

Abstract
The G protein-coupled prostaglandin F2α (PGF2α) receptor [F prostanoid (FP) receptor] has been implicated in many physiological events including cardiovascular, respiratory, immune, reproductive, and endocrine responses. Binding of PGF2α to FP receptor elicits inositol production and protein kinase C-dependent MAPK activation through Gαq coupling. Here we report that AL-8810, previously characterized as an orthosteric antagonist of PGF2α-dependent, Gαq-mediated signaling, potently activates ERK1/2 in a protein kinase C-independent manner. Rather, AL-8810 promoted ERK1/2 activation via an epidermal growth factor receptor transactivation mechanism in both human embryonic kidney 293 cells and in the MG-63 osteoblast-like cells, which express endogenous FP receptors. Neither AL-8810- nor PGF2α-mediated stimulation of FP receptor promoted association with β-arrestins, suggesting that MAPK activation induced by these ligands is independent of β-arrestin's signaling scaffold functions. Interestingly, the spatiotemporal activation of ERK1/2 promoted by AL-8810 and PGF2α showed almost completely opposite responses in the nucleus and the cytosol. Finally, using [3H]thymidine incorporation, we noted differential regulation of PGF2α- and AL-8810-induced cell proliferation in MG-63 cells. This study reveals, for the first time, the signaling biased nature of FP receptor orthosteric ligands toward MAPK signaling. Our findings on the specific patterns of ERK1/2 activation promoted by FP receptor ligands may help dissect the distinct roles of MAPK in FP receptor-dependent physiological responses.
The F prostanoid receptor (FP receptor) is a member of the G protein-coupled receptor (GPCR) family, the endogenous ligand of which is prostaglandin F2α (PGF2α). PGF2α is involved in many physiological responses, and its action is recognized in the regulation of parturition (including luteolysis), smooth muscle contraction, intraocular pressure, cardiac hypertrophy, kidney function, and bone metabolism (1–7). At the cellular level, PGF2α-dependent stimulation of FP receptor triggers signaling cascades that result in activation of the MAPK pathway and cytoskeletal reorganization, mediated by Gαq and Gα12, respectively (8–10). FP receptor-mediated stimulation of Gαq activates ERK1/2 MAPK via phospholipase C (PLC) and protein kinase C (PKC) (11–13), resulting in various physiological effects ranging from gene transcription to cell proliferation. Several other mechanisms of GPCR-induced ERK1/2 activation have been demonstrated, including scaffolding of β-arrestins (14), activation of Src kinases (15), or activation of protein kinase A (16). Another known mechanism of GPCR-mediated MAPK activation involves transactivation of receptor tyrosine kinases, such as the epidermal growth factor receptor (EGFR, for review see Ref. 17).
It is now well recognized that orthosteric ligands, some of which act to antagonize agonist-mediated signaling pathways, can also bias the receptor into other signaling modes (18–22). Numerous selective agonists for the FP receptor have been developed (e.g. fluprostenol, cloprostenol, and bimatoprost), but few antagonists exist at present. AL-8810, which is an 11β-fluoro analog of PGF2α, has been described as a selective FP receptor antagonist for fluprostenol-induced phosphoinositide production (23), although it has also been shown to act as a weak partial agonist on this pathway. Many studies have reported that AL-8810 has minimal or no agonist effects (i.e. acted as an antagonist) in cells and isolated tissues (24–26), but some have reported in vivo activity (27). Considering that we recently showed that the FP receptor could be biased into selective signaling modes by an allosteric ligand (10), we investigated the extent to which an orthosteric PGF2α analog promoted functional selectivity on the FP receptor.
Materials and Methods
Materials
[3H]PGF2α, [3H]thymidine, and ECL reagent were from PerkinElmer (Waltham, MA). PGF2α and AL-8810 were from Cayman Chemical Co. (Ann Arbor, MI). Angiotensin II (AngII), poly l-lysine hydrochloride, N-ethylmaleimide were from Sigma-Aldrich (St Louis, MO). Mouse monoclonal anti-phospho-ERK1/2 (T202/Y204), rabbit polyclonal antitotal ERK1/2, and rabbit polyclonal antiphospho-EGFR (Y1148) antibodies were from Cell Signaling Technology (Danvers, MA). Anti-EEA1 and antihistone 2B were kind gifts from Dr. Barry Posner and Dr. Jason Tanny, respectively (both from McGill University). Paraformaldehyde was from Electron Microscopy Sciences (Hatfield, PA). Dithiobis succinimidyl propionate was from Pierce Chemical Co. (Rockford, IL). MEM and DMEM were from Hyclone Laboratories (Logan, UT). Fetal bovine serum (FBS), l-glutamine, and gentamicin were from Invitrogen (Carlsbad, CA), and puromycin was from Invivogen (San Diego, CA). Anti-HA Affinity Matrix and mouse monoclonal anti-HA (clone 12CA5) antibody were from Roche (Laval, Canada). Phenylmethylsulfonylfluoride, aprotinin, leupeptin, and pepstatin were from Bioshop (Burlington, Canada). Pierce subcellular fractionation kit for cultured cells was purchased from Thermo Scientific (Rockford, IL). BSA Fraction V, Gö6983 and AG1478 were from EMD Chemicals, Inc. (Gibbstown, NJ). Epidermal growth factor (EGF) was from Fitzgerald Industries International (Acton, MA). AS604872 (Merck Serono) was synthesized at L'Institut de Recherche en Immunologie et en Cancérologie (Université de Montréal, Montréal, Canada). Anti-β-arrestin (clone 3978) was generated at the McGill University Animal Facility and described elsewhere (28).
DNA constructs, cell lines, culture, and transfection
PKCβI-green fluorescent protein (GFP) was described in Ref. 10, and yellow fluorescent protein (YFP)-tagged β-arrestins (1 and 2) were described in Ref. 28; and pcDNA3.1-SrcK298R was described in Ref. 29. The ERK activity reporters (EKAR-nucleus and EKAR-cytosol) were purchased from Addgene Inc. (Cambridge, MA) and have been described elsewhere (30). pCMV-EGFR was obtained from Dr. L. M. Luttrell (Medical University of South Carolina, Charleston, SC). Human embryonic kidney (HEK)293 cells stably expressing human hemagglutinin (HA)-tagged FP receptor (FP-R cells) and HA-angiotensin II type 1 receptor (AT1R) were described in Refs. 10 and 28, respectively. MG-63 osteoblasts were kindly provided by Dr. J.-L. Parent (Université de Sherbrooke; Sherbrooke). Cell lines were grown at 37 C in 5% CO2 in MEM (HA-FP, HA-AT1R) or DMEM (MG-63) supplemented with 10% (vol/vol) heat-inactivated FBS, l-glutamine (5 mm), and gentamycin (100 μg/ml). For transient transfection, cells seeded at a density of 1.5 × 106 cells per 100-mm dish, 1.5 × 105 per well in six-well plates or 5 × 104 cells per 35-mm plates for microscopy, were transfected using conventional calcium phosphate coprecipitation. All experiments were performed 48 h after transfection.
Ligand binding experiments
For binding experiments, 100 μg of HEK293 cells stably expressing the FP receptor were incubated with 1 × 105 cpm of [3H]PGF2α (150–240 Ci/mmol) for 1 h in the presence of either cold PGF2α or AL-8810 at varying concentrations at room temperature in 0.5 ml of binding buffer as described previously (10). Binding was stopped by addition of 2 ml cold 50 mm Tris-HCl (pH 7.4), and cells were filtered on GF/B-filters. Incorporated radioactivity was measured by liquid scintillation spectrometry.
Confocal microscopy and fluorescence resonance energy transfer (FRET) experiments
PKC translocation assays were performed as previously described (10). Briefly, FP-R cells transfected with 0.5 μg PKCβI-GFP were serum starved for 30 min and treated with 1 μm PGF2α or 10 μm AL-8810 for 30 min each. For β-arrestin, FP-R cells transfected with 0.5 μg of either β-arrestin1-YFP or β-arrestin2-YFP and GPCR kinase 2, were serum starved for 30 min and treated with either 1 μm PGF2α, 10 μm AL-8810, or 1 μm AngII for 30 min. Images were collected every 30 sec using live-cell microscopy at 37 C on a Zeiss LSM-510 Meta laser scanning microscope (Carl Zeiss, Thornwood, NY) equipped with XL-3 temperature chamber with a ×63 glycerol/water immersion lens in single track mode using excitation at 488 nm for GFP and 514 nm for YFP, measured with LP505 (GFP) and BP530–600 (YFP) filter sets.
For p-ERK1/2 immunofluorescence, FP-R cells were plated on coverslips in six-well plates at a density of 1 × 105 cells per well. Cells were starved 24 h later in 0.5% serum. The next day, cells were stimulated with 1 μm PGF2α or 10 μm AL-8810 and fixed with 4% paraformaldehyde in PBS. Cells were then washed in PBS and blocked/permeabilized in PBS containing 2% BSA and 0.1% Triton-X-100. Antibodies against p-ERK1/2 were added to coverslips (1:300 dilution) for 90 min in the blocking/permeabilizing solution. After PBS washes, cells were blocked again and goat-antimouseAlexa488 secondary antibody was added to coverslips for 60 min (1:1000 dilution). The last 15 min of the incubation, Hoescht dye was added to the cells to label the nucleus. Cells were then mounted and visualized by confocal microscopy using excitation at 488 nm for p-ERK1/2 and 405 nm for Hoescht and emission filter sets BP505–530 and 420–480 for ERK1/2 and Hoescht, respectively. Colocalization was analyzed using the ImageJ program (NIH, colocalization plugin). The p-ERK1/2 and nuclear staining images were background corrected (50-pixel radius), and a composite mask was generated. For the analysis, two pixels (from p-ERK1/2 and Hoescht signals) were considered colocalized if their intensities were higher than the threshold of their respective channels (50/255) and if the intensity ratio was higher than the 50% of the default value.
FRET assays were performed using the ERK activity reporter (EKAR, (30)) an ERK1/2-activity sensor. The core of the sensor is constituted of an ERK1/2 docking domain, a Cdc25C substrate peptide, and a phospho-binding domain, all flanked by a cerulean fluorescent protein (CFP) on the N terminus of a Venus yellow fluorescent protein (YFP) on the C terminus. The two EKAR sensor versions used in this paper contained either a nuclear localization signal (EKAR-nucleus) or a nuclear export signal (EKAR-cytosol), allowing not only the temporal, but the spatial localization of ERK1/2 activity. In the basal state, both the CFP (donor) and YFP (acceptor) are far enough apart that no FRET signal is observed. After ERK1/2 activation, the latter can dock on the sensor and phosphorylate the substrate peptide, which will then allow the phospho-binding domain to bind to the substrate peptide; this leads to a conformational rearrangement of the sensor and of a proximity between the donor and the acceptor, yielding a FRET signal. The dynamic rage of the sensors was evaluated using the potent activator of MAPK, EGF, and found to be approximately 1.4-fold (data not shown). For FRET experiments, FP-R cells were seeded at or 5 × 104 cells per dish in 35-mm microscopy dish and transfected with either different EKAR sensors using calcium phosphate. Cells were serum-starved 24 h later overnight with in 0.2% FBS and challenged the next day with either 1 μm PGF2α or 10 μΜ AL-8810. Images were collected every minute for 60 min using live-cell microscopy at 37 C on a Zeiss LSM-510 microscope equipped with an XL-3 temperature chamber with a ×40 oil or ×63 glycerol/water immersion lenses. FRET (YFP emission) and CFP images were collected using excitation at 405 nm. Emitted images were collected with the following filter sets: BP475–505 for CFP and BP530–600 for FRET. FRET and CFP signals values on single cells were calculated with Zen program, and FRET values were normalized to the CFP signal. For FRET pseudocolor images, the FRET signal was represented using rainbow coarse rendering.
Western blot and immunoprecipitation experiments
For detection of ERK1/2 (phosphorylated and total), cells seeded in six-well plates (1.5 × 105) for 48 h and after 30 min starvation without serum, stimulated for different times with 1 μm PGF2α, 10 μm AL-8810 for indicated times and directly solubilized in 2× Laemmli buffer. For donor cell-mediated EGFR transactivation experiments, FP-R (donor cells) and HEK293 naïve cells (acceptor cells) were plated in 12-well plates and, 48 h later, serum starved. Donor cells were then stimulated with 1 μm PGF2α or 10 μm AL-8810 for 5 and 15 min. After stimulation, the media from FP-R cells were taken and added onto the acceptor cells (final dilution 1:2) and left to stimulate cells for 5 min. Cells were then lysed in Laemmli buffer, and lysates were analyzed by Western blot using anti-phospho-ERK1/2 and anti-total-ERK1/2 antibodies. To detect activated EGFR (p-EGFR), cells were plated at a density of 1 × 105 cells per well in six-well plates and transfected 24 h later with pCMV-EGFR. Cells were starved 24 h after transfection overnight with 0.5% FBS. The next day, cells were pretreated with dimethylsulfoxide (DMSO) or AG-1478 and stimulated with 5 ng/ml of EGF. Cells were lysed in THG buffer [50 mm HEPES (pH 7.4), 1% Triton-X-100 (vol/vol), 50 mm NaCl, 10% glycerol (vol/vol), 5 mm EDTA] containing proteases (25 μg/ml aprotonin, 1 mm pepstatin A, 25 μg/ml leupeptin, and 1 mm phenylmethylsulfonylfluoride) and phosphatase inhibitor (0.1 mm sodium orthovanadate). Proteins were then loaded directly on a 10% SDS-PAGE gel (p-EGFR) or 10% gel (phospho/total-ERK1/2) and transferred/probed for detection. For β-arrestin coimmunoprecipitaion assays, FP-R cells or AT1R expressing cells (both receptors are HA tagged) were seeded at 1.5 × 106 cells in 100-mm dishes and grown for 2 d. Cells were then serum-starved for 30 min and treated with 1 μm PGF2α, 10 μm AL-8810, or 1 μm AngII for 15 min. After stimulation, 2 mm of cross-linking agent dithiobis succinimidyl propionate was added to each plate at room temperature for 30 min. Media were aspirated and washed with 1× PBS containing 50 mm Tris-HCl, pH 7.4. Cells were lysed with glycerol buffer containing protease inhibitors and 10 mm N-ethylmaleimide. Cell lysates were rocked for 30 min at 4 C and centrifuged for 30 min at 4 C, and the supernatant was incubated overnight with anti-HA Affinity Matrix beads. A fraction of the supernatant was kept as the total cell lysate (TCL) before the beads were added. The next day, beads were washed and the immunoprecipitate was resuspended in 2× Laemmli buffer. Samples (immunoprecipitates and TCL) were then loaded on 10% SDS-PAGE gels and transferred to nitrocellulose before being probed with antibodies against proteins of interest (see figure legends for details). For cellular fractionation, FP-R cells were plated at a density of 5 × 105 cells/10-cm dishes and after 48 h, cells were starved for 30 min without serum before stimulation with 0.5 μm of PGF2α or 5 μm AL-8810. After stimulation, fractionation of the different subcellular compartments was done according to the manufacturer's instructions (Pierce subcellular fractionation kit). Samples were loaded on 10% gels and transferred/probed as described in figure legends. Semiquantitative analysis of ERK1/2 and EGFR activation was done using ImageJ program.
[3H]thymidine incorporation experiments
MG-63 osteoblast-like cells were plated at a density of or 1.3 x 104 cells per well in 24-well plates. Cells were starved 24 h later, in 0.2% FBS for 24 h. Then, cells were treated with 1 μm PGF2α or 10 μm AL-8810 for 4, 8, 12, and 24 h. During the last 4 h of the stimulation, 0.5 μCi of [3H]thymidine per well was added. Cells were then washed rapidly once with cold PBS and washed once for 30 min with 5% trichloroacetic acid at 4 C. NaOH (0.2 m) was then added to lyse the cells, and the incorporated radioactivity was counted using a β-counter after addition of scintillation liquid.
Statistical analysis
Statistical tests were performed with GraphPad Prism 5 software (GraphPad Software, San Diego, CA). Assumptions of normality and equal variance were met for all data analyzed. Independent t tests were used to compare vehicle and treated samples (Fig. 1A). One-sample t tests were used when the data were normalized and basal levels were considered in respect to the hypothetical value (1) (Fig. 1E). One-way ANOVA was used for Fig. 3B, Fig. 5D, Fig. 6C, and Fig. 8, A and B. Two-way ANOVA with repeated measures was used in Fig. 8. Two-way ANOVA was used in Fig. 2, B and C, Fig, 5, Fig. 6, A and B, Fig. 7, and Figs. 9 and 10. All ANOVA analyses were followed by a Bonferroni post hoc test. A two-tailed P value <0.05 was considered significant. All results are expressed as mean ± sem. Sample size (n) and P values are given in individual figure legends.
Fig. 1.

AL-8810 binds FP receptor and activates MAPK. A, FP-R cells were incubated for 1 h with vehicle (binding buffer), PGF2α, or AL-8810 at the indicated concentrations, and [3H]PGF2α displacement was measured. B and C, Cells were serum starved for 30 min before stimulation with either 1 μm PGF2α (B) or 10 μm AL-8810 (C) for the indicated times. Cell lysates were analyzed by Western blot using anti-phospho- and anti-total ERK1/2 antibodies. Signals were quantified by densitometry and plotted as fold over basal (i.e. not treated) activation. D, Cells were pretreated with either 1 μm AS604872 or vehicle (ethanol) for 30 min and then stimulated for 5 min with PGF2α (PGF) or AL-8810 (AL). E, MG-63 osteoblasts were serum starved overnight before treatment with either 1 μm PGF2α or 10 μm AL-8810 for the indicated times. Signals were analyzed as in panels B and C. Results are representative of three (panels A, B, and D) and five (panels C and E) independent experiments (*, P < 0.05; **, P < 0.01 compared with nontreated cells). NT, Not treated; Tot, total.
Fig. 3.

PGF2α analogs, except AL-8810, require downstream PKC to activate ERK1/2. A, Chemical structures of PGF2α analogs: bimatoprost (bimat.), cloprostenol (cloprost.), fluprostenol (fluprost.), and AL-8810. B, FP-R cells were serum-starved 30 min before pretreatment with DMSO or 1 μm Go6983 for 30 min. Cells were then stimulated for 5 min with 1 μm PGF2α, bimatoprost, cloprostenol, fluprostenol, or 10 μm AL-8810, and signals were analyzed as in Fig. 2. Results are representative of three (panel B) independent experiments (**, P < 0.01; ***, P < 0.001 compared with DMSO-treated cells). NT, Not treated; Tot, total.
Fig. 5.

AL-8810 activates MAPK through EGFR-dependent transactivation. A–D, FP-R cells (A, B, and C) and MG-63 osteoblasts cells (D) were serum starved for 30 min (A and B) or overnight (C and D) before pretreatment with either DMSO or 125 nm AG1478 for 30 min. Cells were then treated with 1 μm PGF2α (A, C, and D), 10 μm AL-8810 (B, C, and D) or 5 ng/ml EGF (C and D) for the indicated times. Cell lysates were analyzed by Western blot using antiphospho-ERK1/2, antitotal-ERK1/2 (A, B, and D) and anti-phospho EGFR (C) antibodies. Signals were quantified by densitometry and plotted as fold over basal (i.e. not treated). Results are representative of three (D), five (A and B), or six (C) independent experiments. *, P < 0.05; **, P < 0.01, comparing DMSO and AG1478 treatment for each corresponding time points of ligand stimulation (panels A and B); **, P < 0.01; ***, P < 0.0001 compared with DMSO-treated, ligand-unstimulated cells (panels C and D); ††, P < 0.01 compared with DMSO-pretreated, PGF2α-stimulated cells, at 5 and 15 min; $, P < 0.0001 compared with AG1478-pretreated and PGF2α- and AL8810-stimulated cells (panel C); ‡, P < 0.0001 compared with DMSO-pretreated, EGF-stimulated cells (panel C, inset, and panel D). AL, AL-8810; Tot, total.
Fig. 6.
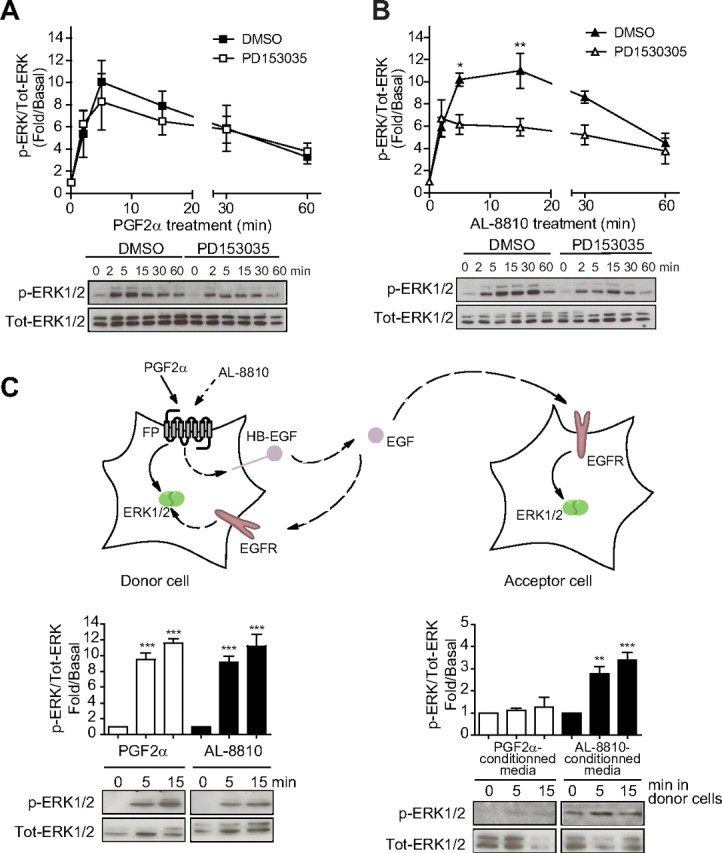
AL-induced EGFR transactivation is through an EGF-shedding signaling pathway. A and B, FP-R cells were plated in six-well plates and 48 h later, cells were serum starved for 30 min before pretreatment with either DMSO or 1 μm PD153035 for 30 min. Cells were then treated with 1 μm PGF2α or 10 μm AL-8810 for the indicated times. C, FP-R (donor cells in the scheme) and HEK293 naïve cells (acceptor cells in the scheme) were plated in 12-well plates and 48 h later, serum starved. FP-R cells were then stimulated with 1 μm PGF2α or 10 μm AL-8810 for 5 and 15 min. After stimulation, the media of FP-R cells were taken and deposited on the acceptor cells (final dilution 1:2) and left for 5 min. Cells were then lysed in Laemmli buffer, and lysates were analyzed by Western blot using antiphospho-ERK1/2 and anti-total-ERK1/2 antibodies. Signals were quantified by densitometry and plotted as fold over basal (i.e. not treated) activation. Results are representative of five (A and B) and three (C) independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with DMSO-treated cells). Tot, Total.
Fig. 8.

ERK1/2 cellular localization is differentially affected by PGF2α or AL-8810 treatment. A and B, FP-R cells were starved 30 min before stimulation with 0.5 μm PGF2α and 5 μm AL-8810 for the indicated times. Cells were then lysed, and the different fractions were extracted (see Materials and Methods for details). Cell lysates were analyzed by Western blot using antitotal ERK1/2, antiearly endosome antigen 1 (EEA1, panel A), and antinucleoporin 62 (Nup62, panel B) antibodies. Panel A shows cytosolic compartment; and panel B shows nuclear compartment; the third panel shows an enrichment control for both cellular compartments. C, FP-R cells were starved 30 min before stimulation with 1 μm PGF2α and 10 μm AL-8810 for the indicated times. Cells were then fixed and activated ERK1/2 was stained with anti-p-ERK1/2 and goat-antimouse-Alexa488 secondary antibodies (green signal). Nuclei were stained with Hoescht (represented as a red signal). Also shown is the overlay between Hoescht and p-ERK1/2 staining and composite mask showing colocalizing pixels between both signals (see Materials and Methods for details). Results are representative of four (panel A and B) and three (panel C) independent experiments. *, P < 0.05 compared with the “0” time-point. Scale bars, 10 μm. NT, Not treated; Tot, total.
Fig. 2.

PGF2α, but not AL-8810, activates MAPK via PKC. A, Images obtained by confocal microscopy showing translocation of PKCβI-GFP to the cell membrane after treatment with 1 μm PGF2α or 10 μm AL-8810. Lower right panel, After 30 min of AL-8810 stimulation, cells were stimulated with an excess of PGF2α to show functional FP receptor-induced PKCβI-GFP translocation. B and C, FP-R cells were serum starved for 30 min before pretreatment with either DMSO or 1 μm Gö6983 for 30 min. Cells were then treated with either 1 μm PGF2α (B) or 10 μm AL-8810 (C) for the indicated times. Cell lysates were analyzed by Western blotting using anti-phospho- and anti-total ERK1/2 antibodies. Signals were quantified by densitometry and plotted as fold over basal (i.e. not treated) activation. Results are representative of (panel A), four (panel C), and five (panel B) independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with DMSO-treated cells). NT, Not treated; Tot, total.
Fig. 7.

Src is involved in AL-8810-induced ERK1/2 activation. A and B, FP-R cells were plated in six-well plates and transfected 24 h later with an empty vector (pcDNA3.1+) or a kinase dead mutant of Src (SrcK298R). After 48 h, cells were serum starved for 30 min and then treated with 1 μm PGF2α or 10 μm AL-8810 for the indicated times. Cells were lysed and analyzed as in Fig. 6. Results are representative of four independent experiments (**, P < 0.01; ***, P < 0.001 compared with empty vector transfected cells). Src, Src kinase; Tot, total.
Fig. 9.
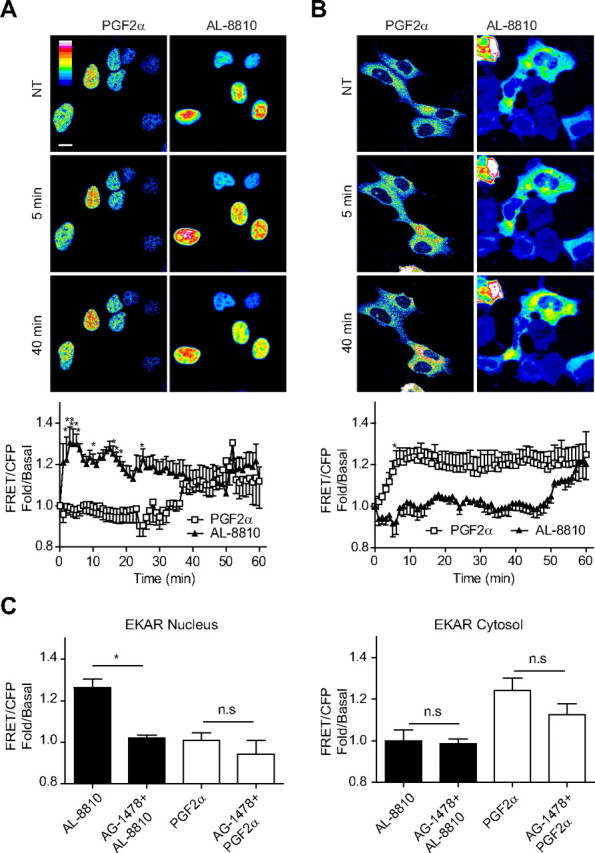
Spatiotemporal control of ERK1/2 activation is differentially affected by PGF2α and AL-8810. A and B, FP-R cells were transfected with EKAR-nucleus (A) or EKAR-cytosol (B) and stimulated with 1 μm PGF2α or 10 μm AL-8810 for a period up to 60 min. FRET and CFP images were collected every minute (see Materials and Methods for details) and the FRET/CFP ratio was calculated and plotted against time. Representative pseudocolor images are shown for EKAR-nucleus (A) or EKAR-cytosol (B). C, FP-R cells were transfected with EKAR-nucleus (left panel) or EKAR-cytosol (right panel) and cells were pretreated with 125 nm AG-1478 for 30 min before stimulation with PGF2α and AL-8810 as in panels A and B. Shown on the graph are the FRET/CFP values for the maximal stimulation at 5 min for EKAR-nucleus and 10 min for EKAR-cytosol. Scale bar, 10 μm. Results are representative of nine (A and B) and three independent experiments (50–83 cells in panel A, 42–50 cells in panel B, and 15–22 cells in panel C). *, P < 0.05 (difference between treatments). n.s. Nonsignificant, P > 0.05; NT, not treated.
Fig. 10.

Different patterns of [3H]thymidine incorporation are observed with PGF2α- and AL-8810-treated cells. MG-63 osteoblast cells were plated in 24-well plates. Cells were starved 24 h later for 24 h before stimulation with 1 μm PGF2α and 10 μm AL-8810 for 4, 8, 12, or 24 h. Inset, Treatment with 10% serum. Results are representative of four independent experiments. *, P < 0.05; and ***, P < 0.0001 (difference between treatments).
Results
AL-8810 binds to the FP receptor and activates MAPK
Initially, we wanted to confirm that AL-8810 binds FP receptor at its orthosteric site. FP-R cells were incubated with radiolabeled [3H]PGF2α, and the displacement of radioligand was measured as a function of increasing concentrations of either AL-8810 or PGF2α. AL-8810 and PGF2α both displaced [3H]PGF2α binding to the FP receptor in a dose-dependent manner, with Ki values of approximately 500 nm (497 ± 63.3 nm), and 3 nm (3.26 ± 0.73 nm; Fig. 1A), respectively. Binding was FP receptor specific, because naïve HEK293 cells showed no displaceable [3H]PGF2α binding (10). We next compared the extent to which PGF2α and AL-8810 promoted MAPK activation. Because of the marked difference between PGF2α and AL-8810 binding affinities, and to ensure that at least 95% of the FP receptor was occupied, we used PGF2α and AL-8810 at concentrations of 1 μm and 10 μm, respectively. Treatment of FP-R cells with PGF2α promoted phosphorylation of ERK1/2 in a time-dependent manner, reaching a maximal response at 5 min of agonist treatment (Fig. 1B). When cells were stimulated with AL-8810, ERK1/2 was also robustly activated, reaching similar levels of ERK1/2 phosphorylation as seen with PGF2α treatment, with peak activation occurring between 5 and 10 min (Fig. 1C). The MAPK activation in FP-R cells was FP receptor specific because both responses induced by AL-8810 and PGF2α were inhibited by the selective FP receptor antagonist, AS604872 (Fig. 1D); in contrast, alone, AS604872 treatment of FP-R cells had no effect on ERK1/2 activation, confirming its antagonistic property on this signaling pathway (Supplemental Fig. 1A published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Because AL-8810 has been often used as an antagonist for FP receptor-mediated responses, using a prolonged pretreatment of cells (more than 30 min), we wondered whether such modality could also lead to inhibition of PGF2α-induced ERK1/2 activation in FP-R cells. Indeed, prolonged pretreatment of cells (30 min) with AL-8810 inhibited such a response (Supplemental Fig. 1B). Finally, we investigated the effects of PGF2α and AL-8810 on MAPK activation in a physiologically relevant cell model expressing endogenous FP receptors using the human osteoblastic cell line (MG-63) (31). Stimulation of MG-63 cells with either PGF2α or AL-8810 promoted rapid and significant phosphorylation of ERK1/2 (Fig. 1E).
AL-8810 promoted MAPK activation independently of PKC
We previously reported that FP receptor-dependent activated ERK1/2 is through the Gαq/PLC/PKC signaling pathway (10). Consistent with the activation of PLC by PGF2α, which generates inositol phosphate production and mobilizes Ca2+, PKCβI-GFP was rapidly translocated from the cytosol to the plasma membrane (Fig. 2A, top panel). On the other hand, AL-8810 stimulation elicited a faint and transient recruitment of PKCβI-GFP to the plasma membrane (Fig. 2A, bottom panel; see arrows). Next, FP-R cells were pretreated with Gö6983, a broad-spectrum PKC inhibitor, and assessed for ERK1/2 activation after either PGF2α or AL-8810 stimulation. In FP-R cells in which PKC was inhibited, PGF2α-mediated MAPK responses were decreased by more than 80% (Fig. 2B), whereas AL-8810-promoted responses were not significantly altered compared with vehicle-treated cells (Fig. 2C). Interestingly, AL-8810 was the only PGF2α analog tested that induced MAPK activation and for which the signal was not significantly affected by PKC inhibition, because bimatoprost-, cloprostenol- and fluprostenol-mediated ERK1/2 activation signals were all inhibited by Gö6983 (Fig. 3).
β-Arrestins are not involved in FP-receptor-mediated MAPK signaling
β-Arrestins are known to activate ERK1/2 after stimulation of many GPCR via scaffolding the different components of the ERK1/2 pathway (8, 31). As an explanation for AL-8810-induced PKC-independent ERK1/2 activation, we considered the potential involvement of β-arrestin's scaffolding function in MAPK signaling. We determined whether PGF2α and AL-8810 promoted complex formation between FP receptor and β-arrestin by assessing receptor-mediated translocation of YFP-β-arrestin2 to the plasma membrane. To ensure that any lack of β-arrestin recruitment to the FP receptor was not due to poor phosphorylation of the receptor, we also coexpressed GPCR kinase 2 with the β-arrestin isoforms. Neither PGF2α nor AL-8810 stimulation of FP-R cells resulted in translocation of either β-arrestin isoforms to the plasma membrane (Fig. 4A). However, under similar conditions, cells stably expressing AT1R, showed rapid and robust translocation of β- arrestin2 to the plasma membrane when stimulated with AngII, as well as internalization into endosomes (Fig. 4A). We also confirmed, using a cross-linking approach, whether agonist-stimulated FP receptor could recruit β-arrestins. Cells expressing the FP receptor or AT1R were stimulated with either PGF2α and AL-8810, or AngII. Receptors were immunoprecipitated and the associated β-arrestin was detected. AngII stimulation promoted complex formation between AT1R and both β-arrestin1 and 2. However, no such complexes were seen between the FP receptor and β-arrestins after either PGF2α or AL-8810 stimulation (Fig. 4B). Therefore, these results suggest that β-arrestin recruitment in FP-R cells is not involved in either PGF2α or AL-8810-induced ERK1/2 activation.
Fig. 4.

β-Arrestin is not recruited to FP receptor in response to either PGF2α or AL-8810. A, Images obtained by confocal microscopy showing redistribution of β-arrestin-2-YFP after 1 μm PGF2α, 10 μm AL-8810 in FP-R cells, or 1 μm AngII treatment in HA-AT1R cells. B, FP-R cells or AT1R cells were serum starved for 1 h before stimulation with 1 μm AngII, 1 μm PGF2α, or 10 μm AL8810 at indicated time points. HA-tagged receptors were immunoprecipitated, and cell lysates were Western blotted using anti-β-arrestin (clone 3978, see Materials and Methods), antiphospho ERK1/2, or anti-HA antibodies (IP indicates immunoprecipitation). Results are representative of three independent experiments. NT, Not treated; WB, Western blot.
AL-8810 activates MAPK through EGFR transactivation
GPCR-mediated activation of MAPK has been shown to occur in murine osteoblasts stimulated with PGF2α (32) via receptor tyrosine kinase transactivation (33). Thus, we next investigated the involvement of FP receptor-promoted epithelial growth factor (EGF) receptor (EGFR) transactivation in FP-R cells. Cells were pretreated or not with AG1478, an EGFR-specific inhibitor, and subsequently challenged with either PGF2α or AL-8810. Cells pretreated with AG1478, showed a reduction, although not statistically significant, of ERK1/2 activation by less than 10% after PGF2α stimulation, as compared with vehicle (Fig. 5A). EGFR inhibition in AL-8810-stimulated cells led to a more robust and significant reduction in ERK1/2 activation, with a greater than 60% inhibition of the response at 5 min (Fig. 5B). We then assessed FP receptor-mediated activation of EGFR by assessing phosphorylation of one of its regulatory sites [i.e. Y1148, (34)]. FP-R cells stimulated with EGF for either 5 or 15 min showed robust activation of EGFR (Fig. 5C, inset). We observed a slight increase for EGFR activation in PGF2α-treated FP-R cells, which however was not statistically significant as compared with untreated cells. On the other hand, stimulation of cells with AL-8810 promoted a significant EGFR activation that was more than 2-fold greater than that seen in unstimulated cells (Fig. 5C). In cells pretreated with AG1478, although we observed a consistent, but not statistically significant, reduction in EGFR activation promoted by PGF2α (Fig. 5C), the responses induced by both AL-8810 and EGF were significantly inhibited after treatment with AG1478 by more than 95% and 75%, respectively. We also determined the involvement of EGFR in the FP receptor-mediated MAPK pathway in MG-63 osteoblast cells. Surprisingly, both AL-8810- and PGF2α-induced ERK1/2 signals were significantly reduced by more than 50%, on average, after EGFR inhibition, whereas the EGF-induced response was blocked by more than 75% (Fig. 5D). Together, these results suggest that AL-8810 preferentially biases FP receptor-induced ERK1/2 through EGFR transactivation.
AL-8810-induced EGF shedding is responsible for EGFR activation
One of the many mechanisms by which GPCR can transactivate tyrosine kinase receptors is by ectodomain shedding of pro-heparin-binding-EGF-like growth factor (pro-HB-EGF) by matrix metalloproteases, leading to autocrine released of mature EGF into the cellular milieu (33). To further investigate the mechanism by which AL-8810 signal could lead to EGFR transactivation and MAPK response, we first used an EGFR antagonist, PD153035, to inhibit binding of autocrine EGF to its receptor and subsequent signaling. PD153035 was found to antagonize EGF-induced ERK1/2 activation (Supplemental Fig. 1C). In cells pretreated with PD153035, no significant effect on PGF2α-induced ERK1/2 activation was detected (Fig. 6A), but significant inhibition (by > 40%) of ERK1/2 activation induced by AL-8810 was observed (at 5 and 15 min; Fig. 6B), consistent with results obtained with AG1478 (Fig. 5B). Our data thus suggest the involvement of pro-HB-EGF shedding and EGFR transactivation in AL-8810-dependent activation of MAPK. To confirm this, we stimulated FP-R cells with AL-8810 or PGF2α and assessed ERK1/2 responses in these cells (defined as “donor cells”; Fig. 6C), and naïve HEK293 cells lacking FP receptor (defined as “acceptor cells”; Fig. 6C) that were stimulated with conditioned medium from the donor cells. Treatment of HEK293 cells with conditioned medium from AL-8810-treated donor cells promoted significant activation of the ERK1/2 response, whereas medium from PGF2α-treated cells had very little effect, consistent with the release of HB-EGF into the media of FP-R cells but only when treated with AL-8810.
Cytosolic nonreceptor tyrosine kinases, such as Src, have also been shown to be involved in EGFR transactivation by GPCR (35). To determine the potential involvement of Src in FP receptor-mediated EGFR transactivation and MAPK activation, we transfected FP-R cells with a Src kinase dead mutant (SrcK298R), which we showed previously to act in a dominant-negative fashion (29). Whereas SrcK298R had no significant effect on PGF2α-induced ERK1/2 activation (Fig. 7A), it inhibited between 60–80% AL-8810-induced ERK1/2 activation (at 2, 5, and 15 min; Fig. 7B). Together, our data suggest the involvement of Src, pro-HB-EGF shedding, and EGFR transactivation in AL-8810-dependent activation of MAPK.
Spatiotemporal activation of ERK1/2 is differentially regulated by PGF2α and AL-8810
Receptor-activated ERK1/2 can remain in the cytosol or translocate to the nucleus, to target specific downstream effectors and transcription factors (36, 37). To assess the extent to which FP receptor activation by either PGF2α or AL-8810 modulates ERK1/2 localization, we performed cellular fractionation of ligand-stimulated FP-R cells at different time points after stimulation. We observed that after PGF2α stimulation, ERK1/2, which was mainly localized in the cytosol, gradually moved away from this cellular compartment over time (Fig. 8A, white bars). On the other hand, ERK1/2 levels increased in the nuclear fraction, which reached significantly different levels after 60 min (Fig. 8B, white bars). A different subcellular localization pattern was observed in AL-8810-stimulated FP-R cells, where ERK1/2 was mainly localized in the nucleus at early time points of ligand treatment (Fig. 8B, black bars), and its presence diminishing over time, at the expense of increasing ERK1/2 levels in the cytosolic fraction (Fig. 8A).
We next used immunofluorescence to further investigate the FP receptor-ligand-induced bias on spatiotemporal regulation of ERK1/2 activation in the nucleus. Cells stimulated with PGF2α showed strong phospho-ERK1/2 signal appearing at longer time points of agonist treatment (30 and 60 min vs. 5 and 15 min; Fig. 8C). This signal colocalized within the nucleus as shown by the pixel composite overlays between both the phospho-ERK1/2 signal and the nucleus staining (as revealed by Hoescht). On the other hand, most of the phospho-ERK1/2 signal induced by AL-8810 appeared at shorter time points of ligand treatment, colocalized with the nucleus (5 and 15 min; Fig. 8C), and disappeared after 30 and 60 min.
Finally, we used the ERK activity reporter (EKAR), a FRET-based sensor that has been modified to selectively assess ERK signaling in either the nucleus (EKARN) or the cytosol (EKARC) (Fig. 9 and Ref. 30) to study ERK1/2 activity in real time in both cellular compartments. FP-R cells were transfected with EKARN or EKARC and challenged with PGF2α or AL-8810, and ERK1/2 activity was monitored using confocal microscopy. AL-8810 stimulation of the FP receptor promoted rapid (∼2 min) and sustained (up to 60 min) ERK activity in the nucleus (Fig. 9A). In marked contrast, ERK activation in this compartment was delayed after PGF2α-mediated stimulation of FP receptor, being most evident only after longer periods of agonist treatment. On the other hand, the converse was observed for ERK1/2 signaling, when assessing activity in the cytosol (Fig. 9B). PGF2α stimulation of the FP receptor induced rapid activation of ERK1/2 in the cytosol, whereas its activity was greatly delayed, if not absent at all, when receptors were challenged with AL-8810. Finally, we observed that AL-8810-mediated ERK1/2 signaling was only significantly inhibited by AG1478 in the nucleus, whereas EGFR inhibition had little effects on ERK signaling promoted by PGF2α, both in the cytosol and nucleus. Taken together, these results imply that PGF2α and AL-8810 can regulate FP receptor-mediated ERK1/2 subcellular localization and activation in a distinct fashion.
AL-8810 and PGF2α induce different patterns of cellular proliferation
GPCR-mediated activation of ERK1/2 can lead to different cellular responses, such as differentiation, development, and proliferation (38, 39). Using [3H]thymidine incorporation into MG-63 osteoblasts cells, we next assessed how PGF2α and AL-8810 induced DNA synthesis, a hallmark of cell growth. As shown in Fig. 10, PGF2α induced a constant increase in cell proliferation, starting after 4 h of stimulation and reaching a maximum response after 24 h. AL-8810, however, induced a stronger increase in [3H]thymidine incorporation a 4 h of stimulation than PGF2α, which was transient and decreased over time. Taken together, these results suggest that, as for the spatiotemporal EKR1/2 redistribution, FP receptor effects on cell proliferation can be biased depending on the ligand used.
Discussion
Here, we describe a previously unappreciated, biased agonist property of AL-8810, which was characterized initially as a selective antagonist of FP receptor-mediated, Gαq-dependent signaling. We show that AL-8810-promoted MAPK activation occurred mainly via EGFR transactivation in both heterologous and homologous FP receptor-expressing cells, and in contrast to PGF2α-mediated ERK1/2 activation, is independent of PKC and involves Src kinase. The consequence of this biased signaling was revealed by distinct spatiotemporal regulation of ERK1/2 activation and cell-proliferative responses.
Consistent with the notion that AL-8810 acts as an orthosteric ligand, it competed for PGF2α binding to the FP receptor. We showed that at concentrations occupying most receptors, AL-8810 robustly activated ERK1/2 with a similar efficacy as PGF2α. This response could not be explained by a nonselective effect of AL-8810 on other receptors, because we did not observe any such responses in HEK293 cells lacking FP receptors. Moreover, activation of ERK1/2 by both PGF2α and AL-8810 was inhibited by another selective FP receptor antagonist, AS604872. We showed in FP-R cells that whereas ERK1/2 activation induced by PGF2α was mostly blocked by PKC inhibition, the response promoted by AL-8810 was insensitive to such treatment, but was largely blocked by inhibition of EGFR and Src. These results suggest that although both PGF2α and AL-8810 direct FP receptor signaling toward MAPK activation, they did so through preferential engagement of distinct downstream signaling mechanisms (Fig. 11). GPCR also recruit β-arrestins to facilitate both their endocytosis and distinct signaling events such as MAPK activation after desensitization of primary cell surface signaling pathways (40, 41). We investigated the potential role of β-arrestin recruitment in both PGF2α- and AL-8810-mediated, FP receptor-dependent signaling. Stimulation with either PGF2α or AL-8810 did not lead to recruitment of β-arrestin-1 or β-arrestin-2 to the FP receptor, suggesting that it does not employ β-arrestins for its internalization, or for activation of MAPK, at least in the cells tested here.
Fig. 11.

Signaling mechanisms for PGF2α and AL-8810 stimulation of FP receptor. PGF2α activates ERK1/2 through the canonical Gαq/11-PKC-dependent pathway, which seems to be mostly EGFR independent in HEK293 cells, and results in activated ERK1/2 remaining mainly in the cytosol at early time points and translocates to the nucleus at later time points. On the other hand, AL-8810 activates ERK1/2 through FP receptor-dependent EGFR transactivation, via Src kinase and EGF shedding, probably via matrix metalloproteases, leading to a rapid and transient activation of ERK1/2 in the nucleus. RTK, Receptor tyrosine kinase.
The concept of ligand-directed signaling, or biased agonism, has been demonstrated for many different GPCR and ligands (for reviews see Refs. 18–22). These ligands include compounds that act at both orthosteric and allosteric binding sites and induce specific receptor conformations that favor selective or specific downstream signaling pathways. Biased GPCR ligands have been shown to differentially activate Gα proteins (10), induce β-arrestin-specific responses (42), and to transactivate EGFR (43). Interesting examples have recently been demonstrated using different competitive β-adrenergic antagonists that promoted MAPK activation in a G protein-independent manner through β-arrestins scaffolding (44, 45). Another example is the oxytocin receptor antagonist atosiban, which was shown to be an antagonist for Gαq-induced signaling, but an agonist for the ERK1/2 pathway (46). Our findings add AL-8810 to this list of orthosteric ligands that act as antagonists for one G protein-dependent signaling pathway, while preferentially activating another pathway. Our findings also underscore the need for caution when using AL-8810 as a competitive antagonist to study PGF2α-mediated responses in complex biological systems (24–27). Indeed, FP receptor could engage MAPK if signaling effectors, such as those involved in autocrine EGFR-dependent transactivation, are also present. Otherwise, if such cellular signaling complements are absent, AL-8810 would act as competitive antagonist on the FP receptor to inhibit PGF2α-mediated MAPK activation. We found that pretreatment of cells with AL-8810 (e.g. for 30 min) inhibited PGF2α-mediated ERK1/2 activation. This antagonistic property, however, most likely reflects the desensitization of this signaling pathway (e.g. dephosphorylation of MAPK) rather than a competition of ligands at the FP receptor level. Such effect may also explain some of the AL-8810 antagonistic properties previously reported by others.
Osteoblasts express endogenous FP receptors, which transactivate EGFR upon PGF2α stimulation (31, 32). We observed that MAPK activation promoted by both PGF2α and AL-8810 in human MG-63 osteoblast cells was highly sensitive to EGFR inhibition, again consistent with the transactivation of this receptor tyrosine kinase by the FP receptor. This contrasts with PGF2α-mediated MAPK signaling in HEK293 cells, which was mostly EGFR independent, highlighting the cell-specific context of biased signaling. Indeed, although we observed consistent, but statistically nonsignificant, effects on some EGFR-dependent responses promoted by PGF2α (e.g. phosphorylation of EGFR), this was insufficient to engage HB-EGF shedding and activation of MAPK.
Little is known about the molecular and structural requirements for ligand binding and FP receptor activation (i.e. conformational rearrangement of the receptor promoted by the binding of ligands). The carboxylic acid group in C1 position of PGF2α has been shown to be important for ligand binding and activity on the receptor (47–51). Substitution of the 15-hydroxyl group in PGF2α or insertion of cyclic groups in the alkyl chain, such as in the case for bimatoprost, cloprostenol, and fluprostenol, are generally well tolerated by the FP receptor in terms of ligand binding and activity. On the other hand, the 9- and 11-hydroxyl groups have been reported to be important for PGF2α binding to FP receptor. Our findings with AL-8810, which bears an 11β-fluoro substitution, are not only consistent with previous binding data, but they also suggest that the hydroxyl group in the cyclopentane moiety is involved in stabilizing a conformation of the FP receptor that engages specific downstream effectors and signaling pathways. Indeed, crystallography studies of GPCR have revealed that even very minor changes in receptor conformation supported by different orthosteric ligands, can promote differential receptor states capable of distinct signaling events (52, 53).
Mechanisms controlling the activity of MAPK and their subcellular localization such as shuttling of ERK1/2 between the cytoplasm and the nucleus are complex and remain incompletely understood (36, 37). Cellular compartmentalization of ERK1/2 seems to be an important component for controlling its activity and function, because it targets specific transcription factors and protein kinases in the nucleus and phosphorylates protein kinases and other structural proteins in the cytosol to regulate distinct cell responses (54). Moreover, the duration of ERK1/2 signals is also a key determinant of the functional and physiological outcomes (55). For instance, nuclear translocation and sustained activation of ERK1/2 promoted by some growth factors like nerve growth factor engender differentiation of pheochromocytoma (PC12) cells and neurite outgrowth, whereas transient activation of ERK1/2 induced by EGF, which remains mostly cytosolic, does not lead to such responses (56, 57). Using cellular fractionation, immunofluorescent labeling, and a FRET-based biosensor, we demonstrated that ERK1/2 is differentially localized and activated in the cytosol and the nucleus, in a time-dependent manner, depending on which FP receptor ligand was used. Whereas the activation of ERK1/2 using biochemical or immunofluorescent approaches appeared transient in nature, our data using a biosensor suggest a more sustained MAPK activation in different cellular compartments (e.g. AL-8810-induced MAPK activation in the nucleus vs. the same response induced by PGF2α is in the cytosol). This latter result perhaps suggests the inability of the biosensor to be deactivated (e.g. dephosphorylated by MAPK phosphatases), and to return to an open inactive conformation. Notwithstanding this possibility, our collective data support a different spatial and temporal regulation of ERK1/2 activation in the nucleus and cytosol by PGF2α and AL-8810.
GPCR are known to either induce or inhibit cell proliferation (58). For instance, osteoblast-like cells have been shown to have dual responses to PTH stimulation, the cellular context being important to either induce or inhibit cell proliferation (59). Despite our finding that PGF2α and AL-8810 can signal to MAPK through similar signaling pathways, our results in MG-63 osteoblast cells using [3H]thymidine incorporation suggest that the FP receptor can be biased into promoting complex physiological responses such as proliferation. This also suggested that other regulatory signaling mechanisms specific to PGF2α- or AL-8810-dependent FP receptor activation must be at play. We have not assessed the spatiotemporal regulation of ERK1/2 activation by PGF2α and AL-8810 in MG-63 osteoblast cells. However, it is tempting to speculate that such differences in the regulation of MAPK between the two ligands could also be responsible for the distinct proliferation pattern observed here. This also opens the question of how PGF2α and AL-8810 differentially control cell proliferation. Future studies will be required to better understand the link between ligand-specific-mediated spatiotemporal activation of MAPK and the differential regulation of cellular responses like proliferation and perhaps other responses such as gene expression and/or cytoskeletal signaling, which may also contribute to the phenotype observed.
Acknowledgments
We thank Dr. Christian LeGouill and Dr. William Lubell (Université de Montréal, Montréal, Canada) for helpful discussions.
This work was supported by a Canadian Institutes of Health Research (CIHR) Team Grant in GPCR Allosteric Regulation (CTiGAR, CTP 79848), in which both T.E.H. and S.A.L. are coinvestigators, and CIHR grants PRG-82673 and MOP-74603 (to S.A.L.). B.Z. holds a Banting and Best Doctoral Fellowship Award from the CIHR. T.E.H. is a Chercheur National of the “Fonds de la Recherche en Santé du Québec.” S.A.L. is Canada Research Chair in Molecular Endocrinology.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AngII
- Angiotensin II
- AT1R
- AngII type 1 receptor
- CFP
- cerulean fluorescent protein
- DMSO
- dimethylsulfoxide
- EGF
- epidermal growth factor
- EGFR
- EGF receptor
- EKAR
- ERK activity reporter
- FBS
- fetal bovine serum
- FRET
- fluorescence resonance energy transfer
- FP receptor
- F prostanoid receptor
- FP-R cells
- FP receptor cells
- GFP
- green fluorescent protein
- GPCR
- G protein-coupled receptor
- HA
- hemagglutinin
- HB-EGF
- heparin-binding EGF
- HEK
- human embryonic kidney
- PGF2α
- prostaglandin F2α
- PKC
- protein kinase C
- PLC
- phospholipase C
- TCL
- total cell lysate
- YFP
- yellow fluorescent protein.
References
- 1. Makino S , Zaragoza DB , Mitchell BF , Robertson S , Olson DM. 2007. Prostaglandin F2α and its receptor as activators of human decidua. Semin Reprod Med 25:60–68 [DOI] [PubMed] [Google Scholar]
- 2. Olson DM , Zaragoza DB , Shallow MC , Cook JL , Mitchell BF , Grigsby P , Hirst J. 2003. Myometrial activation and preterm labour: evidence supporting a role for the prostaglandin F receptor—a review. Placenta 24( Suppl A):S47–S54 [DOI] [PubMed] [Google Scholar]
- 3. Breyer MD , Breyer RM. 2001. G protein-coupled prostanoid receptors and the kidney. Annu Rev Physiol 63:579–605 [DOI] [PubMed] [Google Scholar]
- 4. Lee PY , Shao H , Xu LA , Qu CK. 1988. The effect of prostaglandin F2α on intraocular pressure in normotensive human subjects. Invest Ophthalmol Vis Sci 29:1474–1477 [PubMed] [Google Scholar]
- 5. Lai J , Jin H , Yang R , Winer J , Li W , Yen R , King KL , Zeigler F , Ko A , Cheng J , Bunting S , Paoni NF. 1996. Prostaglandin F2α induces cardiac myocyte hypertrophy in vitro and cardiac growth in vivo. Am J Physiol 271:H2197–H2208 [DOI] [PubMed] [Google Scholar]
- 6. Kamon M , Fujita D , Goto N , Amano H , Sakamoto K. 2008. Prostaglandin F(2α) negatively regulates bone resorption in murine osteoclast development. Prostaglandins Other Lipid Mediat 87:26–33 [DOI] [PubMed] [Google Scholar]
- 7. Hakeda Y , Harada S , Matsumoto T , Tezuka K , Higashino K , Kodama H , Hashimoto-Goto T , Ogata E , Kumegawa M. 1991. Prostaglandin F2α stimulates proliferation of clonal osteoblastic MC3T3–E1 cells by up-regulation of insulin-like growth factor I receptors. J Biol Chem 266:21044–21050 [PubMed] [Google Scholar]
- 8. Chen DB , Westfall SD , Fong HW , Roberson MS , Davis JS. 1998. Prostaglandin F2α stimulates the Raf/MEK1/mitogen-activated protein kinase signaling cascade in bovine luteal cells. Endocrinology 139:3876–3885 [DOI] [PubMed] [Google Scholar]
- 9. Pierce KL , Fujino H , Srinivasan D , Regan JW. 1999. Activation of FP prostanoid receptor isoforms leads to Rho-mediated changes in cell morphology and in the cell cytoskeleton. J Biol Chem 274:35944–35949 [DOI] [PubMed] [Google Scholar]
- 10. Goupil E , Tassy D , Bourguet C , Quiniou C , Wisehart V , Petrin D , Legouill C , Devost D , Zingg HH , Bouvier M , Saragovi HU , Chemtob S , Lubell WD , Claing A , Hebert TE , Laporte SA. 2010. A novel biased allosteric compound inhibitor of parturition, selectively impedes the PGF2α-mediated rho/rock signaling pathway. J Biol Chem 285:25624–25636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davis JS , Weakland LL , Weiland DA , Farese RV , West LA. 1987. Prostaglandin F2α stimulates phosphatidylinositol 4,5-bisphosphate hydrolysis and mobilizes intracellular Ca2+ in bovine luteal cells. Proc Natl Acad Sci USA 84:3728–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jimenez de Asua L , Goin M. 1997. Prostaglandin F2α (PGF2α) triggers protein kinase C (PKC) and tyrosine kinase activity in cultured mammalian cells. Adv Exp Med Biol 400A:531–538 [DOI] [PubMed] [Google Scholar]
- 13. Ito S , Sakamoto K , Mochizuki-Oda N , Ezashi T , Miwa K , Okuda-Ashitaka E , Shevchenko VI , Kiso Y , Hayaishi O. 1994. Prostaglandin F2α receptor is coupled to Gq in cDNA-transfected Chinese hamster ovary cells. Biochem Biophys Res Commun 200:756–762 [DOI] [PubMed] [Google Scholar]
- 14. Daaka Y , Luttrell LM , Ahn S , Della Rocca GJ , Ferguson SS , Caron MG , Lefkowitz RJ. 1998. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem 273:685–688 [DOI] [PubMed] [Google Scholar]
- 15. Dikic I , Tokiwa G , Lev S , Courtneidge SA , Schlessinger J. 1996. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 383:547–550 [DOI] [PubMed] [Google Scholar]
- 16. Daaka Y , Luttrell LM , Lefkowitz RJ. 1997. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390:88–91 [DOI] [PubMed] [Google Scholar]
- 17. Pierce KL , Luttrell LM , Lefkowitz RJ. 2001. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene 20:1532–1539 [DOI] [PubMed] [Google Scholar]
- 18. Kenakin T. 2003. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci 24:346–354 [DOI] [PubMed] [Google Scholar]
- 19. Galandrin S , Oligny-Longpré G , Bouvier M. 2007. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci 28:423–430 [DOI] [PubMed] [Google Scholar]
- 20. Wang L , Martin B , Brenneman R , Luttrell LM , Maudsley S. 2009. Allosteric modulators of g protein-coupled receptors: future therapeutics for complex physiological disorders. J Pharmacol Exp Ther 331:340–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Digby GJ , Conn PJ , Lindsley CW. 2010. Orthosteric- and allosteric-induced ligand-directed trafficking at GPCRs. Curr Opin Drug Discov Dev 13:587–594 [PMC free article] [PubMed] [Google Scholar]
- 22. Conn PJ , Christopoulos A , Lindsley CW. 2009. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Griffin BW , Klimko P , Crider JY , Sharif NA. 1999. AL-8810: a novel prostaglandin F2α analog with selective antagonist effects at the prostaglandin F2α (FP) receptor. J Pharmacol Exp Ther 290:1278–1284 [PubMed] [Google Scholar]
- 24. Ueno T , Fujimori K. 2011. Novel suppression mechanism operating in early phase of adipogenesis by positive feedback loop for enhancement of cyclooxygenase-2 expression through prostaglandin F2α receptor mediated activation of MEK/ERK-CREB cascade. FEBS J 278:2901–2912 [DOI] [PubMed] [Google Scholar]
- 25. Husain S , Jafri F , Crosson CE. 2005. Acute effects of PGF2α on MMP-2 secretion from human ciliary muscle cells: a PKC- and ERK-dependent process. Invest Ophthalmol Vis Sci 46:1706–1713 [DOI] [PubMed] [Google Scholar]
- 26. Sales KJ , Boddy SC , Williams AR , Anderson RA , Jabbour HN. 2007. F-prostanoid receptor regulation of fibroblast growth factor 2 signaling in endometrial adenocarcinoma cells. Endocrinology 148:3635–3644 [DOI] [PubMed] [Google Scholar]
- 27. Woodward DF , Krauss AH , Wang JW , Protzman CE , Nieves AL , Liang Y , Donde Y , Burk RM , Landsverk K , Struble C. 2007. Identification of an antagonist that selectively blocks the activity of prostamides (prostaglandin-ethanolamides) in the feline iris. Br J Pharmacol 150:342–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zimmerman B , Simaan M , Lee MH , Luttrell LM , Laporte SA. 2009. c-Src-mediated phosphorylation of AP-2 reveals a general mechanism for receptors internalizing through the clathrin pathway. Cell Signal 21:103–110 [DOI] [PubMed] [Google Scholar]
- 29. Fessart D , Simaan M , Zimmerman B , Comeau J , Hamdan FF , Wiseman PW , Bouvier M , Laporte SA. 2007. Src-dependent phosphorylation of β2-adaptin dissociates the β-arrestin-AP-2 complex. J Cell Sci 120:1723–1732 [DOI] [PubMed] [Google Scholar]
- 30. Harvey CD , Ehrhardt AG , Cellurale C , Zhong H , Yasuda R , Davis RJ , Svoboda K. 2008. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci USA 105:19264–19269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Samadfam R , Gallant MA , Miousse MC , Parent JL , de Brum- Fernandes AJ. 2006. Implication of prostaglandin receptors in the accumulation of osteoprotegerin in human osteoblast cultures. J Rheumatol 33:1167–1175 [PubMed] [Google Scholar]
- 32. Ahmed I , Gesty-Palmer D , Drezner MK , Luttrell LM. 2003. Transactivation of the epidermal growth factor receptor mediates parathyroid hormone and prostaglandin F2α-stimulated mitogen-activated protein kinase activation in cultured transgenic murine osteoblasts. Mol Endocrinol 17:1607–1621 [DOI] [PubMed] [Google Scholar]
- 33. Prenzel N , Zwick E , Daub H , Leserer M , Abraham R , Wallasch C , Ullrich A. 1999. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402:884–888 [DOI] [PubMed] [Google Scholar]
- 34. Lombardo CR , Consler TG , Kassel DB. 1995. In vitro phosphorylation of the epidermal growth factor receptor autophosphorylation domain by c-src: identification of phosphorylation sites and c-src SH2 domain binding sites. Biochemistry 34:16456–16466 [DOI] [PubMed] [Google Scholar]
- 35. Luttrell LM , Della Rocca GJ , van Biesen T , Luttrell DK , Lefkowitz RJ. 1997. Gβγ subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G protein-coupled receptor-mediated Ras activation. J Biol Chem 272:4637–4644 [DOI] [PubMed] [Google Scholar]
- 36. Pouysségur J , Volmat V , Lenormand P. 2002. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol 64:755–763 [DOI] [PubMed] [Google Scholar]
- 37. Ramos JW. 2008. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol 40:2707–2719 [DOI] [PubMed] [Google Scholar]
- 38. Zhang W , Liu HT. 2002. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12:9–18 [DOI] [PubMed] [Google Scholar]
- 39. Meloche S , Pouysségur J. 2007. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26:3227–3239 [DOI] [PubMed] [Google Scholar]
- 40. DeWire SM , Ahn S , Lefkowitz RJ , Shenoy SK. 2007. β-arrestins and cell signaling. Annu Rev Physiol 69:483–510 [DOI] [PubMed] [Google Scholar]
- 41. Luttrell LM , Gesty-Palmer D. 2010. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev 62:305–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wei H , Ahn S , Shenoy SK , Karnik SS , Hunyady L , Luttrell LM , Lefkowitz RJ. 2003. Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA 100:10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim IM , Tilley DG , Chen J , Salazar NC , Whalen EJ , Violin JD , Rockman HA. 2008. β-Blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA 105:14555–14560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Galandrin S , Oligny-Longpré G , Bonin H , Ogawa K , Galés C , Bouvier M. 2008. Conformational rearrangements and signaling cascades involved in ligand-biased mitogen-activated protein kinase signaling through the β1-adrenergic receptor. Mol Pharmacol 74:162–172 [DOI] [PubMed] [Google Scholar]
- 45. Wisler JW , DeWire SM , Whalen EJ , Violin JD , Drake MT , Ahn S , Shenoy SK , Lefkowitz RJ. 2007. A unique mechanism of β-blocker action: carvedilol stimulates β-arrestin signaling. Proc Natl Acad Sci USA 104:16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reversi A , Rimoldi V , Marrocco T , Cassoni P , Bussolati G , Parenti M , Chini B. 2005. The oxytocin receptor antagonist atosiban inhibits cell growth via a “biased agonist” mechanism. J Biol Chem 280:16311–16318 [DOI] [PubMed] [Google Scholar]
- 47. Powell WS , Hammarstrom S , Samuelsson B , Sjoberg B. 1974. Letter: prostaglandin-F2α receptor in human corpora lutea. Lancet 1:1120. [DOI] [PubMed] [Google Scholar]
- 48. Schaaf TK , Hess HJ. 1979. Synthesis and biological activity of carboxyl-terminus modified prostaglandin analogues. J Med Chem 22:1340–1346 [DOI] [PubMed] [Google Scholar]
- 49. Maddox YT , Ramwell PW , Shiner CS , Corey EJ. 1978. Amide and i-amino derivatives of F prostaglandins as prostaglandin antagonists. Nature 273:549–552 [DOI] [PubMed] [Google Scholar]
- 50. Schuster VL , Itoh S , Andrews SW , Burk RM , Chen J , Kedzie KM , Gil DW , Woodward DF. 2000. Synthetic modification of prostaglandin F2α indicates different structural determinants for binding to the prostaglandin F receptor versus the prostaglandin transporter. Mol Pharmacol 58:1511–1516 [DOI] [PubMed] [Google Scholar]
- 51. Woodward DF , Krauss AH , Chen J , Gil DW , Kedzie KM , Protzman CE , Shi L , Chen R , Krauss HA , Bogardus A , Dinh HT , Wheeler LA , Andrews SW , Burk RM , Gac T , Roof MB , Garst ME , Kaplan LJ , Sachs G , Pierce KL , Regan JW , Ross RA , Chan MF. 2000. Replacement of the carboxylic acid group of prostaglandin F2α with a hydroxyl or methoxy substituent provides biologically unique compounds. Br J Pharmacol 130:1933–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kobilka BK , Deupi X. 2007. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci 28:397–406 [DOI] [PubMed] [Google Scholar]
- 53. Deupi X , Kobilka BK. 2010. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology (Bethesda) 25:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoon S , Seger R. 2006. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21–44 [DOI] [PubMed] [Google Scholar]
- 55. Marshall CJ. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80:179–185 [DOI] [PubMed] [Google Scholar]
- 56. Nguyen TT , Scimeca JC , Filloux C , Peraldi P , Carpentier JL , Van Obberghen E. 1993. Co-regulation of the mitogen-activated protein kinase, extracellular signal-regulated kinase 1, and the 90-kDa ribosomal S6 kinase in PC12 cells. Distinct effects of the neurotrophic factor, nerve growth factor, and the mitogenic factor, epidermal growth factor. J Biol Chem 268:9803–9810 [PubMed] [Google Scholar]
- 57. Traverse S , Gomez N , Paterson H , Marshall C , Cohen P. 1992. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J 288(Pt 2):351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. New DC , Wong YH. 2007. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J Mol Signal 2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fujita T , Meguro T , Fukuyama R , Nakamuta H , Koida M. 2002. New signaling pathway for parathyroid hormone and cyclic AMP action on extracellular-regulated kinase and cell proliferation in bone cells. Checkpoint of modulation by cyclic AMP. J Biol Chem 277:22191–22200 [DOI] [PubMed] [Google Scholar]