

Abstract
Diffuse leptomeningeal glioneuronal tumor is unique for communicating hydrocephalus, diffuse leptomeningeal enhancement, cystic changes, absence of tumor cells in cerebral spinal fluid, and a cell population of both glial and neuronal copositivity. It has likely been misdiagnosed as mixed glioneuronal tumors, oligodendrogliomas, and neuroepithelial tumors. Children with signs of this tumor are often worked up for infection, rheumatologic disease, or disseminated primary malignancy, resulting in unnecessary testing and treatment. We describe a 14-year-old female with recurrent headaches, hydrocephalus, and diffuse leptomeningeal enhancement discovered to be neoplastic 1 year after initial presentation, owing to extensive and unrevealing infectious and immunologic workups. Biopsies revealed atypical cells with markers of both glial and neuronal cells, positivity for OLIG-2, and focal p53 positivity. Great response was seen with temozolomide and craniospinal irradiation. Additionally, we postulate additional diagnostic indicators that may aid in earlier diagnosis and treatment decisions.
Keywords: diffuse leptomeningeal enhancement, intraparenchymal cysts, leptomeningeal glioneuronal tumor, neuroepithelial tumor
Diffuse leptomeningeal glioneuronal tumor is a recently described neoplasm. It has also been termed “diffuse leptomeningeal neuroepithelial tumor.”1 It does not fit into the current World Health Organization classification system, which describes mixed glioneuronal tumors as having elements of both glial and neuronal differentiation in mixed cell populations. Mixed glioneuronal tumors are characteristically biphasic.2 However, diffuse leptomeningeal glioneuronal tumor is comprised of 1 cell population that variably stains for both glial and neuronal components.1 This tumor was described as a distinct entity for the first time in 2010;3 however, it has likely been encountered before, diagnosed as other known tumors. These diagnoses include diffuse leptomeningeal oligodendrogliomatosis, leptomeningeal oligodendroglioma, central neurocytoma with uncharacteristically aggressive features, ganglioneurocytomas, gliomatosis cerebri, and glioneuronal tumors with neuropil-like islands.
Methods
Based on review of our patient’s medical record and current literature, we identified unique commonalities of clinical findings, imaging, and pathology for use in diagnosis and treatment planning. Samples of the patient’s stored tissue were analyzed for immunohistochemical evidence of cytogenetic abnormalities.
Case Summary
A 14-year-old Caucasian female presented to the emergency department with a 2-week history of intermittent headaches associated with nausea and vomiting, becoming persistent and intensified in character. Neurologic examination was negative with the exception of papilledema. There was tetraventricular communicating hydrocephalus on computed tomography (CT) imaging. Initial magnetic resonance imaging (MRI) of the brain and spine was limited by metal artifact from dental braces; however, leptomeningeal enhancement was noted diffusely throughout the brain and spinal cord with particular involvement of the basilar and suprasellar cisterns and distal lumbar nerve roots. The patient was admitted to the pediatric neurology service for evaluation of hydrocephalus and possible meningitis. Multiple consultants evaluated for infectious and rheumatologic etiologies. Four days after admission, her condition deteriorated with development of visual acuity changes and bilateral sixth cranial nerve palsies. An external ventricular drain was placed, but continued progressive hydrocephalus necessitated diversion of cerebrospinal fluid (CSF) via ventriculoperitoneal shunt. Cerebrospinal fluid studies, although repeatedly negative for malignant cells, consistently showed significant protein elevation. Meningeal biopsies revealed only thickened leptomeninges and occasional aggregates of monocytes. They did not demonstrate neoplastic cells, inflammatory cells, nor granulomatous changes. After removal of dental braces, MRI demonstrated continued leptomeningeal enhancement and a new enhancing intramedullary lesion at C7-T2 measuring 4 × 17 mm2 (Figure 1). The radiologic impression suggested a “diffuse meningitic process” and noted the focal signal changes were possibly the result of vascular thrombosis from infection or inflammation. During this initial hospitalization, extensive infectious and inflammatory workups were unrevealing, and the patient was discharged home.
Figure 1.
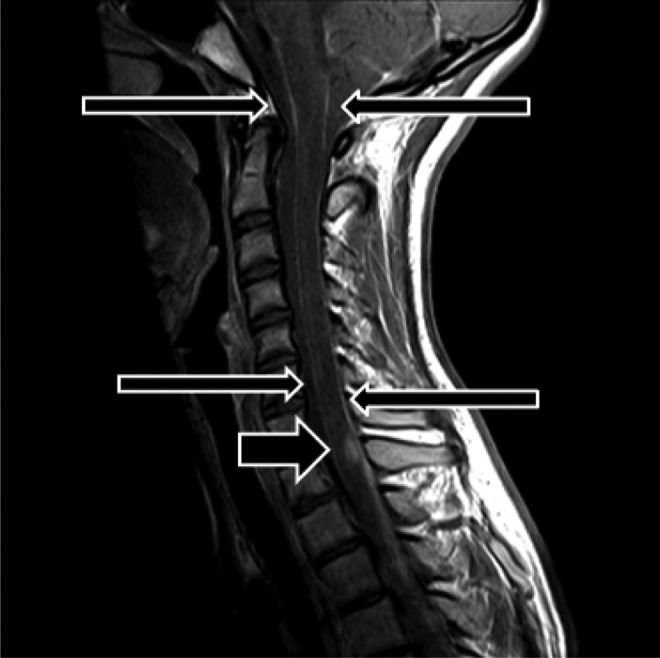
Postcontrast sagittal magnetic resonance imaging C-spine demonstrating dural enhancement (long arrows) and an enhancing intramedullary nodule at C7-T1 (short arrow).
Eight months after presentation, new changes in MRI included multifocal nonenhancing cystic lesions in the white matter. A month later, continued progressive leptomeningeal enhancement prompted repeat meningeal biopsies as well as brain tissue biopsies. The patient also underwent a repeat workup for infectious and inflammatory disease. Whole body bone scan and CT imaging of the chest, abdomen, and pelvis ruled out an occult neoplastic process with leptomeningeal dissemination. The biopsies revealed minimal chronic inflammation of the dura and unidentified atypical cells in the perivascular spaces determined to be of neuroepithelial origin by immunohistochemical staining. There was no infiltration of these cells into the vessels nor was there evidence of infarct. Systemic steroid therapy was considered and, prior to initiation, the patient was referred to pediatric neurooncology for consultation.
The consultation occurred approximately 1 year after initial presentation. Magnetic resonance imaging revealed increased dural thickening and enhancement with a new 1-cm mass-like lesion along the left cavernous sinus. There were also an increased number and size of cysts inferiorly and bilaterally in the brain, thought to represent abnormal dilatation of the perivascular spaces (Figure 2), consistent with previous biopsy findings. Despite emergence of new findings and progression of dural thickening, the spinal lesion at the cervicothoracic junction was stable. A repeat biopsy was recommended.
Figure 2.
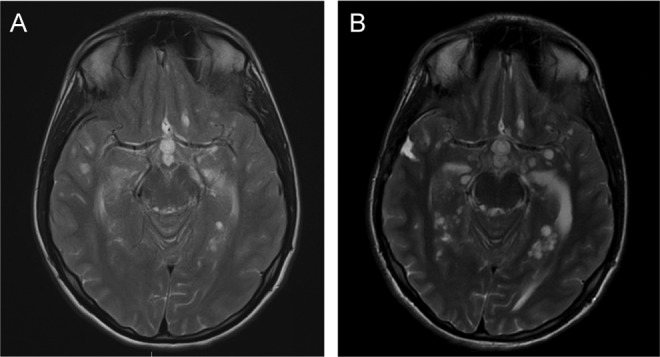
Our patient’s progression of perivascular cystic development from initial presentation (A) to 3 months later (B).
These final biopsies demonstrated cells with moderate pleomorphism and fine cytoplasmic processes, positive for glial fibrillary acid protein (GFAP) and synaptophysin, and negative for isocitrate dehydrogenase 1 (R123h mutation-specific antibody). The tumor cell nuclei were OLIG-2 positive and focally positive for p53. This was now interpreted to be a high-grade glioneuronal tumor due to a high mitotic rate and p53 positivity. Therapy was initiated 13 months after initial presentation.
Magnetic resonance imaging after 6 weeks of temozolomide and craniospinal irradiation revealed a significant decrease in contrast enhancement throughout the brain and spinal cord. The lesions of the parasellar cavernous sinus and the cervicothoracic junction were smaller. The previously noted intraparenchymal cysts were unchanged.
The patient completed 6 weeks of craniospinal irradiation and oral temozolomide, followed by maintenance therapy with temozolomide 5 days each month for 1 year. There was complete resolution of symptoms and all magnetic resonance MRI findings with the exception of persistence of the spinal nodule, which no longer enhanced with contrast administration. At the time of manuscript preparation, she remained in remission and continued excelling in academics and sporting activities.
Review of the Literature
Armao et al4 described a case of an 8-year-old boy who presented with intermittent headaches. Although he had no initial hydrocephalus, over the following 6 months he had fluctuating neurologic signs and symptoms including papilledema, diplopia, ataxia, and upper extremity weakness. Communicating hydrocephalus developed over the first year, and over a 5-year period, the patient had progressive hydrocephalus, seizures, and intermittent encephalopathy. He also underwent CSF studies revealing elevated protein but no malignant cells. On MR imaging, he had diffuse thickening and enhancement of the subarachnoid space and leptomeninges of the brain and spinal cord, more concentrated in the basal cisterns, sylvian fissures, and interhemispheric fissures. He had cystic lesions of the basal cisterns, basal ganglia, medulla, and cerebellar white matter. All cystic lesions noted were reported to have similar signal intensity to CSF. With the development of leptomeningeal enhancement and the appearance of the cystic lesions, the patient was treated for presumed diagnosis of Cryptococcus. He was not treated for malignancy and subsequently died secondary to necrotizing pneumonia and fulminant sepsis. Postmortem examination revealed dense coating of the optic chiasm and interpeduncular fossa. Postmortem biopsy showed tumor cell infiltrates in the perivascular spaces of the basal ganglia and cerebellum. The tumor cells were positive for S100 and Leu-7 markers and negative for GFAP human melanoma black 45, keratin, and CD45. Although the tumor was negative for GFAP (a glial cell marker), Leu-7 is involved in myelination and is often seen in oligodendrogliomas.4 The postmortem diagnosis of this patient was diffuse leptomeningeal oligodendrogliomatosis. The author noted that this type of tumor usually has a primary intra-axial mass, which this case lacked. However, like our patient, he had an 8-mm oval-shaped, enhancing nodule at C6 to C7. This nodule was described by the author as a possible primary tumor.
Gardiman et al3,5 presented 4 relatively similar cases: all childhood/adolescence presenting with neurologic manifestations, communicating hydrocephalus with spinal fluid high in protein and negative for malignant cells, diffuse leptomeningeal enhancement on imaging with cystic lesions thought to represent perivascular infiltration of tumor cells, and none of which had a primary tumor identified. Cases 2 and 3 had intramedullary enhancing lesions of the spinal cord. Biopsies obtained in all 4 cases identified tumor cells with the morphologic appearance of glial differentiation but “a significant number of nuclei were immunopositive for Neu-N,” and tumor cells were diffusely positive for synaptophysin. Only scattered cells were positive for GFAP. All cases initially had <1% MIB-1/KI-67 index. Only case 3 was found to have a 1p deletion on initial fluorescence in situ hybridization testing; however, on his second biopsy, there was a 50% MIB-1 index and further progression to 1p19q codeletion. Cases 1, 2 and 3 received various lengths of chemotherapy with temozolomide. Case 3 received irradiation due to disease progression with development of tumor 2 years after finishing 12 months of temozolamide. Case 4 was not given systemic treatment due to parental refusal. None were disease free after treatment but progressed with a slow indolent course while the child in case 2 subsequently died from disease progression.
In 2012, Gardiman et al published an update on case 4. The patient presented 22 months after the initial presentation and biopsy, with new seizures and personality changes. Magnetic resonance imaging at this time showed multiple intraparenchymal cysts and thick leptomeningeal enhancement with aggregation of tumor deposits in the dural sac. His first biopsy at initial presentation did not clearly provide adequate information for classification of the tumor. It revealed cells with copositivity for both neurocytes and oligodendrocytes, high mitotic index, and no cytogenetic abnormalities were detected. With this second presentation, a repeat biopsy showed a low mitotic index and low MIB-1 in addition to the aforementioned features. The final diagnosis was determined to be a low-grade tumor consistent with diffuse leptomeningeal glioneuronal tumor and he was started on chemotherapy as per a low-grade protocol, which included vincristine and carboplatin. Due to severe neurotoxicity and prolonged myelosuppression, his therapy was initially switched to temozolamide alone. Then, for seizure and behavioral control, as well as the potential augmentation of glial differentiation, daily valproic acid was started and the temozolamide was discontinued (reason for its discontinuation not specified). Three months after initiating the valproic acid, a total of 67 months since initial presentation, the patient’s leptomeningeal involvement and neurologic symptoms (including seizures and behavioral changes) were steadily improving.5
Since the description of diffuse leptomeningeal glioneuronal tumor, Schniederjan et al1 reported 9 additional cases that they termed “diffuse leptomeningeal neuroepithelial tumor.” The ages of the children in this series ranged from 2 to 7 years old. The clinicopathologic presentation of their patients is almost identical to the above-described cases, including that of our patient. Of the 9 patients, 8 had cytogenetic testing for 1p/19q deletions. Of those tested, 6 demonstrated the 1p deletion, and 2 of those 6 also had the 19q codeletion. Of those with only the 1p deletion, 1 did not have sufficient tissue adherence for successful 19q fluorescence in situ hybridization testing. Of the 9 cases tested for deletions, 8 were also tested for isocitrate dehydrogenase 1 mutation and found to be negative. Overall, 2 patients were treated with temozolamide alone, 2 with temozolamide and other chemotherapy agents, 1 with vincristine and carboplatin, 1 with radiation alone, and 1 patient was treated with chemotherapy (drug not specified) and radiation therapy. At the time of their publication, 2 patients were newly diagnosed and had not yet started treatment. The treated patients were alive with stable or slowly progressive disease. (See Table 1 for details and comparisons of all discussed cases.)
Table 1.
Case Comparison.
| Our Patient | Armao | Gardiman 1 | Gardiman 2 | Gardiman 3 | Gardiman 4 | Schniederjan 1 | Schniederjan 2 | Schniederjan 3 | Schniederjan 4 | Schniederjan 5 | Schniederjan 6 | Schniederjan 7 | Schniederjan 8 | Schniederjan 9 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age/sex | 14 yo/F | 8 yo/M | 4 yo/M | 3 yo/F | 3 yo/F | 13 yo/M | 2.5 yo/M | 3 yo/M | 1.5 yo/M | 4 yo/F | 2 yo/F | 5 yo/F | 7 yo/F | 4 yo/F | 6 yo/M |
| Hydrocephalus | + | + | + | + | + | + | + | + | + | + | + | − | − | + | + |
| Bilateral CN 6 palsy | + | − | − | + | − | − | +a | ||||||||
| CSF with elevated protein and no malignant cells | + | + | |||||||||||||
| IP/IM tumor | − | − | − | − | − | − | − | +b | − | +c | − | +d | − | − | − |
| Meningeal nodules | +e | + | − | +e | +e | +f | − | − | |||||||
| Diffuse LM enhancement | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Intraventricular involvement | +g | − | − | +h | − | + | |||||||||
| Thalamic calcifications | − | − | − | − | − | + | |||||||||
| Sella turcica enlargement | − | − | + | − | − | +i | |||||||||
| Coating of optic chiasm | + | + | |||||||||||||
| IP cystsj | + | + | + | + | + | + | + | + | + | + | + | + | |||
| Neuronal and glial markers on same cell | + | +k | + | + | + | + | − | − | − | + | + | − | − | + | + |
| OLIG-2 | + | ||||||||||||||
| p53 | +l | ||||||||||||||
| MIB-1/KI-67 elevated | − | − | − | <3%m | − | − | + | − | − | − | − | + | − | ||
| High mitotic index | + | − | − | + | +/−m | ||||||||||
| 1p/16q deletions | − | − | +/−, +/+n | − | −/− | +/− | +/+ | +/− | −/− | ?/?o | +/?p | +/− | +/+ | ||
| Diagnosis given | DLGNT | DLOq | DLGNT | DLGNT | DLGNT | DLGNT/DLOr | DLNET | DLNET | DLNET | DLNET | DLNET | DLNET | DLNET | DLNET | DLNET |
Abbreviations: LM, leptomeningeal; IP, intraparenchymal; DLGNT, diffuse leptomeningeal glioneuronal tumor; DLO, diffuse leptomeningeal oligodendrogliomatosis; DLNET, diffuse leptomeningeal neuroepithelial tumor; GFAP, glial fibrillary acid protein; CSF, cerebral spinal fluid; FLAIR, fluid-attenuated inversion recovery; yo, years old; F, female; M, male.
aReported as “diplopia.”
bIntramedullary lesion at T8.
cIntramedullary cystic and solid mass at C6 to C7.
dCerebellar lesion.
eCervical meningeal nodules.
fSuprasellar meningeal nodules.
g1 cm mass in left cavernous sinus.
hLeft frontal intraventricular lesion with perivascular pseudorosettes versus pseudopapillary structure.
iInternal auditory canals.
jOur patient’s cysts were nonenhancing; Armao patient’s cyst enhanced like CSF; all 4 Gardiman patients’ cysts were hyperintense on T2 and isohypointense on T1 and FLAIR.
kGFAP negative, Leu-7 positive; Leu-7 is associated with myelin and is present in a majority of oligodendrogliomas.
lFocally positive.
mBiopsy at initial presentation high mitotic index; biopsy at second presentation low mitotic index and <3% MIB-1.
nFirst biopsy—<1% MIB-1 and 1p deletion and second biopsy—50% MIB-1 and 1p/16q codeletion.
oTissue was not available for fluorescence in situ hybridization testing.
pPoor tissue adherence on chromosome 19 fluorescence in situ hybridization testing.
qDLO usually has a primary intra-axial mass—this case did not.
rGardiman diagnosed DLGNT and Rossi diagnosed DLO.
Discussion
Radiologic Findings
It has been believed that central nervous system neoplasms classically disseminate caudally from the origin of the primary tumor. However, of the 15 above-mentioned cases, 7 have a noted tumor or nodules found in the spinal cord with diffuse leptomeningeal enhancement and subsequent cystic changes in the brain.1,3–5 This suggests this tumor can indeed be a primary spinal cord neoplasm demonstrating cranial dissemination.
Additionally, diffuse leptomeningeal enhancement is usually assumed to be evidence of dissemination associated with more progressive disease. The patients described with this unique tumor seem to present with hydrocephalus and diffuse leptomeningeal enhancement and do not usually present with cystic changes. They later develop cystic changes in the leptomeninges and parenchyma, which continue to increase in number and size with progression of the disease. This suggests the leptomeningeal enhancement is not necessarily a marker of advanced disease in these patients, but development and progression of cystic changes may be. Additionally, as the cysts appear to be noncomplex in nature (i.e., without components of protein or blood), these cystic changes are likely perivascular spaces enlarging with collections of extracellular fluid developing secondary to progression of the leptomeningeal disease altering normal drainage pathways.
Histochemical Cytopathology
Mixed glioneuronal tumors and gangliogliomas are defined as consisting of 2 distinct cell populations, one population of neuronal cells mixed with another distinct population of glial cells. The glioneuronal tumors discussed in this article are composed of a singular cell population staining positively for markers of both glial and neuronal cell components. Therefore, histological staining for multiple markers of both types of cells in all samples collected can lead to earlier diagnosis.
In our patient’s case, not all biopsy samples included tumor cells, and different samples revealed varying levels of differentiation and of aggressive features. Gardiman case 4 also demonstrated differing levels of mitotic index at initial and later biopsies, as well as varying degrees of aggressive histological signs.5 Obtaining early and repeated biopsies, each with multiple sample sites, is more likely to provide the most important information leading to an earlier diagnosis of malignancy in patients with diffuse leptomeningeal enhancement and elevated CSF protein without evidence of a primary tumor or malignant cells in the spinal fluid. Additionally, testing these multiple samples for both glial and neuronal cell markers, p53, OLIG-2, isocitrate dehydrogenase 1, and 1p/19q deletion can help with diagnosis, as well as give more information of prognosis and treatment options.
OLIG-2 is a transcription factor specific for oligodendrocytes found in glial progenitor cells.6 However, it has been found in cells of glioneuronal tumors exhibiting both glial and neuronal characteristics.7 This can suggest a common progenitor cell type between glial and neuronal cells, giving support to the theory that these glioneuronal tumors are neoplasms of a common precursor cell for both glial and neuronal cell types.
Isocitrate dehydrogenase 1 is an nicotinamide adenine dinucleotide phosphate-producing cytoplasmic enzyme. Somatic mutations of the IDH1 gene have been associated with several forms of cancer, including astrocytomas, oligodendrogliomas, and hematopoetic dysplasias.8 As it is seen in over 70% of grade II to IV gliomas9 and is not demonstrated in diffuse leptomeningeal glioneuronal tumors, isocitrate dehydrogenase 1 may be a useful marker in testing to differentiate diffuse leptomeningeal glioneuronal tumors from higher grade glial neoplasms. Our patient and the 9 Schniederjan patients all tested negative for isocitrate dehydrogenase 1 (the others did not have isocitrate dehydrogenase 1 status reported).1
Chromosomal analysis for 1p19q codeletion, or even a singular deletion of either 1p or 19q, can be useful in determining tumor aggressiveness and likely response to chemotherapy, especially to temozolomide. This codeletion has been seen in both neurocytomas and adult oligodendrogliomas; and its being seen in both tumor types can also support the idea of a common progenitor cell being the neoplastic culprit in diffuse leptomeningeal glioneuronal tumors. This codeletion has demonstrated correlation with more aggressive disease in extraventricular neurocytomas10 and can be useful in assessment of potentially aggressive behavior of other tumor types, including diffuse leptomeningeal glioneuronal tumors. On the other hand, it has also been highly correlative with increased responsiveness to systemic chemotherapy, especially with temozolomide, with and without adjunctive radiotherapy, as well as disease prognosis.11–13
Treatment
It is clear from the reported cases mentioned herein that this neoplasm is, in fact, treatable. Stable disease has been achieved with chemotherapy alone, as well as with adjunctive irradiation. As evidenced by our patient’s case, remission is also achievable. Therefore, treatment should absolutely be initiated, although the most effective treatment has not yet been identified. Our patient’s remarkable response to her treatment regimen could have been due to the combined temozolamide and radiation therapy, which was allowable given her age. Her p53 positivity was also likely significant in her response to therapy, which can have represented a more aggressive tumor with higher sensitivity to the treatment modalities used. The seemingly significant response to initiation of valproic acid in Gardiman case 4, both with improvement in neurological symptoms and radiographic findings, can indicate valproic acid lends itself to successful adjunctive therapy in cases with seizures and behavioral changes, and possibly even in those without.
The majority of the other cases mentioned did not receive radiotherapy in addition to temozolamide, likely due to patient age, and subsequently had reportedly stable disease. With our patient’s quick and steady response to her combined therapy regimen, it may be advisable to use combined treatment with systemic chemotherapy and craniospinal irradiation from the beginning—more aggressive treatment for a potentially aggressive tumor. However, the risks involved depend on the individual patient and should still be tailored accordingly. Although temozolomide is not the only option for chemotherapy, it certainly has its advantages for the patient—oral administration, less patient toxicity, and it seems to work well for this neoplasm.
Suggestions for future studies, which can provide information leading to more successful therapy, include comparing chemotherapy with temozolomide alone versus chemotherapy with other agents, chemotherapy alone versus chemotherapy in conjunction with craniospinal irradiation, and the efficacy of valproic acid as an adjunctive therapy for patients with and without symptoms including seizures and/or behavioral changes. Additionally, and even more importantly, evaluation of the molecular characteristics of the disease will help targeted therapy.
Conclusions
In conclusion, when patients present with sequelae of nonspecific neurologic signs and symptoms, hydrocephalus, CSF studies significant only for elevated protein without evidence of malignant cells, and diffuse leptomeningeal enhancement on radiographic images, the differential diagnosis should include diffuse leptomeningeal glioneuronal tumor (also described as diffuse leptomeningeal neuroepithelial tumor). Additionally, if cystic changes are seen with the above-mentioned findings, it should prompt aggressive workup for this tumor in addition to the infectious workup usually employed.
OLIG-2 may be a marker implying a common progenitor cell for neurocytes and glial cells in tumors. Although p53 and 1p19q codeletions represent more aggressive tumor activity, 1p19q codeletions have shown significantly increased responsiveness to chemotherapy, especially temozolomide. When this diagnosis is in the differential, it is advisable to obtain earlier (and possibly repeated) biopsies from multiple sites, and these tissue samples should be tested for the following factors: multiple glial cell markers, multiple neuronal cell markers, p53, OLIG-2, isocitrate dehydrogenase 1, and chromosomal deletions (especially of 1p and 19q). Obtaining this information can lead to earlier diagnosis and treatment of diffuse leptomeningeal glioneuronal tumors in patients who present with hydrocephalus and diffuse leptomeningeal enhancement without evidence of a primary tumor.
Chemotherapy with or without craniospinal irradiation should be considered for these patients. Temazolomide seems to be effective and is especially recommended for patients with markers of more aggressive tumor activity. Additionally, valproic acid may be a beneficial adjunctive therapy. Both medications should be considered when appropriate and investigated in more depth in future studies. This is a seemingly low-grade tumor and collective efforts should be undertaken to develop molecular characterization of this entity in hopes of developing targeted therapy.
Acknowledgments
All of the work on this article was completed at the University of Mississippi Medical Center by the 6 authors listed on the title page. Rebecca Dunn Folkerth, MD, assisted in obtaining stored pathological specimens for further analysis.
Footnotes
Author Contributions: Literature research and data interpretation were performed by MRL and BLH with the assistance of JND Radiological interpretation was provided by TAN and histochemical cytopathology analysis and interpretation by JF. BLH acted as lead faculty member and mentor.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval: This case report and review of the literature did not necessitate approval by an institutional review board nor ethics committee.
References
- 1. Schniederjan MJ, Alghamdi S, Castellan-Sanches A, et al. Diffuse leptomeningeal neuroepithelial tumor: 9 pediatric cases with chromosome 1p/16q deletion status and IDH1 (R132H) immunohistochemistry. Am J Surg Pathol. 2013;37 (5):763–771. [DOI] [PubMed] [Google Scholar]
- 2. Ruppert B, Welsh CT, Hannah J, et al. Glioneuronal tumor with neuropil-like islands of the spinal cord with diffuse leptomeningeal neuraxis dissemination. J Neurooncol. 2011;104 (2):529–533. [DOI] [PubMed] [Google Scholar]
- 3. Gardiman MP, Fassan M, Orvieto E, et al. Diffuse leptomeningeal glioneuronal tumors: a new entity? Brain Pathol. 2010;20 (2):361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Armao DM, Stone J, Castillo M, Mitchell KM, Bouldin TW, Suzuki K. Diffuse leptomeningeal oligodendrogliomatosis: radiologic/pathologic correlation. Am J Neuroradiol. 2000;21 (6):1122–1126. [PMC free article] [PubMed] [Google Scholar]
- 5. Gardiman MP, Fassan M, Nozza P, et al. Diffuse leptomeningeal glioneuronal tumours: clinico-pathological follow-up. Pathologica. 2012;104 (6):428–431. [PubMed] [Google Scholar]
- 6. Tanaka Y, Yokoo H, Komori T, et al. A distinct pattern of Olig2-positive cellular distribution in papillary glioneuronal tumors: a manifestation of the oligodendroglial phenotype? Acta Neuropathol. 2005;110 (1):39–47. [DOI] [PubMed] [Google Scholar]
- 7. Matsumura N, Yokoo H, Mao Y, Yin W, Nakazato Y. Olig2-positive cells in glioneuronal tumors show both glial and neuronal characters: the implication of a common progenitor cell? Neuropathology. 2013;33 (3):246–255. [DOI] [PubMed] [Google Scholar]
- 8. Andrulis M, Capper D, Meyer J, et al. IDH1 R132H mutation is a rare event in myeloproliferative neoplasms as determined by a mutation specific antibody. Haematologica. 2010;95 (10):1797–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH2 and IDH2 mutations is a neomorphic enzymatic activity that converts α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17 (3):225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rossi S, Rodriguez F, Dei Tos A, et al. Re: diffuse leptomeningeal glioneuronal tumors: a new entity? Brain Pathology. 2009;19 (4):745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blakeley J, Grossman S. Anaplastic oligodenroglioma. Curr Treat Options Neurol. 2008;10 (4):295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haberer S, Assouline A, Mazeron JJ. Gliomes cérébraux. Cancer/Radiothrapie. 2010;14 (1):S14–S22. [DOI] [PubMed] [Google Scholar]
- 13. Kaloshi G, Everhard S, Laigle-Donadey F, et al. Genetic markers predictive of chemosensitivity and outcome in gliomatosis cerebri. Neurology. 2008;70 (8):590–595. [DOI] [PubMed] [Google Scholar]