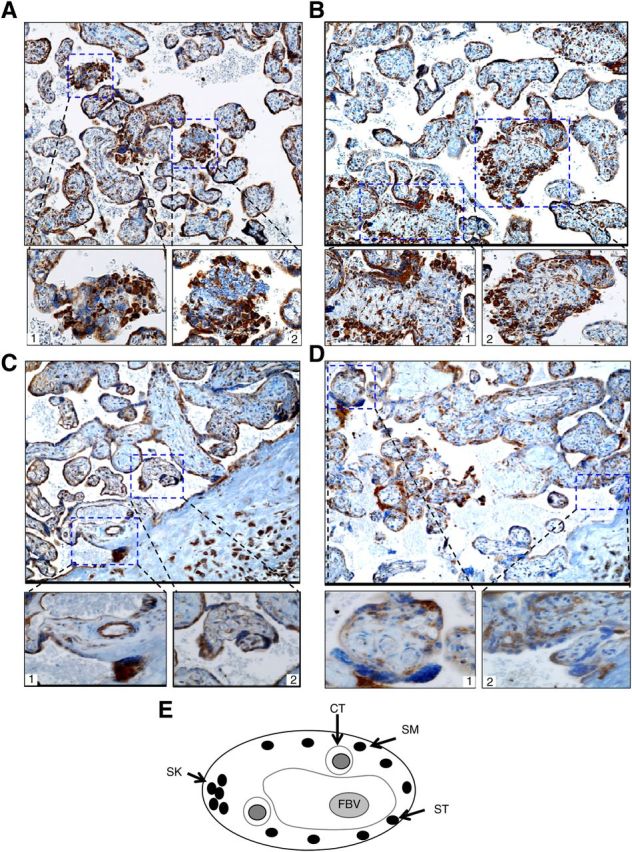
Immunohistochemical analyses of NF-κB components and CRH in human term placenta. Four sections from the same placenta were stained with antibody to human RelB (A), NF-κB2 (B), CRH (C), or Rel A (p65) (D), respectively. Representative images are shown in the top panel; original magnification, ×100. Bottom panels represent insets; original magnification, ×400. E, Schematic presentation of cross-section of human term placental villus. CT, Cytotrophoblasts; ST, syncytiotrophoblast; SM, syncytium; SK, syncytial knot; FBV, fetal blood vessel.
Constitutively activated RelB/NF-κB 2 associates with the CRH gene in the human placenta
All five members of the NF-κB family harbor a highly homologous Rel homology that is responsible for protein-DNA interaction and hetero- or homodimerization. NF-κB dimers share an intrinsic ability of binding to an NF-κB enhancer that is defined as a family of DNA-binding sites of 9–11 bp in length to regulate target genes (37). The consensus NF-κB enhancer, which presents a major source of complexity in NF-κB system, is represented as 5′-GGGRNNYYCC-3′ (R represents purine, N is any nucleotide, and Y represents pyrimidine). The variability of the NF-κB enhancers may allow the usage of specific dimers at specific promoters. As a result, if CRH transcription is regulated by NF-κB in the placenta, the CRH gene promoter should contain NF-κB enhancer(s) that allow NF-κB dimer to bind to for regulation of CRH. Expectedly, an NF-κB motif search at Consite (38) revealed that human the CRH gene promoter contains a highly putative NF-κB enhancer, 5′-GGGAAATCTC-3′ at −295, in addition to regulatory elements previously described, a negative glucocorticoid response element (GRE), a CRE, and a TATA box (Fig. 3A) (39). Consistent with the fact that the NF-κB enhancer selection by the RelB/NF-κB2 dimer uses A:T rich middle segment to permit a broader binding (40), this putative NF-κB enhancer suggests a functional link between RelB/NF-κB2 (p52) and the CRH gene.
Fig. 3.

RelB and NF-κB2 associate with the CRH gene in primary cytotrophoblasts. A, Schematic presentation of the human CRH gene and regulatory elements. The CRH gene contains a putative NF-κB enhancer (κB) at −295, in addition to a negative GRE at −277 and a CRE at −226. B, ChIP analyses were performed to determine occupancy of individual NF-κB family member, p65, NF-κB1 (p50), C-Rel, RelB, and NF-κB2, at the CRH gene promoter. Fold enrichment of CRH with each member was calculated with normalization to a human α-satellite. Rabbit IgG served as a nonspecific control. The bars indicate the average of CRH fold enrichment (y-axis) against each member (x-axis), with error bars representing the sd from three independent experiments. The fold enrichment of CRH with IgG was defined as 1. *, P < 0.001, ***, P > 0.05 (no significant difference, vs. the control, IgG), as determined by one-way ANOVA with Dunnett's test. C, ChIP analyses were performed to determine change in fold enrichment of CRH with RelB or NF-κB2 when cytotrophoblasts were treated with vehicle (−), DEX 1 μm, or progesterone (Prog) 10 μm. Fold enrichment of CRH was determined and presented as described in B. The fold enrichment of CRH with antibody to RelB or NF-κB2 in the presence of vehicle but absence of steroids was defined as 1. *, P < 0.001, **, P < 0.01 (vs. the control cells treated with the vehicle), as determined by one-way ANOVA with Dunnett's test.
To investigate whether the RelB2/NF-κB2 dimer binds to this putative NF-κB enhancer of CRH in cytotrophoblasts to regulate CRH, we performed chromatin immunoprecipitation (ChIP) assays. After 24 h from purification, the primary cytotrophoblasts were subjected to formaldehyde cross-linking. Sonicated cell lysates with sheared DNA fragments of 200–500 bp were then immunoprecipitated with use of a ChIP-grade antibody to an individual member of the NF-κB family, whereas unconjugated rabbit polyclonal antibody served as a nonspecific control. The immunoreprecipitates were reverse cross-linked and analyzed by quantitative real-time PCR to amplify a region encompassing this NF-κB enhancer. We determined the fold change in the occupancy of each individual member at this site by 2(-ΔΔCT) with normalization to usage of primers that amplify a human DNA α-satellite, to which no transcriptional factors should bind. As shown in Fig. 3B, the data generated from three independent experiments demonstrate that the CRH gene promoter was not immunoprecipitated by an antibody to p65, p50 or C-Rel. The CRH promoter was almost equally occupied by RelB and NF-κB2 (p52) in cytotrophoblasts, approximately 5-fold over the nonspecific control. Along with data (not shown) indicating that DNA was sufficiently sheared to 200–500 bp to prevent any coimmunoprecipitated large chromosomal fragments that might interact with p65, p50, or C-Rel at additional contact loci, these results suggest a specific interaction between the CRH gene promoter and the RelB/NF-κB (p52) complex in the human placenta.
Indeed, the transcription of CRH in the placenta is enhanced by glucocorticoid but repressed by progesterone (22) (also see Supplemental Fig. 1). Consequently, we next examined differential effects of these steroids on the occupancy of RelB and NF-κB2 (p52) at the CRH gene promoter. Primary cytotrophoblasts were incubated with dexamethasone (DEX), a potent synthetic glucocorticoid, 1 μm, or progesterone 10 μm for 24 h. After formaldehyde cross-linking, cell lysates were immunoprecipitated with antibody to an individual member of the NF-κB family. As shown in Fig. 3C from three independent experiments, DEX treatment increased RelB or NF-κB (p52) recruitment to the CRH promoter by approximately 2-fold over the baseline in human term cytotrophoblasts. In contrast, progesterone reduced RelB or NF-κB (p52) recruitment to the CRH promoter by more than 60%. In addition, DEX or progesterone treatment had no effects on the recruitment of p65, p50, or C-Rel to the CRH promoter in cytotrophoblasts (Supplemental Fig. 4). These results suggest that the differential effects of glucocorticoid and progesterone on CRH transcription in the placenta are mediated by their effects on the RelB and NF-κB2 (p52) recruitment to the CRH promoter in the human placenta.
The effects of steroids on CRH are mediated by synthesis and/or nuclear translocation of RelB/NF-κB (p52) in the human placenta
Activation of the noncanonical NF-κB pathway depends on phosphorylation-induced p100 processing into p52 to mediate specific functions. Persistent activation of RelB/NF-κB2 requires de novo protein synthesis, in contrast to which the canonical NF-κB pathway relies on otherwise (26). In light of the study that glucocorticoid induced RelB synthesis in the lymphoblastic cell line (41), we hypothesized here that these steroids have an effect on synthesis and/or nuclear localization of RelB/NF-κB2 (p52) to regulate CRH expression in the human placenta. To confirm this hypothesis, we first examined whether glucocorticoid stimulates or progesterone represses synthesis of RelB and NF-κB2 in cytotrophoblasts. Primary cells were treated with DEX or progesterone for 24 h. The total cell lysates were collected and Western blotting analyses were performed. As shown in Fig. 4A, treatment of primary cells with DEX resulted in a significant increase in synthesis of both RelB and NF-κB2. Whereas treatment of cytotrophoblasts with progesterone exerted a negligible effect on production of either of them, it is evident that progesterone significantly inhibited p100 processing.
Fig. 4.

Differential effects of steroids on CRH correspond with their effects on production and/or nuclear translocation of NF-κB2/RelB in cytotrophoblasts. A, Whole-cell lysates of term cytotrophoblasts that were pretreated with vehicle (−) or DEX 1 μm or progesterone (Prog) 10 μm for 24 h were probed with CRH (upper left panel), RelB (lower left panel), or NF-κB2 (right panel) antibodies. β-Actin was used as a loading control. B, Nuclear (top panels) and cytosolic extracts (bottom panels) from the same whole-cell lysates of A were prepared and probed with RelB (left panel) or NF-κB2 (right panel) antibodies. SP1 was used as a loading control for nuclear extracts and β-actin was used as a loading control for both nuclear and cytosolic extracts. These gels are representatives of three repeated experiments performed on three separate placentas. SP, Specificity protein 1.
To test the ability of steroids to modulate the nuclear activity of RelB/NF-κB2, we performed Western blotting analysis on nuclear and cytosolic extracts that were prepared from primary cytotrophoblasts with treatment of DEX or progesterone. As shown in Fig. 4B, representative images from three independent experiments, treatment of cytotrophoblasts with progesterone resulted in a significant decrease in nuclear translocation of both RelB and NF-κB2 (p52) and an increase in cytosolic accumulation of RelB. The inactive form of NF-κB2, p100, was exclusively localized in the cytosolic extracts. DEX treatment of cytotrophoblasts increased the nuclear translocation and cytosolic retention of RelB, which were coupled with the elevated nuclear level of NF-κB2 (p52). Cytosolic accumulation of NF-κB2 (p52) was not observed with progesterone treatment because p100 processing was inhibited.
Taken together, these results suggest that the RelB/NF-κB2 complex may participate in the positive regulation of CRH in the placenta. It is possible that glucocorticoid up-regulates CRH by stimulating synthesis and/or nuclear translocation of RelB/NF-κB2, whereas progesterone inhibits CRH by preventing NF-κB2 processing and their subsequently nuclear translocation in the human placenta.
The NF-κB enhancer of CRH confers transcriptional regulation to a heterologous promoter in primary cytotrophoblasts
Many NF-κB enhancers and other transcriptional regulatory elements confer signal-dependent regulation to a heterologous promoter and subsequent expression of target gene. To investigate gene regulation in a heterologous context, we placed the NF-κB enhancer of the CRH gene upstream of the minimal promoter that drives gene expression of firefly luciferase. Most of the sequence variation in NF-κB enhancers frequently happens in the 3′ half, whereas the 5′ half (GGGAA) is highly conserved (37). As a result, we also cloned a mutant version (5′-GAGCACTCTC-3′, mutant nucleotides underlined) of the NF-κB enhancer as a baseline control (Fig. 5A). After being cultured for 24 h, primary cytrotrophoblasts were transfected with the luciferase reporter plasmids containing one copy of the CRH NF-κB enhancer, four copies of the CRH NF-κB enhancer, or one copy of the mutant CRH enhancer. Plasmid pGL4.32, which harbors five copies of the NF-κB enhancer, was used as the positive control. In the meantime, a plasmid that contains a cytomegalovirus (CMV) promoter, which is not regulated by any NF-κB enhancer, was cotransfected as an internal reference. Twenty-four hours later, cells were harvested and subjected to a dual-luciferase assay. The fold activity of firefly luciferase was obtained with normalization to renilla luciferase activity. As shown in Fig. 5B from three independent experiments, one copy of the CRH NF-κB enhancer conferred NF-κB stimulation to this minimal promoter by more than 2.5-fold over the baseline (the mutant CRH enhancer), whereas four copies of the CRH enhancer generated more than 4-fold of firefly luciefearse activity. Cells transfected with pGL4.32 resulted in approximately 3.8-fold of luciferase activity over the baseline, which was consistent with positive correlation between the copy number of the NF-κB enhancer with the A:T-rich middle segment and the NF-κB -stimulated activity. These results have shown that the NF-κB enhancer of the CRH gene is sufficient for RelB/NF-κB (p52)-dependent regulation to the CRH promoter in the human placenta.
Fig. 5.

The putative NF-κB enhancer of CRH regulates transcription of a heterologous promoter. A, Schematic presentation of luciferase reporter constructs. pGL4.32 (Promega) harbors five copies of the NF-κB enhancer and a minimal promoter (TATA), which regulate expression of firefly luciferase gene (FL). pGL4.32-CRH1X and -CRH4X contain one and four copies of 5′-GGGAAATCTC-3′, respectively. pGL4.32-CRH1XMut that contains one copy of mutant version (mutant residues underlined) was also created. pCMV-RL (renilla luciferase) was used as the internal control. B, Human cytotrophoblasts were cotransfected with pCMV-RL (200 ng) and the constructs (1 μg) as indicated at the bottom. The bars represent the average of fold change of FL with normalization to renilla luciferase (RL), and error bars represent the sd from three experiments. Fold FL activity by pGL-4.32CRH1XMut was defined as 1. *, P < 0.001, **, P < 0.01 (vs. FL activity of control cells transfected with pGL4.32-CRH1XMut), as determined by one-way ANOVA with Dunnett's test. C, Cytotrophoblasts were cotransfected with pGL4.32-CRH1X (1 μg) and pCMV-RL (200 ng) for 24 h and then treated with vehicle (−) or with DEX or progesterone (Prog) for an additional 24 h. FL activity was determined with normalization to RL activity and presented in bars and errors as described in B. Fold FL activity by pGL-4.32CRH1X in the presence of vehicle but absence of steroids was defined as 1. *, P < 0.001, **, P < 0.01 (vs. FL activity of control cells transfected with pGL4.32-CRH1X in the presence of vehicle), as determined by one-way ANOVA with Dunnett's test.
In addition, we examined the differential effects of glucocorticoid and progesterone on NF-κB-stimulated activity mediated by the CRH NF-κB enhancer. After being cotransfected with pGL4.32-CRH1X and pCMV-RL for 24 h, primary cells of cytotrophoblast were incubated with DEX or progesterone for an additional 24 h. Cells were lysated and received a dual-luciferase assay. As indicated in Fig. 5B from three independent experiments, DEX stimulated NF-κB activity by nearly 2-fold, whereas progesterone reduced it by greater than 58%. These results are consistent with the differential effects of steroids on CRH transcription in the human placenta.
RelB/NF-κB2 complex is required for regulation of CRH in the human placenta
We have shown that the noncanonical NF-κB signaling, RelB/NF-κB2, sufficiently drove CRH expression in primary cytotrophoblasts. To determine whether this signaling is essential to CRH expression in the human placenta, we used the approach of loss of function, specifically RNA interference, to inhibit gene expression. Primary cells of cytotrophoblast were transfected with short interfering RNA (siRNA) directed against RelB or NF-κB2 for 24 h. Cells were treated DEX 1 μm or progesterone 10 μm for an additional 24 h before being harvested for Western blotting analysis and RT-PCR for the assay of protein and mRNA levels, respectively. As shown in Fig. 6, A and B, which were representative of three independent experiments, DEX stimulated the transcription and translation of RelB, NF-κB2, and CRH in the absence of targeting siRNA. However, there was an obvious reduction in RelB or NF-κB2 at both translational and transcriptional levels when siRNA to RelB or NF-κB2 was used. CRH was also significantly reduced at both translational and transcriptional levels in the matched assays. Moreover, the silencing of RelB or NF-κB2 prevented DEX-stimulated CRH expression. These results suggest that the expression of CRH in the human placenta requires RelB/NF-κB2 (p52)-dependent transactivation to CRH promoter.
Fig. 6.

The effects of RelB/NF-κB2 gene silencing on transcriptional and translational levels of CRH in primary cytotrophoblasts. A, Human term cytotrophoblasts were transfected with RelB or NF-κB2 siRNA or nontargeting scramble siRNA (SS) for 24 h. The cells were then treated with vehicle (−) or with DEX 1 μm or progesterone (Prog) 10 μm. After another 24 h, the cells were lysed and probed with antibody to human CRH, RelB (left panel), or NF-κB2 (right panel). β-Actin served as a loading control. B, Total RNA were extracted from A, and quantitative RT-PCR was performed with normalization to GAPDH mRNA. The bars represent average of fold change in mRNA levels, and error bars represent sd from three experiments. The mRNA levels of RelB or NF-κB2 in cells treated nontargeting scramble siRNA in the absence of steroids were defined as 100%. *, P < 0.001, **, P < 0.01 (vs. that of control cells transfected with SS and treated with vehicle), as determined by one-way ANOVA with Dunnett's test. C, The cells pretransfected with siRNA indicated in A were cotransfected with pGL4.32-CRH1X (1 μg) and pCMV-RL (200 ng) and with vehicle or DEX or progesterone (Prog) for an additional 24 h. FL activity was determined with normalization to renilla luciferase (RL) activity. The bars represent average of percentage change in FL activity, and error bars represent sd from three experiments. FL activity in cells treated nontargeting scramble siRNA in the presence of vehicle but absence of steroids was defined as 100%. *, P < 0.001, **, P < 0.01 (vs. that of control cells transfected with SS and treated with vehicle), as determined by one-way ANOVA with Dunnett's test.
To determine whether RelB/NF-κB2 is required for the transcriptional regulation by CRH NF-κB enhancer in the heterologous context, we examined the effects of depletion of RelB or NF-κB2 on the expression of transfected firefly luciferase reporter. Primary cytotrophoblasts were transfected with siRNA to RelB or NF-κB2. After 24 h, the cells were cotransfected with pGL4.32-CRH1X and pCMV-RL for 24 h and dual-luciferase assays were performed. As indicated in Fig. 6C, DEX treatment stimulated firefly luciferase (FL) activity in the absence of targeting siRNA. Silencing of either RelB or NF-κB2 reduced FL activity of pGL4.32-CRH1X by more than 62%. DEX treatment lost its ability to stimulate NF-κB activity in primary cytotrophoblasts. These results further support that the constitutively activated RelB/ NF-κB2 (p52) complex transactivates CRH transcription by binding to its NF-κB enhancer and is involved in the positive feedback of CRH-glucocorticoid in the placenta.
Discussion
Maternal plasma CRH level increases exponentially as pregnancy advances, peaking at the time of delivery. A model of the feed-forward loop was first proposed to explain the positive feedback of fetoplacental CRH-glucocorticoid (21). CRH, produced by human placental trophoblasts, enters the fetal circulation to result in release of ACTH from the fetal pituitary, which stimulates the downstream production of cortisol in the fetal adrenal glands. Cortisol proceeds into the placental circulation to activate CRH production, which further stimulates fetal ACTH, thereby completing a cycle. Despite of the observation that in primary cultures of human placenta, cortisol competed with the action of progesterone in the regulation of CRH (22), the molecular mechanism underlying this model has remained elusive. To clarify the mechanism responsible for the positive regulation of placenta CRH, we took advantage of primary cytotrophoblasts before reaching the peak of syncytialization from human term placentas because CRH expression level peaks after 24 h of culture and remains stable during the rest period (21), and syncytialization of cytotrophoblasts does not peak until 72–96 h of culture (42). The present study has shown that the noncanonical NF-κB pathway, RelB/NF-κB2 complex, is activated in the human term placenta. In turn, activated NF-κB acts an endogenous stimulatory signal by binding to the NF-κB enhancer of the CRH promoter to initiate CRH expression. Cortisol may further potentiate NF-κB activity in trophoblasts by promoting synthesis and/or nuclear translocation of RelB and NF-κB2 (p52) to up-regulate CRH production.
In most cells, NF-κB presents as an inactive complex that is sequestered in the cytoplasm but rapidly translocates into the nucleus to activate target genes as soon as the cells receive extracellular stimuli. The RelB/NF-κB2 dimer is activated because of the phosphorylation of p100 at the C terminal by IKKα homodimer, which is triggered by NIK in response to a subset of TNFR signals (26). Coupled with ubiquitination and proteasomal degradation of its C-terminal IKB domain, p100 is processed into active form, p52, and RelB/p52 dimer enters the nucleus to transactivate target genes. Generally, constitutive NF-κB activation is noted in chronic inflammatory disease states such as asthma and rheumatoid arthritis (43) and is an important common pathway in many forms of cancer (44). However, constitutive activation of NF-κB has also been noted in physiological states, such as activated B lymphocytes (45) and neuronal cells (46). Although mechanisms governing constitutive activation are being investigated (47) and may be very different in each of these tissues, it is interesting to speculate that NF-κB activation may contribute to similar functions in all three cell types. Both activated B cells and neurons are capable of memory, so it can be inferred that the NF-κB activation in the placenta may participate in memory or learning of this organ. Our finding that RelB/NF-κB2 nuclear staining occurs in the syncytial knots, which form as the placenta ages, is consistent with this idea.
Our results further support the fact that glucocorticoids induced up-regulation and progesterone caused down-regulation of CRH transcription are mediated by their stimulatory and inhibitory effects on protein synthesis, processing, and nuclear translocation of RelB/NF-κB2 in the human placenta, respectively. Glucocorticoid receptor (GR) usually represents a negative regulator to the NF-κB family either in up-regulating transcription of IκBα (48) or in a manner of cross talk, for instance, physical association between GR and RelA (49), both of which are dependent on ligand activation. DEX treatment has been shown to increase the production of RelB in lymphoblastic cells (41). Our observations suggest that glucocorticoid stimulates expression of RelB and NF-κB2 in the human placenta, likely in a classical manner: the ligand-activated classical GR isoform-α (hGRα) dimerizes to bind to GRE in the promoter region of the target gene, facilitates formation of transcription initiation complex, and promotes transcription rate of the target gene (50). With respect to action by progesterone, there have been conflicting literatures as to whether progesterone receptor (PR) is expressed by the placenta and specifically the syncytiotrophoblast at term. Several studies indicate that the placenta at term have no detectable levels of PR (22, 51, 52), whereas others have detected low levels of PR (53, 54). A more recent study has shown that a short isoform of PR, PR-C, was detected in a relative abundance in the cytoplasma but not in the nucleus of villous syncytiotrophoblasts (55). PR-C is a protein of 60 kDa of 314 amino acids. It is N terminally truncated and short of the first zinger finger (P-box) in the DNA binding domain (DBD) that mediates high affinity recognition of the core half-site of the PR-responsive element. Interestingly, PR-C still retains the second zinc finger (D-box) in the DBD that is responsible for receptor dimerization and the ligand-binding domain (or AF-2) (56, 57). Taken together with the previous finding that DBD and ligand-binding domain were both sufficient as well as essential for direct repression of RelA by ligand-activated PR (58), one possible mechanism for progesterone-induced inhibition of RelB/NF-κB2 in the placenta may be mediated by direct interaction of PR-C and RelB/NF-κB2 in the human placenta.
The molecular mechanisms responsible for constitutive activation of RelB/NF-κB2 in the human placenta and other normal cells are not entirely understood. Constitutive activation of the canonical NF-κB pathway, RelA/p52, is noted in a variety of cancer cells because either the gene encoding IκBα is mutated or defective, such as in B-cell lymphomas (29), and/or genes of the Rel/NF-κB transcriptional factors are amplified, such as in myelomas (30). Constitutive activation of RelB/NF-κB2 in some multiple myelomas results from NIK gene amplification (59). The hallmarks for the noncanonical NF-κB pathway are that it is of persistent kinetics and requires de novo protein synthesis (26). In most cells under physiological circumstance, TNF-associated factor 3 binds to N-terminal of NIK to channel NIK to constant ubiquitination and degradation. After cells receive extracellular signaling, the cross-linking of ligand receptor of the TNFR subset triggers the ubiquitination of TNF-associated factor 3 followed by its degradation to stabilize NIK. Accumulated NIK autophosphorylates and stimulates and/or coordinates with IKKα homodimer to phosphorylate the C terminal of p100 and initiate p100 processing into p52. In addition, IKKα-mediated negative feedback of NIK controls the magnitude of NIK activation. Because of the extremely low abundance of NIK in nonstimulated cells, apparently, the stable level and de novo synthesis of NIK appear to be essential for initiation and persistent activation of RelB/NF-κB2. It has been shown that B cell-activating factor (BAFF) receptor (BAFF-R), which comprises the subset of TNFR, was weakly detected in term villous trophoblasts (∼10%) along with a relatively high level of BAFF in villous mesenchymal cells (60). B cell maturation and survival are dependent on BAFF-R-mediated p100 processing. In the human placenta, mesenchymal BAFF cross-linking to BAFF-R in a paracrine manner might partially contribute to activation of p100 processing in trophoblasts, As far as is known to date, all inducers signal the noncanonical NF-κB through NIK. Provided that protein synthesis is essential to the stabilization and accumulation of NIK for the initiation of p100 processing, an alternative mechanism may be that cortisol, which is continuously secreted from the growing fetus, enters the placental circulation to stimulate NIK synthesis, which results in the accumulation of NIK and initiation of p100 processing. In agreement with this postulation, a preliminary GRE motif search against a consensus GRE database (61) revealed that three central components of the noncanonical NF-κB pathway, NIK, NF-κB2, and RelB, all contain putative GRE(s) in their respective gene promoter within 4 kb of the upstream transcription start site (Supplemental Fig. 5). It is obvious that this hypothesis deserves further validation.
Human labor results from interactions between a series of biological regulatory processes that are precisely coordinated in an integrated system (23). On the one hand, such a precise regulation can be exemplified by our observation that only approximately 27% of villous trophoblasts had a nuclear activity of NF-κB in the term placenta. The magnitude of nuclear activity of NF-κB observed in the human placenta may be a consequence of the effects by multiple regulatory processes, which may include, but not limited to the following: IKKα-mediated negative feedback of NIK activation (26) and the progesterone-PR complex-mediated inhibitory and glucocorticoid-GR-mediated stimulatory effects. On the other hand, our results further support that NF-κB signaling, including RelB/NF-κB2, may play an important role in labor onset (23). Given the fact that DNA binding activity is intrinsic to NF-κB heterodimers, genes containing NF-κB response element in their promoter are likely targeted by this signaling pathway in the human placenta. For instance, prostaglandin-endoperoxide synthase-2 (cyclooxygenase-2), which catalyzes synthesis of prostaglandins, harbors two validated NF-κB enhancers in its gene promoter and constitutes one of the NF-κB target genes (62). It is highly likely that, in addition to regulating CRH, the RelB/NF-κB2-containing complex may also act as a regulator on transcription of cyclooxygenase-2 and the production of prostaglandins in the human placenta.
Overall, RelB/NF-κB2 signaling was shown to be critical for the transcription of CRH in the human placenta. Dysregulation of placental CRH is likely associated with preterm birth in humans. Thus, modulating the activity of the noncanonical NF-κB and the subsequent transcription of CRH may present as a potential therapeutic against preterm birth.
Materials and Methods
Sample collection
The placentas were obtained from healthy pregnant women with a gestational age of 37 and 42 wk who received elective cesarean section.
Purification of cytotrophoblasts
Purification of placental cytototrophoblasts was essentially performed as recently described (63). Briefly, after being dissected, minced, and washed with 1× Hanks' balanced salt solution (Invitrogen, Carlsbad, CA) that is free of calcium and magnesium, villous tissue fragments containing no membranes received enzymatic digestions in a solution containing 0.25% trypsin, 0.2% deoxyribonuclease I, 25 mm HEPES, 2 mm CaCl2, and 0.8 mm MgSO4 in 1× Hanks' balanced salt solution at 37 C. To remove tissues that were not digested, we passed them through gauze and a 100-μm sieve. After three rounds of enzymatic digestion, cell pellets from the second and third digestions were pooled and resuspended in DMEM/F-12 with 10% fetal bovine serum (FBS). To further purify cytotrophoblasts, we used a discontinuous density gradient of Percoll (50/45/35/30%) by centrifuging at 1000 × g at room temperature for 20 min. Target cells at the interface of fractions of 35/45% were collected and further immunopurified by an approach of negative selection with use of human CD9 and CD45 antibodies (BD Biosciences, San Diego, CA) and Dynabeads (Invitrogen). Cells in the supernatant that were separated from Dynabeads with contaminated cells were pelleted, resuspended in DMEM/F-12 plus 10% FBS (charcoal stripped), plated at a density of 2.5–3 × 106/cm2, and maintained at 37 C and 5% CO2 for further analysis.
Enzyme-linked immunoassays
Levels of cAMP in culture media were determined with ELISA (Cell Biolabs, San Diego, CA). Media were added to a 96-well plate coated with cytokine antibody, washed, and treated with secondary antibody according to the manufacturer's instructions. Levels of cytokines were derived and normalized to total cell protein.
Immunohistochemistry
Immunohistochemistry was performed at the Anatomical Tissue Services, Cancer Institute of New Jersey (New Brunswick, NJ). Briefly, as soon as the placenta is received, placental tissue samples were fixed in buffered formalin for 24 h, dehydrated in 70% ethanol, paraffin embedded, and sectioned. To improve the demonstration of antigens, we incubated slides with Protease K for 20 min to help uncover the hidden antigens. After incubation with primary antibodies (RelB at 1:100; Santa Cruz Biotechnology, Santa Cruz, CA; NF-κB 2 at 1:400; Cell Signaling, Beverly, MA; p65 at 1:150, Cell Signaling; and CRH at 1:50, Abnova, Walnut, CA), the sections were washed with Tris-buffered saline and Tween 20 and incubated with biotinylated secondary antirabbit or antimouse antibody diluted in Tris-buffered saline. Sections were washed again with Tris-buffered saline and Tween 20 and secondary antibodies were detected by use of a DAB detection kit (Ventana; Roche, Indianapolis, IN).
Northern blotting analysis
Five micrograms of total RNA were resolved on a 12% urea-polyacrylamide gel and transferred to a Hybond-n + membrane (GE Healthcare, Indianapolis, IN). The membrane was dried and UV cross-linked. The blot was blocked with ULTRAhyb-Oligo hybridization buffer (Ambion, Austin, TX) for 1 h and incubated overnight at 42 C with a probe containing the entire human CRH cDNA. The membrane was washed for 10 min at 42 C in 2× saline sodium citrate and 0.1% sodium dodecyl sulfate (SDS) and for 10 min at 42 C in 0.2× saline sodium citrate and 0.1% SDS, exposed and scanned, and quantified using a Storm PhosphorImaging system (Molecular Dynamics, Sunnyvale, CA). The blots were stripped and then incubated with a probe containing human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA sequence for the loading control.
Chromatin immunoprecipitation
Cells were cross-linked with 1% formaldehyde for 5 min at room temperature, and the reaction was stopped by the addition of 1× glycine. Cells were lysed in ChIP lysis buffer (50 mm HEPES-KCl, pH 7.5; 140 mm NaCl; 1 mm EDTA; 1% Triton X-100; 0.1% sodium deoxycholate; 0.1% SDS) with freshly added 1× protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany) and then sonicated to shear chromatin into 200- to 500-bp fragments. Chromatins were immunoprecipitated with ChIP-grade antibodies (RelB; Santa Cruz Biotechnology; NF-κB2; Abcam, Cambridge, MA; C-Rel; Santa Cruz Biotechnology; p50; Cell Signaling; or p65; Cell Signaling) or normal rabbit IgG antibody (Cell Signaling.) incubated with protein G agarose beads (Roche Applied Science). After immunoprecipitation, cross-linking was reversed with the proteins removed by treatment with Proteinase K and DNA was recovered. DNA was purified by phenol/chloroform extraction and ethanol precipitation. Real-time PCR was performed for the CRH promoter region encompassing the putative NF-κB enhancer. PCR products were quantified by using SYBR green dye (QIAGEN, Valencia, CA) on an ABI 7300 real-time PCR machine (Applied Biosystems, Foster City, CA). Fold enrichment was determined by the method of cycle threshold in the formula of 2-ΔΔCT. Primers used for the amplicon to the CRH promoter containing the NF-κB enhancer were: forward, 5′-GGCCTTTCATAGTAAGAGGTCAA-3′; and reverse, 5′-TCTCACATCCAATTATATCAACAGA-3′. Control primers for the nonbinding site (a human DNA α-satellite) were: forward, 5′-TCTCAGAATCTTCCTTTTGATGTG-3′; and reverse, 5′-CCAGTTGCAGATCCTACAAAGA-3′.
DNA constructs
To create DNA constructs of the luciferase reporter, the expression of which is regulated by the NF-κB enhancer of CRH, we linearized pGL4.32 (Promega, Madison, WI) with KpnI and HindIII, gel purified the backbone, and dephosphorylated with calf intestine phosphatase (New England Biolabs, Beverly, MA). Three sets of DNA oligonucleotides, CRH1X (5′-CACTTGGGAAATCTCATTCA-3′ and 5′-AGCTTGAATGAGATTTCCCAAGTGGTAC-3′), CRH4X (5′-CACTTGGGAAATCTCATTCACACTTGGGAAATCTCATTCA CACTTGGGAAATCTCATTCACACTTGGGAAATCTCATTCA-3′), and 5′-AGCTTGAATGAGATTTCCCAAGTGTGAATGAGATTTCCCAAGTGTGAATGAGATTTCCCAAGTGTGAATGAGATTTCCCAAGTGGTAC-3′), and CRH1XMut (5′-CACTTGAGCACTCTCATTCA-3′ and 5′-AGCTTGAATGAGAGTGCTCAAGTGGTAC-3′) were ordered (Integrated DNA Technology, Coralville, IA) and annealed each other to generate three double-stranded DNA fragments that contained cohesive ends that were compatible with sites of KpnI and HindIII at the 5′ and 3′ ends, respectively. These three double-stranded DNA fragments were phosphorylated by T4 DNA kinase (New England Biolabs) and ligated to KpnI/HindIII-linearized pGL4.32 backbone to generate pGL4.32-CRH1X, pGL4.32-CRH4X, and pGL4.32-CRH1XMut, respectively.
Determining transfection efficiency
To determine the transfection efficiency by Lipofectamine 2000 (Invitrogen) in primary cytotrophoblasts, we used a 24-well plate with 500 μl of DMEM/F-12 and 10% FBS and cell density of 4 × 104 in each well. Specifically, for each transfection with siRNA, we first prepared an siRNA-Lipofectamine 2000 complex. We diluted 25 or 37.5 pmol fluorescently labeled nontargeting siRNA (siGLO Red; Thermo Scientific/Dharmacon, Lafayette, CO) as previously described (64) into 50 μl DMEM/F-12 without serum and incubated for 5 min. In the meantime, we incubated 1.5 μl Lipofectamine 2000 in 50 μl DMEM/F-12 without serum for 5 min. Then diluted siGLO and diluted Lipofectamine 2000 were combined with a total volume of 100 μl and incubated for 20 min at room temperature to allow the siRNA-Lipofectamine 2000 complex to form. Each 100 μl of siRNA-Lipofectamine 2000 complex was added into one well to make the final concentration of siRNA at 50 or 75 nm, respectively. After 48 h, cells were fixed in 4% paraformaldehyde for 30 min, counterstained with 4′,6′-diamino-2-phenylindole (Invitrogen) and visualized with an inverted microscope (Carl Zeiss, New York, NY). Wells supplemented with siRNA but without Lipofectamine 2000 (Invitrogen) were used as a negative control. Transfection efficiency (percentage) was determined as the number of cells containing intracellular fluorescence/number of total cells × 100. We have determined that transfection efficiency of siRNA with Lipofectamine 2000 in primary cytotrophoblasts was 74% with cell survival rate 81% (siRNA at 50 nm) and 85% with cell survival rate 78% (siRNA at 75 nm) (Supplemental Fig. 6). In agreement with previous results, 70% or greater of transfection efficiency was deemed as successful (64).
In an alternate approach, we estimated the transfection efficiency of DNA constructs with Lipofectamine 2000 (Invitrogen) by comparing luciferase activity between Hela cells and primary cytotrophoblasts. Ninety-four percent of the transfection efficiency of DNA constructs with Lipofectamine 2000 has been observed in Hela cells (Invitrogen). Briefly, 8 × 104 of Hela cells or primary cytotrophoblasts were seeded in a 24-well plate. Two hundred nanograms of pCMV-RL were added into 50 μl DMEM/F-12 without serum and incubated for 5 min. In the meantime, we incubated 1.5 μl Lipofectamine 2000 in 50 μl DMEM/F-12 without serum for 5 min. Then diluted pCMV-RL and diluted Lipofectamine 2000 were combined with total volume of 100 μl and incubated for 20 min at room temperature to allow the pCMV-RL-Lipofectamine 2000 complex to form. Each 100 μl of the pCMV-RL-Lipofectamine 2000 complex was added into one well. After 24 h, cells were lysed and luciferase activity was measured as described below (dual luciferase assay). We have estimated the transfection efficiency of DNA construct with Lipofectamine 2000 was 85%.
Dual-luciferase assay
Each well of a 24-well plate contained 8 × 104 of primary cytotrophoblasts. Two hundred nanograms of pCMV-RL along with 1 μg of pGL4.32-CRH FL reporter (as specified in Results) were added into 50 μl DMEM/F-12 without serum and incubated for 5 min in the presence or absence of siRNA (as specified in Results). In the meantime, we incubated 1.5 μl Lipofectamine 2000 (Invitrogen) in 50 μl DMEM/F-12 without serum for 5 min. Then diluted DNA constructs and diluted Lipofectamine 2000 were combined with total volume of 100 μl and incubated for 20 min at room temperature to allow the DNA construct-Lipofectamine 2000 complex to form. Each 100 μl of the DNA construct-Lipofectamine 2000 complex was added into one well. Forty-eight hours later, cells were lysed with 1× passive buffer, and dual-luciferase assays were performed according to the manufacturer's protocol (Promega). FL activity was measured by adding LAR I (30 μl) into one well of a 96-well plate and read in Victor3 V (PerkinElmer, Waltham, MA) for 5 sec. Renilla luciferase activity was measured by adding Stop & Glo (30 μl; Promega) to each well and reread for 5 sec.
Western blotting analysis
Cytosolic and nuclear extracts of cells were prepared by use of a nuclear extraction kit according to the manufacturer's instruction (Millipore, Bedford, MA). The total lysates, or extracts, or immunoprecipitates were resuspended in 1× SDS loading buffer, boiled at 95 C for 5 min, and centrifuged. Supernatants were resolved on SDS-10% PAGE and transferred onto polyvinyl difluoride membranes (Bio-Rad Laboratories, Hercules, CA). Membranes were blocked in 5% nonfat milk powder in 10 mm phosphate buffer (pH 7.2), 150 mm NaCl, and 0.1% Tween 20 (PBST) for 60 min, washed twice with PBST, and incubated with antibodies as indicated in 1% nonfat milk powder-PBST at 4 C overnight. The blots were probed with β-actin antibody (Sigma, St. Louis, MO) as the loading control. Membranes were washed three times with PBST, incubated with horseradish peroxidase-conjugated secondary antibodies at 1:5000 in 1% nonfat milk powder-PBST, and developed by Immun-Star HRP substrate (Bio-Rad Laboratories).
Gene silencing
To determine the role of RelB or NF-κB2 in CRH expression in primary cytotrophoblasts, we used predesigned/synthesized FlexTube siRNA (QIAGEN) with experimentally validated effect and sequences of siRNA attached. Freshly prepared human cytotrophoblast cells (3 × 106 cells/well) were plated in six-well-culture plates in 3 ml of DMEM/F12 (Invitrogen) containing 10% FBS. After being maintained in culture for 24 h, each well was transfected with the complex of FlexTube siRNA targeting RelB or NF-κB2 (225 pmol)-Lipofectamine 2000 (3 μl; Invitrogen) as prepared as described above with a final concentration of siRNA at 75 nm. A nontargeting siRNA or scramble RNA was used as a negative control with the sequences of sense strand and antisense strands, 5′-AACAGUCGCGUUGUCGACUGGUU-3′ and 5′-CCAGUCGCAAACGCGACUGUUUU-3′, respectively. At 48 h after exposure to the targeting or nontargeting siRNA, total RNA were isolated from the cells by Trizol (Invitrogen) and analyzed by RT-PCR or whole-cell lysates were used for Western blotting analysis.
Reverse transcription-polymerase chain reaction
Total RNA from primary cytotrophoblasts was extracted by the Trizol method (Invitrogen). Dried RNA pellets were resuspended in appropriate volumes of diethylpyrocarbonate H2O. The RNA was quantitated by OD260/280 using spectrophotometry and treated with deoxyribonuclease I (Promega). Total cDNA synthesis was prepared by the oligodeoxythymidine primer method using the Superscript II reverse transcription kit (Invitrogen). Quantitative RT-PCR was performed using an ABI 7300 machine (Applied Biosystems). RelB, NF-κB2, and CRH mRNA levels were determined with relative to GAPDH expression using the SYBR Green PCR kit (QIAGEN). The following primers were used: RelB forward, 5′-TCCCAACCAGGATGTCTAGC-3′, RelB reverse, 5′-AGCCATGTCCCTTTTCCTCT-3′, NF-κB2 forward, 5′-GAACAGCCTTGCATCTAGCC-3′, NF-κB2 reverse, 5′-TCCCAGTCGCTATCAGAGG-3′, CRH forward, 5′-GCAGTTAGCACAGCAAGCTCAC-3′, CRH reverse, 5′-CAAATGGCATAAGAGCAGCG-3′, and GAPDH forward, 5′-CTCCCGCTTCGCTCTCTG-3′, GAPDH reverse, 5′-CTGGCGACGCAAAAGAAG-3′.
Statistical analysis
One-way ANOVA analysis was used to test significant differences among groups. When the equality hypothesis was rejected (P < 0.05), Dunnett's test was used for comparisons between the control (reference) group and each test group.
Study approval
All patients signed the written informed consent for their specimen to be used for this study in a protocol approved by the Institutional Review Board of University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School.
Acknowledgments
We thank Seth Guller and Zhonghua Tang for their valuable assistance in the purification of cytotrophoblasts and Lei Cong for technical assistance in immunohistochemistry. We also thank Dr. Susan Shen-Schwarz for analyzing the data of immunohistochemical staining.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BAFF
- B cell-activating factor
- BAFF-R
- BAFF receptor
- CHIP
- chromatin immunoprecipitation
- CMV
- cytomegalovirus
- CRE
- cAMP-response element
- DBD
- DNA binding domain
- DEX
- dexamethasone
- FBS
- fetal bovine serum
- FL
- firefly luciferase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GR
- glucocorticoid receptor
- GRE
- glucocorticoid response element
- IκBα
- inhibitory-κB
- IKB
- inhibitory κB
- IKK
- inhibitory-κB kinase-α
- NF-κB
- nuclear factor-κB
- NIK
- NF-κB inducing kinase
- PBST
- phosphate buffer (pH 7.2), NaCl, and Tween 20
- PR
- progesterone receptor
- PSI
- N-benzyloxycarbonyl-Ile-Glu(O-t-butyl)-Ala-leucinal
- SDS
- sodium dodecyl sulfate
- siRNA
- short interfering RNA
- TNFR
- TNF receptor
- Ucn
- urocortin
- ZLL
- 1,2-di-(N-carboxybenzoyl-l-leucyl)amino acetone.
References
- 1. Petraglia F , Imperatore A , Challis JR. 2010. Neuroendocrine mechanisms in pregnancy and parturition. Endocr Rev 31:783–816 [DOI] [PubMed] [Google Scholar]
- 2. Petraglia F , Sawchenko PE , Rivier J , Vale W. 1987. Evidence for local stimulation of ACTH secretion by corticotropin-releasing factor in human placenta. Nature 328:717–719 [DOI] [PubMed] [Google Scholar]
- 3. Spaziani EP , O'Brien WF , Benoit RR , Gould SF. 2000. Corticotropin-releasing hormone increases the expression of the prostaglandin E(2) receptor subtype EP1 in amnion WISH cells. Biol Reprod 62:23–26 [DOI] [PubMed] [Google Scholar]
- 4. Li W , Challis JR. 2005. Corticotropin-releasing hormone and urocortin induce secretion of matrix metalloproteinase-9 (MMP-9) without change in tissue inhibitors of MMP-1 by cultured cells from human placenta and fetal membranes. J Clin Endocrinol Metab 90:6569–6574 [DOI] [PubMed] [Google Scholar]
- 5. Petraglia F , Florio P , Benedetto C , Marozio L , Di Blasio AM , Ticconi C , Piccione E , Luisi S , Genazzani AR , Vale W. 1999. Urocortin stimulates placental adrenocorticotropin and prostaglandin release and myometrial contractility in vitro. J Clin Endocrinol Metab 84:1420–1423 [DOI] [PubMed] [Google Scholar]
- 6. Clifton VL , Owens PC , Robinson PJ , Smith R. 1995. Identification and characterization of a corticotrophin-releasing hormone receptor in human placenta. Eur J Endocrinol 133:591–597 [DOI] [PubMed] [Google Scholar]
- 7. Clifton VL , Read MA , Leitch IM , Giles WB , Boura AL , Robinson PJ , Smith R. 1995. Corticotropin-releasing hormone-induced vasodilatation in the human fetal-placental circulation: involvement of the nitric oxide-cyclic guanosine 3′,5′-monophosphate-mediated pathway. J Clin Endocrinol Metab 80:2888–2893 [DOI] [PubMed] [Google Scholar]
- 8. Keller PA , Kirkwood K , Morgan J , Westcott S , McCluskey A. 2003. The prevention of preterm labour—corticotropin releasing hormone type 1 receptors as a target for drug design and development. Mini Rev Med Chem 3:295–303 [DOI] [PubMed] [Google Scholar]
- 9. McLean M , Bisits A , Davies J , Woods R , Lowry P , Smith R. 1995. A placental clock controlling the length of human pregnancy. Nat Med 1:460–463 [DOI] [PubMed] [Google Scholar]
- 10. Wadhwa PD , Porto M , Garite TJ , Chicz-DeMet A , Sandman CA. 1998. Maternal corticotropin-releasing hormone levels in the early third trimester predict length of gestation in human pregnancy. Am J Obstet Gynecol 179:1079–1085 [DOI] [PubMed] [Google Scholar]
- 11. Hobel CJ , Dunkel-Schetter C , Roesch SC , Castro LC , Arora CP. 1999. Maternal plasma corticotropin-releasing hormone associated with stress at 20 weeks' gestation in pregnancies ending in preterm delivery. Am J Obstet Gynecol 180:S257–S263 [DOI] [PubMed] [Google Scholar]
- 12. Warren WB , Patrick SL , Goland RS. 1992. Elevated maternal plasma corticotropin-releasing hormone levels in pregnancies complicated by preterm labor. Am J Obstet Gynecol 166:1198–1204; discussion 1204–1207 [DOI] [PubMed] [Google Scholar]
- 13. Florio P , Woods RJ , Genazzani AR , Lowry PJ , Petraglia F. 1997. Changes in amniotic fluid immunoreactive corticotropin-releasing factor (CRF) and CRF-binding protein levels in pregnant women at term and during labor. J Clin Endocrinol Metab 82:835–838 [DOI] [PubMed] [Google Scholar]
- 14. Linton EA , Perkins AV , Woods RJ , Eben F , Wolfe CD , Behan DP , Potter E , Vale WW , Lowry PJ. 1993. Corticotropin releasing hormone-binding protein (CRH-BP): plasma levels decrease during the third trimester of normal human pregnancy. J Clin Endocrinol Metab 76:260–262 [DOI] [PubMed] [Google Scholar]
- 15. Perkins AV , Eben F , Wolfe CD , Schulte HM , Linton EA. 1993. Plasma measurements of corticotrophin-releasing hormone-binding protein in normal and abnormal human pregnancy. J Endocrinol 138:149–157 [DOI] [PubMed] [Google Scholar]
- 16. Perkins AV , Wolfe CD , Eben F , Soothill P , Linton EA. 1995. Corticotrophin-releasing hormone-binding protein in human fetal plasma. J Endocrinol 146:395–401 [DOI] [PubMed] [Google Scholar]
- 17. Alvi SA , Brown NL , Bennett PR , Elder MG , Sullivan MH. 1999. Corticotrophin-releasing hormone and platelet-activating factor induce transcription of the type-2 cyclo-oxygenase gene in human fetal membranes. Mol Hum Reprod 5:476–480 [DOI] [PubMed] [Google Scholar]
- 18. Smith R , Mesiano S , Chan EC , Brown S , Jaffe RB. 1998. Corticotropin-releasing hormone directly and preferentially stimulates dehydroepiandrosterone sulfate secretion by human fetal adrenal cortical cells. J Clin Endocrinol Metab 83:2916–2920 [DOI] [PubMed] [Google Scholar]
- 19. Neulen J , Breckwoldt M. 1994. Placental progesterone, prostaglandins and mechanisms leading to initiation of parturition in the human. Exp Clin Endocrinol 102:195–202 [DOI] [PubMed] [Google Scholar]
- 20. Arbiser JL , Morton CC , Bruns GA , Majzoub JA. 1988. Human corticotropin releasing hormone gene is located on the long arm of chromosome 8. Cytogenet Cell Genet 47:113–116 [DOI] [PubMed] [Google Scholar]
- 21. Robinson BG , Emanuel RL , Frim DM , Majzoub JA. 1988. Glucocorticoid stimulates expression of corticotropin-releasing hormone gene in human placenta. Proc Natl Acad Sci USA 85:5244–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karalis K , Goodwin G , Majzoub JA. 1996. Cortisol blockade of progesterone: a possible molecular mechanism involved in the initiation of human labor. Nat Med 2:556–560 [DOI] [PubMed] [Google Scholar]
- 23. Lappas M , Rice GE. 2007. The role and regulation of the nuclear factor κB signalling pathway in human labour. Placenta 28:543–556 [DOI] [PubMed] [Google Scholar]
- 24. Mendelson CR. 2009. Minireview: fetal-maternal hormonal signaling in pregnancy and labor. Mol Endocrinol 23:947–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayden MS , Ghosh S. 2008. Shared principles in NF-κB signaling. Cell 132:344–362 [DOI] [PubMed] [Google Scholar]
- 26. Sun SC. 2011. Non-canonical NF-κB signaling pathway. Cell Res 21:71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vallabhapurapu S , Karin M. 2009. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol 27:693–733 [DOI] [PubMed] [Google Scholar]
- 28. Dejardin E. 2006. The alternative NF-κB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol 72:1161–1179 [DOI] [PubMed] [Google Scholar]
- 29. Kalaitzidis D , Davis RE , Rosenwald A , Staudt LM , Gilmore TD. 2002. The human B-cell lymphoma cell line RC-K8 has multiple genetic alterations that dysregulate the Rel/NF-κB signal transduction pathway. Oncogene 21:8759–8768 [DOI] [PubMed] [Google Scholar]
- 30. Trecca D , Guerrini L , Fracchiolla NS , Pomati M , Baldini L , Maiolo AT , Neri A. 1997. Identification of a tumor-associated mutant form of the NF-κB RelA gene with reduced DNA-binding and transactivating activities. Oncogene 14:791–799 [DOI] [PubMed] [Google Scholar]
- 31. Rosen T , Krikun G , Ma Y , Wang EY , Lockwood CJ , Guller S. 1998. Chronic antagonism of nuclear factor-κB activity in cytotrophoblasts by dexamethasone: a potential mechanism for antiinflammatory action of glucocorticoids in human placenta. J Clin Endocrinol Metab 83:3647–3652 [DOI] [PubMed] [Google Scholar]
- 32. Schrader EK , Harstad KG , Matouschek A. 2009. Targeting proteins for degradation. Nat Chem Biol 5:815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng YH , Nicholson RC , King B , Chan EC , Fitter JT , Smith R. 2000. Corticotropin-releasing hormone gene expression in primary placental cells is modulated by cyclic adenosine 3′,5′-monophosphate. J Clin Endocrinol Metab 85:1239–1244 [DOI] [PubMed] [Google Scholar]
- 34. Betts JC , Nabel GJ. 1996. Differential regulation of NF-κB2(p100) processing and control by amino-terminal sequences. Mol Cell Biol 16:6363–6371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leitch IM , Boura AL , Botti C , Read MA , Walters WA , Smith R. 1998. Vasodilator actions of urocortin and related peptides in the human perfused placenta in vitro. J Clin Endocrinol Metab 83:4510–4513 [DOI] [PubMed] [Google Scholar]
- 36. Riley SC , Walton JC , Herlick JM , Challis JR. 1991. The localization and distribution of corticotropin-releasing hormone in the human placenta and fetal membranes throughout gestation. J Clin Endocrinol Metab 72:1001–1007 [DOI] [PubMed] [Google Scholar]
- 37. Natoli G , Saccani S , Bosisio D , Marazzi I. 2005. Interactions of NF-κB with chromatin: the art of being at the right place at the right time. Nat Immunol 6:439–445 [DOI] [PubMed] [Google Scholar]
- 38. Sandelin A , Wasserman WW , Lenhard B. 2004. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res 32:W249–W252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nicholson RC , King BR , Smith R. 2004. Complex regulatory interactions control CRH gene expression. Front Biosci 9:32–39 [DOI] [PubMed] [Google Scholar]
- 40. Fusco AJ , Huang DB , Miller D , Wang VY , Vu D , Ghosh G. 2009. NF-κB p52:RelB heterodimer recognizes two classes of κB sites with two distinct modes. EMBO Rep 10:152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Castro-Caldas M , Mendes AF , Carvalho AP , Duarte CB , Lopes MC. 2003. Dexamethasone prevents interleukin-1β-induced nuclear factor-κB activation by upregulating IκB-α synthesis, in lymphoblastic cells. Mediators Inflamm 12:37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kliman HJ , Nestler JE , Sermasi E , Sanger JM , Strauss JF. 1986. Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology 118:1567–1582 [DOI] [PubMed] [Google Scholar]
- 43. Barnes PJ , Karin M. 1997. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 336:1066–1071 [DOI] [PubMed] [Google Scholar]
- 44. Van Waes C. 2007. Nuclear factor-κB in development, prevention, and therapy of cancer. Clin Cancer Res 13:1076–1082 [DOI] [PubMed] [Google Scholar]
- 45. Liu LP , Xia YF , Yang L , DiDonato JA , DiCorleto PE , Zhong CP , Geng JG. 2001. B lymphocytes and plasma cells express functional E-selectin by constitutive activation of NF-κB. Biochem Biophys Res Commun 286:281–291 [DOI] [PubMed] [Google Scholar]
- 46. Kaltschmidt C , Kaltschmidt B , Neumann H , Wekerle H , Baeuerle PA. 1994. Constitutive NF-κB activity in neurons. Mol Cell Biol 14:3981–3992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meffert MK , Baltimore D. 2005. Physiological functions for brain NF-κB. Trends Neurosci 28:37–43 [DOI] [PubMed] [Google Scholar]
- 48. Auphan N , DiDonato JA , Rosette C , Helmberg A , Karin M. 1995. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science 270:286–290 [DOI] [PubMed] [Google Scholar]
- 49. Ray A , Prefontaine KE. 1994. Physical association and functional antagonism between the p65 subunit of transcription factor NF-κB and the glucocorticoid receptor. Proc Natl Acad Sci USA 91:752–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bamberger CM , Schulte HM , Chrousos GP. 1996. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev 17:245–261 [DOI] [PubMed] [Google Scholar]
- 51. Padayachi T , Pegoraro RJ , Rom L , Joubert SM. 1990. Enzyme immunoassay of oestrogen and progesterone receptors in uterine and intrauterine tissue during human pregnancy and labour. J Steroid Biochem Mol Biol 37:509–511 [DOI] [PubMed] [Google Scholar]
- 52. McCormick PD , Razel AJ , Spelsberg TC , Coulam CB. 1981. Absence of high-affinity binding of progesterone (R 5020) in human placenta and fetal membranes. Placenta Suppl 3:123–132 [PubMed] [Google Scholar]
- 53. Shanker YG , Rao AJ. 1999. Progesterone receptor expression in the human placenta. Mol Hum Reprod 5:481–486 [DOI] [PubMed] [Google Scholar]
- 54. Cudeville C , Mondon F , Robert B , Rebourcet R , Mignot TM , Benassayag C , Ferré F. 2000. Evidence for progesterone receptors in the human fetoplacental vascular tree. Biol Reprod 62:759–765 [DOI] [PubMed] [Google Scholar]
- 55. Taylor AH , McParland PC , Taylor DJ , Bell SC. 2009. The cytoplasmic 60 kDa progesterone receptor isoform predominates in the human amniochorion and placenta at term. Reprod Biol Endocrinol 7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wei LL , Hawkins P , Baker C , Norris B , Sheridan PL , Quinn PG. 1996. An amino-terminal truncated progesterone receptor isoform, PRc, enhances progestin-induced transcriptional activity. Mol Endocrinol 10:1379–1387 [DOI] [PubMed] [Google Scholar]
- 57. Bain DL , Heneghan AF , Connaghan-Jones KD , Miura MT. 2007. Nuclear receptor structure: implications for function. Annu Rev Physiol 69:201–220 [DOI] [PubMed] [Google Scholar]
- 58. Kalkhoven E , Wissink S , van der Saag PT , van der Burg B. 1996. Negative interaction between the RelA(p65) subunit of NF-κB and the progesterone receptor. J Biol Chem 271:6217–6224 [DOI] [PubMed] [Google Scholar]
- 59. Annunziata CM , Davis RE , Demchenko Y , Bellamy W , Gabrea A , Zhan F , Lenz G , Hanamura I , Wright G , Xiao W , Dave S , Hurt EM , Tan B , Zhao H , Stephens O , Santra M , Williams DR , Dang L , Barlogie B , Shaughnessy JD , Kuehl WM , Staudt LM. 2007. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 12:115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Langat DL , Wheaton DA , Platt JS , Sifers T , Hunt JS. 2008. Signaling pathways for B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL) in human placenta. Am J Pathol 172:1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Datson NA , Polman JA , de Jonge RT , van Boheemen PT , van Maanen EM , Welten J , McEwen BS , Meiland HC , Meijer OC. 2011. Specific regulatory motifs predict glucocorticoid responsiveness of hippocampal gene expression. Endocrinology 152:3749–3757 [DOI] [PubMed] [Google Scholar]
- 62. Deng WG , Zhu Y , Wu KK. 2003. Up-regulation of p300 binding and p50 acetylation in tumor necrosis factor-α-induced cyclooxygenase-2 promoter activation. J Biol Chem 278:4770–4777 [DOI] [PubMed] [Google Scholar]
- 63. Tang Z , Tadesse S , Norwitz E , Mor G , Abrahams VM , Guller S. 2011. Isolation of hofbauer cells from human term placentas with high yield and purity. Am J Reprod Immunol 66:336–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Forbes K , Desforges M , Garside R , Aplin JD , Westwood M. 2009. Methods for siRNA-mediated reduction of mRNA and protein expression in human placental explants, isolated primary cells and cell lines. Placenta 30:124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]