

Abstract
As one of the nine hereditary neurodegenerative polyQ disorders, spinal and bulbar muscular atrophy (SBMA) results from a polyQ tract expansion in androgen receptor (AR). Although protein aggregates are the pathological hallmark of many neurodegenerative diseases, their direct role in the neurodegeneration is more and more questioned. To determine the early molecular mechanisms causing motor neuron degeneration in SBMA, we established an in vitro system based on the tetracycline-inducible expression of normal (AR20Q), the mutated, 51 glutamine-extended (AR51Q), or polyQ-deleted (AR0Q) AR in NSC34, a motor neuron-like cell line lacking endogenous AR. Although no intracellular aggregates were formed, the expression of the AR51Q leads to a loss of function characterized by reduced neurite outgrowth and to a toxic gain of function resulting in decreased cell viability. In this study, we show that both AR20Q and AR51Q are recruited to lipid rafts in response to testosterone stimulation. However, whereas testosterone induces the activation of the c-jun N-terminal kinase/c-jun pathway via membrane-associated AR20Q, it does not so in NSC34 expressing AR51Q. Phosphorylation of c-jun N-terminal kinase plays a crucial role in AR20Q-dependent survival and differentiation of NSC34. Moreover, c-jun protein levels decrease more slowly in AR20Q- than in AR51Q-expressing NSC34 cells. This is due to a rapid and transient inhibition of glycogen synthase kinase 3α occurring in a phosphatidylinositol 3-kinase-independent manner. Our results demonstrate that the deregulation of nongenomic AR signaling may be involved in SBMA establishment, opening new therapeutic perspectives.
Polyglutamine (polyQ) tract elongation is responsible for nine hereditary neurodegenerative diseases, including Huntington's disease, dentatorubropallidoluy-sian atrophy, several spinocerebellar ataxias (types 1, 2, 3, 6, 7, and 17), and spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease. Although mutant genes are often ubiquitously expressed, these pathologies exclusively affect certain neuronal populations, which vary according to the polyQ disorder (1). SBMA is a rare late-onset neurodegeneration resulting from a loss of motor neurons in the brainstem and spinal cord (2). This is caused by the expansion of more than 40 CAG in the first exon of the gene coding for the androgen receptor (AR) (1) located on the X-chromosome (locus Xq11–12) (3). Besides X-linked recessive transmission, the predominant androgenic environment could explain that males are almost exclusively affected and females rarely show symptoms. Although the mechanisms that lead to the specific loss of motor neurons remain unknown, several pathogenic mechanisms have been suggested.
The formation of nuclear aggregates of mutant AR (neuronal intranuclear inclusions) is considered as the pathological hallmark of the disease (4). It has been shown that the insoluble N-terminal fragments of polyQ-expanded AR accumulate in the cell nucleus in a ligand-dependent manner, are sequestered into inclusion bodies, and deregulate transcription by interfering with transcriptional coregulators (5). Moreover, as in several other polyQ diseases, numerous cellular proteins such as heat-shock proteins (Hsp40, Hsp70, and Hsp90), proteasome pathway elements (ubiquitin and the 19S and 20S proteasome core components) and several coactivators (cAMP response element-binding protein-binding protein and steroid coactivator 1) have been found to be sequestered in these neuronal intranuclear inclusions (6, 7).
However, the toxicity of both insoluble aggregates and nuclear inclusions is more and more questioned (8–11). Recent studies have even shown that the formation of soluble polyQ oligomers suffices to induce degeneration (12), and soluble β-oligomers of the scrapie prion protein were found to be intrinsically toxic for primary hippocampal and cerebellar neurons (13). According to Simeoni et al. (14), motor neuron degeneration is not correlated with the presence of aggregates. Moreover, the loss of endogenous AR by itself has been described as accelerating motor neuron degeneration (15). It is worth noticing that most in vivo SBMA models are based on the overexpression of a mutated AR harboring a polyQ extension of more than 90Q in the presence of normal endogenous AR (15). This overexpression of a long expanded-polyQ AR certainly accelerates the appearance of SBMA symptoms because this disease is normally characterized by a slow, progressive loss of motor neurons. Nonetheless, due to the remaining endogenous AR, such models could emphasize an aggregate phenotype hiding other pathological processes, and results can be distorted, because the polyQ tract in patients suffering from SBMA does not exceed 57 glutamines with a median of 45.7 ± 3.2 copies (16), and male patients do not express an endogenous normal AR.
In several neurodegenerative diseases such as Alzheimer's disease, Huntington's disease or amyotrophic lateral sclerosis, lipid raft disruption, or impairment of membrane-initiated signaling pathways caused by the expression of mutated proteins contribute to the lethal loss of neuronal function (17). Interestingly, the existence of a nonclassic membrane-associated AR has been postulated by several studies based on the existence of other membrane-bound steroid receptors (18) and specific androgen binding to plasma membranes including those of prostate cancer cells (19, 20), macrophages (21), and T lymphocytes (22). Indeed, besides its well-known role as transcription factor, AR has been demonstrated to induce rapid, nongenomic signaling in response to androgen stimulation (23–25). Signaling pathways such as Erk and phosphatidylinositol 3-kinase (PI3K)/Akt have been shown to be modulated by this membrane-associated AR (23, 24, 26, 27). Impaired transcriptional activity of AR due to polyQ tract elongation has already been investigated (28, 29). However, rapid nongenomic actions of androgen were never considered as potential deregulated events in SBMA. In this paper, we investigated the existence of signaling pathways modulated by a membrane-associated AR, in an in vitro motor neuron-like model. We also analyzed the consequences of the expression of polyQ-extended AR on these pathways. For that purpose, mutated AR was expressed in NSC34 cells in an inducible manner. NSC34 cells do not express endogenous AR (14) and present neuronal characteristics (30). We worked with two types of AR mutants. One contains an elongated polyQ (51 glutamines) and has been shown to have lost AR transcription function and to be responsible for motor neuron degeneration. The other is an AR without a polyQ tract (AR0Q) that constitutes a model for AR possessing a short polyQ tract, found to make men more prone to develop prostate cancer (31, 32). The originality of this NSC34/AR model comes from the expression of a mutated AR with a 51-polyQ tract in the absence of an endogenous AR expression. Moreover, the inducibility of the model allows the identification of early molecular events associated with the expression of mutated AR and avoids the selection of cells resistant to the expression of AR51Q. We therefore performed our experiments in an androgenic environment comparing NSC34 expressing different forms of the AR. We showed that contrary to AR20Q and AR0Q, the expression of AR51Q has a deleterious effect on cell viability and neurite outgrowth. Because c-jun has been reported to be involved not only in apoptosis but also in neurite outgrowth and cellular differentiation (33–35), the influence of membrane-associated AR on c-jun activity and stability in response to androgen treatment was analyzed.
Materials and Methods
Cell culture
NSC34, hybrid cells of embryonic mouse motor neurons of spinal cord and neuroblastoma cells, were maintained in DMEM with l-glutamine 2 mM, d-glucose 4.5 g/L and sodium pyruvate 110 mg/L (Invitrogen, Ilkirch, France) supplemented with 5% heat-inactivated fetal bovine serum (FBS) (Invitrogen, Mundolsheim, France), 5 μg/ml ciprofloxacin (Euromedex, France) and 2.5 μg/ml fungizone (Invitrogen, France) and were incubated at 37 C in a 5% CO2 humid atmosphere. At 80% confluence, cells plated on a 75-cm2 cell culture flask (Corning, VWR, France) were washed with 2 ml PBS, followed by incubation with 0.5% trypsin (from bovine pancreas; Sigma, France) supplemented with 0.02% EDTA (Sigma), 1 g/liter d-glucose (Merck, Darmstadt, Germany), 0.06% K2CO3, 0.04% KCl, 0.8% NaCl filtered on 0.2 μm. Doxycycline (Euromedex, France) was prepared in PBS at a concentration of 1 mg/ml, and testosterone (Sigma, Saint-Quentin Fallavier, France) was solubilized in absolute ethanol (10 μm).
Plasmids and constructs
Inducible mammalian expression T-Rex system was purchased from Invitrogen (France). NSC34 were stably transfected with the pcDNA 6/TR for high-level expression of the tetracycline repressor TR protein and selected with 10 μg/ml blasticidin S. Resistant cells were then cloned. The enhanced cyan fluorescent protein, ECFP-ARnQ (n = 0, 20, or 51 Q) (enhanced cyan fluorescent protein fused at the N terminus of the ARnQ). Fusion construct was generated by insertion of the full-length AR cDNA (kindly provided by Prof. S. Alonso, University of Nîmes, Nîmes, France) into XhoI-XbaI sites and the PCR product of ECFP (kindly provided by Dr. P. Brabet, Institut des Neurosciences de Montpellier, Montpellier, France) into BamHI-XhoI sites of the pcDNA 5/TO vector (Invitrogen, France). Transfected cells were selected with 200 μg/ml hygromycin B (Invitrogen, France). Plasmids were amplified in Escherichia coli DH5α and purified by the QIAGEN Plasmid Maxi Kit (QIAGEN, France). Fidelity of all constructs was confirmed by DNA sequencing. The 5/TO ECFP-ARnQ pcDNA was linearized by digestion by Fast Digest FspI (Fermentas, France). NSC34 transfected with pCMV/TetO2/AR20Q expressed the wild-type AR with a polyQ tract of 20 glutamines (NSC34/AR20Q). NSC34 transfected with pCMV/TetO2/AR51Q expressed the mutated AR with an elongated polyQ tract of 51 glutamines (NSC34/AR51Q). NSC34 transfected with pCMV/TetO2/AR0Q expressed an AR without a polyQ tract (NSC34/AR0Q). Noninduced (NI) NSC34/ARnQ refers to NSC34 that were not treated with doxycycline; NI NSC34/ARnQ refers to NSC34 that did not express ARnQ. NI plus testosterone (NI+T) NSC34/ARnQ refers to NSC34 that did not express ARnQ but were treated with testosterone; doxycycline-induced (I) NSC34/ARnQ refers to NSC34 that expressed ARnQ in the absence of testosterone stimulation and I+T NSC34/ARnQ refers to NSC34 that expressed ARnQ in the presence of testosterone.
Transfection
Transfections were achieved by coating a poly-d-lysine/laminin (Sigma, France) network with a mixture of liposomes and pcDNA, called lipoplex, before cell seeding according to the method described by Rakotoarivelo et al., (36). Liposomes used for transfection of NSC34 were composed of N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethyl-ammonium (DOTAP; Sigma) and l-α-phosphatidylcholine (PC; Sigma) in proportions of 1:1. For 200 μl lipoplex, 3 μg plasmid DNA was diluted in 100 μl Hank's buffered saline plus supplements containing 0.75 mm Na2PO4 (Merck), 5 mm KCl (VWR, France), 140 mm NaCl (Prolabo), 6 mm glucose (Merck, Germany), and 25 mm HEPES (Sigma, France). Hank's buffered saline plus supplements (20 μl) was added to 80 μl liposome suspension DOTAP/PC 1:1. After mixing each solution and incubating for 5 min, the two solutions were mixed together and incubated for 20 min at room temperature before coverslip coating. Coverslips were incubated in a 5% CO2 atmosphere with 0.002% (wt/vol) poly-d-lysine (molecular mass > 30 kDa; Sigma, France) overnight at 35 C, then with 0.0003% (wt/vol) laminin (Sigma, France) for 3 h, and finally with lipoplex suspension overnight at 37 C (1.5 ml lipoplex per well in a six-well plate). Cells were then seeded at a density of 70,000 cells per well in 2 ml DMEM supplemented with 5% FBS.
Cell viability assay
The effect of ARnQ (n = 0, 20, or 51 Q) expression on NSC34 proliferation was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) uptake. NSC34 cells were plated on 96-well plates at 3000 cells per well in 200 μl DMEM supplemented with 5% charcoal-stripped heat-inactivated FBS (Invitrogen, France). At time 24 h, cells were treated with vehicle (PBS) or with 1 μg/ml doxycycline for 48 h. Then 10 nm of testosterone (Sigma-Aldrich, France) or vehicle was added. Incorporation of thiazolyl blue tetrazolium bromide (1 mg/ml in PBS; Sigma-Aldrich, France) was performed at various times after treatment. Every 24 h, 20 μl MTT (1 mg/ml in PBS) was added to 200 μl medium into wells being assayed and incubated for 90 min at 37 C in 5% CO2. Medium was then completely removed. After addition of 0.1 ml dimethylsulfoxide, cell viability was determined by OD at 570 nm with a microplate reader (model 550; Bio-Rad, France).
Immunocytochemistry and microscopy
NSC34 cells were seeded in a four-well plate (Nunc) in DMEM supplemented with 5% charcoal-stripped heat-inactivated FBS (without steroids; GIBCO) at a density of 10,000 cells per well, each containing glass coverslips (diameter 12 mm). The expression of AR fused to ECFP was induced by adding 1 μg/ml doxycycline to the medium for 48 h. Then monolayer cells were treated with 10 nm testosterone during 2 h. Cells were then fixed in 4% formaldehyde for 10 min at room temperature and then washed in PBS and incubated with anti-AR (N-20) antibody (1:250 dilutions in PBS supplemented with 1% BSA and 0.05% sodium azide) overnight at 4 C followed by goat antirabbit Alexa 594-conjugated antibody (1:3000 dilutions in PBS containing 5 μg/ml Hoechst 33258, 1% BSA, and 0.05% sodium azide) for 1 h at room temperature. Fluorescence microscopy was carried out with an inverse 1 Zeiss Axioobserver/LSM 5 LIVE DUO, equipped with a ×40 objective. Pixels were quantified using Imaris version 7.0.0 software highlighted the cytoplasm (green) and AR staining with the highest intensity (red points).
Neurite outgrowth
NSC34 cells were seeded in a four-well plate (Nunc, VWR, France) in DMEM supplemented with 5% charcoal-stripped heat-inactivated FBS (without steroids; Invitrogen, France) at a density of 10,000 cells per well, each containing glass coverslips (diameter 12 mm). After 24 h culture, cells were treated with 1 μg/ml doxycycline for 48 h. Then 10 nm testosterone was added for 96 h. For the serum deprivation experiments, cells were gently rinsed with PBS and incubated for 24 h in DMEM without serum supplemented with 1 μg/ml doxycycline and 10 nm testosterone. Cells were fixed in 4% formaldehyde for 10 min at room temperature and then washed in PBS and incubated with antiperipherin antibody (1:100 dilution) followed by goat antirabbit Alexa Fluor 594-conjugated antibody (1:3000 dilution in PBS containing 5 μg/ml Hoechst 33258, 1% BSA, and 0.05% sodium azide). Three coverslips issued from three different cultures were analyzed using an upright Zeiss AxioimagerZ1/apotome equipped with a ×40 oil immersion objective and Axiovion version 4.5 software. To measure neurite elongation, cells were stained with β-III-tubulin antibody revealed by a secondary antibody conjugated to Alexa 488 (green). Three wells resulting from three different cultures of NSC34/AR(n)Q were analyzed, and five mosaics (3 × 3) per well were randomly captured by a Coolsnap HQ2-2 camera connected to an upright Zeiss Axioimager Z1 (objective ×4). Neurite length of cells contained in the central image of each mosaic was measured using the Serf software (http://bram.org/serf). About 1000 cells were analyzed for each cell line.
Western blotting
Cell lysates were prepared in lysis buffer [20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin; Cell Signaling] supplemented with protease inhibitors (Mini Complete; Roche, France). Lysates were centrifuged at 3000 × g for 10 min at 4 C, and protein concentrations of supernatants were determined by the BCA (bicinchoninic acid) method (Thermo Fisher Scientific, France). After denaturation at 95 C for 5 min in Laemmli buffer supplemented with 50 mm dithiothreitol (Bio-Rad, France), lysates were resolved by SDS-PAGE 10% at 100 V. Electrotransfer on polyvinylidene difluoride membranes (Hybond; GE Healthcare, France) was carried out with a Trans-Blot-SD Semi-Dry-Transfer Cell (Bio-Rad, France) at 15 V for 1 h or 1 h 30 min (for AR immunoblotting). Polyvinylidene difluoride membranes were blocked for 90 min in Tris-buffered saline/Tween 0.1% buffer [20 mm Tris-HCl, 270 mm NaCl, 0.1% Tween 20 (pH 7.5)] containing 5% (wt/vol) nonfat milk and incubated with the chosen primary antibodies diluted in the blocking solution overnight at 4 C. After incubation with primary antibodies, membranes were incubated with species-specific secondary antibodies conjugated to horseradish peroxidase (HRP) diluted in the blocking solution for 90 min at room temperature. Proteins were visualized using the Immobilon ECL Western blotting system (Millipore, France) and exposure of membranes to BioMax films (Kodak, France). Bands were quantified by measuring densities and surface areas with Serf software. For determination of testosterone action on kinase activity, 30 μg total proteins was resolved by SDS-PAGE. Antibodies to Akt (pan, rabbit), phospho-Akt(Ser473) (rabbit), glycogen synthase kinase 3α (GSK3α) (rabbit), phospho-GSK3α (rabbit), c-jun (60A8, rabbit), phospho-c-jun(Ser73) (rabbit), SAPK/c-jun (stress-activated protein kinase/c-Jun) N-terminal kinases (JNK) (56G8, rabbit), phospho-SAPK/JNK(Thr183/Tyr185) (98F2, rabbit) (Cell Signaling Technology, Danvers, MA) were purchased and used at a dilution of 1:1000. Anti-flotillin-1 monoclonal antibody (1:500) was purchased from BD Biosciences, anti-AR polyclonal antibodies were from Santa Cruz Technology (Santa Cruz, CA) (N-20, C-19 1:500), and rabbit polyclonal antibody to transferrin receptor (TfR, 1:100) was from Abcam (Cambridge, MA). Goat antirabbit IgG (high and light chains) secondary antibody conjugated to HRP (used at a 1:5000 dilution) was purchased from Cell Signaling and goat antimouse secondary antibody coupled to HRP (used at a 1:20,000 dilution) was from Jackson ImmunoResearch (West Grove, PA).
Detergent-resistant membrane isolation
NSC34/ARnQ (n = 20 or 51) were maintained in DMEM supplemented with 5% steroid-depleted FBS (Invitrogen, France), 5 μg/ml ciprofloxacin (Euromedex, France), and 2.5 μg/ml fungizone, incubated with 1 μg/ml doxycycline for 48 h and treated with 1 μm testosterone for 24 h. Monolayer cells were washed with PBS and scraped on ice in TNET buffer [50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 5 mm EDTA, 1 mm EGTA, 1 mm sodium orthovanadate, 1 mm sodium pyrophosphate, 1% Triton X-100, and complete protease inhibitors]. Lysates were homogenized using a Potter (five times at 500 rpm) on ice, followed by centrifugation at 1000 × g (E818, Universal 16R; Hettich, France) for 10 min at 4 C. Supernatants were then centrifuged at 75,000 × g (TI75 rotor, ultracentrifuge; Beckman, France) for 2 h at 4 C. The pellet was then resuspended in TNET buffer and centrifuged at 75,000 × g for 30 min at 4 C. Supernatants [Triton X-100-soluble fraction (TS)] were combined. The Triton-insoluble (TI) pellet was resuspended in 250 μl n-octylglucoside (wt/vol) (fraction TIn-octyl). Proteins of total lysates and TS and TIn-octyl fractions were determined by the BCA method (Thermo Scientific, France) and 10 μg of total proteins were analyzed by Western blot. Anti-flotillin-1 (mouse; BD Biosciences, France) and anti-AR (N-20, rabbit; Santa Cruz, Germany) antibodies were used at a dilution of 1:500.
Lipid extraction
To measure cholesterol and phospholipid content, methanol and chloroform were added to TI and TS fractions and total lysate to the final concentration CHCl3/MeOH/H2O (4:8:3, vol/vol). Then, 0.2 vol 0.83% KCl was added, and 1 h was necessary to allow the mixture to settle into two phases. After collecting the organic phase, the aqueous phase was reextracted following the same procedure. Both organic phases were combined and evaporated under reduced pressure. Lipids were then resuspended in a mixture of CHCl3/MeOH 1:1. To analyze cholesterol content, lipids in CHCl3/MeOH 1:1 were separated by silica-gel thin-layer chromatography (TLC Silica gel 60; Merck, Germany) in a petroleum ether/ethyl ether/glacial acetic acid 80:15:0.5 mixture. Cholesterol standards (5, 10, 20, and 30 μg; Sigma-Aldrich, France) and each sample were spotted twice. Revelation was carried out using Macala's reagent (8% H3PO4, 3% CuSO4 in milli-Q H2O) at 120 C for 10 min. To measure phospholipid content, extracted lipids of each fraction (TI, TS, and total lysate) were dried and mineralized in 60% HClO4 at 120 C for 90 min. Inorganic phosphates were then quantified using ammonium molybdate/malachite green colorimetric assay. A solution of 4.2% (wt/vol) of ammonium molybdate in 4 m HCl was added to a solution of 0.1% (wt/vol) malachite green in ultrapure H2O. After stirring the mixture for 30 min at room temperature, 1 ml sulfuric acid was added on ice. Standards (0, 2.5, 5, 7.5, 10, 12.5, 15, 17.5, and 20 nmol inorganic phosphate in 50 μl) were achieved using a solution of 60% perchloric acid. Then, 50 μl of sample or standard was incubated with 950 μl malachite green/ammonium molybdate solution for 20 min, and absorbance at 660 nm was then measured.
Gradient density analysis
Cell fractionations were carried out according to the detergent-free method (37). NSC34/ARnQ (n = 20 or 51) were maintained in DMEM supplemented with 5% steroid-depleted FBS, 5 μg/ml ciprofloxacin (Euromedex, France), and 2.5 μg/ml fungizone, incubated with 1 μg/ml doxycycline for 48 h and treated with 1 μm testosterone for 24 h. Monolayer cells were washed with PBS and scraped in base buffer [20 mm Tris-HCl (pH 7.8), 250 mm sucrose] to which were added 1 mm CaCl2 and 1 mm MgCl2. Cells were pelleted by centrifugation at 1000 × g for 15 min and resuspended in 1 ml base buffer containing 1 mm CaCl2, 1 mm MgCl2, and protease inhibitors (Complete Mini; Roche). Cells were then lysed by 20 passages through a 22-gauge needle. After centrifugation at 3000 × g for 10 min at 4 C, 245 μl supernatant was added to 175 μl Optiprep solution (60% wt/vol solution of iodixanol in water; Sigma) resulting in a concentration of 25% Optiprep. This mixture was then poured on a bottom-loaded 2.1-ml discontinuous gradient of 0–20% OptiPrep solutions prepared in base buffer. The gradient was centrifuged for 90 min at 52,000 × g at 4 C (TLS-55 rotor, Optima Max-XP ultracentrifuge; Beckman, France). Gradients were fractionated into 10 fractions of 210 μl each. Total proteins in each fraction were determined using the BCA assay (Thermo Fisher, France). Proteins (10 μg) were resolved by SDS-PAGE. Anti-flotillin-1 (antibody dilution 1:500; BD Biosciences, France) and anti-TfR (1:100; Abcam, France) antibodies were used as Western blotting markers to identify the lipid raft-enriched fractions and the nonraft plasma membrane fractions, respectively.
Total c-jun quantification
After treatment with 1 μg/ml doxycycline for 48 h, cells were incubated in serum-depleted DMEM and treated with 10 nm testosterone for 2 h. Protein synthesis was inhibited by treatment with 0.1 mg/ml cycloheximide (100 mg/ml in dimethylsulfoxide; Sigma, France) for 1, 2, and 6 h. Cells were then rinsed with PBS and scraped in 500 μl lysis buffer supplemented with protease inhibitors (Roche) at 4 C. After centrifugation at 3000 × g for 5 min, proteins contained in supernatants were quantified using the BCA method and analyzed by Western blot.
Lactate dehydrogenase (LDH) release
NSC34/ARnQ (n = 20 or 51) were seeded in a 96-well plate in DMEM supplemented with 5% charcoal-stripped heat-inactivated FBS, 5 μg/ml ciprofloxacin (Euromedex), and 2.5 μg/ml fungizone at a density of 7000 cells per well. At 24 h, cells were treated with 1 μg/ml doxycycline for 48 h. Then cells were maintained in serum-depleted DMEM and incubated for 1 h with 5 μm SP600125 (prepared in milliQ H2O/absolute ethanol 9:1 at a concentration of 5 mm; Sigma) or vehicle. Cells were then treated with 10 nm testosterone for 24 h. For determination of LDH release, four wells per experimental condition were used. Two wells for each condition were treated with 1% Triton X-100 for 30 min. For each well, 100 μl culture medium was collected and centrifuged at 1000 × g for 30 min. According to the manufacturer's instructions, supernatants were incubated at 37 C with the LDH kit (Roche, France) for 90 min. LDH activity was determined by measurement of OD at λ = 655 nm and at λ = 490 nm.
Results
Although neither cytoplasmic aggregates nor nuclear inclusion bodies are observed after the induction of AR51Q, NSC34 viability is affected
In a first step, we validated the expression of the different types of AR(n)Q (n = 0, 20, or 51) in NSC34 by Western blotting and immunofluorescence (Fig. 1A). As shown in Fig. 1A, all cell lines appear to express comparable levels of AR, and the full-length protein size is well correlated to the polyQ size. As expected, no AR(n)Q was detectable in NI NSC34/AR(n)Q (Fig. 1, B and C), confirming the inducibility of the system. Because cells were maintained in a sterol-free medium, nuclear translocation of the AR required addition of testosterone (Fig. 1, D and E). A physiological concentration of 10 nm was used in all our experiments for AR activation. It is worth noticing that neither cytoplasmic aggregates nor nuclear inclusions were detected by immunofluorescence.
Fig. 1.

Characterization of the cellular models. A, Western blot analysis of stably transfected NSC34/AR(n)Q (n = 0, 20, or 51). NSC34/AR(n)Q were treated with 1 μg/ml doxycycline (I) or vehicle (NI) for 48 h and then incubated with 10 nm testosterone (+T) for 24 h. B, Validation of inducible expression of AR(n)Q. Cells were treated with doxycycline or with vehicle for 48 h and then treated with 10 nm testosterone or with vehicle for 24 h. Total proteins (10 μg) were loaded and calibrated against β-actin. Western blots were probed with the AR(N-20) polyclonal antibody or with the AR(C-19) polyclonal antibody. C–E, Expression and localization of AR(51)Q in NSC34 determined by immunofluorescence. Cells were treated with doxycycline (I) or vehicle (NI) for 48 h and then incubated for 24 h with vehicle or 10 nm testosterone (+T). Immunofluorescence were carried out using the AR(N-20) polyclonal antibody (red). Nuclei were stained by Hoechst (blue). Scale bar, 20 μm. MW, Molecular weight.
In NSC34 expressing AR20Q, we observed a growth/survival rate reduction of 40% in the presence of testosterone and of 25% in the absence of testosterone (Fig. 2A) when compared with NI and NI+T NSC34/AR20, respectively. In NSC34 expressing AR51Q, either in the presence or absence of testosterone, we observed a growth/survival reduction of 65% (Fig. 2B) when compared with NI and NI+T NSC34/AR51Q. We also estimate that the cell growth/survival of I+T NSC34/AR51Q compared with I+T NSC34/AR20Q is significantly reduced (Fig. 2, A and B). In conclusion, testosterone treatment had an impact on cell growth/viability of AR20Q-expressing cells but not on AR51Q-expressing cells. In the case of AR51Q-expressing cells, the growth/viability effect was testosterone independent.
Fig. 2.
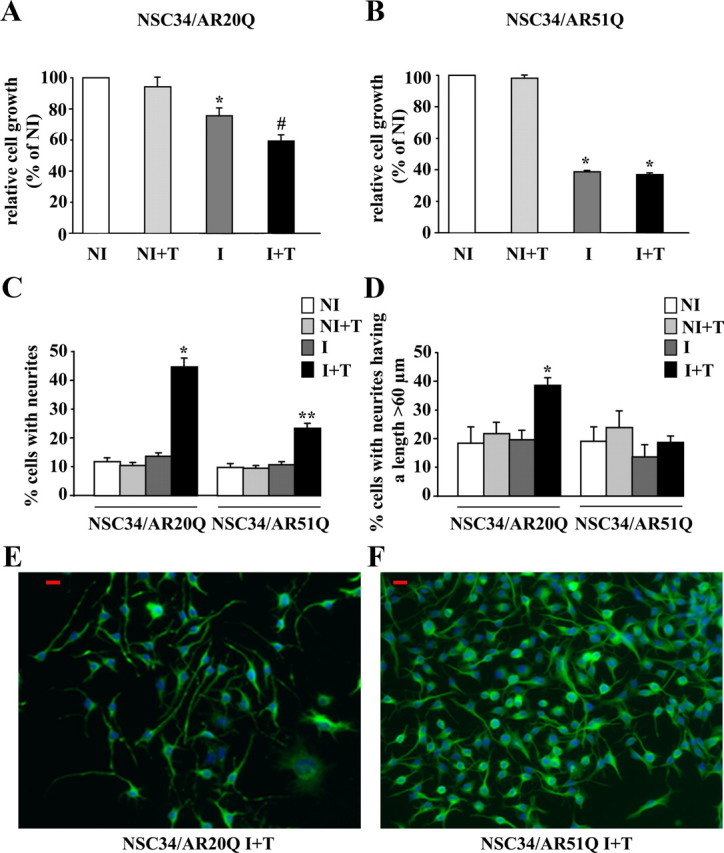
Influence of polyQ tract length on cell viability/growth and neurite outgrowth: AR20Q vs. AR51Q. A and B, Effects of polyQ region length on cell viability measured by MTT assay. NSC34/AR(n)Q were incubated for 48 h with doxycycline or with vehicle. Then cells were treated with 10 nm testosterone or vehicle for 242 h. Cell proliferation and viability are expressed as a percentage relative to NI control cells. Results are presented as means ± sem (n = 8). A, Influence of AR20Q expression on NSC34 viability/growth. *, P < 0.05, I vs. NI and NI+T NSC34/AR20Q ; #, P < 0.01, I+T vs. NI and NI+T NSC34/AR20Q, ANOVA test. B, Influence of AR51Q expression on NSC34 growth. *, P < 0.001, NI vs. I+T NSC34/AR51Q, ANOVA test. C and D, Influence of polyQ tract on neurite outgrowth. After 48 h after exposure to doxycycline (I) or vehicle (NI), NSC34/AR(n)Q (n = 20 or 51) were depleted in serum and treated with 10 nm testosterone (+T) or vehicle for 24 h. Results are represented as means ± sem of pooled data from three separate experiments. C, Influence of AR(n)Q expression and testosterone treatment on percentage of neurite-bearing cells. Asterisks indicate significant difference compared with control (NI and NI+T) or induced cells (I without testosterone): *, P < 0.01; **, P < 0.001 (ANOVA test). D, Influence of AR(n)Q expression and testosterone treatment on cell number with a longest neurite longer than 60 μm. *, P < 0.01, I+T NSC34/AR20Q vs. NI, NI+T, and I NSC34/AR20Q (ANOVA test). E and F, Immunofluorescence of serum-depleted I+T NSC34/AR(n)Q (n = 20 or 51) were performed using the peripherin antibody detected with Alexa 488-conjugated secondary antibody. Nuclei are stained with Hoechst. Peripherin, green; nuclei, blue; red scale bars, 20 μm.
The expression of AR51Q affects neurite outgrowth of NSC34
Because androgens are known to have trophic effects on motor neurons by regulating differentiation and neurite outgrowth (38), we sought to determine whether the reduced cell proliferation of I+T NSC34/AR20Q and I+T NSC34/AR51Q is correlated to cell differentiation. To answer this question, we investigated the influence of the AR(n)Q on neurite outgrowth in normal culture conditions (Fig. 2). Although NI, NI+T, and I NSC34/AR20Q cells do not display a marked neuron-like phenotype (only 10% of cells display short neurites), I+T NSC34/AR20Q cells show a significant increase of the number of bipolarized cells with longer neurites after 96 h testosterone treatment (Fig. 2, C and E). This result indicates that the action of androgen on neurite growth is directly linked to AR expression and its activation by testosterone. Compared with AR20Q, the expression of AR51Q in NSC34 leads to an alteration of neurite outgrowth (Fig. 2C): 26.2 ± 3% of cells have neurites in I+T NSC34/AR51Q vs. 45.2 ± 1.3% in I+T NSC34/AR20Q (Fig. 2C). Moreover, in I+T NSC34/AR51Q, neurites are shorter. Among the I+T NSC34/AR51Q with neurites, 18.7 ± 2.2% have a length longer than 60 μm (Fig. 2D), whereas among I+T NSC34/AR20Q with neurites, 38.5 ± 2.7% have a length longer than 60 μm (Fig. 2D). Moreover, neurites of I+T NSC34/AR51Q are thick and short and numerous per cell. In addition, I+T NSC34/AR51Q cells display multipolar and nonpolarized branching patterns (Fig. 2, F and G). In conclusion, although I+T NSC34/AR51Q and I NSC34/AR51Q do not exhibit a growth/viability, their neurite outgrowth rates differ (Fig. 2, A–D). However, we cannot conclude that untreated AR20Q-expressing cells are equivalent to treated AR51Q. As a matter of fact, Fig. 2 shows that growth reduction is more pronounced for I+T NSC34/AR51Q (65%) than for I NSC34/AR20Q (25%). In addition the percentage of cells with neurites is higher for I+T AR51Q (22%) than for I AR20Q (12%).
To check that the effects on cell growth and neurite outgrowth are the result of an extended polyQ, we used the AR0Q as a control. Contrary to AR20Q and AR51Q, we observed that the expression of AR0Q in presence of its ligand does not affect cell growth/viability (Fig. 3A). Also, the percentage of cells with elongated neurites (Fig. 3B) is lower in induced AR0Q cells (27 ± 3.9%) than in AR20Q-expressing cells (57 ± 2.1%).
Fig. 3.

Effects of AR0Q on cell viability and neurite elongation. A, At 48 h after exposure with doxycycline or vehicle, NSC34/AR0Q were treated with 10 nm testosterone or vehicle for 120 h. *, P < 0.001 (ANOVA test). B, Effect of AR0Q expression at 24 h after testosterone treatment (10 nm) on percentage of neurite-bearing cells in the presence of serum (5%). *, P < 0.01 (ANOVA test); *, significant difference compared with I+T NSC34/AR20Q at P < 0.05 (ANOVA test). C–E, After 48 h after exposure to doxycycline (I), NSC34/AR(n)Q (n = 0 or 20) were depleted in serum and treated with 10 nm testosterone (+T) for 24 h. C and D, Percentage of neurite-bearing cells (C) and the percentage of cells with the longest neurite having a length greater than 60 μm (D). *, P < 0.05, ANOVA test. E, Immunofluorescence of serum-depleted I+T NSC34/AR0Q was performed using the peripherin antibody detected with Alexa 488-conjugated secondary antibody. Nuclei are stained with Hoechst. Peripherin, green; nuclei, blue; red scale bar, 20 μm.
This last observation prompted us to analyze neurite outgrowth in serum-free culture conditions. Interestingly, after 24 h of serum deprivation and treatment with 10 nm testosterone, NSC34/AR0Q growth is reduced and neurite outgrowth appears exacerbated. Among the 32.1 ± 1.5% of I+T NSC34/AR0Q cells with neurites, 53.1 ± 2% have a length longer than 60 μm (Fig. 3, B and C). In the presence of testosterone and in serum deprivation conditions, NSC34/AR0Q and NSC34/AR20Q show similar long polarized unidirectional neurites (Figs. 2F and 3E). These results confirm that the limited number of I+T NSC34/AR0Q with neurites in the presence of serum (Fig. 3C) is due to a greater proliferation rate. Therefore, the reduced proliferation rate observed in NSC34/AR0Q and NSC34/AR20Q could be related to the observed morphological changes, which reflect differentiation. From these data, we conclude that polyQ tract elongation leads to a loss of function characterized by a testosterone-dependent reduction in neurite outgrowth and to the emergence of a testosterone-independent toxic function characterized by a decrease of cell viability.
Both AR51Q and AR20Q are localized in lipid rafts
These last data demonstrate that NSC34/AR51Q degenerate in the presence of testosterone, although no insoluble aggregates are formed. Because the AR can initiate nongenomic signaling in addition to its transcriptional activity and because the loss of AR function is important in SBMA (15), we hypothesized that rapid nongenomic androgen actions through membrane-bound AR could also be disturbed by polyQ elongation.
We therefore determined whether or not normal AR and AR51Q are present in the NSC34/AR(n)Q plasma membrane. Confocal microscopy immunofluorescence revealed that AR20Q and AR51Q stained by anti-AR N-terminal (N-20) antibody not only translocated into the nucleus under testosterone influence but also were localized at the plasma membrane (red arrows, Fig. 4, A and B). The punctuated aspect of immunofluorescence at the plasma membrane (Fig. 4, C and D) as well as the existence of confined areas where staining is the strongest (Fig. 4, C and D) suggest that the AR(n)Q localize within membrane microdomains. To verify the nature of those microdomains, a subcellular fractionation of NSC34/AR(n)Q treated with 1 μm testosterone for 24 h was carried out. To avoid generating artificial clusters of microdomains and proteins (39), subcellular fractions were separated using a detergent-free method (37). Gradients were partitioned into 10 fractions, and each fraction was resolved by Western blot. AR20Q and AR51Q are predominantly present in heavy cytosolic fractions 9 and 10 (Fig. 5, A and B). Both NSC34/AR20Q and NSC34/AR51Q also colocalize with flotillin-rich fractions (Fig. 5, A and B). These fractions corresponding to lipid rafts that do not contain caveolin-1 (data not shown) are not contaminated by plasma membrane, because TfR labeling does not overlap with flotillin-1-rich fractions. These colocalization studies indicate that both AR20Q and AR51Q localize within lipid rafts of NSC34. To determine whether this lipid raft localization is testosterone dependent, we carried out the same analysis using nontreated NSC34/AR20Q and NSC34/AR51Q samples (Fig. 6). As shown in Fig. 6, A and B, the AR are present in fraction 4 of the gradient, but not markedly so, and a correlation present between the AR and flotillin profiles in Fig. 5B is missing in Fig. 6. To confirm the localization of the AR in lipid rafts after testosterone activation, a method based on the insolubility of lipid rafts in 1% Triton X-100 was used to isolate detergent-resistant membranes.
Fig. 4.

Localization of AR(n)Q within plasma membrane by immunofluorescence. Induction of AR(n)Q expression through treatment with doxycycline for 48 h in NSC34/AR(n)Q was followed by incubation with 10 nm testosterone for 2 h. A and B, Immunofluorescence of AR(n)Q (red). Nuclei (blue) are stained with Hoechst 33258. These images were obtained by confocal microscopy. Discontinuous membrane staining is identified using red arrows. C and D, Pixel quantification using Imaris version 7.0.0 software realized from previous confocal images. Points represent zones where AR(n)Q staining shows the highest intensity.
Fig. 5.

Localization of AR(n)Q within lipid rafts. A and B, Analysis of AR(n)Q in density gradient fractions. Both I NSC34/AR20Q and I NSC34/AR51Q were depleted in serum and treated with 1 μm testosterone for 24 h. Ten fractions were collected from the top of the gradient, and 10 μg of proteins of the last nine fractions was resolved by SDS-PAGE. Western blots were probed with anti-AR(C-19), anti-flotillin-1, and anti-TfR antibodies. C and D, Localization of AR(n)Q within lipid rafts by isolation of detergent-resistant membrane. Treatment of NSC34/AR20Q and NSC34/AR51Q with doxycycline for 48 h was followed by exposure with 1 μm testosterone for 24 h. Proteins (10 μg) of the total cell lysate (Tot.) and TS and TIn-octyl fractions were resolved by SDS-PAGE. Western blots were probed with anti-flotillin-1 (C) and anti-AR(N20) (D).
Fig. 6.

Density gradient of I NSC34/AR(n)Q. A and B, AR(n)Q localization in Optiprep density gradient fractions of I NSC34/AR(n)Q (without testosterone). Both I NSC34/AR20Q and I NSC34/AR51Q were depleted in serum and treated with 1 μm vehicle for 24 h. Ten fractions were collected from the top of the gradient, and 10 μg of proteins of the last nine fractions was resolved by SDS-PAGE. Western blots were probed with anti-AR(C-19), anti-flotillin-1, and anti-TfR antibodies.
The TS and TIn-octyl fractions were isolated by ultracentrifugation of NSC34/AR(n)Q treated with 1 μm testosterone for 24 h (27). Cholesterol and phospholipids of these fractions were quantified. The cholesterol/phospholipid ratio of 0.65 ± 0.05 and 3.6 ± 0.2 in the TS and TIn-octyl fractions, respectively, underline that TIn-octyl fractions of both I+T NSC34/AR20Q and I+T NSC34/AR51Q are rich in lipid rafts. The lipid raft nature of the TIn-octyl fractions is confirmed by the enrichment in flotillin-1 (Fig. 5C). Then AR distribution within TS and TIn-octyl fractions was determined by Western blot (Fig. 5D). As expected, the majority of AR is found in the TS fractions. However, in the absence of testosterone, none of the TIn-octyl fractions contains AR20Q or AR51Q, whereas both AR20Q and AR51Q are detected in TIn-octyl fractions after treatment with testosterone (Fig. 5D). Hence, testosterone stimulates the passage of AR(n)Q to lipid rafts. The Optiprep gradient isolation of lipid rafts from I NSC34/AR(n)Q confirmed that AR(n)Q localization in lipid rafts is enhanced by testosterone treatment (data not shown). It is noteworthy that polyQ modification does not prevent AR51Q localization in lipid rafts.
These data suggest that a part of the AR localize to the plasma membrane and migrate to lipid rafts after testosterone treatment.
Activation of JNK/c-jun pathway by membrane-bound AR20Q
The presence of AR in lipid rafts prompted us to analyze which signaling pathways leading to neurite outgrowth and cell survival could be activated by the AR20Q in a testosterone-dependent manner. Subsequently, the ability of raft-bound AR51Q to activate these pathways was estimated. Because c-jun has been reported to be involved not only in apoptosis but also in neurite outgrowth and cell proliferation (33–35), we first examined whether testosterone could promote c-jun activation via membrane-bound AR(n)Q.
NSC34/AR(n)Q were depleted of serum and incubated with 10 nm testosterone for short times, i.e. from 0.5–4 h, to capture early nongenomic events. Significant c-jun phosphorylation on Ser73 was detected in I NSC34/AR20Q as soon as 1 h after testosterone addition (Fig. 7A). On the contrary, no c-jun phosphorylation was observed in I NSC34/AR51Q after testosterone treatment. Because no increase of c-jun phosphorylation occurred in NI+T NSC34/AR20 and NI+T NSC34/AR51Q after testosterone (data not shown), we conclude that upon ligand binding, AR20Q triggers c-jun phosphorylation, whereas AR51Q does not.
Fig. 7.

Influence of testosterone on JNK/c-jun pathway. A and B, Both serum-depleted I NSC34/AR20Q and I NSC34/AR51Q were treated with 10 nm of testosterone at the indicated times. Equal amounts of total proteins (30 μg) per well were resolved by SDS-PAGE, and Western blots were probed with antibodies against phospho-c-jun(Ser73), phospho-SAPK/JNK(Thr183/Tyr185), total c-jun, and total JNK. The c-jun and JNK phosphorylation levels from the autoradiograph were normalized to total c-jun or total JNK. A, Testosterone effects on c-jun(Ser73) phosphorylation levels in I NSC34/AR20Q (left) and I NSC34/AR51Q (right). Histograms represent the mean ratio of phospho-c-jun(Ser73) level to total c-jun level ± sem of three independent experiments at indicated times of exposure with testosterone. #, P < 0.05, t test. B, Testosterone effects on JNK1–3 activity in I NSC34/AR20Q (left) and I NSC34/AR51Q (right). Histograms represent the mean ratio of phospho-SAPK/JNK(Thr183/Tyr185) level to total SAPK/JNK level (n = 3 independent experiments) at indicated times of testosterone treatment. Error bars represent ± sem. #, P < 0.05; *, P < 0.01, t test.
The c-jun is activated by phosphorylation of Ser63 and Ser73 (and also on Thr91 and Thr93) by JNK. Therefore, to gain further insight into the regulation of c-jun, JNK phosphorylation kinetics were analyzed. Sustained JNK activation in NSC34 expressing AR20Q after testosterone induction corroborates with c-jun phosphorylation (Fig. 7). In contrast, the absence of c-jun phosphorylation in I+T NSC34/AR51Q can be explained by the absence of phosphorylated JNK (Fig. 7).
To determine the role of JNK activation in NSC34 phenotypes elicited by AR20Q activation, the effects of JNK inhibition by SP600125 on neurite outgrowth and cell viability were analyzed (Fig. 8). SP600125 (5 μm) leads to peripherin accumulation at dendrite extremities in NSC34/AR20Q (Fig. 8A). However, no significant effect of JNK inhibition on the percentage of cells with neurites was put in evidence (data not shown). SP600125 treatment leads also to a decrease of I+T NSC34/AR20Q viability as determined by MTT assay (Fig. 8B) and LDH release (Fig. 8C). SP600125 has no effect on NSC34/AR51Q viability (Fig. 8, B and C). In conclusion, JNK seems to play a crucial role in the AR20Q-dependent differentiation process in NSC34
Fig. 8.

Influence of JNK inhibition on NSC34/AR(n)Q. A, Influence of JNK inhibition on neurite morphology in NSC34/AR(n)Q. Immunofluorescence of peripherin in serum-depleted I NSC34/AR(n)Q (n = 20 or 51) treated with 10 nm testosterone together with 5 μm SP600125 (I+T+SP) or with vehicle (I+T). Peripherin is in red, and nuclei are stained blue. These images are representative of several images from three different experiments. Red scale bar, 20 μm. B, Influence of JNK inhibition on cell viability in NSC34/AR(n)Q, measured by MTT assay. Serum-depleted I NSC34/AR(n)Q (n = 20 or 51) were treated with 10 nm testosterone for 24 h together with 5 μm SP600125 or with vehicle control. The mean of absorbance (λ = 570 nm) in four wells ± sem is plotted. *, P < 0.05, ANOVA test. C, LDH release measured by absorbance at λ = 490 nm and λ = 655 nm in serum-depleted I NSC34/AR(n)Q (n = 20 or 51) treated with 10 nm testosterone for 24 h together with 5 μm SP600125 or with vehicle control. *, P < 0.05, ANOVA test.
Stability of c-jun is affected by AR51Q expression
Although c-jun is activated by JNK phosphorylation, its stability also plays a regulatory role. To determine the time course of c-jun elimination, I NSC34/AR20Q and I NSC34/AR51Q cells were treated for 2 h with 10 nm testosterone in serum-free conditions and then with cycloheximide to inhibit protein synthesis. In comparison with I+T NSC34/AR20Q, immunoblot analysis of c-jun showed that c-jun is not detectable anymore in I+T NSC34/AR51Q after 2 h cycloheximide treatment (Fig. 9A). Therefore, the c-jun level decreases faster in I+T NSC34/AR51Q than in I+T NSC34/AR20Q. However, c-jun expression seems to actually go up in response to testosterone stimulation of NSC34/AR51Q (Fig. 7A). That increase could be considered as the cell's global transcriptional answer to the reduced c-jun stability. The c-jun expression could be regulated by other MAPK pathways such as P38 or ERK (38).
Fig. 9.

Influence of testosterone on Akt and GSK3α activity. A, Testosterone effects on total c-jun. Serum-depleted I NSC34/AR20Q and I NSC34/AR51Q were treated with 10 nm testosterone for 2 h and then incubated with cycloheximide (CHX, 100 μg/ml) at various times (1, 2, and 6 h). Total proteins (30 μg/well) were subjected to SDS-PAGE, and Western blots were carried out using anti-total c-jun (60A8) antibody. Levels of total c-jun were normalized to β-actin. B, GSK3α and Akt phosphorylation levels. Both serum-depleted I NSC34/AR20Q and I NSC34/AR51Q were treated with 10 nm testosterone (Testo) at the indicated times. Data of three independent experiments are expressed as the ratio of phospho-GSK3α to total GSK3α and the ratio of phospho-Akt (Ser473) to total Akt. *, P < 0.05; **, P < 0.05; #, P < 0.01, t test. C, GSK3α and Akt phosphorylation levels in noninduced cells. Both serum-depleted NI NSC34/AR20Q and NI NSC34/AR51Q were treated with 10 nm testosterone (Testo) at the indicated times. All data of relative activity are expressed in comparison with untreated cells (time zero). Data are representative of two experiments. D, Inhibitory phosphorylation of GSK3α induced by AR20Q is PI3K independent. Serum-depleted I NSC34/AR20Q cells were treated with vehicle (I) or with 5 μm LY294002 alone or together with 10 nm testosterone. Equal amounts of total proteins (30 μg) per well were resolved by SDS-PAGE (B–D), and Western blots were probed with antibodies against phospho-GSK3α(Ser21), phospho-Akt(Ser473), total GSK3α, and total Akt. Histograms represent quantification of phospho-GSK3α and phospho-Akt(Ser473) in each sample.
Stability of c-jun is under the control of GSK3, inducing c-jun phosphorylation of Thr239, which is followed by interaction with the F-box&WD domain repeated 7 (FBW7) ubiquitin ligase, leading to c-jun ubiquitination and proteasome degradation (35). Therefore, in a next step, we analyzed the effect of testosterone on GSK3α activity in I NSC34/AR20Q and I NSC34/AR51Q (Fig. 9, B and C).
At time zero, we observed that expression of the AR20Q alone induced a decrease in Akt and GSK3α phosphorylations with respect to both NI AR20Q and AR51Q (Fig. 9, B and C). After testosterone addition to I NSC34/AR20Q, Akt and GSK3α were phosphorylated on Ser473 and Ser21, respectively, after 1 h (Fig. 9B). Conversely, expression of the AR51Q alone did not decrease Akt phosphorylation with respect to NI (Fig. 9, B and C), and after testosterone addition, a significant decrease of Akt phosphorylation was observed (Fig. 9B). However, GSK3α phosphorylation was reduced after AR51Q expression with respect to NI (Fig. 9, B and C).
In conclusion, c-jun degradation, enhanced by GSK3α, is reduced in I+T NSC34/AR20Q because of the inhibitory phosphorylation of GSK3α.
GSK3α inactivation by membrane-associated AR20Q bypasses PI3K
Because GSK3α is a target of Akt, we asked whether the transient inactivation of GSK3α induced by AR20Q results from an enhanced Akt activity. We observed that PI3K inhibition by LY294002 in I+T NSC34/AR20Q had an effect neither on cell viability nor on neurite outgrowth (data not shown). We therefore checked whether GSK3α phosphorylation induced by AR20Q in presence of testosterone is mediated by PI3K. LY294002 (5 μm) reduced GSK3α phosphorylation in the absence of testosterone, confirming that GSK3α phosphorylation is partly controlled by PI3K and that LY294002 is able to inhibit PI3K during serum deprivation (Fig. 9D). Although PI3K is still inhibited by LY294002, the addition of testosterone (10 nm) results in an increase of GSK3α phosphorylation (Fig. 9D). From these data we conclude that GSK3α phosphorylation mediated by testosterone-activated AR20Q is PI3K independent. Also, the reduction in GSK3α phosphorylation mediated by the expression of AR51Q in the absence of testosterone is not related to a decrease of Akt phosphorylation (Fig. 9, B and C). These data are in accordance with the observation that GSK3α phosphorylation could bypass the PI3k/Akt pathway.
Discussion
Abnormal protein accumulation has been considered to be responsible for polyQ pathology by triggering cell stress responses, such as estrogen receptor stress, mitochondrial stress, and proteasome impairment, inducing apoptosis (40, 41). Large extended polyQ (>90 Q) tracts, expressed in a noninducible manner, have been shown to produce aggregates (42, 43). In our model, the inducible expression of a 51-Q-extended AR, even in serum deprivation conditions, produces neither insoluble cytoplasmic aggregates nor nuclear inclusions. Moreover, none of the cellular stress responses mentioned above were detected as initial molecular events after AR expression (unpublished results).
In contrast with the expression of a normal AR20Q, the inducible expression of the mutated AR51Q is associated with both a decrease in cell survival and an alteration of neurite outgrowth. Interestingly, androgens are known to exert trophic effects on neuronal cells (38). They induce NSC34 cells transfected with AR20Q to adopt a morphology close to those of motoneurons, and they improve cell survival during serum deprivation (44). As confirmed by our work, the activation of the normal AR by its ligand increases in NSC34 the number of neurites that can reach a length five to 10 times the soma length (45). Androgens have also been reported to regulate the expression of several proteins involved in neuritogenesis such as βII and βIII tubulin (46) or neuritin, a glycophosphatidylinositol-anchored protein known to be up-regulated in an early stage of neuronal arbor growth and synaptogenesis (45). Alteration of neuritogenesis has already been reported as the consequence of the polyQ elongation (14, 44, 47). Transgenic animal models of Huntington's disease also showed an alteration of neurites (48, 49).
Nevertheless, only the reduction of neurite outgrowth is dependent on both AR51Q expression and testosterone treatment. As a matter of fact, in the absence of testosterone, growth is lower in AR51Q-expressing cells than in AR20Q-expressing cells (Fig. 2, A and B).
According to these initial data, we wonder whether an alternative deregulation of the AR, different from aggregate formation, occurs in SBMA. Because of the reported nongenomic activities of the AR (50), we asked whether or not rapid AR-mediated nongenomic actions might contribute to neuronal survival and differentiation and account for SBMA onset (Fig. 10).
Fig. 10.
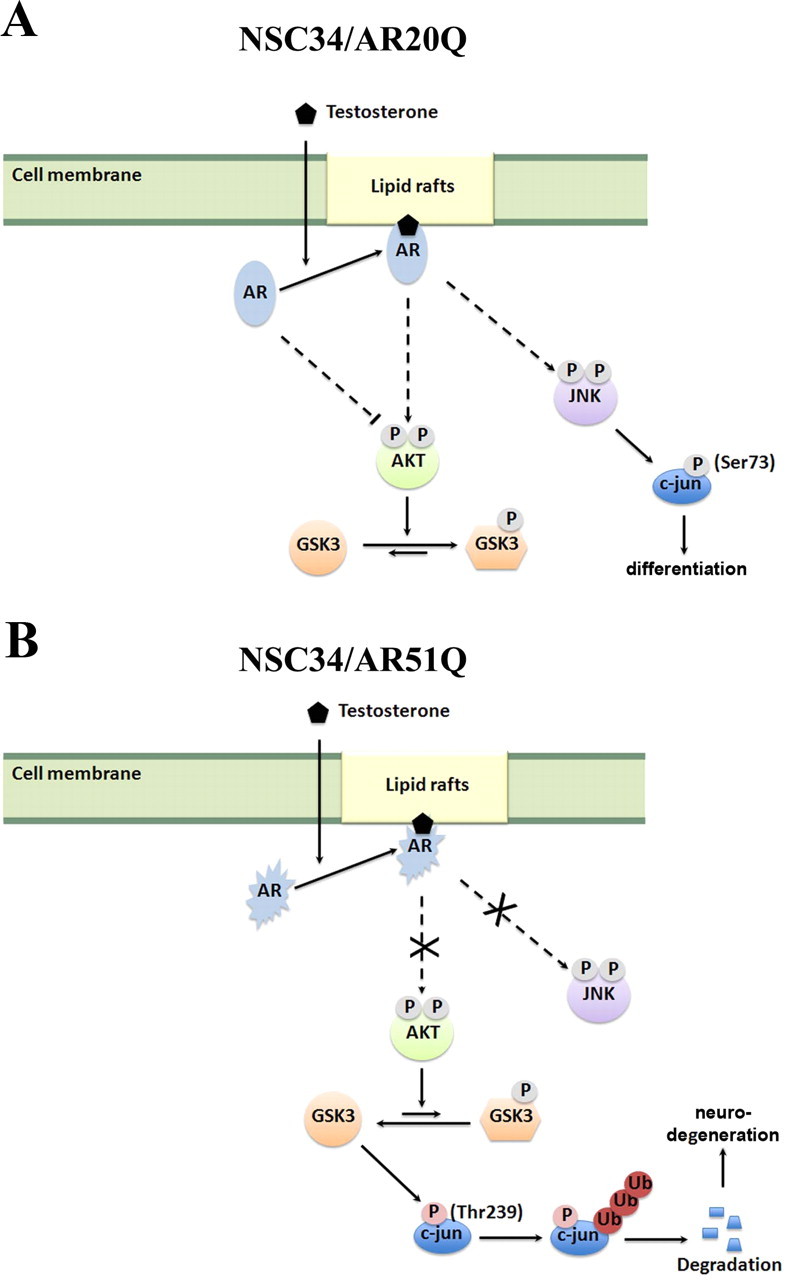
Scheme of nongenomic activation of AR20Q and AR51Q. Solid lines indicate direct action on the target. Dotted lines indicate indirect action. Ub, Ubiquitin.
We first demonstrated that both AR20Q and AR51Q were present within flotillin-1-enriched lipid rafts of NSC34. Regarding these observations, we conclude that polyQ tract elongation does not influence its localization at the membrane. So far, no data put in evidence that AR is anchored to the membrane, and the mechanisms controlling the recruitment of AR to the plasma membrane or to the lipid rafts remain unknown. It has been postulated that recruitment of AR to the plasma membrane might result from its direct interaction with a membrane-bound component such as caveolin-1 (51) and that palmitoylation is required (18). However, in our NSC34 model as well as in prostate cancer cells (LNCaP) (27), AR might be recruited to lipid rafts in a caveolin-independent manner, because neither of these cell lines express caveolin-1. By comparison between I and I+T NSC34/AR(n)Q (n = 20 or 51), we have shown that the recruitment of AR at the plasma membrane is stimulated by androgen. Neither AR20Q nor AR51Q was present in detergent-resistant membrane fractions (TIn-octyl) of I NSC34/AR(n)Q, which were not treated with testosterone. The subcellular fractionations of I and I+T cells by Optiprep density gradient suggest that levels of AR(n)Q (n = 20 or 51) in flotillin-1-positive fractions were enhanced by testosterone treatment. This observation is in agreement with previous studies, which indicated that interaction between AR and Src occurs after androgen exposure (52–54). Moreover, localization of AR in lipid rafts of LNCaP cells was enhanced within minutes in the presence of R1881 (27).
Expression of AR20Q alone reduced growth/viability by 25%, whereas neurite outgrowth was unmodified. In parallel, AR20Q alone induced a decrease in both Akt and GSK3α phosphorylations. These data could be related to a constitutive inhibitory effect of AR20Q on Akt phosphorylation during serum deprivation and to the canonical regulation of cell proliferation by Akt. After testosterone addition, Akt, GSK3α, JNK, and c-jun(Ser73) phosphorylations are increased. We propose that testosterone initiates a cell survival pathway (45% of cell survival) via Akt phosphorylation (55, 56) and differentiation (neurite outgrowth) via GSK3α, JNK, and c-jun activation. The time necessary for AR20Q to fulfill its transcriptional role including dimerization, DNA binding, and recruitment of coregulators and transcriptional machinery is not compatible with the rapid phosphorylation of c-jun (57, 58). We conclude that the activation of the JNK/c-jun pathway is due to membrane-bound AR20Q signaling and not to the transcriptional activity of AR20Q.
Expression of AR51Q alone reduces growth/viability by 65%, whereas neurite outgrowth is unmodified. However, this growth reduction is not associated with reduced Akt phosphorylation. We propose that under serum deprivation, AR51Q fails to control Akt phosphorylation and cell proliferation, resulting in more drastic reduction of cell viability (65%) with respect to AR20Q (25%). After testosterone addition, activated GSK3α phosphorylates c-jun on Ser239 leading to c-jun degradation and inhibition of neurite outgrowth. Because in NI+T NSC34/AR20Q and in NI+T NSC34/AR51Q, a decrease in Akt and GSK3α phosphorylations was observed, it is not possible to conclude whether AR51Q loses function or functions in a way opposite to AR20Q. All these data taken together suggest that the impairment of AR signaling caused by the polyQ expansion is an important event in SBMA.
The inability of AR51Q to activate the JNK/c-jun pathway could explain phenotypes of viability and neurite elongation induced by the expression of AR51Q in NSC34. JNK1, -2, and -3 are known to be involved in neurodegeneration and in neurite outgrowth (59). It has also been shown that JNK might be involved in initiation of neurite outgrowth and axonal regeneration of dorsal root ganglion neurons (60). JNK1−/− mice show hypophosphorylation of MAP2 and MAP1B, leading to a progressive disruption of microtubules within neurites, because it compromises their ability to bind microtubules (61). In our model, both inhibition by SP600125 in I+T NSC34/AR20Q and the absence of JNK phosphorylation in I+T NSC34/AR51Q induce cell death. Moreover, SP600125 does not have any effect on I+T NSC34/AR51Q viability. To visualize neurite outgrowth, we used the peripherin marker known to intercalate with MAP2 (59). We observed that JNK inhibition by SP600125 in I+T NSC34/AR20Q induces impaired peripherin organization, whereas the absence of JNK phosphorylation in I+T NSC34/AR51Q leads to neurite outgrowth reduction. Although both phenotypes show a default in the neurite structure, they were different. Therefore, we conclude that if JNK plays a crucial role in the AR20Q differentiation in NSC34, the observed phenotype difference may reflect a more complex action of AR20Q than only activating JNK.
As a matter of fact, GSK3α phosphorylation on Ser21 in the presence of testosterone was largely insensitive to PI3K inhibition, we conclude that phosphorylation of GSK3α bypasses PI3K. GSK3α should therefore be regulated by other signaling pathways such as Erk, Wnt, AGC kinases (cAMP-dependent protein kinase/protein kinase G/protein kinase C), p70 ribosomal kinase, or p90 ribosomal S6 kinase (62). Enhanced androgen-dependent Akt activity might be a way for membrane-associated AR20Q to regulate the classic genomic AR pathway. Indeed, it has been demonstrated that enhanced Akt phosphorylation induces AR phosphorylation and thereby influences its transcriptional activity (63). In addition, inhibition of protein synthesis by cycloheximide indicated that c-jun protein levels decrease faster in I+T NSC34/AR51Q than in I+T NSC34/AR20Q. According to observations from the literature, c-jun stability depends on GSK3α activity. The c-jun phosphorylated on both Thr239 and Ser243 becomes the target for the FBW7 ubiquitin ligase leading to c-jun degradation via proteasomes (64). Both phosphorylations are required to allow the recruitment of the FBW7 ubiquitin ligase. Recognizing Ser243 phosphorylation as a priming event, active GSK3α (not phosphorylated on its Ser21) mediates c-jun phosphorylation on its Thr239 (65). Inhibition of GSK3α through its phosphorylation on Ser21 by PI3K/Akt or Erk is sufficient to enhance c-jun stability (65). The c-jun ubiquitination by the E3 ligase will therefore be interesting to evaluate as a secondary event after membrane-associated AR activation.
Because nuclear inclusions containing expanded polyQ are considered to be responsible for the pathology and occur in an androgen-dependent manner, therapeutic approaches for SBMA are based on the elimination of protein aggregates by androgen blockade (66). Thus, male AR97Q mice were treated with leuprorelin, an LHRH agonist that reduces testosterone release from the testis. It led to a decrease of insoluble AR fragments, a longer life span, and an amelioration of symptoms (67). However, flutamide, an androgen antagonist that allows nuclear translocation of AR but that does not promote AR-mediated transcription, did not improve muscle atrophy, pathological features, or reduced nuclear AR accumulation (67). Our study demonstrates that the elongated polyQ tract induces a deregulation of signaling pathways initiated by an AR localized in lipid raft microdomains. However, it has been determined that depending on whether a membrane-bound AR or classic AR is activated, androgens might elicit opposing actions. Dihydrotestosterone protected primary cortical astrocytes from iodoacetic acid-induced toxicity, whereas dihydrotestosterone conjugated to BSA inhibited Akt signaling, induced caspase-3/7 activity, and enhanced iodoacetic acid-mediated cell death (24, 25). Moreover, mutated AR can elicit ligand-independent activation (68, 69). These results suggest that both roles of AR must be taken into account to understand the consequences of polyQ tract expansion. This is all the more important given that agonists and antagonists of transcriptional AR do not play the same role upon binding membrane-bound AR (70, 71).
Acknowledgments
We thank Didier Petite and Clovis Rakotoarivelo for expert technical support (Institut Neurosciernces de Montpelleir Unité 1051).
This work was supported by l'Institut National de la Santé et de la Recherche Médicale and l'Ecole Nationale Supérieure de Chimie de Montpellier.
Disclosure Summary: The authors have nothing to disclose.
NURSA Molecule Pages†:
Nuclear Receptors: AR;
Ligands: Testosterone.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- AR
- Androgen receptor
- BCA
- bicinchoninic acid
- ECFP
- enhanced cyan fluorescent protein
- FBS
- fetal bovine serum
- FBW7
- F-box&WD domain repeated 7
- HRP
- horseradish peroxidase
- I
- doxycycline induced
- LDH
- lactate dehydrogenase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NI
- noninduced
- NI+T
- NI plus testosterone
- PI3K
- phosphatidylinositol 3-kinase
- polyQ
- polyglutamine
- SAPK
- stress activated kinase
- SBMA
- spinal and bulbar muscular atrophy
- TfR
- transferrin receptor
- TI
- Triton-insoluble
- TIn-octyl
- TI/n-octylglucoside-soluble
- TS
- Triton X-100-soluble.
References
- 1. Zoghbi HY , Orr HT. 2000. Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23:217–247 [DOI] [PubMed] [Google Scholar]
- 2. Kennedy WR , Alter M , Sung JH. 1968. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology 18:671–680 [DOI] [PubMed] [Google Scholar]
- 3. La Spada AR , Wilson EM , Lubahn DB , Harding AE , Fischbeck KH. 1991. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352:77–79 [DOI] [PubMed] [Google Scholar]
- 4. Li M , Miwa S , Kobayashi Y , Merry DE , Yamamoto M , Tanaka F , Doyu M , Hashizume Y , Fischbeck KH , Sobue G. 1998. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol 44:249–254 [DOI] [PubMed] [Google Scholar]
- 5. McCampbell A , Taylor JP , Taye AA , Robitschek J , Li M , Walcott J , Merry D , Chai Y , Paulson H , Sobue G , Fischbeck KH. 2000. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 9:2197–2202 [DOI] [PubMed] [Google Scholar]
- 6. Cummings CJ , Mancini MA , Antalffy B , DeFranco DB , Orr HT , Zoghbi HY. 1998. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19:148–154 [DOI] [PubMed] [Google Scholar]
- 7. Stenoien DL , Cummings CJ , Adams HP , Mancini MG , Patel K , DeMartino GN , Marcelli M , Weigel NL , Mancini MA. 1999. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet 8:731–741 [DOI] [PubMed] [Google Scholar]
- 8. Paulson HL. 1999. Protein fate in neurodegenerative proteinopathies: polyglutamine diseases join the (mis)fold. Am J Hum Genet 64:339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klement IA , Skinner PJ , Kaytor MD , Yi H , Hersch SM , Clark HB , Zoghbi HY , Orr HT. 1998. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95:41–53 [DOI] [PubMed] [Google Scholar]
- 10. Arrasate M , Mitra S , Schweitzer ES , Segal MR , Finkbeiner S. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810 [DOI] [PubMed] [Google Scholar]
- 11. Li M , Chevalier-Larsen ES , Merry DE , Diamond MI. 2007. Soluble androgen receptor oligomers underlie pathology in a mouse model of spinobulbar muscular atrophy. J Biol Chem 282:3157–3164 [DOI] [PubMed] [Google Scholar]
- 12. Takahashi T , Kikuchi S , Katada S , Nagai Y , Nishizawa M , Onodera O. 2008. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet 17:345–356 [DOI] [PubMed] [Google Scholar]
- 13. Novitskaya V , Bocharova OV , Bronstein I , Baskakov IV. 2006. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem 281:13828–13836 [DOI] [PubMed] [Google Scholar]
- 14. Simeoni S , Mancini MA , Stenoien DL , Marcelli M , Weigel NL , Zanisi M , Martini L , Poletti A. 2000. Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum Mol Genet 9:133–144 [DOI] [PubMed] [Google Scholar]
- 15. Thomas PS , Fraley GS , Damian V , Damien V , Woodke LB , Zapata F , Sopher BL , Plymate SR , La Spada AR. 2006. Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Hum Mol Genet 15:2225–2238 [DOI] [PubMed] [Google Scholar]
- 16. Lund A , Udd B , Juvonen V , Andersen PM , Cederquist K , Davis M , Gellera C , Kölmel C , Ronnevi LO , Sperfeld AD , Sörensen SA , Tranebjaerg L , Van Maldergem L , Watanabe M , Weber M , Yeung L , Savontaus ML. 2001. Multiple founder effects in spinal and bulbar muscular atrophy (SBMA, Kennedy disease) around the world. Eur J Hum Genet 9:431–436 [DOI] [PubMed] [Google Scholar]
- 17. Schengrund CL. 2010. Lipid rafts: keys to neurodegeneration. Brain Res Bull 82:7–17 [DOI] [PubMed] [Google Scholar]
- 18. Pedram A , Razandi M , Sainson RC , Kim JK , Hughes CC , Levin ER. 2007. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem 282:22278–22288 [DOI] [PubMed] [Google Scholar]
- 19. Hatzoglou A , Kampa M , Kogia C , Charalampopoulos I , Theodoropoulos PA , Anezinis P , Dambaki C , Papakonstanti EA , Stathopoulos EN , Stournaras C , Gravanis A , Castanas E. 2005. Membrane androgen receptor activation induces apoptotic regression of human prostate cancer cells in vitro and in vivo. J Clin Endocrinol Metab 90:893–903 [DOI] [PubMed] [Google Scholar]
- 20. Kampa M , Kogia C , Theodoropoulos PA , Anezinis P , Charalampopoulos I , Papakonstanti EA , Stathopoulos EN , Hatzoglou A , Stournaras C , Gravanis A , Castanas E. 2006. Activation of membrane androgen receptors potentiates the antiproliferative effects of paclitaxel on human prostate cancer cells. Mol Cancer Ther 5:1342–1351 [DOI] [PubMed] [Google Scholar]
- 21. Benten WP , Lieberherr M , Stamm O , Wrehlke C , Guo Z , Wunderlich F. 1999. Testosterone signaling through internalizable surface receptors in androgen receptor-free macrophages. Mol Biol Cell 10:3113–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benten WP , Lieberherr M , Giese G , Wrehlke C , Stamm O , Sekeris CE , Mossmann H , Wunderlich F. 1999. Functional testosterone receptors in plasma membranes of T cells. FASEB J 13:123–133 [DOI] [PubMed] [Google Scholar]
- 23. Fix C , Jordan C , Cano P , Walker WH. 2004. Testosterone activates mitogen-activated protein kinase and the cAMP response element binding protein transcription factor in Sertoli cells. Proc Natl Acad Sci USA 101:10919–10924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gatson JW , Kaur P , Singh M. 2006. Dihydrotestosterone differentially modulates the mitogen-activated protein kinase and the phosphoinositide 3-kinase/Akt pathways through the nuclear and novel membrane androgen receptor in C6 cells. Endocrinology 147:2028–2034 [DOI] [PubMed] [Google Scholar]
- 25. Gatson JW , Singh M. 2007. Activation of a membrane-associated androgen receptor promotes cell death in primary cortical astrocytes. Endocrinology 148:2458–2464 [DOI] [PubMed] [Google Scholar]
- 26. Cheng J , Watkins SC , Walker WH. 2007. Testosterone activates mitogen-activated protein kinase via Src kinase and the epidermal growth factor receptor in Sertoli cells. Endocrinology 148:2066–2074 [DOI] [PubMed] [Google Scholar]
- 27. Cinar B , Mukhopadhyay NK , Meng G , Freeman MR. 2007. Phosphoinositide 3-kinase-independent non-genomic signals transit from the androgen receptor to Akt1 in membrane raft microdomains. J Biol Chem 282:29584–29593 [DOI] [PubMed] [Google Scholar]
- 28. Kazemi-Esfarjani P , Trifiro MA , Pinsky L. 1995. Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor: possible pathogenetic relevance for the (CAG)n-expanded neuronopathies. Hum Mol Genet 4:523–527 [DOI] [PubMed] [Google Scholar]
- 29. Lieberman AP , Harmison G , Strand AD , Olson JM , Fischbeck KH. 2002. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet 11:1967–1976 [DOI] [PubMed] [Google Scholar]
- 30. Cashman NR , Durham HD , Blusztajn JK , Oda K , Tabira T , Shaw IT , Dahrouge S , Antel JP. 1992. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn 194:209–221 [DOI] [PubMed] [Google Scholar]
- 31. Giovannucci E , Stampfer MJ , Krithivas K , Brown M , Dahl D , Brufsky A , Talcott J , Hennekens CH , Kantoff PW. 1997. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci USA 94:3320–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giovannucci E. 2002. Is the androgen receptor CAG repeat length significant for prostate cancer? Cancer Epidemiol Biomarkers Prev 11:985–986 [PubMed] [Google Scholar]
- 33. Herdegen T , Waetzig V. 2001. AP-1 proteins in the adult brain: facts and fiction about effectors of neuroprotection and neurodegeneration. Oncogene 20:2424–2437 [DOI] [PubMed] [Google Scholar]
- 34. Lindwall C , Dahlin L , Lundborg G , Kanje M. 2004. Inhibition of c-Jun phosphorylation reduces axonal outgrowth of adult rat nodose ganglia and dorsal root ganglia sensory neurons. Mol Cell Neurosci 27:267–279 [DOI] [PubMed] [Google Scholar]
- 35. Raivich G , Bohatschek M , Da Costa C , Iwata O , Galiano M , Hristova M , Nateri AS , Makwana M , Riera-Sans L , Wolfer DP , Lipp HP , Aguzzi A , Wagner EF , Behrens A. 2004. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43:57–67 [DOI] [PubMed] [Google Scholar]
- 36. Rakotoarivelo C , Petite D , de Weille J , Lumbroso S , Privat A , Sultan C , Mersel M. 2007. Mild surfection of neural cells, especially motoneurons, in primary culture and cell lines. Exp Neurol 204:118–130 [DOI] [PubMed] [Google Scholar]
- 37. Macdonald JL , Pike LJ. 2005. A simplified method for the preparation of detergent-free lipid rafts. J Lipid Res 46:1061–1067 [DOI] [PubMed] [Google Scholar]
- 38. Fargo KN , Galbiati M , Foecking EM , Poletti A , Jones KJ. 2008. Androgen regulation of axon growth and neurite extension in motoneurons. Horm Behav 53:716–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mayor S , Maxfield FR. 1995. Insolubility and redistribution of GPI-anchored proteins at the cell surface after detergent treatment. Mol Biol Cell 6:929–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ross CA , Poirier MA. 2004. Protein aggregation and neurodegenerative disease. Nat Med 10(Suppl):S10–S17 [DOI] [PubMed] [Google Scholar]
- 41. Gatchel JR , Zoghbi HY. 2005. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 6:743–755 [DOI] [PubMed] [Google Scholar]
- 42. Merry DE , Kobayashi Y , Bailey CK , Taye AA , Fischbeck KH. 1998. Cleavage, aggregation and toxicity of the expanded androgen receptor in spinal and bulbar muscular atrophy. Hum Mol Genet 7:693–701 [DOI] [PubMed] [Google Scholar]
- 43. Kobayashi Y , Kume A , Li M , Doyu M , Hata M , Ohtsuka K , Sobue G. 2000. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem 275:8772–8778 [DOI] [PubMed] [Google Scholar]
- 44. Brooks BP , Merry DE , Paulson HL , Lieberman AP , Kolson DL , Fischbeck KH. 1998. A cell culture model for androgen effects in motor neurons. J Neurochem 70:1054–1060 [DOI] [PubMed] [Google Scholar]
- 45. Marron TU , Guerini V , Rusmini P , Sau D , Brevini TA , Martini L , Poletti A. 2005. Androgen-induced neurite outgrowth is mediated by neuritin in motor neurones. J Neurochem 92:10–20 [DOI] [PubMed] [Google Scholar]
- 46. Butler R , Leigh PN , Gallo JM. 2001. Androgen-induced up-regulation of tubulin isoforms in neuroblastoma cells. J Neurochem 78:854–861 [DOI] [PubMed] [Google Scholar]
- 47. Avila DM , Allman DR , Gallo JM , McPhaul MJ. 2003. Androgen receptors containing expanded polyglutamine tracts exhibit progressive toxicity when stably expressed in the neuroblastoma cell line, SH-SY 5Y. Exp Biol Med (Maywood) 228:982–990 [DOI] [PubMed] [Google Scholar]
- 48. DiFiglia M , Sapp E , Chase KO , Davies SW , Bates GP , Vonsattel JP , Aronin N. 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993 [DOI] [PubMed] [Google Scholar]
- 49. Cooper MK , Porter JA , Young KE , Beachy PA. 1998. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280:1603–1607 [DOI] [PubMed] [Google Scholar]
- 50. Michels G , Hoppe UC. 2008. Rapid actions of androgens. Front Neuroendocrinol 29:182–198 [DOI] [PubMed] [Google Scholar]
- 51. Lu ML , Schneider MC , Zheng Y , Zhang X , Richie JP. 2001. Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. J Biol Chem 276:13442–13451 [DOI] [PubMed] [Google Scholar]
- 52. Kim SB , Kanno A , Ozawa T , Tao H , Umezawa Y. 2007. Nongenomic activity of ligands in the association of androgen receptor with SRC. ACS Chem Biol 2:484–492 [DOI] [PubMed] [Google Scholar]
- 53. Kousteni S , Bellido T , Plotkin LI , O'Brien CA , Bodenner DL , Han L , Han K , DiGregorio GB , Katzenellenbogen JA , Katzenellenbogen BS , Roberson PK , Weinstein RS , Jilka RL , Manolagas SC. 2001. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 104:719–730 [PubMed] [Google Scholar]
- 54. Migliaccio A , Castoria G , Di Domenico M , de Falco A , Bilancio A , Lombardi M , Barone MV , Ametrano D , Zannini MS , Abbondanza C , Auricchio F. 2000. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J 19:5406–5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Das M , Scappini E , Martin NP , Wong KA , Dunn S , Chen YJ , Miller SL , Domin J , O'Bryan JP. 2007. Regulation of neuron survival through an intersectin-phosphoinositide 3′-kinase C2beta-AKT pathway. Mol Cell Biol 27:7906–7917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yu F , Sugawara T , Maier CM , Hsieh LB , Chan PH. 2005. Akt/Bad signaling and motor neuron survival after spinal cord injury. Neurobiol Dis 20:491–499 [DOI] [PubMed] [Google Scholar]
- 57. Georget V , Lobaccaro JM , Terouanne B , Mangeat P , Nicolas JC , Sultan C. 1997. Trafficking of the androgen receptor in living cells with fused green fluorescent protein-androgen receptor. Mol Cell Endocrinol 129:17–26 [DOI] [PubMed] [Google Scholar]
- 58. Schaufele F , Carbonell X , Guerbadot M , Borngraeber S , Chapman MS , Ma AA , Miner JN , Diamond MI. 2005. The structural basis of androgen receptor activation: intramolecular and intermolecular amino-carboxy interactions. Proc Natl Acad Sci USA 102:9802–9807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Waetzig V , Zhao Y , Herdegen T. 2006. The bright side of JNKs-Multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog Neurobiol 80:84–97 [DOI] [PubMed] [Google Scholar]
- 60. Barnat M , Enslen H , Propst F , Davis RJ , Soares S , Nothias F. 2010. Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J Neurosci 30:7804–7816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chaves CS , Soares DC , Da Silva RP , Saraiva EM. 2003. Characterization of the species- and stage-specificity of two monoclonal antibodies against Leishmania amazonensis. Exp Parasitol 103:152–159 [DOI] [PubMed] [Google Scholar]
- 62. Frame S , Cohen P. 2001. GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Palazzolo I , Burnett BG , Young JE , Brenne PL , La Spada AR , Fischbeck KH , Howell BW , Pennuto M. 2007. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum Mol Genet 16:1593–1603 [DOI] [PubMed] [Google Scholar]
- 64. Wei W , Jin J , Schlisio S , Harper JW , Kaelin WG. 2005. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 8:25–33 [DOI] [PubMed] [Google Scholar]
- 65. Morton S , Davis RJ , McLaren A , Cohen P. 2003. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J 22:3876–3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Katsuno M , Adachi H , Waza M , Banno H , Suzuki K , Tanaka F , Doyu M , Sobue G. 2006. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA). Exp Neurol 200:8–18 [DOI] [PubMed] [Google Scholar]
- 67. Katsuno M , Adachi H , Doyu M , Minamiyama M , Sang C , Kobayashi Y , Inukai A , Sobue G. 2003. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med 9:768–773 [DOI] [PubMed] [Google Scholar]
- 68. Dehm SM , Tindall DJ. 2006. Ligand-independent androgen receptor activity is activation function-2-independent and resistant to antiandrogens in androgen refractory prostate cancer cells. J Biol Chem 281:27882–27893 [DOI] [PubMed] [Google Scholar]
- 69. Feldman BJ , Feldman D. 2001. The development of androgen-independent prostate cancer. Nature reviews 1:34–45 [DOI] [PubMed] [Google Scholar]
- 70. Estrada M , Espinosa A , Müller M , Jaimovich E. 2003. Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology 144:3586–3597 [DOI] [PubMed] [Google Scholar]
- 71. Lieberherr M , Grosse B. 1994. Androgens increase intracellular calcium concentration and inositol 1,4,5-trisphosphate and diacylglycerol formation via a pertussis toxin-sensitive G-protein. J Biol Chem 269:7217–7223 [PubMed] [Google Scholar]