

Abstract
Background
The survival rate of patients with head and neck squamous cell carcinoma (HNSCC) stands at approximately 50% and this has not improved in decades. This study developed a novel sirtuin-3 (SIRT3) inhibitor (LC-0296) and examined its role in altering HNSCC tumorigenesis.
Materials and Methods
The effect of the SIRT3 inhibitor, LC-0296, on cell survival, proliferation, and apoptosis, and reactive oxygen species levels in HNSCC cells were studied.
Results
LC-0296 reduces cell proliferation and promotes apoptosis of HNSCC cells but not of normal human oral keratinocytes. This inhibitory effect is mediated, in part, via modulation of reactive oxygen species levels. Additionally, LC-0296 works synergistically to increase the sensitivity of HNSCC cells to radiation and cisplatin treatment.
Conclusion
Development of novel SIRT3 inhibitors, such as LC-0296, might enable the development of new targeted therapies to treat and improve the survival rate of patients with head and neck cancer.
Keywords: Sirtuins, sirtuin-3, SIRT3, HNSCC, oral cancer, ROS, sirtuin inhibitor
Head and neck squamous cell carcinoma (HNSCC) originates from the oral and nasal cavities, sinuses, lips, salivary glands, throat, or larynx. Oral squamous cell carcinoma (OSCC) represents the majority of HNSCCs and it is the ninth most common cancer worldwide (1). In certain countries, including India and other south-central Asian countries, oral cancer is among the most commonly occurring cancer. Although other types of cancer, such as breast, prostate and colone, are more prevalent than oral cancer, the 5-year survival rate of patients with this disease is poor, averaging in 50% (2), despite the advancements in therapeutic approaches to treating this devastating disease (3). This underscores the urgent need to explore new areas of research and develop new therapeutic drugs and approaches that can help improve the survival rate of patients with head and neck cancer.
Sirtuins (SIRT1-7) have emerged as important modulators of tumorigenic processes. Sirtuins control cancer cell proliferation and survival, cell-cycle progression, apoptosis, angiogenesis, and metabolism (4–9). Therefore, sirtuins have been implicated as novel potential therapeutic targets to treat cancer (10–12). However, the role of several sirtuins, specifically SIRT1 and SIRT3, in cancer tumorigenesis is controversial (13–18). Thus, expanding the study of sirtuins in this new area of research will help advance the field and help us better understand the mechanisms by which sirtuins can regulate different cancer processes.
We were the first to demonstrate a novel role for SIRT3 in oral cancer tumorigenesis both in vitro and in vivo (19). We reported that out of all seven of the sirtuin family members, SIRT3 is overexpressed in OSCC compared to normal oral tissues, and SIRT3 down-regulation inhibits OSCC cell growth and proliferation in vitro, and reduces tumor burden in vivo (19). Furthermore, SIRT3 down-regulation enhances the sensitivity of radio- and chemoresistant OSCC cells to both radiation and chemotherapeutic drugs. Thus, targeting SIRT3 to induce cytotoxicity to HNSCC cells in patients with high SIRT3-expressing tumors or radio- or chemoresistant tumors may be advantageous, since lower doses of conventional treatment may be required. In this case, SIRT3 would serve as an adjuvant target. In additional studies, we found that SIRT3 and receptor-interacting protein (RIP), a pro-apoptotic protein, are oppositely expressed in human OSCC specimens. Those studies further found that OSCC cells escape anoikis, apoptotic cell death triggered by loss of extracellular matrix contacts, by forming multicellular aggregates or oraspheres to maintain their survival (20). Thus, OSCC oraspheres become anoikis-resistant, a condition defined by a higher SIRT3 and low RIP expression. These anoikis-resistant OSCC cells also induce an increased tumor burden and incidence in mice unlike their adherent OSCC cell counterparts. Furthermore, stable suppression of SIRT3 inhibits anoikis resistance and reduces tumor incidence (20). Lastly, since Sirt3−/−mice are surprisingly healthy (21), targeting SIRT3 in oral cancer may help avoid the toxic side-effects of other treatment approaches. Indeed, our data support this premise, since normal oral keratinocytes exhibit low levels of SIRT3 expression, however, adherent HNSCC cells exhibit higher levels of SIRT3, and anoikis-resistant HNSCC oraspheres in suspension exhibit the highest levels of SIRT3 expression (20). These findings support the concept that SIRT3 promotes anoikis resistance in vivo and enhances tumorigenesis, thus SIRT3 represents a promising therapeutic target for HNSCC.
In this regard, we believe that discovering new drugs that specifically target SIRT3 could enhance the treatment of HNSCC and potentially improve the survival rate of patients. In the present study, we developed a novel sirtuin-3 (SIRT3) inhibitor (LC-0296) and examined its role in altering HNSCC tumorigenesis.
Materials and Methods
Chemical synthesis of SIRT3 inhibitor, LC-0296
The synthesis of compound LC-0296 was straightforward and is depicted in Figure 1A. Commercially available 4-nitro-1H-indole (compound 1) was alkylated to give compound 3, whose nitro group was reduced to the corresponding amine in compound 4 in excellent yields. The methyl ester group in compound 4 was converted into a primary amide with methanolic ammonia. The resulting compound 5 was coupled with L-glutamate derived N-carbobenzyloxy-L-glutamic acid 1-methyl ester (Z-Glu-OMe) in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and 1-hydroxybenzotriazole (HOBt) to produce LC-0296 (compound 6) in good yields (Figure 1A). More detailed information about the chemical synthesis is described below.
Figure 1.
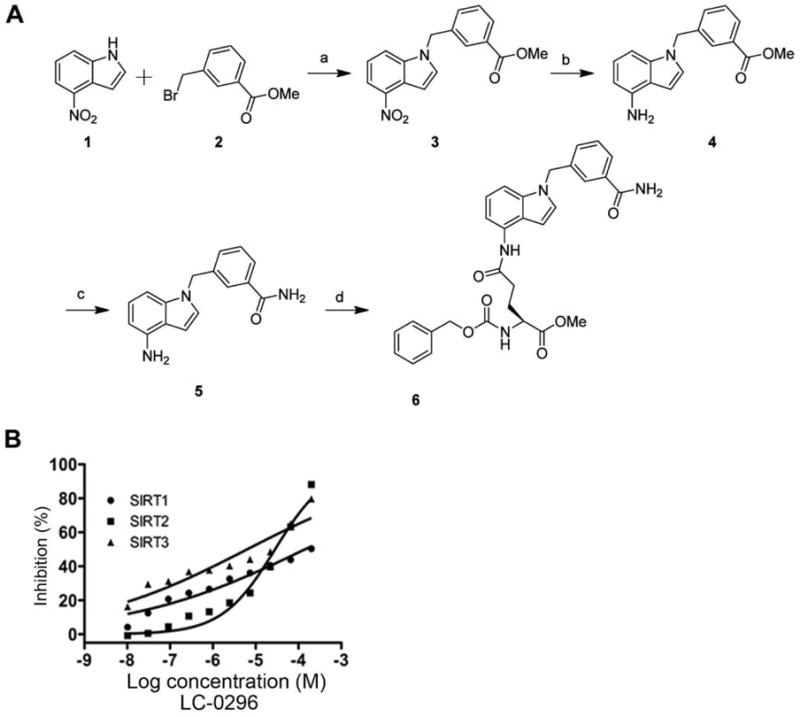
A: Schematic chemical synthesis of the sirtuin-3 (SIRT3) inhibitor (LC-0296). Commercially available 4-nitro-1H-indole (1) was alkylated to give compound 3, whose nitro group was reduced to the corresponding amine in compound 4 in excellent yields. The methyl ester group in compound 4 was converted into a primary amide with methanolic ammonia. The resulting compound 5 was coupled with L-glutamate derived Z-Glu-OMe in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and 1-hydroxybenzotriazole (HOBt) to yield LC-0296 (6) in good yields. Reaction conditions were: (a) NaH, DMF, yield 68%; (b) NiCl2·6H2O, NaBH4, MeOH, yield 89%; (c) methanolic NH3, CaCl2, 70°C, yield 85%; (d) Z-Glu-OMe, EDC, HOBt, NMM, CH2Cl2, yield 60%. B: Effect of the sirtuin-3 (SIRT3) inhibitor, LC-0296 on sirtuin-1 (SIRT1), sirtuin-2 (SIRT2) and sirtuin-3 (SIRT3) in an in vitro enzymatic assay.
All commercial reagents (Sigma-Aldrich, St. Louis, MO, USA) were used as provided unless otherwise indicated. An anhydrous solvent dispensing system (J. C. Meyer) using two packed columns of neutral alumina was used for drying tetrahydrofuran (THF), Et2O, and CH2Cl2, while two packed columns of molecular sieves were used to dry dimethylformamide (DMF). Solvents were dispensed under argon. Flash chromatography was performed with Ultra Pure silica gel (Silicycle, Quebec city, Quebec, Canada) with the indicated solvent system. Nuclear magnetic resonance spectra were recorded on a Varian 600 MHz (Varian Inc., Lexington, MA, USA) with Me4Si or signals from residual solvent as the internal standard for 1H. Chemical shifts are reported in ppm, and signals are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad singlet (bs), and double doublet (dd). Values given for coupling constants are first order. High-resolution mass spectra were recorded on an Agilent TOF II time-of-flight mass spectrometry instrument equipped with either an electrospray ionization or atmospheric pressure chemical ionization interface.
Methyl 3-((4-nitro-1H-indol-1-yl)methyl)benzoate (3)
To a solution of 4-nitro-1H-indole (1, 2.43 g, 15.0 mmol) in dry DMF (50 ml) was added NaH (60% dispersion, 0.72 g, 18 mmol) in several portions. The reaction mixture turned dark red. After 40 min, a solution of methyl 3-(bromomethyl)benzoate (2; 3.72 g, 15.8 mmol) in dry DMF (10 ml) was added via a syringe. After the reaction mixture was stirred at room temperature for 12 h, it was concentrated and the residue was treated with ethyl acetate (200 ml) and saturated NH4Cl solution (150 ml). The organic phase was washed with brine, dried with Na2SO4 and concentrated. The desired product was isolated by chromatography on silica gel using ethyl acetate/hexanes (1:5 to 1:1) as eluent to give compound 3 as a yellow solid (3.71 g, 68%). 1H nuclear magnetic resonance (NMR) (DMSO-d6, 600 MHz) δ 8.08 (d, J=8.4 Hz, 1H), 8.03 (d, J=7.2 Hz, 1H), 7.98 (d, J=2.4 Hz, 1H), 7.88–7.83 (pseudo s, 2H), 7.49–7.62 (m, 2H), 7.34 (dd, J=8.4, 8.4 Hz, 1H), 7.12 (d, J=2.4 Hz, 1H), 5.68 (s, 2H), 3.81 (s, 3H). HRMS (ESI+) calcd for C17H15N2O4 (M+H)+ 311.1032, found 311.1027.
Methyl 3-((4-amino-1H-indol-1-yl)methyl)benzoate (4)
NiCl2·6H2O (943 mg, 4.00 mmol) was added to a solution of compound 3 (620 mg, 2.00 mmol) in anhydrous methanol (150 ml), and the mixture was submerged into an ice bath. NaBH4 (302 mg, 8.00 mmol) was added in several portions with lots of gas formed and the reaction mixture turned black. After 1 h, the reaction was quenched with saturated NH4Cl solution and methanol was removed in vacuo. The residue was treated with ethyl acetate (150 ml) and water (100 ml). The organic phase was washed with brine, dried with Na2SO4 and concentrated. The desired product was isolated by chromatography on silica gel using ethyl acetate/hexane (1:5 to 1:1) as eluent to give compound 4 (500 mg, 89%). 1H NMR (DMSO-d6, 600 MHz) δ 7.82 (d, J=7.8 Hz, 1H), 7.76 (s, 1H), 7.47–7.40 (m, 2H), 7.25 (d, J=2.4 Hz, 1H), 6.77 (dd, J=7.8, 7.8 Hz, 1H), 6.60–6.55 (m, 2H), 6.16 (d, J=7.8 Hz, 1H), 5.38 (s, 2H), 5.22 (s, 2H), 3.81 (s, 3H). HRMS (ESI+) calcd for C17H17N2O2 (M+H)+ 281.1290, found 281.1281.
3-((4-Amino-1H-indol-1-yl)methyl)benzamide (5)
To a reaction vessel equipped with a magnetic stir bar was added compound 4 (500 mg, 1.79 mmol), CaCl2 (198 mg, 1.79 mmol), and 7 N NH3 in MeOH (5 ml, about 35 mmol). The reaction vessel was sealed and heated at 70°C for 24 h. The reaction mixture was concentrated and purified by chromatography on silica gel using MeOH/CH2Cl2 (0% to 10%) as eluent to give compound 4 (404 mg, 85%). 1H NMR (DMSO-d6, 600 MHz) δ 7.93 (s, 1H), 7.75 (s, 1H), 7.73 (d, J=7.8 Hz, 1H), 7.37–7.32 (m, 2H), 7.26–7.22 (m, 2H), 6.77 (dd, J=8.1, 8.1 Hz, 1H), 6.60–6.55 (m, 2H), 6.15 (d, J=7.8 Hz, 1H), 5.32 (s, 2H), 5.19 (s, 2H). HRMS (ESI+) calcd for C16H16N3O (M+H)+ 266.1293, found 266.1283.
(S)-Methyl 2-(((benzyloxy)carbonyl)amino)-5-((1-(3-carbamo-ylbenzyl)-1H-indol-4-yl)amino)-5-oxopentanoate (6; LC-0296)
A solution of Z-Glu-OMe (58 mg, 0.20 mmol) in CH2Cl2 was cooled with an ice bath, and EDC (38 mg, 0.20 mmol), HOBt (27 mg, 0.20 mmol), N-methylmorpholine (57 mg, 0.57 mmol) and compound 5 (50 mg, 0.20 mmol) were added. The mixture was stirred at room temperature overnight and the organic solvent was evaporated. The resulting crude product was purified by chromatography to give product compound 6, SIRT3 inhibitor LC-0296 (61 mg, 60%) as a pale solid. 1H NMR (DMSO-d6, 600 MHz) δ 9.59 (s, 1H), 7.94 (s, 1H), 7.80 (d, J=7.2 Hz, 1H), 7.78 (s, 1H), 7.74 (d, J=7.8 Hz, 1H), 7.61 (d, J=7.2 Hz, 1H), 7.45 (d, J=3.0 Hz, 1H), 7.40–7.29 (m, 7H), 7.26 (d, J=8.4 Hz, 1H), 7.17 (d, J=8.4 Hz, 1H), 7.01 (dd, J=7.8, 7.8 Hz, 1H), 6.78 (s, 1H), 5.43 (s, 2H), 5.04 (s, 2H), 4.14 (dd, J=13.2, 8.4 Hz, 1H), 3.66 (s, 3H), 2.55 (t, J=7.8 Hz, 2H), 2.14–2.08 (m, 1H), 1.92–1.84 (m, 1H). HRMS (ESI+) calcd for C30H29N4O6 541.2087 (M-H)−, found 541.2087.
Biochemical in vitro assays to determine the specificity of LC-0296 S1RT3 inhibitor
The biochemical assays for human SIRT1-3 were performed by the Reaction Biology Incorporation (RBC) (Malvern, PA, USA, http://www.reactionbiology.com) as described previously (22).
Cell lines and culture
Human HNSCC cell lines, UM-SCC-1 and UM-SCC-17B, originated from the floor of the mouth and larynx, respectively, and were provided by Tom Carey (University of Michigan) (23). HNSCC cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin. Primary normal human oral keratinocytes (Cat# 2610; ScienCell, Carlsbad, CA, USA) were maintained in human oral keratinocyte medium (#2611; ScienCell).
Cell viability and colony-formation assays
To determine the effect of the SIRT3 inhibitor LC-0296 on cell viability we used the QUANT Cell Proliferation Assay Kit according to the manufacturer’s instructions (Invitrogen). In order to determine the synergistic effect of LC-0296, cells were either treated with LC-0296 alone or combined with ionizing radiation or cisplatin. For colony-formation assays, HNSCC cells and normal human oral keratinocytes were treated with and without the inhibitor and cultured for one week. Colonies were fixed with methanol then stained with 0.5% crystal violet. Colonies containing more than 50 cells were counted.
Apoptosis staining and cell death detection assay-enzyme-linked immunosorbent assay (ELISA)
To measure apoptosis in vitro, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) and a DNA fragmentation enzyme-linked immunosorbent assay (ELISA) was used according to manufacturer’s instructions (Roche Diagnostics, Indianapolis, IN, USA).
Combination index (CI)
To determine synergistic or additive effects of the drug combinations (LC-0296 plus ionizing radiation or cisplatin), a CI was used following the procedure developed by Fischel et al. (24), with the equation adapted from the method developed previously by Chou and Talalay (25).
According to Fischel et al. (24), if: R<0.8, then the association is considered to be synergistic; 0.8<R<1.2, then the association is considered to be additive; and if R>1.2, then the association is considered to be antagonistic.
Determination of S1RT3 de-acetylation activity in HNSCC cells
To determine the effect of LC-0296 on SIRT3 deacetylation activity within a cellular context, HNSCC cells were treated with LC-0296 (50 μM) or control (DMSO) for 24 h. Corresponding lysates were then immunoprecipitated with specific antibodies to the SIRT3 deacetylation target, NADH dehydrogenase 1 alpha sub-complex subunit 9 (NDUFA9; mouse monoclonal antibody #Sc-58392; Santa Cruz Biotechnology, St. Louis, MO, USA) and glutamate dehydrogenase (GDH; goat polyclonal #Sc-160383; from Santa Cruz Biotechnology), and immunoblotted with an antibody detecting acetylated lysine residues (#944 1;Cell Signaling, Danvers, MA, USA).
Mitochondrial isolation
Mitochondria were isolated from HNSCC cells using a mitochondrial/cytosol fractionation kit (Cat no: MIT1000; Millipore) according to the manufacturer’s instructions. Briefly, cells were resuspended in an isotonic mitochondrial buffer containing a protease inhibitor cocktail and homogenized with a Dounce homogenizer (50 strokes) on ice. The lysate was centrifuged at 600 × g for 10 min at 4°C to remove the nuclei and unbroken cells. The supernatant was centrifuged at 10000 × g for 30 min at 4°C. The resulting pellet was collected as the enriched mitochondrial fraction and resuspended in mitochondrial lysis buffer containing a protease inhibitor cocktail. Mitochondrial purity was evaluated by immunoblotting for the mitochondrial and cytosolic protein markers VDAC and GAPDH, respectively.
Immunoblot analysis
Western blotting was performed as previously described (19) using antibodies against SIRT3 (#2627) and acetylated-lysine (AC-K) (#9441) from Cell Signaling; voltage-dependent anion channel (VDAC) (SC-32063) from Santa Cruz Biotechnology; and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (MAB374) Millipore, Billerica, MA, USA. The NDUFA9 mouse monoclonal antibody (#ab55521) was from Abcam, Cambridge, MA, USA. To demonstrate equal protein loading, membranes were stripped and reprobed with an anti-β-actin antibody (sc-1615; Santa Cruz Biotechnology).
Reactive oxygen species (ROS) detection assay
To measure the intracellular ROS levels, the fluorogenic marker for ROS, carboxy-2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and N-acetyl-cysteine (NAC), a scavenger for ROS were used according to the manufacturer’s instructions (Invitrogen).
Statistical analysis
Values are expressed as means±SD. Comparisons between groups were determined by one-way analysis of variance (ANOVA) followed by Tukey-HSD multiple-comparison test. Statistical significance was defined as p≤0.05. All experiments were repeated three times and at least in triplicates when applicable.
Results
Defining the chemical structure and enzymatic selectivity of the SIRT3 inhibitor (LC-0296) in vitro
The schematic diagram for the chemical synthesis of LC-0296 and the chemical structure are shown in Figure 1A. The SIRT3 inhibitor, LC-0296, possesses a selective inhibitory effect, about ~20- and 10-fold greater inhibition of SIRT3 enzymatic activity compared to SIRT1 and SIRT2 (Figure 1B, Table I).
Table I.
Sirtuins 1–3 (SIRT1-3) inhibition in vitro by LC-0296.
| Inhibitor | MW | IC50 (μM)
|
||
|---|---|---|---|---|
| SIRT1 | SIRT2 | SIRT3 | ||
| LC-0296 | 542.58 | 67 | 33 | 3.6 |
Inhibitory concentration 50% (IC50).
LC-0296 inhibits HNSCC cell survival without affecting normal human oral keratinocytes
To assess the effect of the SIRT3 inhibitor LC-0296 on HNSCC cell survival, we first performed dose- and time-dependent experiments using a wide range of doses (0.001, 0.01, 0.1, 1, and 10 μM, at 24, 48, and 72 h), and compared these data to those for normal human oral keratinocytes (Figure 2). Since we did not find significant inhibitory effects on HNSCC cell survival under these doses, we tested higher doses of LC-0296. At doses in the micromolar range, LC-0296 had significant inhibitory and dose-dependent effects on HNSCC cell viability, without affecting normal human oral keratinocytes (Figure 3B). A representative image of HNSCC cells and normal human oral keratinocytes treated with LC-0296 (75 μM) is presented in Figure 3C. Figure 3C further demonstrates the absence of morphological changes in normal human oral keratinocytes compared to HNSCC cells treated with LC-0296 (75 μM).
Figure 2.
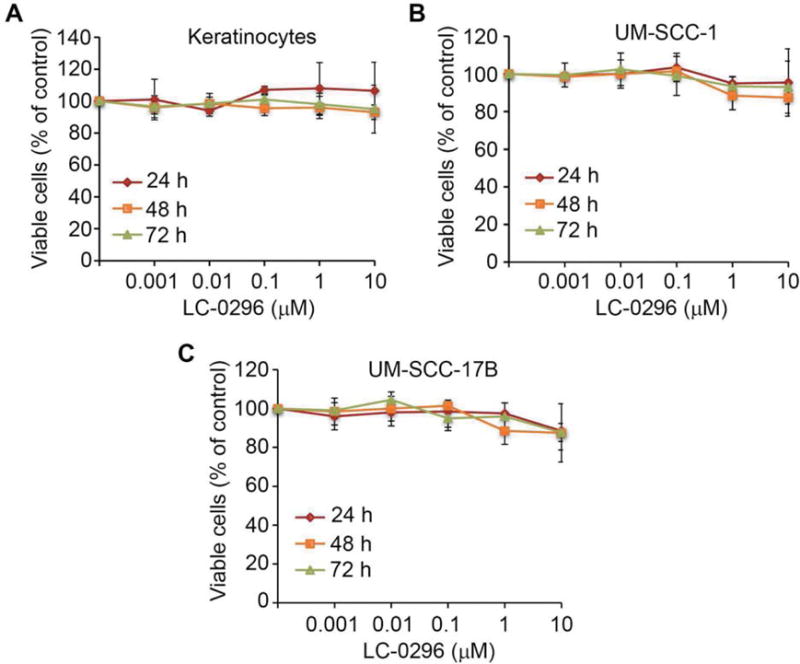
The effect of the sirtuin-3 (SIRT3) inhibitor, LC-0296, on head and neck squamous cell carcinoma cells and keratinocytes using low doses. Normal human oral keratinocytes (A), UM-SCC-1 (B), and UM-SCC-17B (C) cells were seeded in 96-well plates at 5×103 cells/well, then treated with dimethyl sulfoxide (control) or LC-0296, as indicated for 24, 48, and 72 h. Cell viability was then determined by the QUANT Cell Proliferation Assay Kit. The graphs illustrate the percentage of viable cells following treatment.
Figure 3.
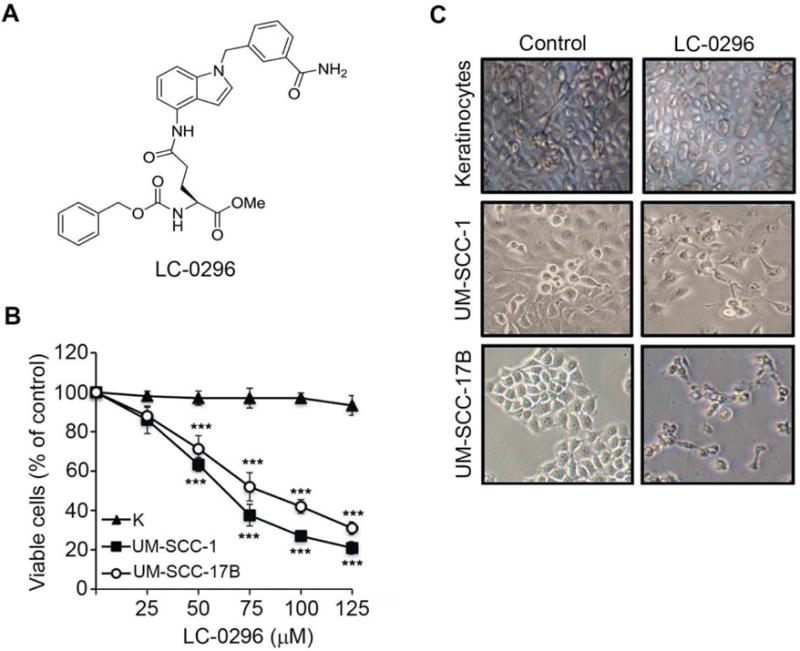
Sirtuin-3 (SIRT3) inhibitor, LC-0296, inhibits head and neck squamous cell carcinoma (HNSCC) cell survival without affecting normal human oral keratinocytes. A: The chemical structure of the SIRT3 inhibitor LC-0296 is shown. B: HNSCC cells (UM-SCC-1 and UM-SCC-17B) and normal human oral keratinocytes (K) were seeded in 96-well plates at 5×103 cells/well then treated with LC-0296 as indicated for 24 h. Cell viability was determined by the QUANT Cell Proliferation Assay Kit (Invitrogen). C: Phase-contrast images show the morphology of HNSCC cells (UM-SCC-1 and UM-SCC-17B) and normal human oral keratinocytes after treatment with vehicle dimethyl sulfoxide (control) or LC-0296 (75 μΜ) for 24 h. ***p≤0.001 for LC-0296 vs. control.
LC-0296 inhibits cell growth and proliferation and promotes apoptosis of HNSCC cells
To further evaluate the effect of the SIRT3 inhibitor LC-0296 on HNSCC cell growth and proliferation, colony-formation assays were performed using different doses of LC-0296. These data demonstrate that in addition to inhibiting cell viability, LC-0296 also blocked HNSCC cell colony formation in a dose-dependent manner (Figure 4A). Notably, the 75 μM dose had an even greater effect on colony formation ability than on cell viability as shown in Figure 3B. This may be explained by the fact that single-cell colony growth assays are devoid of growth signals from neighboring cells and cell–cell contact, thereby promoting a less robust survival environment. In addition, LC-0296 also induced apoptosis of HNSCC cells in a dose-dependent manner (Figure 4C and E). Importantly, LC-0296, did not exert significant effects on cell growth and proliferation or apoptosis of normal human oral keratinocytes at any of the inhibitor doses (Figure 4B, D and F).
Figure 4.
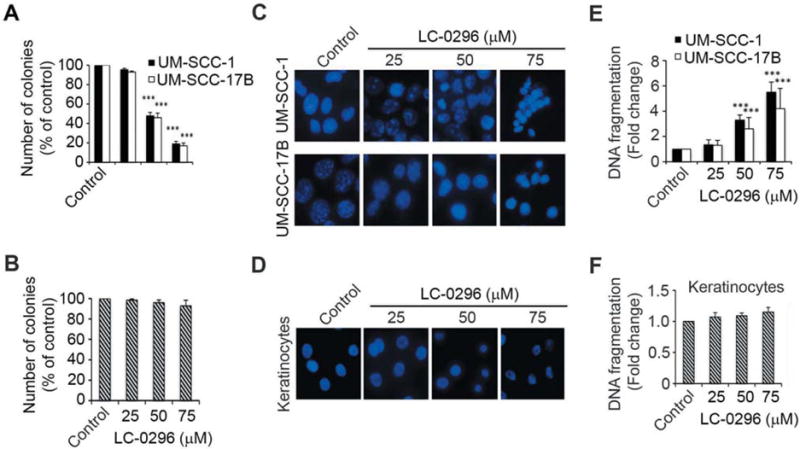
Sirtuin-3 (SIRT3) inhibitor LC-0296 inhibits cell growth and proliferation and promotes apoptosis of head and neck squamous cell carcinoma cells. A and B: The histograms show the quantification of the colony-forming assays for HNSCC cells (UM-SCC-1 and UM-SCC-17B) and normal human oral keratinocytes treated with LC-0296 or the vehicle dimethyl sulfoxide (control). The number of colonies are presented as the percentage of colonies obtained relative to controls. C and D: Representative images showing HNSCC cells (UM-SCC-1 and UM-SCC-17B) and normal human oral keratinocytes stained with 4′,6-diamidino-2-phenylindole after treatment with LC-0296 or the vehicle DMSO (control) for 24 h. Culture media were collected and centrifuged to collect floating cells, which were added back to their respective wells prior to assessment. E and F: The graphs show the fold change in DNA fragmentation for HNSCC cells and normal human oral keratinocytes after treatment with LC-0296 or vehicle DMSO (control) for 24 h. ***p≤0.001for LC-0296 vs. control.
SIRT3 inhibitor, LC-0296, enhances the sensitivity of HNSCC cells to both radiation and chemotherapeutic drugs
Thus far, our data indicate that LC-0296 can be used as a single agent to inhibit HNSCC cell growth and survival, and promote apoptosis-mediated cell death. However, we further asked whether LC-0296 could function as an effective adjuvant treatment for cases that do not respond well to either radiation or chemotherapeutic approaches. Both UM-SCC-1 and UM-SCC-17B cells are very aggressive HNSCC cell lines, which are highly resistant to radiation (26, 27), and the former is also resistant to cisplatin (data not shown). Interestingly, LC-0296 worked synergistically to enhance the sensitivity of HNSCC cell lines to both treatments compared to untreated controls or cells treated with radiation or cisplatin alone (Figure 5A and B). The synergistic effect was assessed using the CI.
Figure 5.
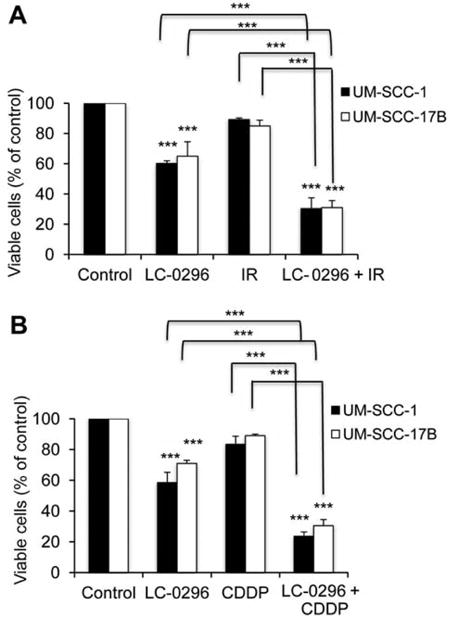
Sirtuin-3 (SIRT3) inhibitor LC-0296 enhances the sensitivity of head and neck squamous cell carcinoma cells to both radiation and chemotherapeutic drug. The graphs shows the percentage of viable HNSCC cells after treatment with LC-0296 (50 μΜ), with or without ionizing radiation (IR; 2.5 Gy) (A) or cisplatin (CDDP; 20 μΜ) (B) for 24 h, and assessment of cytotoxicity using the QUANT Cell Proliferation Assay Kit. The control was treated with dimethyl sulfoxide diluting agent, for 24 h. ***p≤0.001 for LC-0296 vs. control, or comparison indicated.
LC-0296 inhibits de-acetylation by SIRT3 and increases mitochondrial protein acetylation in HNSCC cells
Previously, we showed that down-regulation of SIRT3 significantly limited OSCC cell survival and tumorigenesis both in vitro and in vivo (19). Although our novel SIRT3 chemical inhibitor selectively inhibits enzymatic activity of SIRT3 in vitro (Figure 1B, Table I), it was not known whether LC-0296 functions by inhibiting de-acetylation by SIRT3 in a cellular context. Therefore, we assayed deacetylation by SIRT3 in cell lysates from HNSCC cell lines treated with 50 μM LC-0296 or vehicle control (DMSO). To demonstrate that LC-0296 specifically targets de-acetylation by SIRT3 in the mitochondria, mitochondrial fractions were assessed for global mitochondrial protein acetylation. Our data show that LC-0296 blocks de-acetylation by SIRT3 within the mitochondria compared to vehicle control (DMSO) (Figure 6A and B). Furthermore, LC-0296 specifically inhibited de-acetylation by SIRT3, thus preventing deacetylation of SIRT3 target proteins, such as NDUFA9 and GDH in the mitochondria (Figure 6C). In addition, we performed western blot analyses to assess the effect of LC-0296 on SIRT3 protein levels in HNSCC cells. Interestingly, our results showed that LC-0296 inhibits the de-acetylation function of SIRT3 in cells without affecting SIRT3 protein levels (Figure 6D).
Figure 6.
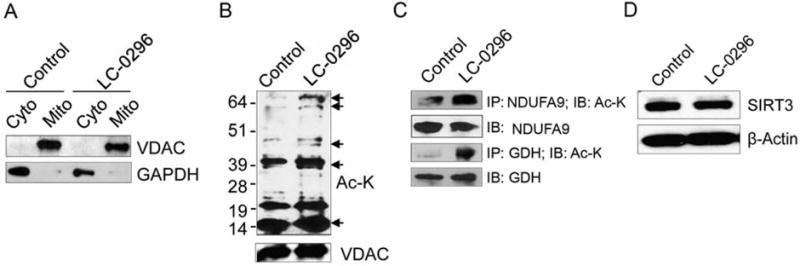
Sirtuin-3 (SIRT3) inhibitor LC-0296 inhibits deacetylation by SIRT3 and increases global mitochondrial protein acetylation in head and neck squamous cell carcinoma cells. A: Immunoblots reveal the voltage-dependent anion channel and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) protein levels present within mitochondrial (mito) and cytoplasmic (cyto) fractions isolated from HNSCC cells (UM-SCC-17B) treated with control dimethyl sulfoxide (DMSO) or LC-0296 (50 μM) for 24 h. B: Immunoblots reveal mitochondrial protein acetylation in UMSCC-17B cells after treatment with DMSO (vehicle; control) or LC-0296 (50 μM) for 24 h. VDAC served as a mitochondrial loading control. C: Immunoblots show the levels of acetylated mitochondrial protein NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 9 (NDUFA9) and glutamate dehydrogenase (GDH) after treatment of HNSCC cells (UM-SCC-17B) with DMSO (control) or LC-0296 (50 μM) for 24 h, then lysates were immunoprecipitated with NDUFA9 or GDH antibodies and immunoblotted with an acetylated-lysine (Ac-K) antibody. D: Immunoblots show SIRT3 expression levels in HNSCC cells after treatment with vehicle DMSO (control) or LC-0296 (50 μM). β-Actin served as the loading control. IP: immunoprecipitation; IB: immunoblotting.
LC-0296 inhibits cell survival and enhances apoptosis via modulating ROS levels in HNSCC cells
Several studies have found that normal cells have lower ROS levels compared to cancer cells (28, 29), and it is well known that SIRT3 plays a key regulatory role in controlling ROS levels in mitochondria (5, 30). Therefore, we hypothesized that the SIRT3 inhibitor LC-0296 blocks de-acetylation by SIRT3, which affects ROS levels in these cells, and thereby negatively affects their survival and promotes their apoptosis. Furthermore, since ROS levels are higher in cancer cells compared to normal cells, this may explain why LC-0296 was more effective on HNSCC cells compared to normal human oral keratinocytes. To test this hypothesis, we evaluated ROS levels in HNSCC cells compared to normal human oral keratinocytes. In agreement with previous studies in other cancer cells (28, 29), ROS levels in HNSCC cells were significantly higher than those in normal human oral keratinocytes (Figure 7A). Next, we measured ROS levels in HNSCC cells following LC-0296 treatment. Interestingly, we found a significant increase in ROS levels in LC-0296-treated HNSCC cells compared to untreated vehicle controls (Figure 7B), indicating that SIRT3 works by suppressing ROS levels in HNSCC cells. In addition, to further confirm that LC-0296 works in HNSCC cells by modulating ROS levels, we next used NAC, a scavenger for ROS in this context. Indeed, addition of NAC abrogated the effect of LC-0296 on HNSCC cells, thus reducing ROS levels, increasing cell viability and reducing apoptosis (Figure 7C and D). These data show that the SIRT3 inhibitor LC-0296 has a specific inhibitory effect on HNSCC cell viability, and enhances apoptosis, at least in part, by modulating ROS levels.
Figure 7.
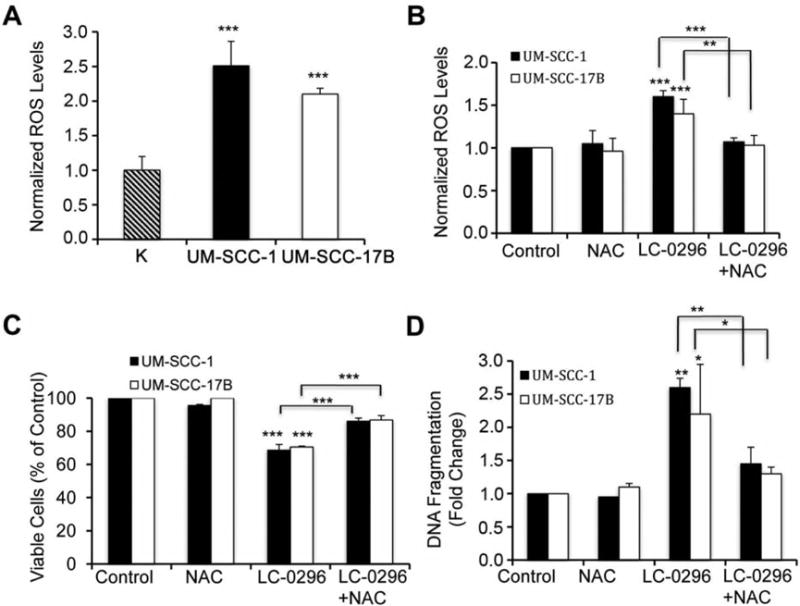
Sirtuin-3 (SIRT3) inhibitor LC-0296 inhibits cell survival and enhances apoptosis via modulating reactive oxygen species (ROS) levels in head and neck squamous cell carcinoma (HNSCC) cells. A: Normal human oral keratinocytes (K), and HNSCC cells (UM-SCC-1 and UM-SCC17B) were seeded in 96 well-plates at 5×103 cells/well, then ROS levels were measured after 24 h using the fluorogenic marker carboxy-2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA). The graph shows the levels of ROS in these HNSCC cells normalized to the levels of ROS in the keratinocytes. B: HNSCC cells were pre-treated with N-acetyl-cysteine (NAC, 20 mM), a scavenger for ROS, or dimethyl sulfoxide (DMSO) (control) for 2 h, then the pre-treatments were removed, and cells were subsequently treated with either DMSO (control) or LC-0296 (50 μΜ) for 10 h, and finally stained with carboxy-H2DCFDA for ROS assays. The graph shows the levels of ROS in these HNSCC cells, that were normalized to the levels of ROS in control cells. C: HNSCC cells were treated as described for panel B for 12 h, and then cell viability was determined by the QUANT Cell Proliferation Assay. The graph shows the percentage of viable treated HNSCC cells normalized to control cells. D: HNSCC cells were treated as described for panel B for 12 h, and then apoptosis was determined using DNA fragmentation enzyme-linked immunosorbent assay. The graph shows the fold change in DNA fragmentation for treated HNSCC cells normalized to that of control cells. *p≤0.05, **p≤0.01 and ***p≤0.001 vs. control, or comparison indicated.
Discussion
The poor survival rates for patients with head and neck cancer urgently requires for development of new areas of research that might identify new strategies and approaches for drug development or the discovery of new targets or markers that could aid in the early diagnosis and treatment of such patients.
The study of sirtuins is an exciting area that seems to hold great promise toward enhancing our understanding of and aiding in the development of treatments for age-related diseases such as diabetes, neurodegenerative disorders, heart disease, and cancer (6, 31). However, although this field has been extensively studied for over a decade, there is still a great deal to be learned on sirtuin biology. One such area is the controversy over the role of several sirtuins in cancer biology. SIRT1 and SIRT3 are at the center of this controversy (13–18). In agreement with our aforementioned studies, others also found that SIRT3 is overexpressed in esophageal squamous cell carcinoma and colonic cancer, and its high expression is correlated with a poor overall survival and prognosis (32–35).
In the present study, we report that a novel SIRT3 inhibitor, LC-0296, has enhanced selectivity toward inhibition of de-acetylation by SIRT3. Importantly, LC-0296 shows specificity toward retarding HNSCC cell survival and enhancing apoptosis, without affecting normal human oral keratinocytes. Furthermore, this inhibitor functioned both as a single agent and in combination with either radiation or cisplatin treatment to reduce HNSCC cell viability, especially in cell lines that were derived from patients with resistance to these treatments. These attributes favor the development of this type of SIRT3 inhibitor as a potential future HNSCC therapeutic.
The role of ROS in carcinogenesis/tumorigenesis is well-documented (36,37). ROS are responsible for normal cell transformation, thereby promoting tumorigenesis (36, 37). However, it is important to keep in mind that normal cells still need low levels of ROS for physiological functions. ROS can modulate both cell survival and apoptotic pathways (38, 39). Thus, the balance between ROS production and anti-oxidants seems to be a key factor in controlling normal cellular processes and abnormal cellular transformation (40, 41). SIRT3 is a key regulatory protein in mitochondria, maintaining mitochondrial integrity and protecting them from increased ROS levels. Thus, SIRT3 may respond to ROS-mediated cell transformation to cancer via deacetylation and activation of mitochondrial antioxidants, such as manganese superoxide dismutase (MnSOD) and SOD2 (42–44). However, ROS levels are lower in normal cells in general compared to cancer cells (28,29). Thus, it seems that following cellular transformation, cancer cells adapt to increased ROS levels as part of their abnormal genomic dysregulation. Interestingly, ROS levels seem to be a sensor for cellular survival and proliferation, and responsible for the increased aggressive phenotype and resistance to conventional cancer treatments (37, 40, 41,45). Therefore, because SIRT3 is overexpressed in HNSCC cells (19), for a reason that is yet unknown, and as part of the abnormal genomic dysregulation in these cells, we believe that SIRT3 may, in part, be responsible for controlling the balance of ROS levels in these cells. SIRT3 may maintain ROS levels at a threshold that promotes cancer cell survival, thereby promoting a more aggressive phenotype that resists conventional cancer treatments. Given these ideas, we investigated whether LC-0296 works by modulating ROS levels in HNSCC cells. Interestingly, because ROS levels are higher in HNSCC cells compared to normal keratinocytes, LC-0296 seems to alter the balance of ROS in cancer cells toward compromising cell survival and enhancing apoptosis. This may also explain, at least in part, the increased sensitivity of radiation- and cisplatin-resistant HNSCC cells to this compound, especially, when used in combination treatments. Taking advantage of increased ROS levels in cancer cells is one strategy for achieving more potent and selective effects of chemotherapeutic drugs. There exist several ROS-based chemical inhibitors that have been developed and tested in clinical trials with promising results (46, 47).
Because of the growing importance of sirtuins in cancer tumorigenesis, numerous studies have developed activators and inhibitors for this family of proteins (11, 12, 48). Unfortunately, lack of selectivity of these compounds is still one of the major drawbacks hindering the advancement of these compounds into clinical trials. Most of these inhibitors possess greater specificity toward SIRT1 and SIRT2 (49). The lack of specific SIRT3 inhibitors, and the important roles of SIRT3 in controlling the hallmarks of cancer (9), underscores the importance of developing such compounds (50–53). One SIRT3 inhibitor that has been introduced is compound 2, a bioisosteric analog of nicotinamide. The inhibitory concentration by 50% (IC50) of this compound was reported to be 38±5 μM. Its selectivity for SIRT3 over SIRT1 and SIRT2 was reported to be approximately 6-fold greater. At a dose of 100 μM, compound 2 reduced ATP levels and increased superoxide production in human melanoma cells compared to controls (54). In addition, treatment of HeLa cells with 1 mM of compound 2 led to increased acetylation of mitochondrial proteins. In comparison to compound 2, LC-0296 has an IC50 of 3.6 μM and its selectivity for SIRT3 over SIRT1 and SIRT2 is 18.6- and 9.2-fold greater, respectively. When tested for biological efficacy here, LC-0296 inhibited cell proliferation and colony formation and increased DNA fragmentation dose-dependently in HNSCC cells. Effective biological doses started at 25 μM. Furthermore, LC-0296 sensitized HNSCC cells to cisplatin and radiation treatment when tested at a 50 μM dose. Lastly, LC-0296 treatment promoted global acetylation of mitochondrial proteins when tested at a 50 μM dose. These processes were mediated, in part, by regulation of ROS levels. Thus, LC-0296 appears to be a more potent inhibitor with greater selectivity than compound 2, and the biological efficacy of LC-0296 supports its use against HNSCC tumorigenesis. Further in vivo testing of these two compounds to compare their activity and selectivity is warranted.
In addition, new SIRT3 inhibitors were generated via a virtual screening approach (55). The 2,4-dichloro substitution pattern (compound 9) showed the highest inhibitory effect (71%) compared to others within in vitro settings. However, the biological effects of compound 9 need to be tested in cancer cell lines (55). Similarly, other SIRT3 inhibitors have recently been reported, however, their biological activity on cancer cell lines or in vivo has yet to be tested, or they lack selectivity for SIRT3 (50–53).
To our knowledge, there are no published reports on clinical trials using class III histone deacetylase inhibitors of sirtuins to treat cancer. In agreement with the few and recently published reports (54, 55), our current study also represents a proof-of-principle wherein treatment with SIRT3 inhibitors may be advantageous for overcoming HNSCC aggressiveness and drug-resistant phenotypes. Our novel SIRT3 inhibitor, LC-0296, is the first generation of this type of inhibitor. Our group is currently working on developing more potent and suitable versions of this compound, for use at the nanomolar range, which may be more applicable for testing within an in vivo setting and for potential future clinical trials.
In summary, development of novel SIRT3 inhibitors, such as LC-0296, may open new avenues for the discovery and development of targeted therapies for HNSCC by taking advantage of increased ROS levels in HNSCC cells. This may result in better treatment outcomes with fewer side-effects, a better quality of life, and improved survival rates for patients with head and neck cancer.
Acknowledgments
This work was supported by the National Institutes of Health research grants RO1DEO14429 and R56DE014429 to YLK. Funds for TYA were obtained from King Abdulaziz City for Science and Technology Riyadh, grant No. (165-34-TA). We thank Dr. Tom Carey (University of Michigan) for providing the human OSCC cell lines, UM-SCC-1 and UM-SCC-17B, Takayuki Hayami for helping with statistical analysis, and Drs. Nisha D’Silva, Jacques Nör, and Jill Macoska for their helpful comments during the manuscript preparation. The research work in the laboratory of Liqiang Chen was supported by the Center for Drug Design in the Academic Health Center of the University of Minnesota.
Footnotes
This article is freely accessible online.
References
- 1.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v 1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No 11 [Internet] Lyon, France: International Agency for Research on Cancer; 2013. [Google Scholar]
- 2.http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf
- 3.http://www.cancer.ca/en/cancer-information/cancer-101/cancer-statistics-at-a-glance/?region=ab
- 4.McGuinness D, McGuinness DH, McCaul JA, Shiels PG. Sirtuins, bioageing, and cancer. J Aging Res. 2011;40:235754. doi: 10.4061/2011/235754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010;35:669–675. doi: 10.1016/j.tibs.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sebastian C, Satterstrom FK, Haigis MC, Mostoslavsky R. From sirtuin biology to human diseases: an update. J Biol Chem. 2012;287:42444–42452. doi: 10.1074/jbc.R112.402768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan H, Su L, Chen WY. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. Onco Targets Ther. 2013;6:1399–1416. doi: 10.2147/OTT.S37750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roth M, Chen WY. Sorting out functions of sirtuins in cancer. Oncogene. 2014;33:1609–1620. doi: 10.1038/onc.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alhazzazi TY, Kamarajan P, Verdin E, Kapila YL. Sirtuin-3 (SIRT3) and the Hallmarks of Cancer. Genes Cancer. 2013;4:164–171. doi: 10.1177/1947601913486351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balcerczyk A, Pirola L. Therapeutic potential of activators and inhibitors of sirtuins. BioFactors. 2010;36:383–393. doi: 10.1002/biof.112. [DOI] [PubMed] [Google Scholar]
- 11.Villalba JM, Alcain FJ. Sirtuin activators and inhibitors. Biofactors. 2012;38:349–359. doi: 10.1002/biof.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruzzone S, Parenti MD, Grozio A, Ballestrero A, Bauer I, Del Rio A, Nencioni A. Rejuvenating sirtuins: the rise of a new family of cancer drug targets. Curr Pharm Des. 2013;19:614–623. doi: 10.2174/138161213804581954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5:147–152. doi: 10.7150/ijbs.5.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim CS. SIRT1: tumor promoter or tumor suppressor? Med Hypotheses. 2006;67:341–344. doi: 10.1016/j.mehy.2006.01.050. [DOI] [PubMed] [Google Scholar]
- 15.Alhazzazi TY, Kamarajan P, Verdin E, Kapila YL. SIRT3 and cancer: Tumor promoter or suppressor? Biochim Biophys Acta. 2011;1816:80–88. doi: 10.1016/j.bbcan.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang Y, Nicholl MB. A dual role for sirtuin 1 in tumorigenesis. Curr Pharm Des. 2014;20:2634–2636. doi: 10.2174/13816128113199990488. [DOI] [PubMed] [Google Scholar]
- 17.Bosch-Presegue L, Vaquero A. The dual role of sirtuins in cancer. Genes Cancer. 2011;2:648–662. doi: 10.1177/1947601911417862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y, Huang JS. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 5:e1047. doi: 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alhazzazi TY, Kamarajan P, Joo N, Huang JY, Verdin E, D’Silva NJ, Kapila YL. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer. 2011;117:1670–1678. doi: 10.1002/cncr.25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamarajan P, Alhazzazi TY, Danciu T, D’Silva NJ, Verdin E, Kapila YL. Receptor-interacting protein (RIP) and sirtuin-3 (SIRT3) are on opposite sides of anoikis and tumorigenesis. Cancer. 2012;118:5800–5810. doi: 10.1002/cncr.27655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr, Weissman S, Verdin E, Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui H, Kamal Z, Ai T, Xu Y, More SS, Wilson DJ, Chen L. Discovery of potent and selective sirtuin 2 (SIRT2) inhibitors using a fragment-based approach. J Med Chem. 2014;57:8340–8357. doi: 10.1021/jm500777s. [DOI] [PubMed] [Google Scholar]
- 23.Carey TE. Head and neck tumor cell lines. In: Hay AGR, Park J-G, editors. Atlas of Human Tumor Cell Lines. Academic Press Inc./Harcourt. Brace Jovanovich Publishers; New York City, NY: 1994. pp. 79–120. [Google Scholar]
- 24.Fischel JL, Formento P, Milano G. Epidermal growth factor receptor double targeting by a tyrosine kinase inhibitor (Iressa) and a monoclonal antibody (cetuximab). Impact on cell growth and molecular factors. Br J Cancer. 2005;92:1063–1068. doi: 10.1038/sj.bjc.6602428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv enzyme reg. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 26.Grenman R, Burk D, Virolainen E, Wagner JG, Lichter AS, Carey TE. Radiosensitivity of head and neck cancer cells in vitro. A 96-well plate clonogenic cell assay for squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 1988;114:427–431. doi: 10.1001/archotol.1988.01860160071024. [DOI] [PubMed] [Google Scholar]
- 27.Carey TE, Van Dyke DL, Worsham MJ, Bradford CR, Babu VR, Schwartz DR, Hsu S, Baker SR. Characterization of human laryngeal primary and metastatic squamous cell carcinoma cell lines UM-SCC-17A and UM-SCC-17B. Cancer Res. 1989;49:6098–6107. [PubMed] [Google Scholar]
- 28.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 29.Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS lett. 1995;358:1–3. doi: 10.1016/0014-5793(94)01368-b. [DOI] [PubMed] [Google Scholar]
- 30.Huang JY, Hirschey MD, Shimazu T, Ho L, Verdin E. Mitochondrial sirtuins. Biochim Biophys Acta. 2010;1804:1645–1651. doi: 10.1016/j.bbapap.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 31.Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins-novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- 32.Liu C, Huang Z, Jiang H, Shi F. The sirtuin 3 expression profile is associated with pathological and clinical outcomes in colon cancer patients. Biomed Res Int. 2014;871263 doi: 10.1155/2014/871263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y, Yang H, Wang X, Zhang R, Wang C, Guo Z. Sirtuin-3 (SIRT3) expression is associated with overall survival in esophageal cancer. Ann Diagn Pathol. 2013;17:483–485. doi: 10.1016/j.anndiagpath.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Yan SM, Han X, Han PJ, Chen HM, Huang LY, Li Y. SIRT3 is a novel prognostic biomarker for esophageal squamous cell carcinoma. Med Oncol. 2014;31:103. doi: 10.1007/s12032-014-0103-8. [DOI] [PubMed] [Google Scholar]
- 35.Yang M, Yang C, Pei Y. Effects of downregulation of SIRT3 expression on proliferation and apoptosis in esophageal squamous cell carcinoma EC9706 cells and its molecular mechanisms. Biomed Mater Eng. 2014;24:3883–3890. doi: 10.3233/BME-141219. [DOI] [PubMed] [Google Scholar]
- 36.Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Meta Rev. 2006;25:695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 37.Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Society Trans. 2003;31:1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 38.Fleury C, Mignotte B, Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141. doi: 10.1016/s0300-9084(02)01369-x. [DOI] [PubMed] [Google Scholar]
- 39.Gamaley IA, Klyubin IV. Roles of reactive oxygen species: signaling and regulation of cellular functions. Int Rev cytol. 1999;188:203–255. doi: 10.1016/s0074-7696(08)61568-5. [DOI] [PubMed] [Google Scholar]
- 40.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug resist updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Pervaiz S, Clement MV. Tumor intracellular redox status and drug resistance-serendipity or a causal relationship? Curr pharma design. 2004;10:1969–1977. doi: 10.2174/1381612043384411. [DOI] [PubMed] [Google Scholar]
- 42.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 43.Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D. SIRT3 Is a Mitochondria-Localized Tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1–13. doi: 10.1016/j.gene.2004.04.032. [DOI] [PubMed] [Google Scholar]
- 46.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms:A radical therapeutic approach? Nature rev Drug discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 47.Montero AJ, Jassem J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs. 2011;71:1385–1396. doi: 10.2165/11592590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 48.Sinclair DA, Guarente L. Small-molecule allosteric activators of sirtuins. Ann Rev Pharmacol Toxicol. 2014;54:363–380. doi: 10.1146/annurev-pharmtox-010611-134657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu J, Jing H, Lin H. Sirtuin inhibitors as anticancer agents. Future Med Chem. 2014;6:945–966. doi: 10.4155/fmc.14.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahajan SS, Scian M, Sripathy S, Posakony J, Lao U, Loe TK, Leko V, Thalhofer A, Schuler AD, Bedalov A, Simon JA. Development of pyrazolone and isoxazol-5-one cambinol analogues as sirtuin inhibitors. J Med Chem. 2014;57:3283–3294. doi: 10.1021/jm4018064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen B, Wang J, Huang Y, Zheng W. Human SIRT3 tripeptidic inhibitors containing N(epsilon)-thioacetyl-lysine. Bioorg Med Chem Lett. 2015;25:3481–3487. doi: 10.1016/j.bmcl.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 52.Kokkonen P, Kokkola T, Suuronen T, Poso A, Jarho E, Lahtela-Kakkonen M. Virtual screening approach of sirtuin inhibitors results in two new scaffolds. Eur J Pharm Sci. 2015;76:27–32. doi: 10.1016/j.ejps.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 53.Pate K, Sherrill J, Mrksich M, Scholle MD. Discovery of SIRT3 inhibitors using SAMDI mass spectrometry. J Biomol Screen. 2015;20:842–848. doi: 10.1177/1087057115588512. [DOI] [PubMed] [Google Scholar]
- 54.Galli U, Mesenzani O, Coppo C, Sorba G, Canonico PL, Tron GC, Genazzani AA. Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2. Eur J Med chem. 2012;55:58–66. doi: 10.1016/j.ejmech.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 55.Salo HS, Laitinen T, Poso A, Jarho E, Lahtela-Kakkonen M. Identification of novel SIRT3 inhibitor scaffolds by virtual screening. Bioorg Med Chem Lett. 2013;23:2990–2995. doi: 10.1016/j.bmcl.2013.03.033. [DOI] [PubMed] [Google Scholar]