

Abstract
Wistar rat pups had the left common carotid artery cut, and they were exposed to 8% oxygen with free access to food and water until they were killed at 1, 12, 24, and 48 hours after the hypoxia–ischemia (HI) insult. Connexin 43 (Cx43), hemichannel (HC1), and caspase 3 (Casp3) in cerebral HI tissues were examined by immunohistochemistry and Western blot analyses. Astrocytes cell line, astrocytes transduced with a retroviral empty vector (Psup astrocyte), or a Cx43-specific small hairpin RNA (shRNA) construct (shRNA astrocytes) was treated with oxygen–glucose deprivation (OGD) insult. The viability of astrocytes was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. The results showed the expression of Cx43, HC1, and Casp3 in rats’ brain, and astrocytes and Psup astrocytes increased significantly after 24 hours of HI/OGD insult. Cell viability decreased after 24 hours of the insult. The results suggest that Cx43 and hemichannel are likely to mediate the astrocytic death after HI insult.
Keywords: connexin 43, gap junction hemichannel, astrocytes, hypoxia–ischemia (HI), oxygen–glucose deprivation (OGD), neuroprotection
Perinatal hypoxia–ischemia (HI) is the most common cause of various neurological disabilities in children with a high societal cost.1 Approximately 15% to 25% of the affected newborns die in the postnatal period and 25% develop severe and permanent neuropsychological sequelae, including cerebral palsy, seizures, visual impairment, mental retardation, learning impairment, and epilepsy.2,3 Hypoxic–ischemic encephalopathy (HIE) is an evolving process, and ample evidence suggests distinct difference between the immature and mature brain in the pathology and consequences of brain injury.4 Therefore, it is of utmost importance to better understand the mechanisms underlying the HI injury in neonatal brain to devise effective therapeutic strategies. Nonetheless, the mechanisms involved in this pathology in the developing brain remain inadequately understood.
Hence, it is necessary to rescue the immature brain from asphyxia and to assure development without neurologic sequelae. In recent years, many researchers have demonstrated that gap junctions (GJs), the channels that connect the cytoplasm of neighboring cells, play a role in the communication of cell death messages between cells, in particular in the context of apoptotic cell death.5–8
Gap junction channels and hemichannels (HCls) formed of connexin (Cx) subunits are found in most cell types in vertebrates. Gap junctions connect cells via channels not open to the extracellular space and permit the passage of ions and molecules of ~1 kilo Dalton (kDa).9 Single Cx HC1s, which are Cx hexamers, are present in the surface membrane before docking with a HC1 in an apposed membrane.9 Because of their high conductance and permeability in cell–cell channels, it had been thought that Cx HC1s remained closed until docking to form a cell–cell channel. Now it is clear that at least some HC1s can open to allow passage of molecules between the cytoplasm and extracellular space.10 The GJ channels can allow intercellular diffusion of necrotic or apoptotic signals but can also allow diffusion of ions and substances from healthy to injured cells, thereby contributing to cell survival.11,12 Moreover, opening of GJ HC1s can exacerbate cell injury or mediate paracrine or autocrine signaling.10 In addition to the cell-specific features of an ischemic insult, propagation of cell damage and death within affected tissues may be affected by expression and regulation of GJ channels and HC1s formed by Cxs.11–15
In this study, we explore the effect of Cx43 and HC1s in neonatal rats’ brain and astrocytes at different times after HI/oxygen–glucose deprivation (OGD) injury and investigate the molecular mechanisms underlying the action of Cx43.
Materials and Methods
Reagents
Rabbit Cx43 antibody was from Sigma-Aldrich (C6219, aminal acid residues 363-382; Poole, Dorset, United Kingdom), rabbit Cx43 HC1 antibody was designed and made by Custom Hybridoma/Pickcell Laboratory and the epitope sequence is ESAWGDEQSAFRCNTQQPGC. Rabbit caspase 3 (Casp3) antibody was from Sigma-Aldrich (HPA002643; Poole). Mouse monoclonal anti–glial fibrillary acidic protein (GFAP), a marker of the astrocyte antibody, was from Abcam (GF5, ab10062; Abcam, Cambridge, United Kingdom). All other reagents and chemicals were from Sigma-Aldrich (Poole) unless stated otherwise.
Animals
Forty unsexed Wistar rat pups (12-17 g) on postnatal day 7 (P7) were obtained from the Experimental Animal Center of Shandong University, Shandong, People’s Republic of China. The use of animal tissue was in accordance with the tenets of the Declaration of Helsinki and with the permission for the research from Shandong University, People’s Republic of China.
Establishment of Neonatal HIE Rat Model
Forty Wistar rats were randomly arranged into the following 5 groups (n = 8 for each group): control and HI groups with 1, 12, 24, and 48 hours (T1, T12, T24, and T48 hours) after HI.
A model of neonatal HIE was developed at P7.16 Under anesthesia using lidocaine (Tianjin Pharma Group, Tianjin, China) topically, the left common carotid artery was isolated, double ligated, and cut. After allowing for recovery for 2 hours with their mothers, the pups were exposed to 8% oxygen at an ambient temperature of 35°C for 3 hours and then returned to their mothers. The pups were conditioned at 23°C ± 1°C and a constant humidity of 60% ± 10% in a room (12-/12-hour light/dark cycle). They had free access to food and water until they were killed. The same mother littermates as age-matched controls underwent neither surgical procedure nor hypoxic exposure were killed at postnatal day 9.
Tissue Preparation
Rat pups were killed at 1, 12, 24, and 48 hours after the HI insult for histopathological and Western blot analyses. Decapitated brains were dissected coronally at the level of the third plane of thalamus. The whole brain was separated equally into left and right sides, only the left sides (damaged cortex) were used for histopathological and Western blot analyses.
Immunofluorescent Detection of Cx43 + Glial Fibrillary Acidic Protein, HC1 + Glial Fibrillary Acidic Protein, Cx43, HC1, and Casp3 in Rats’ Brain Tissue
The brains were removed, and the brain sections with subventricular zone (SVZ) were fixed for 15 minutes at room temperature with 4% paraformaldehyde and permeabilized for 15 minutes in 0.2% Triton X-100 (t-octylphenoxypolyethoxyethanol). After 3 washes with phosphate-buffered saline, the sections were incubated overnight with primary antibodies, diluted in PBS, against Cx43 + glial fibrillary acidic protein (1:2000 rabbit and 1:100 mouse), HC1 + glial fibrillary acidic protein (1:200 rabbit and 1:100 Mouse), Cx43 (1:2000), HC1 (1:200), and Casp3 (HPA002643, 1:250, rabbit), followed by incubation for 2 hours in appropriate Alexa Fluor 488/594 (1:1000; Molecular Probes, United Kingdom) and Alexa Fluor 488 (1:1000, Molecular Probes) secondary antibodies. After an additional 3 washes with PBS, the sections were mounted in Hydromount-containing bis-benzimide (Hoechst 33342, 1/500; BDH, United Kingdom). Substitution of the primary antibodies with the same-species serum served as a negative control. The images were visualized and captured using a Leica microscope (SP2UV; Milton Keynes, United Kingdom).
All images were obtained as a stack in which each image had an optical thickness of 1 μm, and each image was analyzed with ImageJ (National Institutes of Health). Afterward, the ImageJ feature for analysis of particles was used, and the Feret diameter was measured to quantify the diameter of positive immunoreactive puncta. The percentages of cells with Casp3 staining were obtained by using ImageJ. The total number of cells was obtained by counting the Hoechst-labeled nuclei.
Western Blot Analysis of Brain Tissue
The corresponding areas of brain tissue in SVZ was washed twice with PBS, lysed in 500 μL of lysis buffer (50 mmol/L Tris-HCl, pH 7.6, 5 mmol/L EDTA, 25% sucrose, 0.6 μL of 100 μg/mL phenylmethylsulphonyl fluoride, and 2.4 μL β-mercaptoethanol) for 30 minutes, and then centrifuged at 90 000 rpm for 15 minutes at 4°C. Whole tissue extracts equivalent to 100 μg protein were separated by 8% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) with the running buffer (25 mmol/L Tris, 200 mmol/L glycine, and 1% w/v SDS, pH 8.3), and transferred to nitrocellulose membranes (Gibco BRL, USA). The membranes were blocked with 5% skimmed milk in TBST (20 mmol/L Tris-HCl at pH 8.0, 50 mmol/L NaCl, and 0.05% Tween 20) at 4°C overnight, followed by incubation with primary antibodies (same as mentioned earlier) for 2 hours and again incubated for 1 hour at room temperature with 1:10 000 peroxidase-conjugated goat antirabbit immunoglobulin G. The bound antibody was detected with chemiluminescence (SuperSignaI West Pico Stable Peroxide Solution, PIERCE, USA), and the tublin served as the internal standard. The intensity of the band was calculated with Bio-Rad, USA imaging system. Resulting immunoblots were scanned with a ScanJet 3c flat bed scanner. The densitometric analysis was performed by assessing pixel number and obtaining final numerical data using SCION IMAGE software (Hewlett Packard HP, C2527A, China).
Cell Culture, Retroviral Constructs, and Transfection
Human cerebral cortex astrocyte cell line purchased from ScienCell Research Laboratories was transduced with a Cx43-specific small hairpin RNA (shRNA) construct (shRNA astrocytes; gift from W. H. Moolenaar. sequence GGTGTGGCTGTCAGTGCTC) or a retroviral empty vector (Psup astrocytes; pSuppressor; Imgenex Co, San Diego, California),17 essentially as described in Carr and Whitmore.18 The GP2-293 packaging cell line (Clontech, USA) was transfected by calcium phosphate precipitation with retroviral plasmid (15 g) containing the Cx43 shRNA insert or empty vector, 5 g pMD.G envelope plasmid, and 10 g pBSII SK-carrier plasmid (Stratagene, USA). Selection for transduced cells was via resistance to 2 g/mL puromycin (Cx43 shRNA) or 500 g/mL geneticin (empty vector).
In this study, astrocytes cell line and astrocytes transfected with Cx43 shRNA were used to detect the roles of Cx43 and its HC1 in astrocytic death and apoptosis. Astrocytes transfected with an empty vector were used as a control to compare with Cx43 shRNA. After the transfection, the suppression of the Cx43 expression was confirmed by Western blotting (the technique used was the same as detailed in the “Western blot analysis of astrocytes” section). All experiments were carried out when the cells reached near to confluence.
Oxygen–Glucose Deprivation Techniques
Cell cultures were subjected to the OGD astrocytes media (previously bubbled with 5% CO2/95% N2 for 30 minutes). Hypoxia was induced by placing the cultures inside of an incubator chamber and removing the air with a CO2/N2 (5, 95%) flow for 15 minutes, after which the chamber was closed for 3 hours as described previously.18 Recovery was allowed in normal media (minimum essential medium with 5 mmol/L glucose and 10% fetal bovine serum) at 37°C in a 5% CO2/95% air atmosphere at nearly 100% relative humidity with different time points (1, 12, 24, and 48 hours).
Cell Death Quantification—3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Assay
The 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay was used as an index of cell viability and was performed according to the manufacturer’s instructions (Catalog # 4890-050-K, R & D Systems Ltd. Europe). Briefly, astrocytes were cultured in 96-well plates at a density of 5000 cells/well with culture medium for 24 hours to allow the cells attach stably. After the treatment, 0.4 mg/mL MTT was added into the culture medium. After 3 hours, the MTT solution was aspirated and dimethylsulfoxide (0.3 mL/well) was added. Optical density of the supernatant was read at 570 nm using a microplate spectrophotometer (Spectra Max 340; Molecular Devices, USA). Absorbances were normalized to the untreated control cultures which represented 100% viability:
% viability = mean absorbance of sample/mean absorbance of control × 100
Western Blot Analysis of Astrocytes
The cell cultures were rinsed twice with a saline solution (Hank), pH 7.4, and the cells were scraped off the dish with a rubber policeman and harvested in a solution containing inhibitors of proteases (200 mg/mL soybean trypsin inhibitor, 1 mg/mL benzamidine, 1 mg/mL e-aminocaproic acid, and 2 mmol/L phenylmethylsulfonyl fluoride) and phosphatases (20 mmol/L Na4P2O7 and 100 mmol/L NaF) for 30 minutes. The suspension was centrifuged (14 000 rpm for 2 minutes at 4°C), and the pellet was resuspended in 50 mL of the solution containing protease inhibitors and sonicated (Ultrasonic cell disrupter; Microson). Proteins were measured in aliquots of cell lysates with the Bio-Rad (Catalog # 40000-616, China) protein assay. Samples were mixed with Laemmli buffer (Catalog # 161-0737, Bio-Rad Laboratories, Inc) and resolved in 8% SDS/PAGE. The following steps were the same as described for brain tissue analysis.
Statistical Evaluation
All cell biology experiments were based on triplicate wells, and each experiment was repeated at least 3 times. All numerical results were expressed as the mean ± standard deviation. Student’s unpaired t test (GraphPad Prism version 4.0) was used to assess the differences in positive plaque pixels between cells treated with and without ischemia reperfusion/OGD injury. One-way analysis of variance (Prism 4.0) was used to assess any differences between sets of data obtained when cells were grown with different treatment. Statistical significance was defined as *P < .05.
Results
The Expression of Cx43, HC1, and Glial Fibrillary Acidic Protein in the Astrocytes From Rats’ Brain
To confirm that Cx43 and HC1 were expressed in the astrocytes with glial fibrillary acidic protein, an astrocytic marker, double immunostaining of Cx43 + glial fibrillary acidic protein, and HC1 + glial fibrillary acidic protein were performed. The results showed Cx43 and HC1 expressed in the astrocytes in which glial fibrillary acidic protein was detected at the same time (Figure 1A and B). There appears to be a markedly lower level of labeling for HC1 compared to Cx43.
Figure 1.
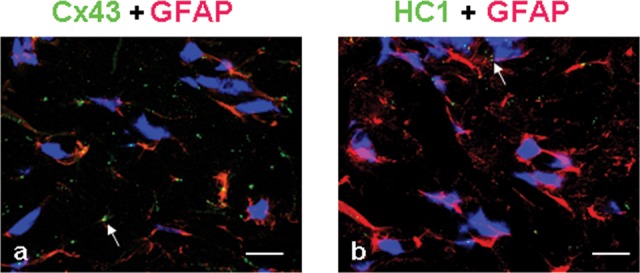
Double immunostaining for the expression of Cx43 + glial fibrillary acidic protein and HC1 + glial fibrillary acidic protein in the astrocytes from rat’s brain after HI insult. Cx43 and HC1 were stained green, glial fibrillary acidic protein was stained red, and nuclei were stained blue. The arrows show the positive staining of Cx43/HC1. Bar = 50 µm. Cx43 indicates connexin 43; GFAP, glial fibrillary acidic protein; HI, hypoxia–ischemia; HCl, hemichannel.
The Expression of Cx43, HC1, and Casp3 in Rats’ Brain Following HI Insult
To detect the expression of Cx43, HC1, and Casp3 in rats’ brain, immunostaining was undertaken following HI with appropriate time points. The results showed that the expression of Cx43, HC1, and Casp3 significantly increased in SVZ of rat’s brain after 24 hours of HI (Figure 2c1-3, d1-3, e1-3, and f1-2; P < .05), whereas no significant change was observed on 1 and 12 hours after HI (Figure 2b1-3 and f1-2).
Figure 2.

Immunostaining for the expression of Cx43, HC1, and Casp3 in rat’s brain after HI insult. Cx43, HC1, and Casp3 were stained green, and nuclei were stained blue. Bar = 25 µm. a1-3, The expression of Cx43, HC1, and Casp3 in SVZ from rat without the insult of HI (Cont.); b1-3, The expression of Cx43, HC1, and Casp3 in SVZ from rat 1 hour after HI (T1 hour); c1-3, The expression of Cx43, HC1, and Casp3 in SVZ from rat 12 hours after HI (T12 hour); d1-3, The expression of Cx43, HC1, and Casp3 from rat 24 hours after HI (T24 hour); e1-3, The expression of Cx43, HC1, and Casp3 in SVZ from rat 48 hours after HI (T48 hour); f1, The plaque area (pixels) of Cx43 and HC1 staining per cell with the time points. f2, The percentage of Casp3 positive cells (%) with the time points. *P < .05. Cx43 indicates connexin 43; Casp3, caspase 3; HCl, hemichannel; HI, hypoxia–ischemia; SVZ, subventricular zone.
Western blotting was performed to further confirm the above-mentioned results. The elevated expression of Cx43, HC1, and xasp3 from brain tissue in SVZ appeared after 24 hours of HI (Figure 3A and B; P < .05).
Figure 3.

Western blot analysis of Cx43, HC1, and Casp3 expression in subventricular zone of rat’s brain after HI insult. A, Protein expression of Cx43, HC1, Casp3, and tublin was identified from the tissue extracts in subventricular zone of rat’s brain. B, Bar histograms show the mean ± SD corresponding to data displayed in part A. *P < .05. Cx43 indicates connexin 43; Casp3, caspase 3; HCl, hemichannel; HI, hypoxia–ischemia; SD, standard deviation.
Effect of Transfection With a Retroviral Empty Vector and Null Cx43 Gene in Astrocytes
Western blotting was performed to confirm the effects of the transfection. The results showed there was expression of Cx43 and HC1 in both astrocytic cell line and Psup astrocytes but not in shRNA astrocytes after the transfection (Figure 4).
Figure 4.
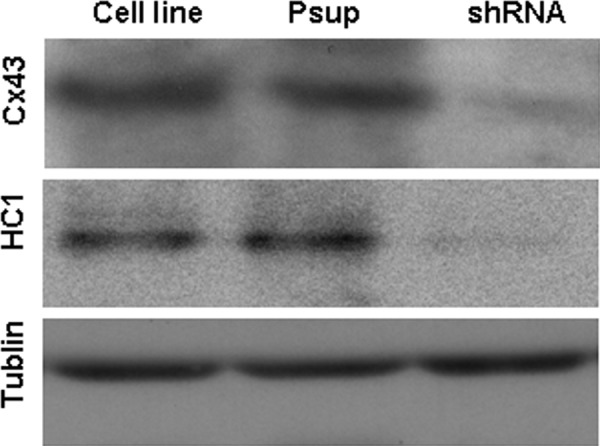
Western blot analysis to confirm the effect of transfection with null Cx43 gene shRNA in astrocytes. After 24 hours of the gene transfection, the lysate from astrocytes was resolved by SDS-PAGE, and immunopositive bands were semiquantified by scanning laser densitometry. Protein expression of Cx43, HC1, and tublin was identified, respectively. Cx43 indicates connexin 43; shRNA, small hairpin RNA; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Viability of Psup and shRNA Astrocytes After OGD
To investigate the viability of astrocytes after OGD, the MTT assay was performed. The results showed there were significant decreases in viability for astrocytes cell line (Figure 5A; P < .05) and Psup astrocytes (Figure 5B; P < .05) on 24 hours after OGD (Figure 5A and B). In contrast, there were no changes in the viability for shRNA astrocytes after OGD (Figure 5C; P > .05).
Figure 5.

Viability of astrocytes after OGD treatment. Astrocytes were cultured in 96-well plates at a density of 5000 cells/well and treated with OGD. Cells that did not undergo OGD treatment act as a control. Cell viability was assessed by the MTT assay (MTT measured in units of optical density at 570 nm). A, Cell line astrocytes; B, psup astrocytes; C, shRNA astrocytes. *P < .05. OGD indicates oxygen–glucose deprivation; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; OGD, oxygen–glucose deprivation; shRNA, small hairpin RNA.
Oxygen–glucose deprivation Increases the Expression of Cx43, HC1, and Casp3 in Cell Line and Psup Astrocytes But Not in shRNA Cells
The expression of astrocytes after OGD insult was detected using Western blotting. The results showed the elevated expression of Cx43, HC1, and Casp3 from cell line (Figure 6A1 and A2; P < .05) and Psup astrocytes (Figure 6B1 and B2; P < .05) at 24 hours after OGD (Figure 6A1-2 and B1-2). There were no changes in the expression of Cx43, HC1, and Casp3 for shRNA cells after OGD (Figure 6C1 and C2; P > .05).
Figure 6.

Western blot analysis of Cx43, HC1, and Casp3 expression from astrocytes following OGD treatment. Astrocytes were treated with OGD techniques. The lysates were resolved by SDS-PAGE, and immunopositive bands were semiquantified by scanning laser densitometry. Data are expressed as mean ± SD corresponding to data displayed, * P < .05. A1-2, Astrocytes cell line; B1-2, Psup astrocytes; C1-2, shRNA astrocytes; D1, Tublin was detected as a control. Cx43 indicates connexin 43; Casp3, caspase 3; HCl, hemichannel; OGD, oxygen–glucose deprivation; shRNA, small hairpin RNA; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; SD, standard deviation.
Discussion
Transient HI is characterized by a striking, progressive evolution of damage over days to weeks after the insult.3,4 Hypoxia–ischemia lesion has been shown to result in earlier acute necrotic neuronal death in the core of the infarction, followed by a delayed and prolonged apoptotic-like neuronal death in the surrounding zone.19,20 Astrocytic functions are essential for normal neuronal activity, which provide metabolic and structural support to neurons; they control the extracellular concentrations of glutamate, K+, and H+ and regulate the volume of extracellular space.21 Numerous studies have demonstrated that astrocytic GJs modulate neuronal survival during and after brain ischemia. Gap junctions are also found between activated microglia, between many types of neurons, between astrocytes and oligodendrocytes, and in a few somewhat controversial instances between astrocytes and neurons.
Gap junction channels can be acutely regulated in response to various stimuli, including changes in voltage, pH, and Cx phosphorylation. Phosphorylation has been implicated in the regulation of gap junctional communication at several stages of the cell cycle and the Cx “life cycle,” such as trafficking, assembly/disassembly, degradation as well as in the gating of HC1s or intact GJ channels. Based on several studies, Cx43 undergoes multiple phosphorylation events22,23 including mitogen-activated protein kinase and protein tyrosine phosphatase μ. They phosphorylate Cx43 on the majority of the 21 serine and tyrosine residues, respectively, which are generally correlated with the marked disruption in intercellular junctional communication.24,25
Our study suggests that upregulation of Cx43 expression can mediate the delayed and prolonged apoptotic-like astrocytic death with the rise of the Casp3 and the decreased cell viability after the insult. Also, in our study we confirmed that downregulation of Cx43 contributed to astrocytic survival. These results further confirm that Cx43 accelerates the astrocytic death which is consistent with previous reports.26 How does ischemia affect junction channels? It has been reported that in both brain slices and primary astrocyte cultures, gap junctional communication is reduced, but not abolished, during ischemia.27–30 The reduction in cellular communication could be caused by several gating stimuli including cellular acidosis, rise in free intracellular Ca2+ concentration, and changes in phosphorylation of Cxs. This was also proved in our previous study.31
In contrast with the GJ, Cx43 HC1s have little distribution and very low open probability under physiological conditions.32,33 Cx43 HC1s are the biogenetic precursors of GJs, which is the main Cx expressed in astrocytes and have long been thought to exist as half of a GJ.32–34 Recent evidence suggests that they can also function as stand-alone channels and have shown the involvement of functional HC1s in diverse physiological and pathological conditions.34–37 Our data show that Cx43 HC1s have lower expression than that of Cx43 and are associated with cell death with an increase in Casp3 and decrease in cell viability. These are consistent with other’s reports.15 Also, in our study the authors confirmed that downregulation of Cx43, which also caused the downregulation of HC1s, contributed to astrocytic survival. The data shown here reinforce the idea that opening of HC1s can be increased by some Cx mutations, which can promote cell death by this novel gain of function.
Oxygen deprivation during hypoxia and ischemia causes intracellular accumulation of toxic metabolites and adenosine triphosphate depletion, which can lead to cell death. In numerous studies, metabolic inhibition (MI) has been used as a model to elucidate the effect of hypoxia with or without substrate deprivation on cells in culture or in ex vivo preparations. In these preparations, HC1 opening induced by MI or ischemia is thought to accelerate cell death.33 In this manner, Cx HC1s could have an influence on the progression of the cell death process as has been demonstrated in HeLa cells (ATCC® CCL-2TM, USA) transfected with Cx43 exposed to various apoptotic agents38 and to trigger apoptosis in Marshall and L2 lung epithelial cells in response to oxidative cell stress.11
Recent studies, in astrocytes as well as in other cell types, have demonstrated that HC1s may open in response to various triggers.9 The list of conditions and intracellular signals triggering HC1 opening and consecutive messenger release is extensive and includes depolarization of the membrane potential,11 reactive oxygen species,11 redox status,12 phosphorylation status,13 mechanical stimulation,14 ischemia/hypoxia,38,39 and also certain Cx mutations.40
In summary, this study shows that Cx43 and its HC1s significantly mediated Casp3 in brain tissues and astrocytes. They can play important roles in HI-induced astrotic death. They transform each other in all process of the cell death. In the early stages of cell death, increased expression of Cx43 increases the intercellular communication; before the cell’s rupture, both Cx43 and HC1s show increased expression, enabling the gates of cell membrane to easily exchange materials in the surrounding environment. Connexin 43 and its HC1s may become the signals to predict early stroke and HI injury. The blockage or inhibition of Cx43 and its HC1s has the potential for the development of novel neuroprotective therapy.
Footnotes
Author Contributions: Conceived and designed the experiments: JW, AM, BZ. Performed the experiments: JW, AM, JX. Analyzed the data: JW, AM, JX, YW. Contributed reagents/materials/analysis tools: AM, YW, BZ. Wrote the paper: JW, AM, BZ.
Authors’ Note: Jingwei Wang and Aihua Ma are cofirst authors of this article.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Shandong Natural Foundation (ZR2010HM098), Shandong Province Science and Technology Development Plan (2010G0020256), and the Shandong Province Medical Development Plan (2011HZ072), China, to Profs B. Zhao and A. Ma.
References
- 1. Vannucci RC. Hypoxic-ischemic encephalopathy. Am J Perinatol. 2000;17(3):113–120. [DOI] [PubMed] [Google Scholar]
- 2. Lai MC, Yang SN. Perinatal hypoxic-ischemic encephalopathy. J Biomed Biotechnol. 2011;2011:609813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vannucci RC, Perlman JM. Interventions for perinatal hypoxic-ischemic encephalopathy. Pediatrics. 1997;100(6):1004–1014. [DOI] [PubMed] [Google Scholar]
- 4. Edwards AD, Brocklehurst P, Gunn AJ, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and metaanalysis of trial data. Br Med J. 2010;340:c363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cusato K, Bosco A, Rozental R, et al. Gap junctions mediate bystander cell death in developing retina. J Neurosci. 2003;23(16):6413–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krutovskikh VA, Piccoli C, Yamasaki H. Gap junction intercellular communication propagates cell death in cancerous cells. Oncogene. 2002;21(13):1989–1999. [DOI] [PubMed] [Google Scholar]
- 7. Krysko DV, Leybaert L, Vandenabeele P, D’Herde K. Gap junctions and the propagation of cell survival and cell death signals. Apoptosis. 2005;10(3):459–469. [DOI] [PubMed] [Google Scholar]
- 8. Vinken M, Vanhaecke T, Papeleu P, Snykers S, Henkens T, Rogiers V. Connexins and their channels in cell growth and cell death. Cell Signal. 2006;18(5):592–600. [DOI] [PubMed] [Google Scholar]
- 9. Evans WH, De Vuyst E, Leybaert L. The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J. 2006;397(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Decrock E, De Vuyst E, Vinken M, et al. Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 2009;16(1):151–163. [DOI] [PubMed] [Google Scholar]
- 11. Ramachandran S, Xie LH, John SA, Subramaniam S, Lal R. A novel role for connexin hemichannel in oxidative stress and smoking-induced cell injury. PLoS One. 2007;2(8):e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Retamal MA, Schalper KA, Shoji KF, Bennett MV, Saez JC. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc Natl Acad Sci U S A. 2007;104(20):8322–8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Vuyst E, Decrock E, De Bock M, et al. Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol Biol Cell. 2007;18(1):34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gomes P, Srinivas SP, Van Driessche W, Vereecke J, Himpens B. ATP release through connexin hemichannels in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2005;46(4):1208–1218. [DOI] [PubMed] [Google Scholar]
- 15. Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312(5775):924–927. [DOI] [PubMed] [Google Scholar]
- 16. Vannucci RC, Vannucci SJ. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev Neurosci. 2005;27(2-4):81–86. [DOI] [PubMed] [Google Scholar]
- 17. Van Zeijl L, Ponsioen B, Giepmans BN, et al. Regulation of connexin43 gap junctional communication by phosphatidylinositol 4,5-bisphosphate. J Cell Biol. 2007;177(5):881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carr AJ, Whitmore D. Imaging of single light-responsive clock cells reveals fluctuating free-running periods. Nat Cell Biol. 2005;7(3):319–321. [DOI] [PubMed] [Google Scholar]
- 19. Nakajima W, Ishida A, Lange MS, et al. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci. 2000;20(21):7994–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis. 2001;8(2):207–219. [DOI] [PubMed] [Google Scholar]
- 21. Derugin N, Wendland M, Muramatsu K, et al. Evolution of brain injury after transient middle cerebral artery occlusion in neonatal rats. Stroke. 2000;31(7):1752–1761. [DOI] [PubMed] [Google Scholar]
- 22. Cooper CD, Solan JL, Dolejsi MK, Lampe PD. Analysis of connexin phosphorylation sites. Methods. 2000;20(2):196–204. [DOI] [PubMed] [Google Scholar]
- 23. Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim et Biophys Acta. 2005;1711(2):154–163. [DOI] [PubMed] [Google Scholar]
- 24. Vikhamar G, Rivedal E, Mollerup S, Sanner T. Role of Cx43 phosphorylation and MAP kinase activation in EGF induced enhancement of cell communication in human kidney epithelial cells. Cell Adhes Commun. 1998;5(6):451–460. [DOI] [PubMed] [Google Scholar]
- 25. Warn-Cramer BJ, Cottrell GT, Burt JM, Lau AF. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J Biol Chem. 1998;273(15):9188–9196. [DOI] [PubMed] [Google Scholar]
- 26. Zvalova D, Cordier J, Mesnil M, Junier MP, Chneiweiss H. p38/SAPK2 controls gap junction closure in astrocytes. Glia. 2004;46(3):323–333. [DOI] [PubMed] [Google Scholar]
- 27. Penrice J, Cady EB, Lorek A, et al. Proton magnetic resonance spectroscopy of the brain in normal preterm and term infants, and early changes after perinatal hypoxia-ischemia. Pediatr Res. 1996;40(1):6–14. [DOI] [PubMed] [Google Scholar]
- 28. Williams CE, Gunn A, Gluckman PD. Time course of intracellular edema and epileptiform activity following prenatal cerebral ischemia in sheep. Stroke. 1991;22(4):516–521. [DOI] [PubMed] [Google Scholar]
- 29. Inder TE, Volpe JJ. Mechanisms of perinatal brain injury. Semin Neonatol. 2000;5(1):3–16. [DOI] [PubMed] [Google Scholar]
- 30. Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4(2):193–205. [DOI] [PubMed] [Google Scholar]
- 31. Wang X, Ma A, Zhu W, et al. The role of connexin 43 and hemichannels correlated with the astrocytic death following ischemia/reperfusion insult. Cell Mol Neurobiol. 2013;33(3):401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83(4):1359–1400. [DOI] [PubMed] [Google Scholar]
- 33. Thimm J, Mechler A, Lin H, Rhee S, Lal R. Calcium-dependent open/closed conformations and interfacial energy maps of reconstituted hemichannels. J Biol Chem. 2005;280(11):10646–10654. [DOI] [PubMed] [Google Scholar]
- 34. Bennett MV, Contreras JE, Bukauskas FF, Sáez JC. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 2003;26(11):610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spray DC, Ye ZC, Ransom BRGlia. Functional connexin “hemichannels”: a critical appraisal. 2006;54(7):758–773. [DOI] [PubMed] [Google Scholar]
- 36. Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23(9):3588–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rana S, Dringen R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci Lett. 2007;415(1):45–48. [DOI] [PubMed] [Google Scholar]
- 38. Kalvelyte A, Imbrasaite A, Bukauskiene A, Verselis VK, Bukauskas FF. Connexins and apoptotic transformation. Biochem Pharmacol. 2003;66(8):1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li F, Sugishita K, Su Z, Ueda I, Barry WH. Activation of connexin-43 hemichannels can elevate [Ca(2+)]i and [Na(+)]i in rabbit ventricular myocytes during metabolic inhibition. J Mol Cell Cardiol. 2001;33(12):2145–2155. [DOI] [PubMed] [Google Scholar]
- 40. Stong BC, Chang Q, Ahmad S, Lin X. A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope. 2006;116(12):2205–2210. [DOI] [PubMed] [Google Scholar]