

Abstract
Our previous studies have shown that microRNA-383 (miR-383) is one of the most down-regulated miRNA in TGF-β1-treated mouse ovarian granulosa cells (GC). However, the roles and mechanisms of miR-383 in GC function during follicular development remain unknown. In this study, we found that miR-383 was mainly expressed in GC and oocytes of mouse ovarian follicles. Overexpression of miR-383 enhanced estradiol release from GC through targeting RNA binding motif, single stranded interacting protein 1 (RBMS1). miR-383 inhibited RBMS1 by affecting its mRNA stability, which subsequently suppressed the level of c-Myc (a downstream target of RBMS1). Forced expression of RBMS1 or c-Myc attenuated miR-383-mediated steroidogenesis-promoting effects. Knockdown of the transcription factor steroidogenic factor-1 (SF-1) significantly suppressed the expression of Sarcoglycan zeta (SGCZ) (miR-383 host gene), primary and mature miR-383 in GC, indicating that miR-383 was transcriptionally regulated by SF-1. Luciferase and chromatin immunoprecipitation assays revealed that SF-1 specifically bound to the promoter region of SGCZ and directly transactivated miR-383 in parallel with SGCZ. In addition, SF-1 was involved in regulation of miR-383- and RBMS1/c-Myc-mediated estradiol release from GC. These results suggest that miR-383 functions to promote steroidogenesis by targeting RBMS1, at least in part, through inactivation of c-Myc. SF-1 acts as a positive regulator of miR-383 processing and function in GC. Understanding of regulation of miRNA biogenesis and function in estrogen production will potentiate the usefulness of miRNA in the control of reproduction and treatment of some steroid-related disorders.
The functional unit of the mammalian ovary, the follicle, comprises an oocyte surrounded by one or more layers of somatic granulosa cells (GC). As follicles develop, the somatic cells proliferate and differentiate, which GC acquire the ability to secrete or respond to sex steroids, including estrogens, androgens, and progestogens (1, 2). Estrogens play a central role in regulating female reproduction (3, 4). For example, estradiol (E2) promotes GC proliferation, antrum formation, gap-junction formation, and enhances FSH action in GC (5–7).
Ovarian follicle development is a complex process that requires tightly regulated expression and interaction of a multitude of genes (8, 9). For example, the transcription factor steroidogenic factor-1 (SF-1) plays a key role in steroidogenesis, which regulates transcription of Cyp19a1 gene (the rate-limiting enzyme for estrogen synthesis) through interactions with coactivators/corepressors and other transcription factors (10). The transcription factor RNA binding motif, single stranded interacting protein 1 (RBMS1), which plays important roles in regulation of DNA replication, transcription, apoptosis, and cell cycle progression by interacting with the c-Myc protein (11, 12), is also of importance to both the growth of the embryo and the hormone synthesis in adult female mouse (13). Recently, microRNA (miRNA) are also indicated a critical role in ovarian follicle development by targeting genes involved in folliculogenesis (14–16).
miRNA are small noncoding RNA of 19–25 nucleotides in length, which are endogenously expressed in most eukaryotes. miRNA posttranscriptionally regulate gene expression through base pairing with the 3′-untranslated region (UTR) of target mRNA, leading either mRNA cleavage or translational repression (17–19). Many studies have shown that miRNA play important roles in diverse biological processes, such as development, cell differentiation, proliferation, and apoptosis (20–23). Their biological importance, initially demonstrated in cancer, is also more recently discovered in ovarian development and functions (24–28). For example, Dicer (an ribonuclease III endonuclease essential for miRNA biogenesis) and its product miRNA are required for meiotic maturation of mouse oocytes and normal development of the female reproductive system (14, 15, 25, 29, 30). miRNA function is inactivated during oocyte development, and the suppression of miRNA function is critical for reprogramming gene expression during the transition of a differentiated oocyte to pluripotent blastomere of the embryos (31, 32). Some researchers also investigated the miRNA expression profiles in normal (33) or growth factor-treated (16) ovarian cells and in ovarian carcinoma cells (34). Ro et al. (33) identified miRNA in mouse ovaries and found some miRNA critical to regulate genes essential for ovarian folliculogenesis and endocrine function. We profiled the miRNA signature of TGF-β1-treated mouse preantral GC (16). TGF-β superfamily members have been demonstrated to exert important effects on early follicle development (35) and GC proliferation and differentiation (36). TGF-β1 is usually suggested an inhibitory role in preantral follicle development and progression (37). Our miRNA profiles in GC identified 16 miRNA that were differentially expressed (three up-regulated and 13 down-regulated) during TGF-β1 treatment (16). miRNA-224, the second most significantly up-regulated miRNA, is shown to regulate preantral GC proliferation and hormone secretion through targeting Smad4 (SMAD family member 4, a critical component of TGF-β-signaling pathway) (16). These studies suggest that miRNA as well as their processing play an important role in the regulation of ovarian follicle growth and female fertility. Although miRNA profiles of ovarian cells have been characterized in several studies, including our own (16, 33), as mentioned above, the precise regulatory mechanisms by which miRNA and miRNA processing itself affect ovarian function, such as E2 synthesis, remain largely unknown.
miRNA-383 (miR-383) is one of the most down-regulated miRNA in TGF-β1-treated GC (16). Our previous studies have shown that miR-383 functions as a negative regulator of NTERA-2 (testicular embryonal carcinoma) cell proliferation by targeting interferon regulatory factor-1, indicating that miR-383 acts as a tumor suppressor gene (38). However, the roles and mechanisms of miR-383 in GC function during follicular development remain unknown. In this study, miR-383 was functionally characterized in primary GC by identifying its target genes and its upstream regulatory factors.
Materials and Methods
Animals
ICR female mice were obtained from the Animal Center, University of Science and Technology of China (USTC) and maintained under a 14-h light, 10-h dark cycle at room temperature (23 ± 2 C). Mice were provided food and water ad libitum. This study received ethical approval from the institutional review boards of the USTC.
Vectors
pEGFP-C1 vector and psiCHECK-2 dual luciferase reporter vector were kindly provided by Mian Wu (USTC) and Biliang Zhang (Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, People's Republic of China), respectively. For construction of expression vectors, total RNA isolated from mouse liver tissue was reverse transcribed to cDNA. The full-length cDNA was amplified by PCR utilizing primers indicated in Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org.
The RBMS1, c-Myc expression constructs were generated by cloning the mouse cDNA into the pEGFP-C1 vector at the EcoRI and SalI sites whereas pEGFP-SF-1 at the XhoI and EcoR I sites. For construction of luciferase reporter plasmids, wild-type RBMS1 3′-UTR was obtained by amplifying a 985-bp 3′-UTR fragment of RBMS1 harboring the miR-383 binding site predicted by the TargetScan (http://www.targetscan.org/), miRBase Targets (http://microrna.sanger.ac.uk/targets/v5/), miRGen (http://www.diana.pcbi.upenn.edu/miRGen.html), and PicTar (http://pictar.mdc-berlin.de/). Mutated RBMS1 3′-UTR was generated through PCR-based site-directed mutagenesis. Wild-type and mutated RBMS1 3′-UTR, were inserted into the XhoI and NotI sites of psiCHECK-2 reporter vector immediately downstream of the stop codon of an simian virus 40 promotor-driven Renilla luciferase gene. A 303-bp fragment containing the 3′-UTR of c-Myc gene was cloned into the psiCHECK-2 reporter vector using NotI and PmeI restriction sites. The mouse Sarcoglycan zeta (SGCZ) 5′-flanking fragments of different lengths were amplified by PCR using mouse liver genomic DNA as template, and oligonucleotide primers introduced a KpnI and XhoI site at the 5′- and 3′-end, respectively. The PCR products were digested with KpnI and XhoI and then cloned into KpnI and XhoI sites of the luciferase reporter plasmid pGL3-Basic (Promega, Madison, WI). All SGCZ promoter mutants were generated by using a mutated forward primer and a reverse primer binding to the luciferase gene within the reporter. Primer sequences are listed in Supplemental Table 1. All constructs generated were confirmed by sequencing.
Reagents and oligonucleotides
TGF-β1 (R&D Systems, Minneapolis, MN) was reconstituted in acidified buffer [4 mm HCl (pH 5.7) and 1 mg/ml BSA] (16) to prepare 1 mg/ml stock solutions, and FSH (The National Hormone and Peptide Program, Torrance, CA) was reconstituted in PBS. Pregnant mare serum gonadotropin (PMSG) and human chorionic gonadotropin (hCG) were purchased from Sigma Chemical Co. (St. Louis, MO). miR-383 mimics and inhibitors, as well as scramble negative control (NC), were synthesized and purified by Shanghai Gene-Pharma Co. (Shanghai, People's Republic of China). The miR-383 mimics are chemically synthesized, double-stranded RNA that mimic mature endogenous miR-383, whereas the miR-383 inhibitors are single-stranded 2-O-methyl-modified oligoribonucleotide fragments exactly antisense to miR-383. Small interfering (si)RNA (si-RBMS1, si-c-Myc, and si-SF-1) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). For in situ hybridization (ISH) assay, the Mus musculus-miR-383 locked nucleic acid (LNA)-modified oligonucleotides labeled with digoxigenin and scramble NC (LNA scrambled) were purchased from Exiqon A/S (Vedbaek, Denmark). Oligonucleotide sequences are provided in Supplemental Table 2.
Immunohistochemistry and ISH
Immunohistochemical analysis was performed to determine the cellular localization of RBMS1, c-Myc proteins in the ovary, as described by Yao et al. (16). Briefly, mouse ovarian sections were incubated overnight at 4 C, with the primary antibodies [anti-RBMS1 (Millipore, Bedford, MA) or anti-c-Myc (Cell Signaling Technology, Beverly, MA), both diluted at 1:200], followed by incubation with biotinylated secondary antibody (Abcam, Cambridge, MA) for 2 h at room temperature. Immunosignals were visualized using streptavidin peroxidase with 3, 3′-diaminobenzidine (Maixin Bio, Fuzhou, People's Republic of China) as the chromogen. A NC was included where the primary antibody was omitted.
Expression of miR-383 in the mouse ovary was performed by ISH using LNA-modified DNA probes, as described by Lian et al. (39). The probe sequences are listed in Supplemental Table 2. Briefly, 10-μm ovarian cryosections from postnatal day (PND) 21 mice and PND 21 mice after PMSG treatment were prehybridized for 6 h at 52 C with 700 μl of prehybridization buffer [50% formamide, 5× saline sodium citrate, 5× Denhardt's, 200 μg/ml yeast RNA, 500 μg/ml salmon sperm DNA, 2% Roche blocking reagents (Roche, Basel, Switzerland), and diethylpyrocarbonate-treated water]. Sections were then covered in 150 μl of denatured hybridization buffer (prehybridization buffer containing 1 pmol LNA probes) and incubated overnight at 52 C in a humidified chamber. The hybridization signals were detected with antidigoxigenin-alkaline phosphotase fragments of antigen binding fragments (1:250; Roche) and nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate solution (Roche).
Follicle isolation, cell, cell culture, and transfection
Preantral, antral, and large antral follicles were isolated from PND 10, 25, and 27 mouse ovaries, respectively. Preantral follicles with 60–80 μm in diameter (no. of follicles isolated, n = 150), 90–110 μm in diameter (n = 100), 130–150 μm in diameter (n = 100), and 290–310 μm in diameter (n = 30) were mechanically isolated from PND 8, 10, 14, and 21 mouse ovaries, respectively. Primary GC were isolated from preantral follicles (90–110 μm in diameter) of PND 10–12 mouse ovaries, as described previously by Liang et al. (40) and Yao et al. (16). Briefly, the ovaries were excised, and the follicles were isolated with no. 5 fine needles (Sigma Chemical Co.). The follicles were then treated with type IV collagenase (Sigma Chemical Co.) and Tryple Express (Life Technologies, Inc., Gaithersburg, MD), and the GC were washed and collected by brief centrifugation. The isolated GC were grown in DMEM/F12 (1:1; Life Technologies, Inc.) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Inc.) and 1% antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin; Life Technologies, Inc.) at 37 C with 5% CO2. After 2 h, nonadherent cells were removed by a culture medium wash. Culture media were changed every 2 d thereafter, and the cells were used for experiments at two to three passages. HEK293T, NIH3T3, and MCF-7 cells were cultured in DMEM supplemented with 10% FBS (Life Technologies, Inc.) and 1% antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin; Life Technologies, Inc.) at 37 C under 5% CO2 atmosphere.
Primary GC were transfected with oligonucleotides and plasmids using HiPerFect Transfection Reagent (QIAGEN GmbH, Hilden, Germany). Transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) for HEK293T and MCF-7 cells, and Lipofectamine LTX (Invitrogen) for NIH3T3 cells. The transfection procedure was performed following the manufacturer's instructions. Unless otherwise mentioned, cells were transfected with 50 nm indicated small interfering RNA, 120 nm miR-383 mimics, or 150 nm miR-383 inhibitors.
Real-time PCR analysis
Total RNA was extracted with TRIzol (Invitrogen) and reverse transcribed into cDNA using a PrimeScript RT reagent kit (TaKaRa Bio, Inc., Otsu, Japan) according to the manufacturer's instructions. Real-time PCR was performed on ABI Step One System (Applied Biosystems, Foster City, CA) using the SYBR Premix Ex Taq II kit (TaKaRa Bio, Inc.), as described previously (16). Expression levels were normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. Primer sequences are listed in Supplemental Table 3. For mature mmu-miR-383 detection, TaqMan MicroRNA Assays (Applied Biosystems) was used according to the manufacturer's instructions. U6 snRNA (Applied Biosystems) was used for normalization.
Western blotting
Cells were lysed in radioimmunoprecipitation assay buffer [50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Triton X-100, 1% sodium dodecyl sulfate, 1% sodium deoxycholate, and 1 mm EDTA] containing proteinase inhibitors (10 μg/ml each of aprotinin, pepstatin, and leupeptin) and 1 mm phenylmethylsulfonyl fluoride. Protein lysates were resolved by SDS-PAGE, transferred to Hybond ECL Nitrocellulose membrane (Amersham Biosciences, Freiburg, Germany), immunoblotted with antibodies, and visualized using enhanced chemiluminescence (Kodak, Rochester, NY). The primary antibodies used for immunoblotting were as follows: anti-RBMS1 (clone 4D11; Millipore), anti-c-Myc (Cell Signaling Technology), anti-SF-1 (Millipore), and anti-GAPDH (Cell Signaling Technology).
Luciferase reporter assay
Luciferase reporter assays were performed using the vector psiCHECK2 or pGL3-Basic, as described by Lian et al. (39). HEK293T cells were transiently transfected with reporter constructs together with indicated plasmids or miR-383 mimics/inhibitors. Cells were harvested 30 h after transfection and assayed for reporter activity using the Dual-Luciferase Reporter Assay System (Promega).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed basically using ChIP Assay kit (Millipore), as described by Liang et al. (40). Briefly, primary GC were cross-linked, lysed, and sonicated to obtain DNA fragments of 500-1000 bp. The sonicated cell supernatant was diluted 10-fold with ChIP dilution buffer [0.01% sodium dodecyl sulfate, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl (pH 8.1), 167 mm NaCl, and protease inhibitor cocktail (Roche)], and 1% of the diluted cell supernatant was kept as input. The remaining supernatants were immunoprecipitated overnight at 4 C with anti-SF-1 antibody (Millipore). IP with normal mouse IgG was also performed as a NC. Eluted protein-DNA complexes were digested with ribonuclease A and proteinase K. DNA fragments were purified by using Spin Columns, and a 193-bp fragment of mouse SGCZ promoter region was amplified by PCR using the following primers: forward, 5′-TTATCTGAGCAAGAAGCTGC-3′ and reverse, 5′- ACCACCAGGTGACTGACTTC-3′. The PCR products were resolved by 2% agarose gel electrophoresis and visualized with ethidium bromide staining.
Hormone analysis
To determine the effects of miR-383 and its target on GC function, GC were transfected with oligonucleotides or plasmids. After 24 h, the culture medium was changed and replaced with DMEM/F12 supplemented with 10% FBS and testosterone (Sigma Chemical Co.) at 10−6 m, followed by continued culture for another 24 h. E2 and progesterone concentrations in culture medium were measured according to the manufacturer's specifications using the Access Immunoassay System (Beckman Coulter, Inc., Brea, CA), an automated random-access chemiluminescence-based assay. The intra- and interassay coefficients of variation did not exceed 10 and 15%, respectively.
Statistics
Data represent mean ± sem of at least three independent experiments. Student's t test was used to examine the differences among variables. A P value less than 0.05 was considered to be statistically significant.
Results
miR-383 promotes E2 release from GC
Follicle growth is characterized by an increase in the oocyte size concomitant with the surrounding GC proliferation (1). This growth is accompanied by differentiation of granulosa and theca cells, leading to antrum formation and increased steroidogenic function (2, 41). During ovarian folliculogenesis, E2 is produced in increasing quantities before ovulation and drops quickly just before ovulation (41). TGF-β1 could augment FSH-stimulated E2 production from rodent GC (42). In addition, TGF-β1 is indicated to play an inhibitory role in preantral follicle growth, which involves an increase in apoptosis at the primary and preantral stages of follicle development (37). In our previous studies on miRNA expression profiles in mouse GC (mGC) treated with TGF-β1, miR-383 was one of the most significantly down-regulated miRNA (the other two are miR-500 and miR-320) (16). To confirm the microarray data, the changes in miR-383 expression were validated by stem-loop primer-based real-time PCR. Indeed, TGF-β1 significantly decreased miR-383 expression in GC (Fig. 1A). Expression pattern of miR-383 in the mouse ovary was detected by real-time PCR and ISH. Because the follicle diameter positively correlated with GC proliferation, follicular growth, and E2 production (43), the expression levels of miR-383 in mouse ovarian follicles with different diameters were first examined by real-time PCR. The results revealed that miR-383 levels in antral follicles were higher than that in preantral follicles but decreased sharply after ovulation (Fig. 1B). However, miR-383 levels were not altered with increasing diameters of preantral follicles (Supplemental Fig. 1A). Treatment of immature female mice with PMSG results in enhanced E2 secretion and maturation of the preovulatory follicles (Supplemental Fig. 1, B and C). In addition, the administration of PMSG followed by hCG mimics the stimulatory effect of LH on progesterone secretion and inhibitory effect of LH on E2 secretion (44, 41). In this study, the pattern of changes in miR-383 levels in the ovaries was broadly similar to the changes in plasma E2 levels in PMSG-treated immature mice at all time points, with the exception of the 48-h time point (Supplemental Fig. 1D). In addition, hCG injection (12 h, 24 h) after PMSG treatment (24 h) caused a rapid decrease in miR-383 expression levels (Fig. 1C). The down-regulation of ovarian miR-383 expression in mice 48 h after PMSG treatment (Supplemental Fig. 1D) may indicate that the transactivating factors of miR-383 start to function at this time point. Next, the expressions of miR-383 in the PND 21 mouse ovary and PND 21 mouse ovary after PMSG treatment were performed by ISH using LNA-modified probes. miR-383 expression was predominantly found in GC and oocytes of follicles at various stages (Fig. 1D). Taken together, these results indicate that miR-383 may be involved in GC proliferation, GC function, such as E2 secretion, oocyte development, and maturation during follicular development.
Fig. 1.

miR-383 promotes E2 synthesis in mouse ovarian GC. A, TGF-β1 suppressed miR-383 expression in GC. mGC were treated with TGF-β1 (5 ng/ml) for 6, 12, and 24 h and subjected to real-time PCR analysis. B, miR-383 expression levels in preantral, antral, and ovulated follicles. Total RNA was extracted from follicles at different stages and then subjected to real-time PCR. Data obtained from the preantral follicles were arbitrarily set at 1.0. C, Changes in miR-383 expression in ovaries of PMSG/hCG-treated immature mice. Total RNA was extracted from ovaries of PMSG-treated mice at 24 h and PMSG/hCG-treated mice at indicated post-hCG time points and then subjected to real-time PCR. D, miR-383 is mainly localized in GC (arrow) and oocytes (arrowhead) of ovarian follicles at various developmental stages. miR-383 expression was performed on 10-μm frozen ovarian sections by ISH using LNA-modified DNA probe complementary to miR-383. Left panel, Positive ISH signals (purple) of miR-383. Middle panel, NC of ISH (nonspecific LNA-modified DNA oligonucleotide). Right panel, Hematoxylin and eosin (HE) staining of mouse ovarian tissues. Scale bar, 100 μm. E and F, Overexpression and knockdown of miR-383 significantly stimulated and inhibited, respectively, E2 release from GC in a dose-dependent manner. GC were transfected with different concentrations (100, 150, and 200 nm) of miR-383 mimics and mimics NC, or of miR-383 inhibitors and inhibitors NC for 48 h. After culture, media were collected for measurement of E2 levels. G, Overexpression and knockdown of miR-383 did not affect progesterone release in GC. Primary GC were transfected with 120 nm each of miR-383 mimics and mimics NC, or 150 nm each of miR-383 inhibitors and inhibitors NC for 48 h. Progesterone levels were evaluated in the medium after 48 h of culture. H, miR-383 mimics enhanced Cyp19a1 mRNA expression, whereas knockdown of endogenous miR-383 suppressed Cyp19a1 mRNA levels in GC. GC were transfected with oligonucleotides for 48 h as described above. Total RNA was extracted from GC, and Cyp19a1 mRNA was analyzed by real-time PCR. In A–C and E–H, data shown represent the mean ± sem of three independent experiments performed in triplicate. *, P < 0.05; **, P < 0.01. Con, Concentration.
We next examined the effect of miR-383 on steroidogenesis by transfection of either miR-383 mimics or inhibitors into the mGC for 48 h. Transfection efficiency of miR-383 was monitored by real-time PCR. The results showed that the intracellular miR-383 levels were increased by approximately 120-fold in GC transfected with miR-383 mimics (Supplemental Fig. 1E), whereas miR-383 inhibitors caused a more than 50% decrease in mature miR-383 levels in GC (Supplemental Fig. 1F). miR-383 mimics significantly enhanced E2 release from GC in a dose-dependent manner (Fig. 1E). In contrast, knockdown of miR-383 resulted in decreased E2 biosynthesis in a dose-dependent manner (Fig. 1F). However, miR-383 mimics/inhibitors had no effect on GC progesterone production (Fig. 1G), GC proliferation (Supplemental Fig. 1G), and apoptosis (Supplemental Fig. 1H).
To address the mechanisms responsible for the steroidogenic effects of miR-383, we detected Cyp19a1 mRNA and protein expression levels in GC transfected with miR-383 mimics or inhibitors. Real-time PCR and immunoblotting results showed that forced expression of miR-383 increased Cyp19a1 mRNA (Fig. 1H) and protein (Supplemental Fig. 1, I and J) levels, whereas inhibition of endogenous miR-383 decreased expression of Cyp19a1 in GC. However, miR-383 did not affect Cyp11a1 (a key enzyme in progesterone biosynthesis) mRNA levels in GC (Supplemental Fig. 1K). In this study, we noted that knockdown of endogenous miR-383 in GC resulted in statistically significant decrease in E2 biosynthesis and Cyp19a1 expression levels but with small amplitude. This could be explained by very low levels of endogenous miR-383 expression in primary GC (Supplemental Fig. 1L).
RBMS1 is a direct target of miR-383
Because miR-383 is mainly expressed in GC/oocytes of different stage follicles and significantly promotes E2 release, potential targets that modulate hormone synthesis, cell proliferation, or apoptosis were predicted by miRanda, PicTar, and TargetScan databases. Among the candidate miR-383 targets, we found that RBMS1 gene seems to be the most appropriate candidate, because RBMS1 is found to induce apoptosis in some types of cancer cells (12) and regulate hormone synthesis in the female mice (13). The 3′-UTR of RBMS1 mRNA contains an evolutionarily conserved miR-383 putative binding site in vertebrates (Supplemental Fig. 2A). To determine whether RBMS1 is a true target of miR-383, mGC or MCF-7 cells (an estrogen-responsive breast cancer cell line) were transfected with miR-383 mimics/control or miR-383 inhibitors/control. Immunoblotting and real-time PCR analysis showed that both RBMS1 protein (Fig. 2, A and B, and Supplemental Fig. 2, B and C) and RBMS1 mRNA (Fig. 2C and Supplemental Fig. 2D) expressions were significantly decreased in miR-383-transfected cells compared with the control cells, whereas the knockdown of miR-383 was able to increase RBMS1 expression. These results demonstrate that miR-383 down-regulates RBMS1 expression through destabilizing its mRNA.
Fig. 2.
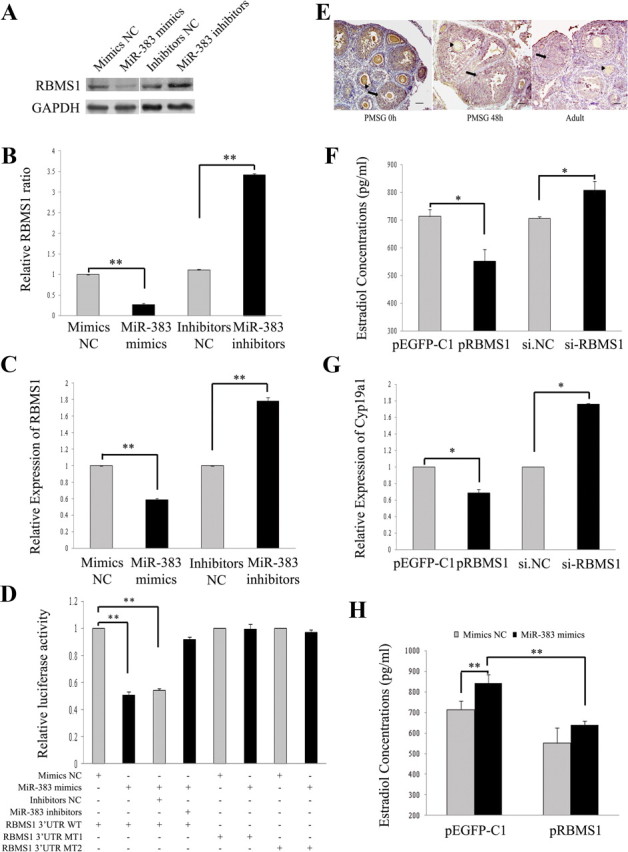
RBMS1 is a direct target of miR-383. A–C, miR-383 reduces RBMS1 protein (A and B) and mRNA (C) expression levels in mGC. GC were transfected with oligonucleotides as described in Fig. 1. The mRNA and protein expression of RBMS1 were determined by real-time PCR and Western blotting, respectively. Representative Western blotting for RBMS1 (upper bands) and GAPDH (lower bands) (A) and the corresponding densitometric analysis (B) are shown. The ratio of the RBMS1 band intensity over the GAPDH band intensity in controls was arbitrarily set at 1.0. D, Effect of miR-383 on RBMS1 3′-UTR luciferase activity. miR-383 mimics inhibit RBMS1 3′-UTR luciferase activity. In contrast, knockdown of miR-383 or seed mutant forms of miR-383 could no longer inhibit reporter luciferase activity. Cells were cotransfected with wild-type (WT) RBMS1 3′-UTR or mutant RBMS1 3′-UTR reporter constructs and miR-383 mimics/control, or miR-383 inhibitors/control. After 30 h of transfection, Firefly luciferase activity was measured and normalized to Renilla luciferase activity. E, Immunohistochemical staining for RBMS1 in the ovaries from immature mice that were either untreated (PMSG 0 h) or treated with PMSG for 48 h (PMSG 48 h), and from the adult mice. RBMS1 proteins (stained brown) are mainly located in GC (arrow) and oocytes (arrowhead) at different stages of follicular development. Scale bar, 50 μm. F–H, RBMS1 mediates miR-383-stimulated steroidogenesis. Ectopic expression or knockdown of RBMS1 in GC attenuated or potentiated, respectively, E2 synthesis (F) and Cyp19a1 mRNA levels (G). GC were transfected with si-RBMS1/si.NC or pEGFP-RBMS1 vector/control vector pEGFP-C1 for 48 h. After culture, media were collected for measurement of E2 levels, and GC were subjected to real-time PCR analysis of Cyp19a1 mRNA expression. Overexpression of RBMS1 in GC partially decreased miR-383-stimulated E2 production (H). GC were cotransfected with pEGFP-RBMS1/control vector and miR-383 mimics/mimics NC for 48 h. The culture medium was collected for hormone analysis. In B–D and F–H, data were presented as mean ± sem for at least three independent experiments. *, P < 0.05; **, P < 0.01. +, Present; −, absent.
To validate whether the RBMS1 gene is a direct target of miR-383, we constructed Renilla luciferase reporter vectors containing the wild-type full-length of RBMS1 3′-UTR as well as mutant forms of the miR-383 seed sites (Supplemental Fig. 2A). HEK293T cells were cotransfected with the reporter plasmids and miR-383 mimics/control, or miR-383 inhibitors/control, and harvested 30 h after transfection for dual luciferase assay. As shown in Fig. 2D, the luciferase activity from the wild-type RBMS1 3′-UTR was reduced by 50% with miR-383 mimics (also see Supplemental Fig. 2E), whereas miR-383 inhibitors were able to rescue the inhibition of reporter activity by miR-383. However, mutant forms of seeding sites abolished the silencing effects of miR-383 on RBMS1 3′-UTR reporter activities (Fig. 2D). These results indicate that miR-383 suppresses RBMS1 expression through direct binding to its 3′-UTR.
We next ask whether RBMS1 mediates the effects of miR-383 on steroidogenesis in GC. Firstly, the localization of RBMS1 protein in the mouse ovary was determined by immunohistochemistry. The results showed that RBMS1 was predominantly expressed in GC and oocytes at various stages of follicular development (Fig. 2E). The similar expression patterns between RBMS1 and miR-383 indicate that they might interact with each other in mGC. Secondly, RBMS1 was knockdown by using RNA interference in mGC (Supplemental Fig. 2F). As expected, knockdown of RBMS1 (si-RBMS1) and enforced RBMS1 expression (Supplemental Fig. 2F) significantly increased and suppressed E2 release (Fig. 2F and Supplemental Fig. 2G) and Cyp19a1 mRNA levels (Fig. 2G) in GC, respectively. However, RBMS1 had no effect on GC proliferation (Supplemental Fig. 2H) and apoptosis (Supplemental Fig. 2I). Furthermore, overexpression of RBMS1 partially attenuated miR-383-stimulated E2 release from mGC (Fig. 2H). These results further verify that RBMS1 is a real target of miR-383 and mediates, at least in part, the miR-383's effects on GC function.
miR-383 inhibits the expression of RBMS1 target gene c-Myc
RBMS1 is a transcription factor that activates a set of target genes, such as α-smooth muscle actin (45), c-Myc (11), Fas (46), etc. We subsequently examined the effects of miR-383 on the expression of known RBMS1 target genes involving in regulation of cell cycle and apoptosis. The results showed that miR-383 significantly decreased c-Myc mRNA (Fig. 3A) and protein (Fig. 3, B and C) expression levels, whereas knockdown of miR-383 increased its protein expression (Fig. 3, B and C). This finding leads us to ask whether c-Myc is also a target of miR-383. As shown in Supplemental Fig. 3, A and B, miR-383 mimics/inhibitors did not change the luciferase activity of the reporter vector containing the full-length c-Myc 3′-UTR. Expectedly, si-RBMS1 and overexpression of RBMS1 decreased and increased c-Myc protein levels in GC, respectively (Supplemental Fig. 3, C and D). Furthermore, forced expression of RBMS1 partly rescued c-Myc protein expression levels suppressed by miR-383 mimics (Fig. 3, B and C).
Fig. 3.
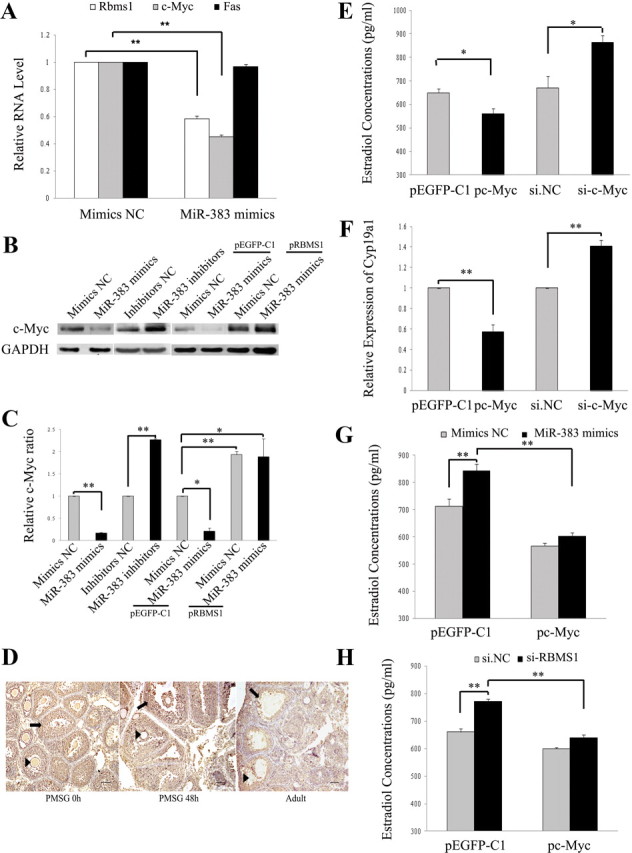
miR-383 regulates c-Myc expression in GC. A–C, Effects of miR-383 and RBMS1 on mRNA (A) and protein (B and C) expression levels of RBMS1 targets in GC were determined using real-time PCR and Western blot analysis, respectively. c-Myc expression in GC was inhibited by miR-383 mimics, whereas miR-383 inhibitors and RBMS1 overexpression partially reversed these inhibitory effects (B and C). Representative Western blot analysis for c-Myc (upper bands) and GAPDH (lower bands) (B) and the corresponding densitometric analysis (C) are shown. The ratio of the c-Myc band intensity over the GAPDH band intensity in controls was arbitrarily set at 1.0. D, Immunohistochemical staining for c-Myc in the ovaries from immature mice that were either untreated (PMSG 0 h) or treated with PMSG for 48 h (PMSG 48 h), and from the adult mice, showing similar expression patterns to RBMS1. Arrow, GC; arrowhead, oocytes. Scale bar, 50 μm. E–H, c-Myc mediates miR-383/RBMS1-stimulated steroidogenesis. Ectopic expression or knockdown of c-Myc in GC inhibited or promoted, respectively, E2 synthesis (E) and Cyp19a1 mRNA levels (F). Forced expression of c-Myc abrogated miR-383 mimics-induced (G) and si-RBMS1-induced (H) effects on GC steroidogenesis. GC were cotransfected with pEGFP-c-Myc/control vector and miR-383 mimics/mimics NC, or pEGFP-c-Myc/control vector and si-RBMS1/si.NC for 48 h. In A, C, and E–H, data were presented as means ± sem for at least three independent experiments. *, P < 0.05; **, P < 0.01.
In the ovary, c-Myc protein was also mainly localized in GC at various follicular stages (Fig. 3D), displaying a very similar expression pattern to that of miR-383 and RBMS1. Silence of c-Myc (Supplemental Fig. 3E) in GC also up-regulated the levels of E2 and Cyp19a1 mRNA (Fig. 3, E and F), whereas overexpression of c-Myc (Supplemental Fig. 3E) exerted the opposite effect (Fig. 3, E and F). In addition, both miR-383 mimics- and si-RBMS1-induced effects on steroidogenesis were abrogated by forced expression of c-Myc in GC (Fig. 3, G and H). These results indicate that c-Myc mediates the effects of both miR-383 and its target RBMS1 on E2 release in GC. In this study, miR-383 had no effect on GC proliferation and apoptosis. This could be explained by miR-383-mediated down-regulation of RBMS1/c-Myc, which is involved in cell cycle regulation and cell proliferation (11, 12, 47).
SF-1 transcriptionally regulates miR-383 expression and function in GC
Although the expression and function of miR-383 were first investigated in the mouse ovary in this study, further understanding of the regulation of miRNA biogenesis will provide important insights into normal and pathological conditions (14, 16, 29, 48). According to miRBase, mouse miR-383 gene is located in the first intron of SGCZ gene on chromosome 8A4 (Supplemental Fig. 4A). We then examined whether miR-383 was coexpressed with its host gene. Our results showed similar gene expression patterns for SGCZ and primary miR-383 (pri-miR-383) in different mouse tissues (Fig. 4A). In addition, TGF-β1 treatment caused a simultaneous decrease in expression levels of SGCZ, pri-miR-383, and miR-383 in GC (Fig. 4B). These results suggest that miR-383 transcription is driven by its host gene SGCZ.
Fig. 4.

SF-1 transactivates miR-383 through direct binding to the promoter of miR-383 host gene SGCZ. A, Expression patterns of SGCZ and pri-miR-383 in different mouse tissues. B, RNA expression levels of SGCZ, pri-miR-383, and miR-383 in TGF-β1-treated GC. C, Knockdown of SF-1 decreased the RNA levels of SGCZ, pri-miR-383, and mature miR-383. GC were transfected with si-SF-1 (30 nm), and the RNA levels was determined after 48 h of transfection by real-time PCR. D, Identification of the minimal promoter region of the mouse SGCZ gene. Left panel, Schematic representation of 5′- or 3′-deleted fragments of the mouse SGCZ promoter in conjunction with the luciferase gene (LUC) in pGL3-Basic vector. The nucleotides are numbered from the transcriptional starting site that was assigned +1. Right panel, Luciferase activity of the deleted promoter constructs. NIH3T3 cells were transfected with serial mouse SGCZ promoter-luciferase reporter deletion constructs. Relative luciferase activity was calculated as a ratio of luciferase activity of the SGCZ deletion constructs driven by SF-1 over the pGL3-Basic empty vector, which was arbitrarily set at 1. E, Mutational analysis of SGCZ promoter-luciferase reporter constructs. Mutated SGCZ promoter constructs (−128/+56) were cloned into the luciferase reporter vector pGL3-Basic. F, ChIP analysis reveals in vivo binding of SF-1 to the SGCZ promoter. ChIP assays were performed on GC extracts using antibodies against IgG and SF-1, followed by PCR amplification with primers sequences corresponding to the mouse SGCZ promoter region between nucleotides −200 and −8. G, Western blot analysis of the effect of si-SF-1/SF-1 overexpression (pSF-1) on the expression of RBMS1 and c-Myc. H and I, miR-383 mimics abrogated si-SF-1-induced RBMS1/c-Myc expression. Representative Western blotting for c-Myc (upper bands), RBMS1 (middle bands), and GAPDH (lower bands) (H) and the corresponding densitometric analysis (I) are shown. The ratio of the c-Myc/RBMS1 band intensity over the GAPDH band intensity in controls was arbitrarily set at 1.0. J, Luciferase activity of wild-type RBMS1 3′-UTR in NIH3T3 cells cotransfected with either pEGFP-SF-1 vector or control vector and miR-383 inhibitors/inhibitors NC. Left panel, Luciferase activity was measured 24 h after transfection. Right panel, Validation of SF-1 overexpression in NIH3T3 cell line. In A–E, I, and J, data were presented as means ± sem for at least three independent experiments. **, P < 0.01.
Two putative SF-1 binding sites at −419 and −980 bp in the SGCZ promoter spanning 1500 bp upstream of translation start site are predicted using bioinformatic analysis. In this study, knockdown and overexpression of SF-1 simultaneously inhibited and enhanced the gene expression of SGCZ, pri-miR-383, and mature miR-383 (Fig. 4C and Supplemental Fig. 4B), respectively. Here, we noted that the amplitude of SF-1 overexpression-induced increase in pri-miR-383 levels was greater than that of SGCZ, mature miR-383, and especially pre-miR-383 levels (Supplemental Fig. 4B). One possible explanation is that overexpression of SF-1 in GC causes relatively slow processing of pri-miRNA to pre-miRNA. Because FSH can enhance the expression of SF-1 in GC (49, 50), we also examined the effect of FSH on miR-383 expression. Real-time PCR results showed that FSH increased the expression of miR-383, SGCZ, and SF-1 in GC (Supplemental Fig. 4C). The developmental changes of miR-383 and SF-1 expression in the mouse ovary were investigated by real-time PCR and immunoblotting analysis. Very similar expression patterns were observed between them (Supplemental Fig. 4, D and E). For example, both miR-383 and SF-1 expression levels increased to a peak at PND 4 and then gradually declined. We also found that SF-1 mRNA expression (Supplemental Fig. 4, F and G) showed a similar pattern to that of miR-383 in follicles at different developmental stages (Fig. 1B) and in ovaries of immature mice treated sequentially with PMSG and hCG (Supplemental Fig. 4G), whereas miR-383 mimics/inhibitors had no effect on SF-1 protein expression (Supplemental Fig. 4H). These results indicate that SF-1 regulates miR-383 expression at the transcriptional level.
To determine the effects of SF-1 overexpression on the activity of the SGCZ gene promoter, an SF-1 expression vector was cotransfected with SGCZ promoter reporter plasmids in NIH3T3 cells. As shown in Supplemental Fig. 4I, SF-1 overexpression increased the activity of SGCZ promoter region (−1489/+56), which contains two putative SF-1 binding sites located at −419 bp (5′-TGACCT-3′) and −980 bp (5′-TCAAGGTCA-3′) by 5.8 fold, suggesting that SF-1 acts as a transcriptional activator of the SGCZ promoter. To evaluate the contributions of the putative SF-1 sites at −419 and −980 bp, these sequences were mutated. The results showed that overexpression of SF-1 did not decrease the promoter activities of SGCZ (−1489/+56) mutant 1 (mutated site at −980 bp) and SGCZ (−1489/+56) mutant 2 (mutated site at −419 bp) (Supplemental Fig. 4I), demonstrating that the two putative SF-1 binding sites did not mediate transcriptional activation of the SGCZ promoter. To further investigate the transcriptional regulation of mouse SGCZ gene by SF-1, a series of deletion mutant constructs from −1489 to +56 nucleotides of the SGCZ promoter region were generated by cloning the relevant regions into the pGL3-Basic vector and transfected in NIH3T3 cells. With the exception of the SGCZ promoter construct containing the shortest fragment (−100 to +56), overexpression of SF-1 resulted in an approximately 4- to 5-fold increase in luciferase activities of almost all constructs compared with vector controls (Fig. 4D). In addition, forced expression of SF-1 failed to increase luciferase activity of SGCZ (−128/+56) plasmid containing mutations between −128 and −100 nucleotides (Fig. 4E). These results indicate that the SGCZ promoter region from −128 to −100 nucleotides might contain a critical SF-1-responsive regulatory element. ChIP assay with mGC and NIH3T3 cells further revealed the binding of SF-1 to the promoter elements in the 5′-flanking region of the SGCZ gene (Fig. 4F and Supplemental Fig. 4J). Antibodies to normal IgG failed to immunoprecipitate SGCZ promoter (Fig. 4F and Supplemental Fig. 4J).
We next asked whether RBMS1 and its downstream target c-Myc were also regulated by SF-1. Our results demonstrated that overexpression and knockdown of SF-1 reduced and enhanced RBMS1/c-Myc levels (Fig. 4G), respectively. In addition, si-SF-1-induced RBMS1/c-Myc expression was abrogated by miR-383 mimics (Fig. 4, H and I); overexpression of SF-1 (Fig. 4J, right panel) caused a decrease in luciferase activity of the RBMS1 3′-UTR reporter constructs, whereas miR-383 inhibitors partly rescued this effect (Fig. 4J, left panel). These results demonstrated that SF-1 could regulate the transcription of miR-383 and then down-regulate the expression of RBMS1 and its downstream c-Myc.
It is indicated that SF-1 promotes E2 release in mGC (40, 51). In this study, overexpression of SF-1 in mGC partly rescued E2 production and Cyp19a1 mRNA levels suppressed by miR-383 inhibitors (Fig. 5A and Supplemental Fig. 5A), overexpression of RBMS1 (Fig. 5C and Supplemental Fig. 5C), or of c-Myc (Fig. 5D and Supplemental Fig. 5D), whereas si-SF-1 inhibited miR-383-mediated E2 release and Cyp19a1 mRNA levels in mGC (Fig. 5B and Supplemental Fig. 5B). Furthermore, compared with GC transfected with miR-383 inhibitors, cotransfection of si-SF-1 and miR-383 inhibitors into GC resulted in a much more decrease in E2 biosynthesis and Cyp19a1 mRNA levels (Fig. 5E and Supplemental Fig. 5E). These studies indicate that SF-1 is able to induce much more E2 production via transcriptional regulation of miR-383 expression.
Fig. 5.

SF-1 mediates the effects of miR-383/RBMS1/c-Myc on E2 release from GC. A, C, and D, SF-1 overexpression in mGC increased the attenuated E2 production by miR-383 inhibitors (A), overexpression of RBMS1 (C), or of c-Myc (D). B, si-SF-1 abrogated miR-383-mediated E2 release in mGC. E, miR-383 inhibitors-induced suppression of E2 release from GC was further decreased by si-SF-1. GC were cotransfected with pEGFP-SF-1 and indicated expression vectors or miR-383 inhibitors, or with si-SF-1 and miR-383 mimics/inhibitors for 48 h. The culture medium was collected for hormone analysis. Data were presented as means ± sem for at least three independent experiments. *, P < 0.05; **, P < 0.01.
Discussion
In this study, we have found that miR-383 promotes E2 release in mGC by direct targeting RBMS1. RBMS1 and its downstream c-Myc also mediate the steroidogenic actions of miR-383 in GC. On the other hand, the orphan nuclear receptor SF-1 transcriptionally induces miR-383, which then represses RBMS1 and c-Myc expression, and is involved in the regulation of miR-383 function in GC. Importantly, SF-1 transcriptionally actives the expression of miR-383 through binding to a potential SF-1 response element in the SGCZ promoter. Here, we provide mechanistic insights into the role of miR-383 in ovarian follicle development and reveal a new steroidogenic pathway through SF-1-mediated transcription of miR-383 gene for the first time.
Gonadal steroids and steroid-mediated signaling in ovarian GC are critical for normal ovarian processes, such as follicle growth, oocyte maturation, and ovulation (52, 53). Abnormalities in steroid production or action are associated with the development of some reproductive and nonreproductive pathology, including polycystic ovarian syndrome (54) and estrogen-related cancers in women (55). Thus, deeper understanding of the molecular mechanisms controlling steroid production within GC may help discover new therapeutic targets for these diseases. Recently, some miRNA were identified to affect steroid hormone release in human primary ovarian GC by a genome-scale screen (56). However, the molecular mechanisms behind this effect are unclear. In our previous study, we have found that miRNA-224, one of the most significantly up-regulated miRNA in TGF-β1-stimulated mGC, promotes TGF-β1-induced mGC E2 production by targeting the transcription factor Smad4 (16). miR-383 was the third most down-regulated miRNA in mGC treated with TGF-β1 (16). In this study, we found that miR-383, which was expressed predominantly in GC of preovulatory follicles at different developmental stages, also stimulated E2 release in mGC by enhancing Cyp19a1 (a key enzyme in E2 biosynthesis) expression levels, without affecting GC proliferation and apoptosis. miR-383 expression in isolated mouse follicles increased from the preantral to antral phase. In addition, almost similar patterns of ovarian miR-383 expression and plasma E2 were observed in PMSG-treated mice. Meanwhile, PMSG/hCG-induced ovulation caused a rapid decline in miR-383 expression, which was similar to the E2 secretion profiles during ovulation (44, 41). Taken together, these data further support the concept that miR-383 exerts effects on steroidogenesis during follicular development. In this study, TGF-β1 decreased miR-383 expression in preantral GC. In addition, miR-383 expression levels were not altered in preantral follicles but increased in antral follicles. TGF-β1 is suggested to have important regulatory functions in ovarian follicular development (37). Furthermore, TGF-β1 promotes E2 release by mGC (16) and enhances FSH-dependent E2 synthesis in the rat ovary (42). However, some studies have shown that TGF-β1 counteracts E2 biosynthesis in FSH-stimulated bovine GC (57). These results suggest that TGF-β1 may play a role in steroidogenesis during the preantral to early antral transition, by regulating miR-383 expression. Here, we add another miRNA to be a potent regulator of ovarian functions. Furthermore, our other studies show that down-regulation of miR-383 is associated with male infertility by regulating germ cell proliferation at early stages of spermatogenesis (38, 39).
miRNA are known to influence diverse biological outcomes through targeting genes during normal development and pathological responses (58–60). Here, miR-383 mediated E2 release in GC by direct targeting the transcription factor RBMS1 [also known as c-Myc single strand-binding protein (MSSP)-1]. Five highly homologous MSSP cDNA clones (MSSP-1, MSSP-2, suppressor of cdc2 with RNA binding motif 2, suppressor of cdc2 with RNA binding motif 3, and human YC1) have been reported (61–63). Each of these five proteins of the MSSP family contains two RNP (RNA-binding protein) consensus motifs (11). Through an interaction with the c-Myc protein, RBMS1 mediates c-Myc-induced apoptosis (12), promotes ras/myc cooperative cell transforming (11), and stimulates DNA replication by interacting with a catalytic subunit of DNA polymerase α (64). Moreover, disruption of MSSP leads to growth defect in the embryo and the hormonal defect in the adult female mouse (13). These reports indicate that MSSP exhibit pleiotropic biological functions on different target cells. In this study, RBMS1 is also mainly located in GC of various follicle stages. Knockdown of RBMS1 in GC promoted E2 release. In addition, RBMS1 mediated the effect of miR-383 on GC function. These results indicate that RBMS1 functions as a potent regulator of steroidogenesis during ovarian development. Whether miR-383 targets the homologs of RBMS1 gene and participates in the processes attributable to RBMS1, such as embryonic development and DNA replication, remains to be elucidated.
The transcription factor RBMS1 is also discovered to bind a multitude of target genes, including c-Myc and Fas in different cell lines (11, 46, 12). In GC, miR-383 only decreased c-Myc mRNA and protein levels. However, c-Myc was not a target of miR-383 by luciferase assay. The c-Myc protooncogene encodes a transcription factor involved in cell proliferation, differentiation, and apoptosis (47, 65–67). For example, c-Myc stimulates immature GC in early folliculogenesis (68, 69) and plays a role in cell death during follicular atresia (70, 71). In this study, c-Myc showed similar expression patterns to miR-383 and RBMS1 in the mouse ovary. Overexpression of c-Myc in GC caused the suppression of E2 release. In addition, c-Myc mediated RBMS1- and miR-383-induced effects on E2 release in GC. These results indicate that c-Myc is a downstream effector target of miR-383-regulated RBMS1 in GC. Altogether, our results demonstrate a cascade of events where an miRNA, single-handedly or in cooperation with other factors, suppresses the expression of a transcription factor, thereby inhibiting transcription of downstream target genes responsible for physiological processes. Further elucidation of miRNA biogenesis and functionality in ovarian follicle cells will provide important insights into normal follicular development and pathological conditions, such as polycystic ovarian syndrome and cancer.
miR-383 is located within the first intron of SGCZ gene. The intronic miRNA usually derive from the common transcript with their host genes (72) or are independently transcribed with its own RNA polymerase II-like promoter (73). In this study, we observed that miR-383 was coordinately expressed with its host gene SGCZ in mouse tissues and in TGF-β1-treated GC, indicating that they are cotranscribed. In addition, the transcription factor SF-1 regulated the promoter activity of the mouse SGCZ gene. SF-1 [also known as nuclear receptor subfamily 5, group A, member 1; adrenal 4-binding protein; or fushi tarazu factor homolog 1 (OMIM 184757)] plays a critical role in the development and differentiation of steroidogenic tissues (74, 75). For example, many genes encoding steroidogenic enzymes, such as Cyp19a1 and Cyp11a1, or regulators of endocrine function were controlled by SF-1 for their expression and contained SF-1 response elements within their proximal promoters (51, 76, 77); SF-1 is required for the expression of Mullerian inhibiting substance in Sertoli cells (78). Silencing or overexpression of SF-1 in mGC induced a parallel suppression or increase of RNA levels of SGCZ, pri-miR-383, and mature miR-383, suggesting a transcriptional regulation of miR-383 by SF-1. A putative SF-1 transcriptional regulatory element was identified in the region between nucleotides −128 and −100 of promoter region of SGCZ gene, which is not a canonical SF-1 consensus element in the SGCZ promoter. SF-1 binding to the noncanonical response elements may facilitate the recruitment and activation of other transcription factors involved in steroidogenesis, such as specificity protein 1 (79, 80), which may coordinately enhance miR-383-mediated estrogen synthesis. Actually, specificity protein 1, estrogen receptors α and β are also predicted in the mouse SGCZ promoter region spanning from nucleotides −128 to −100 by bioinformatic analysis. These results provide further evidence that miR-383 is directly regulated by SF-1. Further studies will be required to more precisely determine the modulation of SF-1 in miR-383 processing through deletion analysis and site-directed mutagenesis.
The functional consequences of transactivation of miR-383 by SF-1 were examined. The results showed that knockdown of SF-1 increased protein expression levels of miR-383's target RBMS1 and its downstream c-Myc. A report showed that transient repression of SF-1 expression by LH surge also induced c-Myc expression in GC during the periovulatory period (81). Whether LH-induced events are mediated by miR-383 remains unknown. SF-1 plays key roles in steroidogenesis in both adrenal cortical and gonadal cells (50). In the present study, SF-1 promoted much more E2 release in GC via enhancing endogenous miR-383 expression. In addition, both RBMS1 and c-Myc were involved in SF-1-mediated steroidogenic effects. Taken together, our results indicated that, except that SF-1 regulates expression of multiple steroidogenic enzymes, including Cyp19a1, Cyp11A, and Cyp17, through interactions with their promoters (40, 51), SF-1 also influences E2 release by transcriptional regulation of miR-383 expression and its targets. In addition, SF-1 is able to promote more E2 production via the new steroidogenic pathway. It is well known that healthy developing follicles are characterized as increased E2 secretion (7, 82). Whether miR-383 regulation is involved in increasing pool of healthy follicles is unclear. Here, we have defined a novel mechanism mediating transactivation of miR-383 expression by SF-1, a process involved in the regulation of steroidogenesis in GC. It will be of interest to extend these studies to determine the extent of miR-383 regulated via this mechanism and study the functional consequences of miR-383 activation by SF-1 under normal and pathological conditions, for example, the described pathway in response to growth factor stimulation.
In summary, we demonstrate that miR-383 functions as a positive regulator of E2 release in GC through inhibiting expression of RBMS1 and its downstream c-Myc. Moreover, SF-1 is able to transactivate the promoter of miR-383 host gene SGCZ and mediates the effects of miR-383/RBMS1/c-Myc on GC steroidogenesis (Fig. 6). miR-383 thereby potentiates a stimulator for female fertility. These effects of miR-383 on physiological processes within the ovary could be potentially useful for regulating reproduction and treating some reproductive or steroid-related disorders.
Fig. 6.

A model for the steroidogenic pathway through SF-1-mediated transcription of miR-383 in GC. Transactivation of miR-383 by SF-1 inhibits the expression of RBMS1 and its downstream c-Myc, and subsequently promotes E2 release in GC.
Acknowledgments
We thank Dr. Mian Wu for the HEK293T and MCF-7 cell lines and Dr. Tao Zhu (Hefei, People's Republic of China) for the NIH3T3 cell line.
This work was supported by National Natural Science Foundation of China Grants 81125005 and 31171379 (to F.S.), the National Basic Research Program of China Grant 2009CB941700 (to F.S.), the Chinese Academy of Sciences Knowledge Creative Program Grant KSCX2-EW-R-07 (to F.S.), and the Fundamental Research Funds for the Central Universities Grant WK2070000008 (to F.S.).
Disclosure Summary: The authors have nothing to disclose.
NURSA Molecule Pages†:
Nuclear Receptors: SF-1;
Ligands: 17β-estradiol.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- ChIP
- Chromatin immunoprecipitation
- E2
- estradiol
- FBS
- fetal bovine serum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GC
- granulosa cell
- hCG
- human chorionic gonadotropin
- ISH
- in situ hybridization
- LNA
- locked nucleic acid
- mGC
- mouse GC
- miR-383
- miRNA-383
- miRNA
- microRNA
- MSSP
- c-Myc single strand-binding protein
- NC
- negative control
- PMSG
- pregnant mare serum gonadotropin
- PND
- postnatal day
- pri-miR-383
- primary miR-383
- RBMS1
- RNA binding motif, single stranded interacting protein 1
- SF-1
- steroidogenic factor-1
- SGCZ
- Sarcoglycan zeta
- si
- small interfering
- Smad4
- SMAD family member 4
- USTC
- University of Science and Technology of China
- UTR
- untranslated region.
References
- 1. Sasson R , Dantes A , Tajima K , Amsterdam A. 2003. Novel genes modulated by FSH in normal and immortalized FSH-responsive cells: new insights into the mechanism of FSH action. FASEB J 17:1256–1266 [DOI] [PubMed] [Google Scholar]
- 2. Vanderhyden BC , Cohen JN , Morley P. 1993. Mouse oocytes regulate granulosa cell steroidogenesis. Endocrinology 133:423–426 [DOI] [PubMed] [Google Scholar]
- 3. Lubahn DB , Moyer JS , Golding TS , Couse JF , Korach KS , Smithies O. 1993. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA 90:11162–11166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Richards JS , Jonassen JA , Rolfes AI , Kersey K , Reichert LE. 1979. Adenosine 3′,5′-monophosphate, luteinizing hormone receptor, and progesterone during granulosa cell differentiation: effects of estradiol and follicle-stimulating hormone. Endocrinology 104:765–773 [DOI] [PubMed] [Google Scholar]
- 5. Hrabia A , Ha Y , Shimada K. 2004. Expression of estrogen receptor α mRNA in theca and granulosa layers of the ovary in relation to follicular growth in quail. Folia Biol 52:191–195 [DOI] [PubMed] [Google Scholar]
- 6. Robker RL , Richards JS. 1998. Hormone-induced proliferation and differentiation of granulosa cells: a coordinated balance of the cell cycle regulators cyclin D2 and p27Kip1. Mol Endocrinol 12:924–940 [DOI] [PubMed] [Google Scholar]
- 7. Adashi EY , Hsueh AJ. 1982. Estrogens augment the stimulation of ovarian aromatase activity by follicle-stimulating hormone in cultured rat granulosa cells. J Biol Chem 257:6077–6083 [PubMed] [Google Scholar]
- 8. Mazerbourg S , Klein C , Roh J , Kaivo-Oja N , Mottershead DG , Korchynskyi O , Ritvos O , Hsueh AJ. 2004. Growth differentiation factor-9 signaling is mediated by the type I receptor, activin receptor-like kinase 5. Mol Endocrinol 18:653–665 [DOI] [PubMed] [Google Scholar]
- 9. Shimasaki S , Zachow RJ , Li D , Kim H , Iemura S , Ueno N , Sampath K , Chang RJ , Erickson GF. 1999. A functional bone morphogenetic protein system in the ovary. Proc Natl Acad Sci USA 96:7282–7287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parakh TN , Hernandez JA , Grammer JC , Weck J , Hunzicker-Dunn M , Zeleznik AJ , Nilson JH. 2006. Follicle-stimulating hormone/cAMP regulation of aromatase gene expression requires β-catenin. Proc Natl Acad Sci USA 103:12435–12440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Niki T , Izumi S , Saëgusa Y , Taira T , Takai T , Iguchi-Ariga SM , Ariga H. 2000. MSSP promotes ras/myc cooperative cell transforming activity by binding to c-Myc. Genes Cells 5:127–141 [DOI] [PubMed] [Google Scholar]
- 12. Iida M , Taira T , Ariga H , Iguchi-Ariga SM. 1997. Induction of apoptosis in HeLa cells by MSSP, c-myc binding proteins. Biol Pharm Bull 20:10–14 [DOI] [PubMed] [Google Scholar]
- 13. Fujimoto M , Matsumoto K , Iguchi-Ariga SM , Ariga H. 2001. Disruption of MSSP, c-myc single-strand binding protein, leads to embryonic lethality in some homozygous mice. Genes Cells 6:1067–1075 [DOI] [PubMed] [Google Scholar]
- 14. Otsuka M , Zheng M , Hayashi M , Lee JD , Yoshino O , Lin S , Han J. 2008. Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J Clin Invest 118:1944–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagaraja AK , Andreu-Vieyra C , Franco HL , Ma L , Chen R , Han DY , Zhu H , Agno JE , Gunaratne PH , DeMayo FJ , Matzuk MM. 2008. Deletion of Dicer in somatic cells of the female reproductive tract causes sterility. Mol Endocrinol 22:2336–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yao G , Yin M , Lian J , Tian H , Liu L , Li X , Sun F. 2010. MicroRNA-224 is involved in transforming growth factor-β-mediated mouse granulosa cell proliferation and granulosa cell function by targeting Smad4. Mol Endocrinol 24:540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wightman B , Ha I , Ruvkun G. 1993. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75:855–862 [DOI] [PubMed] [Google Scholar]
- 18. Seggerson K , Tang L , Moss EG. 2002. Two genetic circuits repress the Caenorhabditis elegans heterochronic gene lin-28 after translation initiation. Dev Biol 243:215–225 [DOI] [PubMed] [Google Scholar]
- 19. Zeng Y , Wagner EJ , Cullen BR. 2002. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell 9:1327–1333 [DOI] [PubMed] [Google Scholar]
- 20. Lee YS , Kim HK , Chung S , Kim KS , Dutta A. 2005. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J Biol Chem 280:16635–16641 [DOI] [PubMed] [Google Scholar]
- 21. Calin GA , Sevignani C , Dumitru CD , Hyslop T , Noch E , Yendamuri S , Shimizu M , Rattan S , Bullrich F , Negrini M , Croce CM. 2004. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 101:2999–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brennecke J , Hipfner DR , Stark A , Russell RB , Cohen SM. 2003. Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113:25–36 [DOI] [PubMed] [Google Scholar]
- 23. Sun W , Shen W , Yang S , Hu F , Li H , Zhu TH. 2010. miR-223 and miR-142 attenuate hematopoietic cell proliferation, and miR-223 positively regulates miR-142 through LMO2 isoforms and CEBP-β. Cell Res 20:1158–1169 [DOI] [PubMed] [Google Scholar]
- 24. Shih KK , Qin LX , Tanner EJ , Zhou Q , Bisogna M , Dao F , Olvera N , Viale A , Barakat RR , Levine DA. 2011. A microRNA survival signature (MiSS) for advanced ovarian cancer. Gynecol Oncol 121:444–450 [DOI] [PubMed] [Google Scholar]
- 25. Hong X , Luense LJ , McGinnis LK , Nothnick WB , Christenson LK. 2008. Dicer1 is essential for female fertility and normal development of the female reproductive system. Endocrinology 149:6207–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iorio MV , Visone R , Di Leva G , Donati V , Petrocca F , Casalini P , Taccioli C , Volinia S , Liu CG , Alder H , Calin GA , Ménard S , Croce CM. 2007. MicroRNA signatures in human ovarian cancer. Cancer Res 67:8699–8707 [DOI] [PubMed] [Google Scholar]
- 27. Nam EJ , Yoon H , Kim SW , Kim H , Kim YT , Kim JH , Kim JW , Kim S. 2008. MicroRNA expression profiles in serous ovarian carcinoma. Clin Cancer Res 14:2690–2695 [DOI] [PubMed] [Google Scholar]
- 28. Davis BN , Hilyard AC , Lagna G , Hata A. 2008. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454:56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lei L , Jin S , Gonzalez G , Behringer RR , Woodruff TK. 2010. The regulatory role of Dicer in folliculogenesis in mice. Mol Cell Endocrinol 315:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez G , Behringer RR. 2009. Dicer is required for female reproductive tract development and fertility in the mouse. Mol Reprod Dev 76:678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma J , Flemr M , Stein P , Berninger P , Malik R , Zavolan M , Svoboda P , Schultz RM. 2010. MicroRNA activity is suppressed in mouse oocytes. Curr Biol 20:265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murchison EP , Stein P , Xuan Z , Pan H , Zhang MQ , Schultz RM , Hannon GJ. 2007. Critical roles for Dicer in the female germline. Genes Dev 21:682–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ro S , Song R , Park C , Zheng H , Sanders KM , Yan W. 2007. Cloning and expression profiling of small RNAs expressed in the mouse ovary. RNA 13:2366–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Corney DC , Flesken-Nikitin A , Godwin AK , Wang W , Nikitin AY. 2007. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res 67:8433–8438 [DOI] [PubMed] [Google Scholar]
- 35. Moore RK , Shimasaki S. 2005. Molecular biology and physiological role of the oocyte factor, BMP-15. Mol Cell Endocrinol 234:67–73 [DOI] [PubMed] [Google Scholar]
- 36. Mondschein JS , Canning SF , Hammond JM. 1988. Effects of transforming growth factor-β on the production of immunoreactive insulin-like growth factor I and progesterone and on [3H]thymidine incorporation in porcine granulosa cell cultures. Endocrinology 123:1970–1976 [DOI] [PubMed] [Google Scholar]
- 37. Rosairo D , Kuyznierewicz I , Findlay J , Drummond A. 2008. Transforming growth factor-β: its role in ovarian follicle development. Reproduction 136:799–809 [DOI] [PubMed] [Google Scholar]
- 38. Lian J , Tian H , Liu L , Zhang XS , Li WQ , Deng YM , Yao GD , Yin MM , Sun F. 2010. Downregulation of microRNA-383 is associated with male infertility and promotes testicular embryonal carcinoma cell proliferation by targeting IRF1. Cell Death Dis 1:e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lian J , Zhang X , Tian H , Liang N , Wang Y , Liang C , Li X , Sun F. 2009. Altered microRNA expression in patients with non-obstructive azoospermia. Reprod Biol Endocrinol 7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liang N , Xu Y , Yin Y , Yao G , Tian H , Wang G , Lian J , Wang Y , Sun F. 2011. Steroidogenic factor-1 is required for TGF-β3-mediated 17β-estradiol synthesis in mouse ovarian granulosa cells. Endocrinology 152:3213–3225 [DOI] [PubMed] [Google Scholar]
- 41. Vanderhyden BC , Macdonald EA. 1998. Mouse oocytes regulate granulosa cell steroidogenesis throughout follicular development. Biol Reprod 59:1296–1301 [DOI] [PubMed] [Google Scholar]
- 42. Adashi EY , Reshick CE. 1986. Antagonistic interactions of transforming growth factors in the regulation of granulosa cell differentiation. Endocrinology 119:1879–1881 [DOI] [PubMed] [Google Scholar]
- 43. Vitt UA , Kloosterboer HJ , Rose UM , Mulders JW , Kiesel PS , Bete S , Nayudu PL. 1998. Isoforms of human recombinant follicle-stimulating hormone: comparison of effects on murine follicle development in vitro. Biol Reprod 59:854–861 [DOI] [PubMed] [Google Scholar]
- 44. Shao R , Ljungström K , Weijdegård B , Egecioglu E , Fernandez-Rodriguez J , Zhang FP , Thurin-Kjellberg A , Bergh C , Billig H. 2007. Estrogen-induced upregulation of AR expression and enhancement of AR nuclear translocation in mouse fallopian tubes in vivo. Am J Physiol Endocrinol Metab 292:E604–E614 [DOI] [PubMed] [Google Scholar]
- 45. Kimura K , Saga H , Hayashi K , Obata H , Chimori Y , Ariga H , Sobue K. 1998. c-Myc gene single-strand binding protein-1, MSSP-1, suppresses transcription of α-smooth muscle actin gene in chicken visceral smooth muscle cells. Nucleic Acids Res 26:2420–2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nomura J , Matsumoto K , Iguchi-Ariga SM , Ariga H. 2005. Positive regulation of Fas gene expression by MSSP and abrogation of Fas-mediated apoptosis induction in MSSP-deficient mice. Exp Cell Res 305:324–332 [DOI] [PubMed] [Google Scholar]
- 47. Lin L , Zhang JH , Panicker LM , Simonds WF. 2008. The parafibromin tumor suppressor protein inhibits cell proliferation by repression of the c-myc proto-oncogene. P Natl Acad Sci USA 105:17420–17425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yao N , Lu CL , Zhao JJ , Xia HF , Sun DG , Shi XQ , Wang C , Li D , Cui Y , Ma X. 2009. A network of miRNAs expressed in the ovary are regulated by FSH. Front Biosci 14:3239–3245 [DOI] [PubMed] [Google Scholar]
- 49. Shapiro DB , Pappalardo A , White BA , Peluso JJ. 1996. Steroidogenic factor-1 as a positive regulator of rat granulosa cell differentiation and a negative regulator of mitosis. Endocrinology 137:1187–1195 [DOI] [PubMed] [Google Scholar]
- 50. Falender AE , Lanz R , Malenfant D , Belanger L , Richards JS. 2003. Differential expression of steroidogenic factor-1 and FTF/LRH-1 in the rodent ovary. Endocrinology 144:3598–3610 [DOI] [PubMed] [Google Scholar]
- 51. Yang HJ , Shozu M , Murakami K , Sumitani H , Segawa T , Kasai T , Inoue M. 2002. Spatially heterogeneous expression of aromatase P450 through promoter II is closely correlated with the level of steroidogenic factor-1 transcript in endometrioma tissues. J Clin Endocrinol Metab 87:3745–3753 [DOI] [PubMed] [Google Scholar]
- 52. Smyth CD , Miró F , Howles CM , Hillier SG. 1995. Effect of luteinizing-hormone on follicle-stimulating hormone-activated paracrine signaling in rat ovary. Hum Reprod 10:33–39 [DOI] [PubMed] [Google Scholar]
- 53. Smyth CD , Miró F , Whitelaw PF , Howles CM , Hillier SG. 1993. Ovarian thecal interstitial androgen synthesis is enhanced by a follicle-stimulating hormone-stimulated paracrine mechanism. Endocrinology 133:1532–1538 [DOI] [PubMed] [Google Scholar]
- 54. Eagleson CA , Gingrich MB , Pastor CL , Arora TK , Burt CM , Evans WS , Marshall JC. 2000. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocr Metab 85:4047–4052 [DOI] [PubMed] [Google Scholar]
- 55. Gibson LJ , Dawson CL , Lawrence DJ , Bliss JM. 2007. Aromatase inhibitors for treatment of advanced breast cancer in postmenopausal women. Cochrane Db Syst Rev CD003370. [DOI] [PubMed] [Google Scholar]
- 56. Sirotkin AV , Ovcharenko D , Grossmann R , Lauková M , Mlyncek M. 2009. Identification of microRNAs controlling human ovarian cell steroidogenesis via a genome-scale screen. J Cell Physiol 219:415–420 [DOI] [PubMed] [Google Scholar]
- 57. Zheng X , Price CA , Tremblay Y , Lussier JG , Carrière PD. 2008. Role of transforming growth factor-β1 in gene expression and activity of estradiol and progesterone-generating enzymes in FSH-stimulated bovine granulosa cells. Reproduction 136:447–457 [DOI] [PubMed] [Google Scholar]
- 58. Johnnidis JB , Harris MH , Wheeler RT , Stehling-Sun S , Lam MH , Kirak O , Brummelkamp TR , Fleming MD , Camargo FD. 2008. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451:1125–1129 [DOI] [PubMed] [Google Scholar]
- 59. Petrocca F , Visone R , Onelli MR , Shah MH , Nicoloso MS , de Martino I , Iliopoulos D , Pilozzi E , Liu CG , Negrini M , Cavazzini L , Volinia S , Alder H , Ruco LP , Baldassarre G , Croce CM , Vecchione A. 2008. E2F1-regulated microRNAs impair TGFβ-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell 13:272–286 [DOI] [PubMed] [Google Scholar]
- 60. Huang Q , Gumireddy K , Schrier M , le Sage C , Nagel R , Nair S , Egan DA , Li A , Huang G , Klein-Szanto AJ , Gimotty PA , Katsaros D , Coukos G , Zhang L , Puré E , Agami R. 2008. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol 10:202–210 [DOI] [PubMed] [Google Scholar]
- 61. Negishi Y , Nishita Y , Saëgusa Y , Kakizaki I , Galli I , Kihara F , Tamai K , Miyajima N , Iguchi-Ariga SM , Ariga H. 1994. Identification and cDNA cloning of single-stranded DNA binding proteins that interact with the region upstream of the human c-myc gene. Oncogene 9:1133–1143 [PubMed] [Google Scholar]
- 62. Takai T , Nishita Y , Iguchi-Ariga SM , Ariga H. 1994. Molecular cloning of MSSP-2, a c-myc gene single-strand binding protein: characterization of binding specificity and DNA replication activity. Nucleic Acids Res 22:5576–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kanaoka Y , Nojima H. 1994. SCR: novel human suppressors of cdc2/cdc13 mutants of Schizosaccharomyces pombe harbour motifs for RNA binding proteins. Nucleic Acids Res 22:2687–2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Niki T , Galli I , Ariga H , Iguchi-Ariga SM. 2000. MSSP, a protein binding to an origin of replication in the c-myc gene, interacts with a catalytic subunit of DNA polymerase α and stimulates its polymerase activity. FEBS Lett 475:209–212 [DOI] [PubMed] [Google Scholar]
- 65. Chen Z , Zeng H , Guo Y , Liu P , Pan H , Deng A , Hu J. 2010. miRNA-145 inhibits non-small cell lung cancer cell proliferation by targeting c-Myc. J Exp Clin Cancer Res 29:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Harrington EA , Bennett MR , Fanidi A , Evan GI. 1994. C-Myc-induced apoptosis in fibroblasts is inhibited by specific cytokines. EMBO J 13:3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Packham G , Cleveland JL. 1994. Ornithine decarboxylase is a mediator of C-Myc-induced apoptosis. Mol Cell Biol 14:5741–5747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li S , Maruo T. 1997. [Expression of transforming growth factor-α and myc oncoprotein in the human ovary during follicular growth, atresia and corpus luteum formation regression]. Zhonghua Fu Chan Ke Za Zhi 32:593–596 (Chinese) [PubMed] [Google Scholar]
- 69. Maruo T. 1995. [Expression of oncogenes, growth factors and their receptors in follicular growth, regression and atresia: their roles in granulosa cell proliferation and differentiation]. Nippon Sanka Fujinka Gakkai Zasshi 47:738–750 (Japanese) [PubMed] [Google Scholar]
- 70. Nandedkar TD , Dharma SJ. 2001. Expression of bcl(xs) and c-myc in atretic follicles of mouse ovary. Reprod Biomed Online 3:221–225 [DOI] [PubMed] [Google Scholar]
- 71. Kole AR. 1999. Mechanism of induction of follicular atresia after equine chorionic gonadotrophin (eCG) antisera treatment in hypophysectomized eCG-injected hamster model: possible involvement of c-myc and cdc25. Med Hypotheses 52:265–267 [DOI] [PubMed] [Google Scholar]
- 72. Kim YK , Kim VN. 2007. Processing of intronic microRNAs. EMBO J 26:775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xiong H , Qian J , He T , Li F. 2009. Independent transcription of miR-281 in the intron of ODA in Drosophila melanogaster. Biochem Biophys Res Commun 378:883–889 [DOI] [PubMed] [Google Scholar]
- 74. Noël JC , Anaf V , Borghese B , Vaiman D , Fayt I , Chapron C. 2011. The steroidogenic factor-1 protein is not expressed in various forms of endometriosis but is strongly present in ovarian cortical or medullary mesenchymatous cells adjacent to endometriotic foci. Fertil Steril 95:2655–2657 [DOI] [PubMed] [Google Scholar]
- 75. Noel JC , Borghese B , Vaiman D , Fayt I , Anaf V , Chapron C. 2010. Steroidogenic factor-1 expression in ovarian endometriosis. Appl Immunohisto M M 18:258–261 [DOI] [PubMed] [Google Scholar]
- 76. Jenkins C , Michael D , Mahendroo M , Simpson E. 1993. Exon-specific northern analysis and rapid amplification of cDNA ends (RACE) reveal that the proximal promoter II (PII) is responsible for aromatase cytochrome P450 (CYP19) expression in human ovary. Mol Cell Endocrinol 97:R1–R6 [DOI] [PubMed] [Google Scholar]
- 77. Gurates B , Amsterdam A , Tamura M , Yang S , Zhou J , Fang Z , Amin S , Sebastian S , Bulun SE. 2003. WT1 and DAX-1 regulate SF-1-mediated human P450arom gene expression in gonadal cells. Mol Cell Endocrinol 208:61–75 [DOI] [PubMed] [Google Scholar]
- 78. Giuili G , Shen WH , Ingraham HA. 1997. The nuclear receptor SF-1 mediates sexually dimorphic expression of Mullerian inhibiting substance, in vivo. Development 124:1799–1807 [DOI] [PubMed] [Google Scholar]
- 79. Venepally P , Waterman MR. 1995. Two Sp1-binding sites mediate cAMP-induced transcription of the bovine CYP11A gene through the protein kinase A signaling pathway. J Biol Chem 270:25402–25410 [DOI] [PubMed] [Google Scholar]
- 80. Urban RJ , Bodenburg YH , Jiang J , Denner L , Chedrese J. 2004. Protein kinase Cι enhances the transcriptional activity of the porcine P-450 side-chain cleavage insulin-like response element. Am J Physiol Endocrinol Metab 286:E975–E979 [DOI] [PubMed] [Google Scholar]
- 81. Agarwal P , Peluso JJ , White BA. 1996. Steroidogenic factor-1 expression is transiently repressed and c-myc expression and deoxyribonucleic acid synthesis are induced in rat granulosa cells during the periovulatory period. Biol Reprod 55:1271–1275 [DOI] [PubMed] [Google Scholar]
- 82. Schomberg DW , Couse JF , Mukherjee A , Lubahn DB , Sar M , Mayo KE , Korach KS. 1999. Targeted disruption of the estrogen receptor-α gene in female mice: characterization of ovarian responses and phenotype in the adult. Endocrinology 140:2733–2744 [DOI] [PubMed] [Google Scholar]