

Abstract
The glucocorticoid-induced leucine zipper (Tsc22d3-2) is a widely expressed dexamethasone-induced transcript that has been proposed to be important in immunity, adipogenesis, and renal sodium handling based on in vitro studies. To address its function in vivo, we have used Cre/loxP technology to generate mice deficient for Tsc22d3-2. Male knockout mice were viable but surprisingly did not show any major deficiencies in immunological processes or inflammatory responses. Tsc22d3-2 knockout mice adapted to a sodium-deprived diet and to water deprivation conditions but developed a subtle deficiency in renal sodium and water handling. Moreover, the affected animals developed a mild metabolic phenotype evident by a reduction in weight from 6 months of age, mild hyperinsulinemia, and resistance to a high-fat diet. Tsc22d3-2-deficient males were infertile and exhibited severe testis dysplasia from postnatal d 10 onward with increases in apoptotic cells within seminiferous tubules, an increased number of Leydig cells, and significantly elevated FSH and testosterone levels. Thus, our analysis of the Tsc22d3-2-deficient mice demonstrated a previously uncharacterized function of glucocorticoid-induced leucine zipper protein in testis development.
Glucocorticoids are involved in the physiological regulation of a variety of processes including immune responses, metabolism, cell growth, and development. Due to their antiinflammatory and immunosuppressive roles, they are widely used in the clinic to treat inflammation, allergy, or malignancies (reviewed in Ayroldi and Riccardi, Ref. 1). The X-linked glucocorticoid-induced leucine zipper (GILZ, Tsc22d3-2) was originally identified as a dexamethasone-induced transcript protecting T lymphocytes from T cell receptor (TCR)/CD3-activated cell death (2). TSC22D3-2 encodes a new member of the TSC22-domain leucine zipper family and is expressed in a variety of different organs and tissues. Members of this family (TSC22D1 to TSC22D4) share a highly conserved TSC (tuberous sclerosis complex) and a PDZ (postsynaptic density protein; PSD95) box domain (reviewed in Ayroldi and Riccardi, Ref. 1). TSC22D3-2 homo- or heterodimerizes by means of its leucine zipper domain (3). In total, four isoforms have been characterized as splice variants of the Tsc22d3-2 gene, named GILZ1–4 (4). Although these four isoforms are present with varying abundance in mouse and rat tissues, they are not functionally redundant. They are involved in distinct aspects of cellular physiology and may therefore modulate different signaling pathways (4). Moreover, due to multiple protein interactions in a variety of cell types, TSC22D3-2 has been implicated not only in apoptosis and cell proliferation but also in the modulation of T lymphocyte activation and IL-2 production (5–7) and in dendritic cell function (8, 9). Furthermore, TSC22D3-2 was shown to inhibit Ras-induced cell proliferation (6) and to mediate renal sodium transport (10, 11) or adipogenesis (12).
In the present report, we have generated a mouse model constitutively lacking all main isoforms of TSC22D3-2 (4). Male mice lacking TSC22D3-2 are viable and show no abnormalities in the immune system, in adipogenesis, or in sodium reabsorption. However, mating and histological analyses revealed that loss of TSC22D3-2 leads to male sterility.
Materials and Methods
Animals
All animals were housed in a controlled environment with a 14-h light (0500–1900 h), 10-h dark cycle with free access to water and a standard laboratory diet. Males were aged from 3–6 months and were backcrossed to C57BL/6N mice (N4-N6). The control group consisted of age-matched Tsc22d3-2+/y and Tsc22d3-2lox/y littermates. Mouse experiments were conducted under the approval of local authorities and followed Swiss guidelines.
Generation of conditional and null mutant Tsc22d3-2 mice
The Tsc22d3-2 gene (NM_010286.3) was cloned from a 129/Sv mouse genomic BAC library (Incite Genomics, Inc., Wilmington, DE). The following fragments were then subcloned into a modified lox-targeting vector containing two loxP and two frt sites. First, a 10-kb (5′ region, containing exon 3) and a 5-kb EcoRI fragment (3′ region, containing exons 4–6), were subcloned into pBSII KS(−) and extracted using PacI and StuI restriction enzyme recognition sites. The neomycin resistance gene cassette was flanked by frt sites and followed by one loxP site (pAT-FRT-K13) (13) and cloned into an EcoRV site (3′ of the polyadenylation sequences). The second loxP sequence was introduced in a SmaI restriction site created by PCR-based mutagenesis, along with a new diagnostic EcoRV restriction site 5′ of the third exon. Finally, the thymidine kinase cassette (HSV-tk) was inserted 3′ of the homologous region. Additional details of cloning are available on request. The targeting vector was linearized with PacI and transfected into A2 embryonic stem (ES) cells of the 129Sv/EV background (14, 15) as described previously (16). G418 and ganciclovir-resistant colonies were expanded and screened by PCR using the following primers: 5′ recombination, sense 5′-ATAGCCTGTGCTCTGGAACT-3′ and antisense 5′-TTATGGCGCGGGGATATCTA-3′, and 3′ recombination, sense 5′-GCCTCCGAGGTTGCAGTGTTT-3′ and antisense 5′-TCGCCTTCTTGACGAGTTCTTC-3′. Targeted clones were confirmed by Southern blot analysis using two external probes (3′ probe: a 315-bp fragment isolated from the 3′ EcoRI clone was isolated by StuI and BglII and used as a probe on EcoRI-digested genomic DNA, and 5′ probe: a 1085-bp fragment isolated from the 5′ EcoRI clone by DraI was used on EcoRV-digested genomic DNA). PCR-amplified neomycin sequences were used as an internal probe. Correctly targeted clones were injected into C57BL/6N blastocysts as described (17). Breeding of Tsc22d3-2loxneo mice with Flp mice (18) allowed the excision of the neomycin cassette and with nestin-CRE mice (19) to generate mice harboring the Δ allele. To obtain knockout males (Tsc22d3-2Δ/y), heterozygous mutant Tsc22d3-2Δ/+ females were crossed with wild-type males. Tsc22d3-2Δ/+ and Tsc22d3-2+/+ females as well as Tsc22d3-2+/y and Tsc22d3-2Δ/y males were genotyped by PCR [sense (s1), 5′-CAGGTCTGAGTAACTTGTCC-3′; antisense (as), 5′-CAGTCTGTGGTGACCGTTTC-3′; and sense (s2), 5′-TGACAGCTGCGTTTCTCAGTG-3′]; s1, s2, and as were used for genotyping of lox and wild-type alleles, and s1 and as for the Δ allele. For homologous recombination of the Tsc22d3-2 gene, the targeting vector was electroporated into ES cells, and five independent correctly targeted clones were obtained. Supplemental Fig. 1B (published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) shows representative Southern blot analyses from targeted ES cell clones digested with EcoRV (5′ probe) and EcoRI (3′ probe) that revealed diagnostic fragments of 7.8 and 2 kb, respectively. Three recombined ES cell clones were injected into blastocysts of C57BL/6N mice. Chimeric mice that transmitted the floxed allele to their offspring (Tsc22d3-2loxneo/+; Supplemental Fig. 1C) were further crossed with the Flp-deleter mice to obtain mice harboring a floxed (Tsc22d3-2lox/+) allele (Supplemental Fig. 1D). After breeding Tsc22d3-2loxneo/+ mice with the germline deleter strain Nestin-Cre (19), we obtained a Tsc22d3-2 allele lacking exons 3–6 (Tsc22d3-2Δ; Supplemental Fig. 1D). Supplemental Fig. 1E illustrates Tsd22d3-2 expression from the Tsc22d3-2lox allele, whereas the Tsc22d3-2Δ allele did not show any expression (Supplemental Fig. 2).
Breeding strategy
Because the Tsc22d3-2 gene is X-linked, and males were sterile, we did not obtain homozygous mutant females. For all additional experiments, to obtain male Tsc22d3-2 knockout animals, we crossed heterozygous mutant females to wild-type males to get age-matched knockout and littermate controls.
Histology and immunohistochemistry
Mice were dissected and organs fixed with buffered 10% formalin (pH 7.2) for 12 h (see also Supplemental Fig. 3). Tissues were embedded in paraffin (Leica Microsystems, Wetzlar, Germany) and 2- to 3-mm sections stained with hematoxylin and eosin (H&E). Testis cellular proliferation was assessed by Ki-67 antibody (monoclonal rat antimouse; Dako M7249, Carpinteria, CA; dilution 1:50) with goat antirat horseradish peroxidase-conjugated secondary antibody (Biosource ALI 3404, Camarillo, CA). Apoptosis was analyzed using terminal transferase and biotin-16-dUTP [terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) enzyme, Roche 11 767 305 001, dilution 1:250, and biotin-16-dUTP, dilution 1:250, Roche 11 093 070 910; Roche Diagnostics GmbH, Mannheim, Germany]. Sertoli and Leydig cells were labeled with the rabbit anti-GATA4 (GATA-binding protein 4) (ab84593; Abcam plc, Cambridge, UK; dilution 1:140) with EnVision (DakoCytomation, Glostrup, Denmark) used as secondary antibody (20). Pancreas sections were incubated with antibodies against insulin (Linco 4011-01; Linco Research Inc., St. Charles, MO; dilution 1:6000) and glucagon (Linco 4030-01F, Linco; dilution 1:6000) and then revealed with Fuchsin (Kit DAKO K0625; Dako, Glostrup, Denmark; dilution 1:50) and diaminobenzidine (Kit DAKO K3468; Dako; dilution 1:50), respectively. GILZ1 and GILZ2 proteins were labeled with the polyclonal TSC22D3-2 antibody as described (4).
Quantitative real-time PCR
Tissues were collected and quickly snap-frozen in liquid nitrogen and kept at −80 C until use. Total RNA was extracted from tissues with the RNeasy Mini Kit or QIAzol (QIAGEN, Valencia, CA). Total RNA (1 μg) was subjected to reverse transcription using SuperScript II (Invitrogen/Life Technologies Corp., Carlsbad, CA) following the manufacturer's instructions. The resulting cDNA was used as a template for quantitative PCR. Results were normalized using qRT-PCR for glyceraldehyde 3-phosphate dehydrogenase or RNA polymerase II. Quantitative real-time analysis of selected genes was performed using the ABI Prism 7500 Fast Real-Time PCR System with SYBR Green as DNA-binding dye for the detection of PCR products or the TaqMan probe technology (Applied Biosystems/Life Technologies Corp., Carlsbad, CA). Intron-spanning primers were designed (Microsynth AG, Balgach, Switzerland) (see supplemental data). The cycling conditions were 95 C for 10 min followed by 40 cycles of 95 C for 15 sec and 60 C for 1 min. To detect and eliminate possible primer-dimer artifacts, the dissociation curve was generated by adding a cycle of 95 C for 15 sec, 60 C for 1 min, and 95 C for 15 sec. All primer sets produced amplicons of the expected size, and their identity was also verified by migration of the PCR products on agarose gel. Quantification was done by measuring the ΔΔCt (2(-Δ Δ C(T)) normalized to the reference gene. All measurements were done at least in triplicate, with n = 3 mice per group, and data represent average ± sem.
Western blot analyses
Tissue samples were collected from dexamethasone-treated (55 μg/100 g body weight) and nontreated control and knockout mice. The dexamethasone was administered ip 3–4 h before euthanasia. Tissue lysis was performed in ice-cold urea (8 m) using the TissueLyser (QIAGEN). Protein levels were quantified using the Pierce BCA protein assay reagent (Thermo Fisher Scientific, Waltham, MA) with BSA as a standard. Equal amounts of protein extracts were resolved by 12% SDS-PAGE and electrotransferred onto a Protran nitrocellulose membrane (Whatman/GE Healthcare, Piscataway, NJ). Blots were blocked in Tris-buffered saline containing 0.1% Tween supplemented with 4% milk powder and immunoblotted using anti-TSC22D3-2 antibody, as described previously (4). The blots were stripped and reprobed with glyceraldehyde 3-phosphate dehydrogenase antibody (Chemicon/Millipore, Billerica, MA) as a loading control; n = 2 mice per genotype.
Hormone measurements and metabolic parameters
Plasma aldosterone, corticosterone, and testosterone levels were assayed using commercially available kits as described previously (21–23). To avoid stress-induced increases in plasma hormone levels, animals were trained in a 1- to 2-wk period with handling (habituation) before euthanasia. For plasma aldosterone and corticosterone measurements, mice (n ≥ 7, 3–4 months old) were killed by rapid decapitation between 1600 and 1800 h. Blood was collected in EDTA tubes, immediately centrifuged, and stored at −20 C until assayed for hormone level determination. Plasma aldosterone levels were measured according to standard procedure using the Coat-A-Count RIA kit (Siemens Medical Solutions Diagnostics, Ballerup, Denmark). Mice samples exhibiting values greater than 1200 pg/ml were previously diluted using a serum pool with a low aldosterone concentration (<50 pg/ml). Corticosterone levels were measured by RIA (IBL, Hamburg, Germany), the rat FSH immunoradiometric assay kit (no. AHR004) and the rat LH RIA (no. AHR002) were obtained from IDS (Lüttich, Belgium). Total testosterone levels in nonextracted serum were assayed using a kit from ICN Biomedicals, Inc., Costa Mesa, CA (catalog item 07-189102; 100 tubes; Testo DA Kit, now MP Biomedicals, Eschwege, Germany). The standard curves ranged between 2 and 200 ng/ml for FSH, 0.25 and 15 ng/ml for LH, and 0.1 and 10 ng/ml for testosterone. Intra- and interassay coefficients of variation of all three assays were less than 5 and 10%, respectively. Body weight (weekly) and body composition on individual 2.5- and 15-month-old mice were analyzed as described (see Supplemental Fig. 4 and Supplemental Table 1). Plasma glucose and insulin levels were determined, and glucose and insulin tolerance tests were determined as described (see also Supplemental Table 2).
Metabolic parameters
Body weight was measured on a weekly basis over the whole study. Body composition was analyzed on individual 2.5- and 15-month-old mice under light gas anesthesia (1–2% isoflurane), by quantitative nuclear magnetic resonance using an EchoMRI whole body composition analyzer (EchoMedical Systems, Houston, TX). Data for individual mice were obtained by averaging results from two consecutive measurements. Plasma glucose levels in the fed, 15-h fasted or 6-h refed states were measured with a glucometer (Ascensia Breeze2; Bayer, Zürich, Switzerland). Plasma insulin levels were determined from tail-tip bleedings by ELISA (ultrasensitive mouse insulin ELISA; Mercodia AB, Uppsala, Sweden).
Glucose tolerance test
After a 15-h overnight fasting period, 6-month-old mice were injected ip with 1.5 mg/g glucose. Plasma glucose levels were measured 30 min before and at several precise timepoints during 120 min after administration (n ≥ 6 per group).
Insulin tolerance test
After 4 h of food removal (0900–1300 h), age-matched 7-month-old mice were injected ip with 0.8 mU/g insulin. Blood samples were obtained from tail-tip bleedings, and plasma glucose levels were measured with a glucometer (Ascensia Breeze 2; Bayer, Switzerland).
Adipocyte differentiation
Mouse embryonic fibroblasts (MEF) were generated as described (24–26) from 13.5-d-old embryos obtained from heterozygous mutant female Tsc22d3-2+/Δ mated to wild-type males. Briefly, after removal of head and visceral organs (used for genotyping), embryos were minced and trypsinized for 30 min at 37 C. Embryonic fibroblasts were then plated and maintained in DMEM [with 10% fetal calf serum (FCS) (Life Technologies), 100 U/ml penicillin, and 100 μg/ml streptomycin] at 37 C in an atmosphere of 5% CO2. All experiments were performed with Tsc22d3-2 wild-type and knockout MEF after 15–20 passages. For adipocyte differentiation, 2-d-postconfluent cells (d 0) were transferred to DMEM supplemented with 10% FCS, 0.5 mm 3-isobutyl-1-methylxanthine, 1 μm dexamethasone, and 1 μg/ml insulin (all from Sigma-Aldrich, Buchs, Switzerland) and 1 μm rosiglitazone (Alexis Biochemicals, San Diego, CA) for 2 d. Medium was renewed every 2 d with DMEM containing 10% FCS, 1 μg/ml insulin, and 1 μm rosiglitazone. To visualize lipid accumulation, cells were stained on d 8 with Oil Red O (27). Briefly, cells were washed with PBS, fixed with 3.7% formaldehyde solution for 1 h, and stained with Oil Red O for 1 h using a 60:40 (vol/vol) dilution in water of a 0.5% stock solution (in isopropanol). Cells were then washed twice with PBS and twice with water. MEF were pooled from n = 2 per genotype. Experiments were performed in triplicate.
Flow cytometry and monoclonal antibodies
Thymocyte, lymph node, and spleen single-cell suspensions were prepared by standard methods and stained for fluorescence-activated cell sorting (FACS) analysis as described. Briefly, thymocyte, lymph node, and spleen single-cell suspensions were preincubated in 50% antimouse FcR (CD16/32; clone 2.4.G.2) culture supernatant and then stained with the following monoclonal antibody conjugates: CD4 (RM-4.5 or GK1.5)-fluorescein isothiocyanate (FITC), -PE (phycoerythrine)-Cy5, or -PE-Cy7; CD8a (53.6.7)-FITC or Alexa 647; CD11b (M1/70)-FITC, PE-Cy5 or Alexa 647; CD24 (M1/69)-PE; CD25 (PC61.5)-Alexa 700 or APC-Alexa 750; CD41 (MWReg30)-FITC or -PE; CD44 (IM781)-PE-Cy7 or Pacific Blue; CD45R/B220 (RA3-6B2)-FITC, PE-Texas Red, or PE-Cy7; CD62L (Mel14)-FITC; CD71 (R17217)-PE; CD122 (5H4)-PE; CD117 (2B8)-APC (allophycocyanine); F4/80 (BM8)-APC Alexa750, Gr1 (RB6-8C5)-FITC, Alexa 647 or Alexa 700; Ter119-FITC or APC-Alexa 750; TCRb (H57)-FITC, PE-Cy5, or APC-Alexa750; TCRgd (GL3)-FITC or PE-Cy5. DN1 (CD117+CD44+CD25−), DN2 (CD117+CD44+CD25+), DN3 (CD117−CD44−CD25+), DN4 (CD117−CD44−CD25−), ISP (CD4−CD8+TCRb−), gdT cells, double positive (CD4+CD8+), CD8 single positive (CD4−CD8+), and CD4 single positive (CD4−CD8+) were labeled. All FITC conjugates were purified and conjugated in the Ludwig Institute with the exception of CD41-FITC, which was purchased from BD Biosciences (San Jose, CA). All Alexa 647 conjugates were prepared using the Alexa 647 conjugation kit from Invitrogen (Carlsbad, CA). All other monoclonal antibody conjugates were purchased from eBioscience (San Diego, CA). Intracellular staining for TCRb was performed after first surface staining with all monoclonal antibodies. After fixation and permeabilization in Cytofix/Cytoperm (BD Biosciences), the cells were incubated overnight at 4 C in 1× Permwash solution (BD Biosciences) containing TCRb-PE-Cy5 and then washed in 1× Permwash and resuspended in PBS/3% FCS for FACS analysis. Samples were analyzed on either a FACS Canto or a FACS LSR II (both from Becton Dickinson, San Jose, CA) and the data analyzed with FlowJo software (TreeStar, Ashland, OR).
Inflammation experiments
Three-month-old mice were killed (n = 3, each group) and bone marrow-derived dendritic cells (BMDC), bone marrow-derived macrophages (BMDM), and thioglycolate-elicited peritoneal macrophages were prepared and stimulated as previously described (28–32). Briefly, lipopolysaccharide (LPS), MSU (monosodium urate), ATP, Candida, and CpG stimuli were obtained from InvivoGen (San Diego, CA), and ELISA kits were from R&D Systems (Minneapolis, MN). BMDM were obtained as previously described (33, 34) and cultured in IMDM (Iscove's Modified Dulbecco's Medium) containing 10% FCS (Sigma-Aldrich), 100 IU/ml penicillin, 100 μg/ml streptomycin, and 50 μm 2-mercaptoethanol. Cells were preincubated for 1 h with dexamethasone (Sigma-Aldrich) before stimulation with 10 ng/ml Salmonella minnesota Ultra Pure LPS (List Biological Laboratories, Campbell, CA), 10 μg/ml Pam3CSK4 lipopeptide (EMC Microcollections, Tübingen, Germany), 5 × 107 heat-inactivated Escherichia coli J5 or 0.1 mm CpG oligodeoxynucleotide (CpG ODN; Coley Pharmaceutical Group, Wellesley, MA). Cell culture supernatants were collected to measure the concentration of IL-12p40 by ELISA (BD Biosciences), and the concentration of TNF and IL-6 by bioassay (35). Splenocytes (5 × 105 cells in 200 μl) were seeded in 96-well plates coated with 1 μg/ml anti-CD3 and anti-CD28 antibodies (BD Biosciences) or stimulated with 2.5 μg/ml concanavalin A (Sigma-Aldrich) in the presence or absence of dexamethasone. IL-2 levels in cell culture supernatants collected after 24 h were measured by ELISA (BD Biosciences).
Sodium-restriction diet and water deprivation
Mice (20–24 wk old) were fed with a standard salt diet (0.17% sodium; ssniff Spezialdiäten GmbH, Soest, Germany) followed by 12 d with a sodium-deficient diet (<0.01% sodium; ssniff Spezialdiäten). The 23-h water deprivation was performed under a normal-salt diet. Body weight and food and water intake were measured during the experiment. Spot urine and serum/blood samples were collected before and after each experiment, and osmolarity as well as sodium, potassium, and creatinine composition were analyzed at the Laboratory of Clinical Chemistry of the University Hospital (Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland).
Statistical analysis
Data were analyzed using the Student's t test for comparison between the groups. Results are expressed as the mean ± sem. A P value <0.05 was considered as statistically significant.
Results
Generation of Tsc22d3-2 knockout mice
We generated mice with a Tsc22d3-2 knockout allele (Supplemental Fig. 1). When heterozygous mutant females were bred to wild-type males, we did not observe any abnormalities in litter size. We obtained 7.7 ± 2.5 pups (n = 20 litters), which was not different in comparison with other parallel breeding settings done at the same time in our animal facility. From such matings, deficient male mice (Tsc22d3-2Δ/y) were born according to the expected Mendelian distribution (male y/+ crossed with female Δ/+; from a total of 181 offspring analyzed: males, 47 +/+ and 52 Δ/+; females, 41 +/+ and 41 Δ/+). The heterozygous mutant females were mated at the age of about 10 wk and kept up to 6 months, and up to six consecutive litters were registered. In contrast, when three mating cages were set with one knockout male (3–6 months old) and two C57BL/6N females (8–12 wk, replaced every 3 wk), we never observed pregnancies and/or born litters. This strongly suggested a male sterility problem. In wild-type mice, levels of Tsc22d3-2 were highest in kidney, brain, lung, and heart, with moderate expression in thymus, liver, and skin and relatively low levels in testis and spleen (Supplemental Fig. 2A). In knockout mice, Tsc22d3-2 mRNA and protein expression was lacking (Supplemental Fig. 2B). The GILZ1 isoform is normally present in thymus, spleen, and lung, whereas GILZ2 is detected in testis, liver, skin, and brain; both of these isoforms were missing in the knockout (Supplemental Fig. 2C). This demonstrates that the Tsc22d3-2 gene locus was efficiently deleted in Tsc22d3-2Δ/y mice, resulting in the absence of the major GILZ isoforms.
Tsc22d3-2Δ/y mice show mild metabolic alterations with age
Knockout mice were born with normal body weight and gained weight normally until about 6 months of age but then slowed significantly (Fig. 1A and Supplemental Table 1; P < 0.05). Body length and body fluids were unaltered, but fat values were significantly reduced in old knockout mice (Supplemental Table 1; P < 0.05). Plasma insulin and glucose levels measured in knockout and control mice kept under fed, fasted, and refed states revealed a significantly higher plasma insulin level upon normal feeding (Supplemental Table 2; P < 0.05), whereas plasma glucose levels were unchanged. Upon fasted and refed conditions, both groups exhibited no significant changes in plasma insulin levels (Supplemental Table 2) or impairment in the glucose and insulin tolerance test, although recovery of plasma glucose concentration was slowed (Fig. 1, B and C). Interestingly, pancreas sections stained for insulin and glucagon levels revealed significantly more glucagon-secreting α-cells in the knockout group per islet despite unchanged plasma glucagon levels (Fig. 1, D and E; P < 0.01). After 18 wk on a high-fat diet, knockout mice showed significantly less weight and body fat content (Echo MRI analysis) (Fig. 1G and Supplemental Table 1). Insulin levels were similar in all groups, with the exception of higher insulin levels in the knockout group fed with a normal diet (Supplemental Table 2 and Fig. 1G). Interestingly, after a high-fat diet, liver histology revealed that the knockout mice appear protected from developing hepatic steatosis (Fig. 1H).
Fig. 1.

Tsc22d3-2Δ/y mice show reduction in weight, mild hyperinsulinemia, and resistance to high-fat diet. Panel A, Body weight values of control mice (●) and knockout mice (○; n ≥ 8 per group) *, P < 0.05. Panels B and C, Glucose tolerance test (GTT) (1.5 mg/g body weight) (panel B) and insulin tolerance test (ITT) (0.8 mU/g) (panel C) (n ≥ 6 per group). Panel D, Glucagon (brown, α cells) and insulin (rose, β cells) expression in pancreatic islets of Langerhans from control (left) and knockout (right) mice; (n = 3 mice per group; bar, 50 mm). Panel E, Ratio of glucagon-positive Langerhans cells per islet (left) and plasma glucagon levels (right) (n ≥ 15 images; n = 3 mice per group) in control (black bars) and knockout mice (white bars). **, P < 0.001. F, Body weight values of control (●) and knockout mice after 18 wk of a high-fat diet (○) (n ≥ 11 per group). Panel G, Plasma insulin concentration in control and knockout mice upon normal-chow (black, control; white, knockout) and high-fat (gray, control; red, knockout) diet in fed, fasted, and refed conditions (n ≥ 10 per group). Panel H, Representative pictures of H&E-stained liver sections from control (left) and knockout (right) mice upon chow (top) and high-fat diet (bottom) after 18 consecutive weeks (n = 3 mice per group; bar, 100 mm). Panels I and J, Two days after confluence, MEF cells were induced (D, differentiated) or not (ND, not differentiated) to differentiate into adipocytes; overview (panel I) and higher-magnification view (panel J) of Oil Red O-stained cells at 8 d after induction. Panels K and L, Quantitative RT-PCR of Tsc22d3-2, Pparγ2, Pparγ, and Fabp4 from nondifferentiated (ND) and differentiated (D) MEF cells from control and knockout mice (panel K) and Pparγ2, Pparγ, Tsc22d1, adiponectin, aP2 (Fabp4), and Klf15 in fat tissues isolated from control (black bars) and knockout (white bars) (n = 3 animals) (panel L). Experiments were performed in triplicate. *, P < 0.05; ***, P < 0.001. ctrl, Control; ko, knockout.
We next exposed MEF to an adipogenic cocktail. After Oil Red O staining, the percentage of MEF differentiating into adipocytes was similar in the knockout and control (Fig. 1, I and J). In parallel, quantitative RT-PCR measurements revealed no difference in basal Pparγ2 mRNA expression in nondifferentiated MEF, whereas the basal fatty acid-binding protein 4 (Fabp4, aP2) mRNA transcript expression level was already significantly increased in the knockout mice (Fig. 1K). Upon differentiation into adipocytes, Pparγ2 and Fabp4 mRNA transcript expression levels were increased in both groups and significantly elevated in the knockout group (Fig. 1K). In the controls, Tsc22d3-2 (Gilz) mRNA transcript expression level was increased 4-fold upon differentiation (Fig. 1K). Quantitative RT-PCR analyses on adipose tissue from control and knockout mice revealed no differences in the expression of Pparγ, Pparγ2, Tsc22d1, adiponectin, Fabp, and Krüppel-like factor 15 (Klf15) (Fig. 1L). In summary, Tsc22d3-2-deficient mice exhibited a relatively mild metabolic phenotype despite the observed higher insulin level. Our data demonstrated that in vitro adipogenesis was not affected by the absence of TSC22D3-2. However, mature adipocytes were unable to accumulate lipids normally, an effect that was further accentuated upon a high-fat diet.
Tsc22d3-2 is not required for the development of the immune system, inflammatory response, or sepsis
To test the role of Tsc22d3-2 in the immune system, we first measured thymus weight at P10, P20, and after puberty. Although thymuses were significantly smaller in the oldest knockout group, younger animals had normal absolute cell numbers (Fig. 2, A and B; P < 0.05). FACS analysis of thymocyte subsets revealed no major changes in the mature single-positive CD4 or CD8 populations. However, a reduced number of DN3 (double negative, CD4−CD8−), (CD25+CD44−) immature thymocytes and a corresponding increase in DN4 (CD25−CD44−) thymocytes was observed (Fig. 2B; P < 0.05). The glucocorticoid-sensitive thymocyte double-positive (CD4+ CD8+) subset of T cells is unaffected in Tsc22d3-2-knockout mice (Fig. 2B). Similarly, with the exception of an increased number of erythroblasts, the absolute cell number of spleen subsets and lymph node subsets is not altered in the knockout groups (Fig. 2, C and D; P < 0.05). Overall, the architecture and cell distribution in the thymus and spleen from knockout mice was preserved (Supplemental Fig. 3).
Fig. 2.
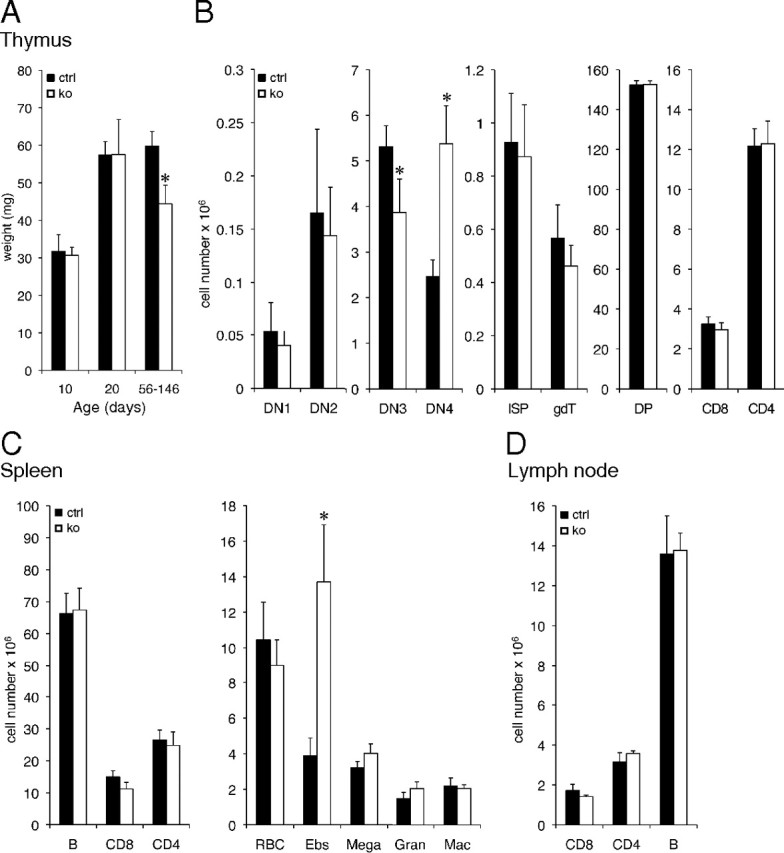
Tsc22d3-2Δ/y mice exhibit minor deficiencies in thymus, spleen, and lymph nodes. A, Thymus size in 10-, 20-, and 56- to 146-d-old mice (n ≥ 7). B, Absolute cell numbers (×106) for indicated thymocyte subsets. C, Absolute cell numbers (×106) for indicated splenocyte subsets. D, Absolute cell numbers (×106) for indicated lymph node subsets. Black bars, control (ctrl); white bars, knockout (ko) mice. Data represent average ± sem (n ≥ 3 mice per group, 10-wk-old males). *, P < 0.05 when compared with control.
To further test whether the inflammatory immune response is altered in mice lacking TSC22D3-2, we measured TNF and MIP-2 (macrophage inflammatory protein-2) cytokine secretion from BMDM upon stimulation with various inflammatory stimuli with or without pretreatment with dexamethasone (Fig. 3, A–C). Under all conditions tested, we observed normal cytokine production in TSC22D3-2-deficient cells (Fig. 3, A–C). When BMDC were treated with various stimuli (LPS, MSU, ATP, Candida, and CpG), we found no differences in the secretion of IL-1β, TNF, or RANTES (Regulated upon Activation, Normal T-cell Expressed, and Secreted) (Fig. 3, E–G). Dexamethasone treatment or pretreatment with heat-inactivated Candida cells led to a dose-dependent reduction in cytokine production that was unaffected by the loss of TSC22D3-2 (Fig. 3, E–G). Furthermore, we tested whether Tsc22d3-2 is implicated in a sepsis model and analyzed cytokine secretion of IL-6, IL-12, TNF, and IL-2 in BMDM upon various stimuli (Fig. 4). No significant differences were observed, and BMDM responded to dexamethasone administration in a dose-dependent manner (Fig. 4, A–C). Splenocytes, when stimulated by anti-CD3 plus anti-CD28 or concanavalin A, exhibited a dose-dependent response independent of TSC22D3-2 (Fig. 4, D and E). Altogether, our experiments suggest that TSC22D3-2 is not of major importance in immunological processes or inflammatory responses.
Fig. 3.

Tsc22d3-2Δ/y mice show no impairment of inflammatory responses. Peritoneal and bone marrow-derived cytokine secretion. (A and B), ELISA for TNF (A and C), and MIP-2 (B and D) after stimulation or not of peritoneal macrophages with LPS supplemented by indicated stimuli. C and D, Stimulation of BMDM (see A and B) with or without dexamethasone (DEX) (0.2 mm). Data are shown as mean values of duplicated stimulations ± sem from three mice per group. E–G, Quantification of IL-1β (E), TNF and Rantes (G) production in bone marrow-derived cells primed with 10 ng/ml LPS with or without dexamethasone (Dex) for indicated periods before inflammasome activation by 100 ng/ml MSU for 5 h or 5 mm ATP for 1 h) (E), heat-inactivated Candida cells (F) or 1 μm CpG for 6 h (G). Data are shown as mean values of triplet stimulations ± sem and representative for three independent experiments with a total of six control and six knockout mice.
Fig. 4.
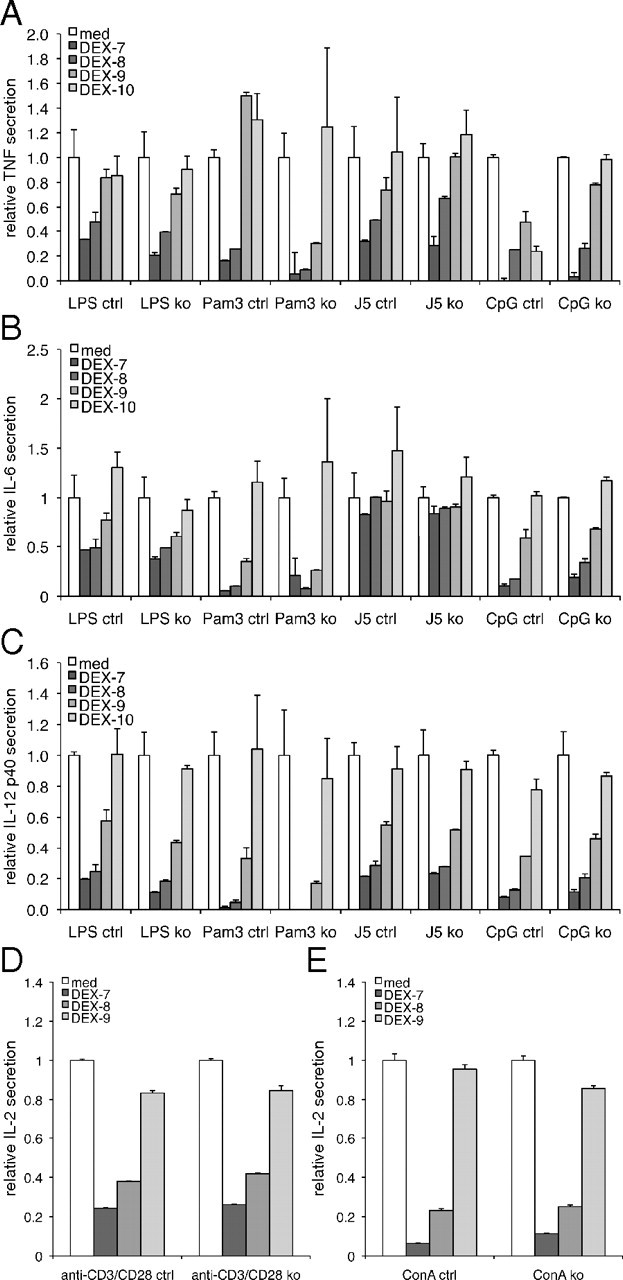
Tsc22d3-2-deficient BMDM are not susceptible to the immunosuppressive effects of dexamethasone. Control and knockout (A–C) and splenocytes (D and E) are incubated with or without increasing concentrations of dexamethasone (DEX, 10−10–10−7 m) and indicated stimuli. (A–C) IL-2 concentrations collected 4 h (TNF) or 18 h (IL-6, IL-12p40, and IL-2) after stimulation. Cytokine concentrations in cells stimulated without dexamethasone are set at 1 for normalization. Data are means ± sd of triplicate samples from one experiment comprising two mice per experimental condition and are representative of two experiments. ctrl, Control; ko, knockout.
Tsc22d3-2-deficient mice present slightly impaired renal sodium and water handling
Upon a standard salt diet, the plasma potassium level was significantly increased in Tsc22d3-2-deficient mice, even though plasma sodium, urinary electrolytes, and aldosterone levels are unaltered (Supplemental Table 3). Plasma and urinary osmolarity was also conserved upon standard and sodium-deprived diets and after water deprivation (Supplemental Table 3). After feeding a sodium-deprived diet for 10 consecutive days, knockout mice exhibited the same water and food intake with no change in body weight (Supplemental Fig. 3). However, these mice showed significantly increased plasma sodium and decreased potassium levels (Supplemental Table 3; P < 0.05), although plasma aldosterone levels were unaffected. In summary, Tsc22d3-2 knockout mice were generally able to adapt to deprivation of sodium or water even though they developed a subtle deficiency in renal sodium and water handling.
Tsc22d3-2 deficiency causes male sterility
Heterozygous females did not show any obvious fertility problems (see above). In contrast, breeding of knockout males (hemizygous, Tsc22d3-2Δ/y, 3–6 months old) to wild-type females (C57BL/6N) never revealed any offspring. We observed the presence of vaginal plugs in spontaneously cycling or hormone-stimulated females (three plugged of four knockout males vs. two plugged of four wild-type males), indicating that the mating behavior of the male knockouts was not affected. Because the Tsc22d3-2 gene is X-linked, we could not obtain homozygous mutant females. Gross histopathological examination of all organs revealed differences only in testis (Supplemental Fig. 4). In adult mice, the testicular mass and size were strikingly reduced (age-matched wild type was 0.123± 0.011 g vs. knockout littermates, 0.023 ± 0.002 g, n = 7 per group, mean age 19.3 ± 1.7 wk, P < 0.001; Fig. 5A). Until the age of 10 d, testicular histology was nearly indistinguishable between the knockout and control groups. From P 20 onward, spermatogenesis is progressively affected and in 2- to 6-month-old knockouts, no germ cells or mature spermatozoa could be identified (Fig. 5, B and C). Plasma level of corticosterone was significantly decreased, whereas FSH and testosterone were significantly increased, and LH was unchanged (Fig. 5D). In 20-d-old knockout mice, the total cell number per seminiferous tubule decreased by about 60% mainly due to the increased number of TUNEL-positive cells, whereas the portion of proliferating cells, as measured by Ki-67 staining, remained constant (Fig. 5E; P < 0.001). At 2 months of age, the number of Sertoli cells per tubule is increased, although at 6 months of age, this difference is no longer evident (Fig. 5F). We found an increased number of interstitial Leydig cells as shown by GATA4 immunostaining and quantification (Fig. 5, C and G). The mRNA transcript levels of somatic (Fshr and Gata4) markers were about 28- and 5-fold increased, respectively, whereas the germ cell marker (Mvh) mRNA transcript expression was nearly abolished (Fig. 5H). This finding was not confirmed in other Pparγ- and Pparγ2-expressing organs (Supplemental Fig. 5C). Expression of genes in testis, e.g. Pparγ, Nr3c1 (glucocorticoid receptor), and Klf5 were significantly down-regulated, whereas levels of Tsc22d1, Nr3c4 (androgen receptor), and Klf15 were significantly up-regulated (Fig. 5I). The observed transcriptional down-regulation of the direct TSC22D3-2 target gene Pparγ2 was accompanied by a reduced protein expression level (Supplemental Fig. 5A). Altogether, we have identified a novel and crucial role for TSC22D3-2 in testis development and fertility.
Fig. 5.

Tsc22d3-2 knockout males are sterile. A, Representative pictures from 2- and 6-month-old control (top) and knockout (bottom) adult testes; B, H&E-stained paraffin sections; C, immunohistochemistry of Sertoli and Leydig cells counterstained with H&E-stained paraffin sections from control and knockout testes. In B and C, note absence of mature spermatozoa (white arrowhead) and apparent hyperproliferation of Leydig cells (black arrow) in the knockouts. Panel C is a higher-magnification image (rabbit anti-GATA4 antibody) with counterstaining with hematoxylin (blue nuclei); elongated spermatids (white arrowhead) are seen in controls. D, Corticosterone (cort), FSH, LH, and testosterone (testo) hormone levels in plasma (n ≥ 10 animals per group, 3–4 months old). E, Total number of cells per seminiferous tubule (n ≥ 20 tubules per group; left), ratio of Ki-67-positive cells divided by total cell number, middle), and ratio of TUNEL-positive cells divided by total cell number per seminiferous tubule, n = 18 tubules, right). F, Quantification of Sertoli cells per tubule (F) (n ≥ 25 tubules) and Leydig cells in the intertubular space (G) (n ≥ 27) at 2 months (n = 2) and 6 months (n = 4 animals). H, qRT-PCR of Fshr, Gata4, and Mvh control (black bars) and knockout (white bars) animals. I, qRT-PCR of Pparγ2, Tsc22d1, Gr, Ar, Klf5, and Klf15 in testis from control (black bars) and knockout (white bars) animals. Scale bar, 1 mm (A), 100 μm (B), and 50 μm (C). *, P < 0.05; **, P < 0.01; ***, P < 0.001. ctrl, Control; ko, knockout.
Discussion
Tsc22d3-2 is an X-linked gene, which is constitutively expressed in a variety of mouse, rat, and human tissues. The encoded protein, GILZ, was suggested to play roles in the immune system and in adipogenesis and sodium homeostasis. Although TSC22D3-2 was reported to interact with signaling molecules like nuclear factor (NF)-κB, c-Fos, Raf-1, Ras, ERK1/2, CCAAT/enhancer-binding protein (C/EBP), and histone deacetylase 1 (HDAC1) (see Ayroldi and Riccardi for review, Ref. 1), little was known about its regulation and in vivo physiological roles. In this study, we analyzed a knockout model of TSC22D3-2 with respect to the immune system and immune responses, growth, and metabolism, and for the capacity to regulate ENaC (epithelial sodium channel)-mediated sodium reabsorption in the kidney after salt- and/or water-deprivation. The observed data only partially support the suggested role for TSC22D3-2/GILZ in metabolism or sodium handling, whereas they revealed no important role in the immune system. In contrast, we have shown a crucial novel role for TSC22D3-2/GILZ in male fertility.
Mild phenotype in metabolism, adipogenesis, and sodium handling in knockout mice
Glucocorticoids are thought to be implicated in the differentiation of mesenchymal progenitor cells, and factors that block adipogenesis favor osteogenic lineage commitment (33). Despite less body fat in older mice, Tsc22d3-2-deficient mice generally do not present severe metabolic alterations or obvious bone abnormalities (Supplemental Fig. 3 and Supplemental Tables 1 and 3). The presence of an increased number of glucagon-positive Langerhans cells in the pancreas may be a compensatory mechanism for the higher plasma insulin levels. This suggests an altered insulin sensitivity (Fig. 1 and Supplemental Table 2), although knockout mice react in the normal range to glucose and insulin tolerance tests and to fasted and refed conditions (Fig. 1). Furthermore, adipocyte differentiation is not severely modified in the Tsc22d3-2-knockout mice (Fig. 1). This is surprising because TSC22D3-2 was reported to bind to the tandem repeat of CCAAT/enhancer-binding protein (C/EBP) binding sites in the Pparγ2 promoter, thereby blocking Pparγ2 transcription and consequently inducing adipogenesis (12). Indeed, in differentiated knockout adipocytes, we find significantly increased Pparγ2 and Fabp4 mRNA transcript expression (Fig. 1H). Our findings are also in contrast to studies where ectopic expression of TSC22D3-2 inhibited glucocorticoid-induced adipocyte differentiation in 3T3-L1 preadipocyte cells (12, 34). Even more surprisingly, in liver and testis from Tsc22d3-2 knockout mice, the purported increase in Pparγ2 promoter activation as a consequence of Tsc22d3-2 deficiency was not confirmed, and instead, Pparγ2 mRNA transcript and protein expression levels are down-regulated in the testis (Supplemental Fig. 5A). This down-regulation in the testis could be biased by a complete loss of germ cells in relation to somatic cells. Alternatively, these observations suggest that additional tissue-specific factors may modulate the transcriptional and translational levels of Pparγ2. Indeed, after a high-fat diet for 18 wk, Tsc22d3-2-knockout mice not only were protected from weight gain but also developed less hepatic steatosis (Fig. 1L). This is in contrast to in vitro data that predict an increase in hepatic steatosis in the absence of TSC22D3-2 and an induction of ENaC-mediated transport in a cortical collecting duct cell line (mpkCCDc14) (11). Even upon various challenges like salt and water deprivation, we were unable to observe significant changes compared with the control group, suggesting that TSC22D3-2 does not play an important role in water or ENaC-mediated sodium handling and in the control of blood pressure and blood volume, which is in contrast to previous publications (36, 37). In this context, it has been shown that fluid retention is independent of collecting duct ENaC activity when induced by thiazolidinedione, an agonist of Pparγ (peroxisome proliferator-activated receptor gamma) (38). In contrast, Pparγ agonists induce sodium and water retention in type 2 diabetes-treated patients (see for review Ref. 39). Although we cannot exclude compensatory mechanisms, we did not find an up-regulation of Tsc22d1 mRNA transcript expression levels; the αENaC mRNA transcript expression level was unchanged (Supplemental Fig. 5C). We suggest that ENaC regulation by GILZ does not play a crucial role in kidney homeostasis.
Immune system and immune responses are not dependent on TSC22D3-2
Glucocorticoids are potent modulators of the immune system and are therefore used as antiinflammatory and immunosuppressive drugs. They induce apoptosis of CD4+CD8+ double-positive cells, which requires gene transcription and coordinated activation of caspases (36, 37). TSC22D3-2 is strongly up-regulated by glucocorticoids in the thymus (2) and might have a dual role in inducing apoptosis of thymocytes and rescuing them from TCR-induced cell death (see for review Ashwell et al., Ref. (40). Surprisingly, despite the predicted Tsc22d3-2-dependent negative regulation of T-cell function, we found no severe impairment of thymus and spleen development (Fig. 2). In addition, our data do not support an antiproliferative activity of TSC22D3-2 on T lymphocytes, as found in concanavalin A-activated T lymphocytes (6), or a negative regulation of erythroid progenitor differentiation (41) but, instead, a significant increase in erythroblast number (Fig. 2).
After stimulation of peritoneal and BMDM from Tsc22d3-2 knockout mice, secretion of TNF, RANTES, MIP-2, and IL-6 was unaltered (Fig. 3, A–D). This is in contrast to transgenic mice that overexpress GILZ in all thymocyte subsets and mature peripheral CD4+ and CD8+ T cells (42). Tsc22d3-2 mRNA transcript expression levels were up-regulated in spleen and peritoneal macrophages of mice that received restraint stress (43). In the treatment of severe sepsis and septic shock, low-dose glucocorticoids exhibit antiinflammatory effects as seen by a decrease in the inflammatory response and an increase in antiinflammatory cytokines (44, 45). Unexpectedly, BMDM from Tsc22d3-2-deficient mice did not secrete less proinflammatory cytokines upon treatment with LPS or Pam3 (N-palmitoyl-S-(2,3-bis(palmitoyl)-(2RS)-propyl)-(R)) or after E. coli infection and were not more resistant to dose-dependent dexamethasone treatment (Fig. 4). Our findings thus suggest that glucocorticoid action is not mediated or dependent on TSC22D3-2 in this in vitro sepsis model.
TSC22D3-2 plays a crucial and novel role in testis development and fertility
The most striking and obvious phenotype in Tsc22d3-2- knockout mice was the male sterility manifested by the significantly reduced testis size and weight, the absence of germ cells, and the increased apoptosis in adult seminiferous tubules (Fig. 5E). Normal spermatogenesis requires both pituitary gonadotropins, namely LH and FSH, and the testicular androgen testosterone (see for review Steinberger, Ref. (46). With normal LH and elevated FSH levels (Fig. 5D), we did not observe the anticipated down-regulation of FSH and LH secretion (46), suggesting that lack of TSC22D3-2 impairs feedback inhibition of gonadotropin secretion by the pituitary.
Our phenotype strikingly resembles that of Six5- and c-kit-deficient mice (47, 48). Sarkar and colleagues (48) proposed that Six5, via reduced c-kit levels contributed to the male reproductive defects in myotonic dystrophy 1, although we did not find changes in Six5 mRNA transcript expression level (Supplemental Fig. 5B). This is a multisystem disorder characterized by endocrine defects that include testicular and tubular atrophy, oligospermia, Leydig cell hyperproliferation, and increased FSH levels, although LH and testosterone levels were unchanged (49). Interestingly, in addition to the significantly lower plasma corticosterone levels, high testosterone levels in the Tsc22d3-2-knockout mice may further impair corticosterone activity by down-regulation of glucocorticoid receptor expression (Fig. 5) (50, 51). At the same time, the complete loss of germ cell-specific Mvh expression and the significant increase in Fshr and Gata4 as somatic markers might well explain the deregulation of genes such as Klf15 or Tsc22d1 in the testis (Fig. 5, H and I). It has been proposed that testosterone exerts its regulating function on spermatogenesis by modifying cortisol-dependent apoptosis (52), coinciding with the significantly higher cell apoptosis in the knockout mice (Fig. 5). In this context, it is interesting to note that androgen receptor knockout mice show a severe disruption of spermatogenesis with the failure of germ cells to progress beyond the early stages of meiosis (53). Although the underlying mechanism is not yet fully understood, Leydig cell hyperproliferation is proposed to present a compensatory mechanism to increase testicular steroidogenesis induced by testosterone insufficiency (54). Because we do not find a higher proliferation rate of the Leydig cell population in the Tsc22d3-2 knockouts, our Leydig cell hyperplasia per intertubular space is only apparent and seems not to be altered per tubule. LH seems to be most effective in increasing the number of interstitial cells (55), but FSH might exert a proliferative effect on precursor mesenchymal cells that form Leydig cells postnatally (56–58). In summary, TSC22D3-2 is essential for male gametogenesis and exerts a crucial role in the interplay between endocrine stimulation, somatic cell activity, and spermatogenesis. The use of germ-cell- and Sertoli-cell-specific conditional knockouts will further help to dissect this crucial role of TSC22D3-2 in germ via somatic cells. In addition, use of conditional knockouts might also allow the generation of mice lacking TSC22D3-2 in females and thus to directly address effects of this protein in female fertility and physiology.
In summary, mice lacking TSC22D3-2 are viable and exhibit a significant decrease in plasma cortisol. Our data did not confirm a crucial involvement of TSC22D3-2 in immunity, inflammatory responses, or adipogenesis. Functional redundancy by other members of the same family might compensate for this loss, as suggested by the increased Tsc22d1 mRNA transcript expression in the testis. All other glucocorticoid-dependent organs and tissues analyzed in this study exhibit only minor defects, if any, or a default that might only become visible under challenge conditions. Tsc22d3-2-deficient mice might therefore be useful to dissect these glucocorticoid-dependent and -independent processes in the future.
Acknowledgments
We thank Jürg Tschopp (who passed away during the preparation of this manuscript) for his active contribution to this study. We thank the Transgenic Animal Facility, the Mouse Metabolic Evaluation Facility, the Mouse Pathology Platform, the Cellular Imaging Facility, and the Flow Cytometry Facility (all at the University of Lausanne) for their help and services.
This work was supported by the Swiss National Science Foundation to E.H. (3100A0-128658/1), Association Institute for Arthritis Research, and Louis-Jeantet Prize for Medicine to Jürg Tschopp and his collaborators O.G. and V.P. This work has been furthermore supported by the Fondation Emma Muschamps, the Swiss National Science Foundation to T.R. (310030–132744), and the Leducq Foundation to E.H. and B.C.R. V.P. is recipient of a Marie Curie fellowship.
Present address for P.E.S.: Division of Molecular Pediatrics, CHUV, Clinique Infantile, Lausanne, Switzerland.
Present address for V.P.: Université de Lyon; Inserm U1052; CNRS UMR5286, Centre de Recherche en Cancérologie de Lyon; 69000 Lyon, France.
Addendum: After submission of our manuscript, a knockout of the long-glucocorticoid-induced leucine zipper (L-GILZ) was published (59) that exhibits a similar but not identical testis phenotype compared with our complete knockout.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- APC
- Allophycocyanin
- BMDC
- bone marrow-derived dendritic cells
- BMDM
- bone marrow-derived macrophages
- ENaC
- epithelial sodium channel
- FACS
- fluorescence-activated cell sorting
- FCS
- fetal calf serum
- FITC
- fluorescein isothiocyanate
- GATA
- GATA-binding protein
- GILZ
- glucocorticoid-induced leucine zipper
- H&E
- hematoxylin and eosin
- LPS
- lipopolysaccharide
- MEF
- mouse embryonic fibroblasts
- MIP-2
- macrophage inflammatory protein-2
- MSU
- monosodium urate
- PE
- phycoerythrine
- TCR
- T cell receptor
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling.
References
- 1. Ayroldi E , Riccardi C. 2009. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J 23:3649–3658 [DOI] [PubMed] [Google Scholar]
- 2. D'Adamio F , Zollo O , Moraca R , Ayroldi E , Bruscoli S , Bartoli A , Cannarile L , Migliorati G , Riccardi C. 1997. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity 7:803–812 [DOI] [PubMed] [Google Scholar]
- 3. Mittelstadt PR , Ashwell JD. 2001. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem 276:29603–29610 [DOI] [PubMed] [Google Scholar]
- 4. Soundararajan R , Wang J , Melters D , Pearce D. 2007. Differential activities of glucocorticoid-induced leucine zipper protein isoforms. J Biol Chem 282:36303–36313 [DOI] [PubMed] [Google Scholar]
- 5. Ayroldi E , Migliorati G , Bruscoli S , Marchetti C , Zollo O , Cannarile L , D'Adamio F , Riccardi C. 2001. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor κB. Blood 98:743–753 [DOI] [PubMed] [Google Scholar]
- 6. Ayroldi E , Zollo O , Bastianelli A , Marchetti C , Agostini M , Di Virgilio R , Riccardi C. 2007. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest 117:1605–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Delfino DV , Agostini M , Spinicelli S , Vito P , Riccardi C. 2004. Decrease of Bcl-xL and augmentation of thymocyte apoptosis in GILZ overexpressing transgenic mice. Blood 104:4134–4141 [DOI] [PubMed] [Google Scholar]
- 8. Cohen N , Mouly E , Hamdi H , Maillot MC , Pallardy M , Godot V , Capel F , Balian A , Naveau S , Galanaud P , Lemoine FM , Emilie D. 2006. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood 107:2037–2044 [DOI] [PubMed] [Google Scholar]
- 9. Hamdi H , Bigorgne A , Naveau S , Balian A , Bouchet-Delbos L , Cassard-Doulcier AM , Maillot MC , Durand-Gasselin I , Prévot S , Delaveaucoupet J , Emilie D , Perlemuter G. 2007. Glucocorticoid-induced leucine zipper: A key protein in the sensitization of monocytes to lipopolysaccharide in alcoholic hepatitis. Hepatology 46:1986–1992 [DOI] [PubMed] [Google Scholar]
- 10. Muller OG , Parnova RG , Centeno G , Rossier BC , Firsov D , Horisberger JD. 2003. Mineralocorticoid effects in the kidney: correlation between αENaC, GILZ, and Sgk-1 mRNA expression and urinary excretion of Na+ and K+. J Am Soc Nephrol 14:1107–1115 [DOI] [PubMed] [Google Scholar]
- 11. Soundararajan R , Zhang TT , Wang J , Vandewalle A , Pearce D. 2005. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem 280:39970–39981 [DOI] [PubMed] [Google Scholar]
- 12. Shi X , Shi W , Li Q , Song B , Wan M , Bai S , Cao X. 2003. A glucocorticoid-induced leucine-zipper protein, GILZ, inhibits adipogenesis of mesenchymal cells. EMBO Rep 4:374–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trumpp A , Refaeli Y , Oskarsson T , Gasser S , Murphy M , Martin GR , Bishop JM. 2001. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414:768–773 [DOI] [PubMed] [Google Scholar]
- 14. Hummler E , Mérillat AM , Rubera I , Rossier BC , Beermann F. 2002. Conditional gene targeting of the Scnn1a (αENaC) gene locus. Genesis 32:169–172 [DOI] [PubMed] [Google Scholar]
- 15. Reis LF , Ruffner H , Stark G , Aguet M , Weissmann C. 1994. Mice devoid of interferon regulatory factor 1 (IRF-1) show normal expression of type I interferon genes. EMBO J 13:4798–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hummler E , Barker P , Gatzy J , Beermann F , Verdumo C , Schmidt A , Boucher R , Rossier BC. 1996. Early death due to defective neonatal lung liquid clearance in α-ENaC-deficient mice. Nat Genet 12:325–328 [DOI] [PubMed] [Google Scholar]
- 17. Porret A , Mérillat AM , Guichard S , Beermann F , Hummler E. 2006. Tissue-specific transgenic and knockout mice. Methods Mol Biol 337:185–205 [DOI] [PubMed] [Google Scholar]
- 18. Rodríguez CI , Buchholz F , Galloway J , Sequerra R , Kasper J , Ayala R , Stewart AF , Dymecki SM. 2000. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet 25:139–140 [DOI] [PubMed] [Google Scholar]
- 19. Buchholz F , Refaeli Y , Trumpp A , Bishop JM. 2000. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep 1:133–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jelinic P , Stehle JC , Shaw P. 2006. The testis-specific factor CTCFL cooperates with the protein methyltransferase PRMT7 in H19 imprinting control region methylation. PLoS Biol 4:e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Christensen BM , Perrier R , Wang Q , Zuber AM , Maillard M , Mordasini D , Malsure S , Ronzaud C , Stehle JC , Rossier BC , Hummler E. 2010. Sodium and potassium balance depends on αENaC expression in connecting tubule. J Am Soc Nephrol 21:1942–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cederroth CR , Zimmermann C , Beny JL , Schaad O , Combepine C , Descombes P , Doerge DR , Pralong FP , Vassalli JD , Nef S. 2010. Potential detrimental effects of a phytoestrogen-rich diet on male fertility in mice. Mol Cell Endocrinol 321:152–160 [DOI] [PubMed] [Google Scholar]
- 23. Romero Y , Meikar O , Papaioannou MD , Conne B , Grey C , Weier M , Pralong F , De Massy B , Kaessmann H , Vassalli JD , Kotaja N , Nef S. 2011. Dicer 1 depletion in male germ cells leads to infertility due to cumulative meiotic and spermiogenic defects. PLoS One 6:e25241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hansen JB , Petersen RK , Larsen BM , Bartkova J , Alsner J , Kristiansen K. 1999. Activation of peroxisome proliferator-activated receptor γ bypasses the function of the retinoblastoma protein in adipocyte differentiation. J Biol Chem 274:2386–2393 [DOI] [PubMed] [Google Scholar]
- 25. Yang YC , Hsu HK , Hwang JH , Hong SJ. 2003. Enhancement of glucose uptake in 3T3-L1 adipocytes by Toona sinensis leaf extract. Kaohsiung J Med Sci 19:327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lukas J , Bartkova J , Rohde M , Strauss M , Bartek J. 1995. Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of Cdk4 activity. Mol Cell Biol 15:2600–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ramírez-Zacarías JL , Castro-Muñozledo F , Kuri-Harcuch W. 1992. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry 97:493–497 [DOI] [PubMed] [Google Scholar]
- 28. Gross O , Gewies A , Finger K , Schäfer M , Sparwasser T , Peschel C , Förster I , Ruland J. 2006. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442:651–656 [DOI] [PubMed] [Google Scholar]
- 29. Gross O , Poeck H , Bscheider M , Dostert C , Hannesschläger N , Endres S , Hartmann G , Tardivel A , Schweighoffer E , Tybulewicz V , Mocsai A , Tschopp J , Ruland J. 2009. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459:433–436 [DOI] [PubMed] [Google Scholar]
- 30. Muruve DA , Pétrilli V , Zaiss AK , White LR , Clark SA , Ross PJ , Parks RJ , Tschopp J. 2008. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452:103–107 [DOI] [PubMed] [Google Scholar]
- 31. Roger T , Lugrin J , Le Roy D , Goy G , Mombelli M , Koessler T , Ding XC , Chanson AL , Reymond MK , Miconnet I , Schrenzel J , François P , Calandra T. 2011. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 117:1205–1217 [DOI] [PubMed] [Google Scholar]
- 32. Roger T , Froidevaux C , Le Roy D , Reymond MK , Chanson AL , Mauri D , Burns K , Riederer BM , Akira S , Calandra T. 2009. Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci USA 106:2348–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beresford JN , Bennett JH , Devlin C , Leboy PS , Owen ME. 1992. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. J Cell Sci 102 (Pt 2):341–351 [DOI] [PubMed] [Google Scholar]
- 34. Batchvarova N , Wang XZ , Ron D. 1995. Inhibition of adipogenesis by the stress-induced protein CHOP (Gadd153). EMBO J 14:4654–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roger T , David J , Glauser MP , Calandra T. 2001. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 414:920–924 [DOI] [PubMed] [Google Scholar]
- 36. Cifone MG , Migliorati G , Parroni R , Marchetti C , Millimaggi D , Santoni A , Riccardi C. 1999. Dexamethasone-induced thymocyte apoptosis: apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase C, acidic sphingomyelinase, and caspases. Blood 93:2282–2296 [PubMed] [Google Scholar]
- 37. Marchetti MC , Di Marco B , Cifone G , Migliorati G , Riccardi C. 2003. Dexamethasone-induced apoptosis of thymocytes: role of glucocorticoid receptor-associated Src kinase and caspase-8 activation. Blood 101:585–593 [DOI] [PubMed] [Google Scholar]
- 38. Vallon V , Hummler E , Rieg T , Pochynyuk O , Bugaj V , Schroth J , Dechenes G , Rossier B , Cunard R , Stockand J. 2009. Thiazolidinedione-induced fluid retention is independent of collecting duct αENaC activity. J Am Soc Nephrol 20:721–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blazer-Yost BL. 2010. PPARγ agonists: blood pressure and edema. PPAR Res 2010:785369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashwell JD , Lu FW , Vacchio MS. 2000. Glucocorticoids in T cell development and function. Annu Rev Immunol 18:309–345 [DOI] [PubMed] [Google Scholar]
- 41. Kolbus A , Blázquez-Domingo M , Carotta S , Bakker W , Luedemann S , von Lindern M , Steinlein P , Beug H. 2003. Cooperative signaling between cytokine receptors and the glucocorticoid receptor in the expansion of erythroid progenitors: molecular analysis by expression profiling. Blood 102:3136–3146 [DOI] [PubMed] [Google Scholar]
- 42. Cannarile L , Fallarino F , Agostini M , Cuzzocrea S , Mazzon E , Vacca C , Genovese T , Migliorati G , Ayroldi E , Riccardi C. 2006. Increased GILZ expression in transgenic mice up-regulates Th-2 lymphokines. Blood 107:1039–1047 [DOI] [PubMed] [Google Scholar]
- 43. Wang Y , Lu Y , Yu D , Wang Y , Chen F , Yang H , Zheng SJ. 2008. Enhanced resistance of restraint-stressed mice to sepsis. J Immunol 181:3441–3448 [DOI] [PubMed] [Google Scholar]
- 44. Briegel J , Kellermann W , Forst H , Haller M , Bittl M , Hoffmann GE , Büchler M , Uhl W , Peter K. 1994. Low-dose hydrocortisone infusion attenuates the systemic inflammatory response syndrome. The Phospholipase A2 Study Group. Clin Investig 72:782–787 [DOI] [PubMed] [Google Scholar]
- 45. Prigent H , Maxime V , Annane D. 2004. Science review: mechanisms of impaired adrenal function in sepsis and molecular actions of glucocorticoids. Crit Care 8:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Steinberger A. 1991. Effects of temperature on the biochemistry of the testis. Adv Exp Med Biol 286:33–47 [DOI] [PubMed] [Google Scholar]
- 47. Rothschild G , Sottas CM , Kissel H , Agosti V , Manova K , Hardy MP , Besmer P. 2003. A role for kit receptor signaling in Leydig cell steroidogenesis. Biol Reprod 69:925–932 [DOI] [PubMed] [Google Scholar]
- 48. Sarkar PS , Paul S , Han J , Reddy S. 2004. Six5 is required for spermatogenic cell survival and spermiogenesis. Hum Mol Genet 13:1421–1431 [DOI] [PubMed] [Google Scholar]
- 49. Vazquez JA , Pinies JA , Martul P , De los Rios A , Gatzambide S , Busturia MA. 1990. Hypothalamic-pituitary-testicular function in 70 patients with myotonic dystrophy. J Endocrinol Invest 13:375–379 [DOI] [PubMed] [Google Scholar]
- 50. Chen S , Wang J , Yu G , Liu W , Pearce D. 1997. Androgen and glucocorticoid receptor heterodimer formation. A possible mechanism for mutual inhibition of transcriptional activity. J Biol Chem 272:14087–14092 [DOI] [PubMed] [Google Scholar]
- 51. Kerr JE , Beck SG , Handa RJ. 1996. Androgens modulate glucocorticoid receptor mRNA, but not mineralocorticoid receptor mRNA levels, in the rat hippocampus. J Neuroendocrinol 8:439–447 [DOI] [PubMed] [Google Scholar]
- 52. Russell LD , Clermont Y. 1977. Degeneration of germ cells in normal, hypophysectomized and hormone treated hypophysectomized rats. Anat Rec 187:347–366 [DOI] [PubMed] [Google Scholar]
- 53. De Gendt K , Swinnen JV , Saunders PT , Schoonjans L , Dewerchin M , Devos A , Tan K , Atanassova N , Claessens F , Lécureuil C , Heyns W , Carmeliet P , Guillou F , Sharpe RM , Verhoeven G. 2004. A Sertoli cell-selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc Natl Acad Sci USA 101:1327–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mylchreest E , Sar M , Wallace DG , Foster PM. 2002. Fetal testosterone insufficiency and abnormal proliferation of Leydig cells and gonocytes in rats exposed to di(n-butyl) phthalate. Reprod Toxicol 16:19–28 [DOI] [PubMed] [Google Scholar]
- 55. Dombrowicz D , Sente B , Reiter E , Closset J , Hennen G. 1996. Pituitary control of proliferation and differentiation of Leydig cells and their putative precursors in immature hypophysectomized rat testis. J Androl 17:639–650 [PubMed] [Google Scholar]
- 56. Benton L , Shan LX , Hardy MP. 1995. Differentiation of adult Leydig cells. J Steroid Biochem Mol Biol 53:61–68 [DOI] [PubMed] [Google Scholar]
- 57. Hardy MP , Zirkin BR , Ewing LL. 1989. Kinetic studies on the development of the adult population of Leydig cells in testes of the pubertal rat. Endocrinology 124:762–770 [DOI] [PubMed] [Google Scholar]
- 58. Saez JM. 1994. Leydig cells: endocrine, paracrine, and autocrine regulation. Endocr Rev 15:574–626 [DOI] [PubMed] [Google Scholar]
- 59. Bruscoli S , Velardi E , Di Sante M , Bereshchenko O , Venanzi A , Coppo M , Berno V , Mameli MG , Colella R , Cavaliere A , Riccardi C. 2012. Long-Glucocorticoid-induced leucine Zipper (L-GILZ) interacts with Ras pathway and contributes to spermatogenesis control. J Biol Chem 287:1242–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]