

ABSTRACT
A 63-year-old woman presented to her ophthalmologist complaining of reading difficulties for two years. Ophthalmological examination revealed a homonymous hemianopsia. Brain magnetic resonance imaging (MRI) scan was interpreted as normal, but positron emission tomography (PET) showed areas of posterior brain hypometabolism. This case highlights the high diagnostic suspicion that ophthalmologists should have regarding posterior cortical atrophy (including the visual variant of Alzheimer disease) in patients complaining of reading difficulties in the setting of a normal ophthalmic examination.
KEYWORDS: Brain PET, homonymous hemianopsia, posterior cortical atrophy, visual variant of Alzheimer disease
Introduction
A homonymous hemianopsia generally localises to a lesion of the contralateral, retrochiasmal optic pathway (e.g., optic tract, optic radiations, or occipital cortex). When a homonymous hemianopsia is present, cranial magnetic resonance imaging (MRI) typically demonstrates the causative structural lesion. If, however, the neuroimaging is negative, then the differential diagnosis includes nonorganic visual loss, the Heidenhain variant of Creutzfeldt-Jacob disease (CJD), neurodegenerative disease (e.g., posterior cortical atrophy, visual variant Alzheimer disease), subtle occipital ischaemia, seizure, or non-ketotic hyperglycaemia.1 We describe a patient with a homonymous hemianopsia with normal cranial MRI in whom further clinical evaluation and cranial positron emission tomography (PET) ultimately led to a final diagnosis of posterior cortical atrophy (PCA).
Case report
A 63-year-old woman presented to her ophthalmologist complaining of reading difficulties for the prior 2 years. Eight months prior to presentation, she had been prescribed multiple pairs of glasses without improvement. Automated static perimetry (Figure 1) showed a left homonymous hemianopsia. Brain contrast-enhanced MRI was reported as normal. She was referred to neuro-ophthalmology for a second opinion.
Figure 1.
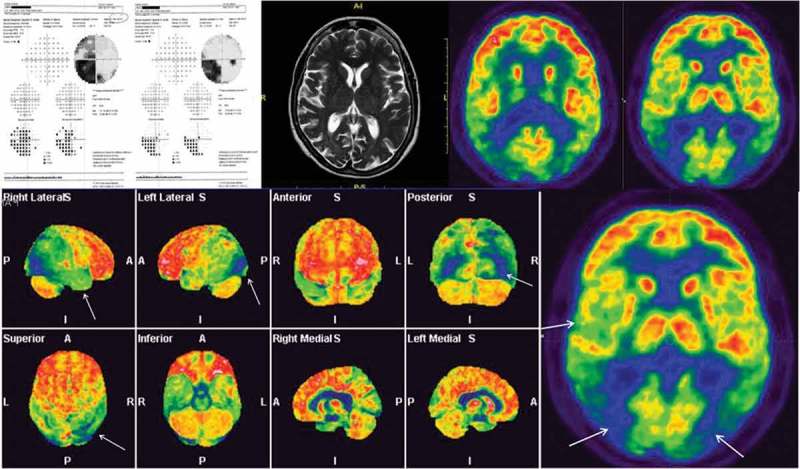
Top left: Automated static perimetry shows left homonymous hemianopsia, denser inferiorly, with 10° central sparing in the right visual field. Top center: Brain MRI image of the patient. Top right and bottom: Patient’s brain PET shows hypometabolism of the occipital, temporal, and posterior parietal cortex, more on the right.
The patient was not taking any medications. Past ocular, surgical, and medical histories were non-contributory. Her diet was normal; she denied alcohol consumption and she did not smoke. Family history was relevant for a stroke in her brother. She did not complain of visual hallucinations and had no extrapyramidal signs, myoclonus, or ataxia.
The patient’s husband reported that the patient had increased difficulty dressing and cooking, specifying that she could not pour water into a pot. He also noticed that in the last few months, she showed no interest in using her makeup.
Best-corrected visual acuity (BCVA) was 20/20 OU. Her reading speed was slow, and she read with difficulty with many pauses. Intraocular pressure measured 14 mm Hg OU. She correctly identified 7 out of 12 charts in OD and 5 in OS on Ishihara testing. The remainder of the examination was normal OU. Review of her MRI scan showed no signs of parieto-occipital stroke but disclosed a mild posterior cortical atrophy.
PET with [18F]fluorodeoxyglucose ([18F]FDG) showed bilateral hypometabolism of the occipital, temporal, and posterior parietal cortex, more on the right. On neuropsychological examination, the patient had a Mini-Mental Status Examination (MMSE) score of 26/30, but she had severe difficulties describing the complex scene in the “Cookie Theft Picture,” consistent with simultanagnosia). Lumbar puncture was positive for an increased level of tau phosphorylated at threonine 181 (p-Tau181) of 85 pg/mL (normal range: 25–55 pg/mL) and a decreased amyloid Aβ 42 level of 66 pg/mL (normal range: 660–1075 pg/mL). An electroencephalography (EEG) was performed and was negative for periodic rhythmic discharges (suggestive of CJD). The patient was diagnosed with PCA and treated with memantine and rivastigmine.
Discussion
In 1988, Benson et al.2 reported a series of 5 patients with prominent visual complaints and no evident ophthalmic cause. In this series, all patients exhibited Balint syndrome (e.g., optic ataxia, simultanagnosia, oculomotor apraxia) and Gerstmann syndrome (acalculia, agraphia, finger agnosia, right-left confusion). Posterior cortical atrophy (PCA) was observed on cranial MRI in most of these patients. Subsequent histopathological studies identified Alzheimer disease (AD) as the most common underlying pathology, leading to the synonymous use of other terms such as visual variant of AD (VVAD), biparietal AD, progressive agnosia and apraxia, and Benson syndrome. However, PCA is also associated with non-AD pathology, which has led to suggestions for PCA to be considered a distinct clinical syndrome. The definition of PCA has evolved over time and has expanded to include progressive dementing syndromes characterised by higher visual disorders. There is a progressive, and relatively selective, decline in visual processing skills and other function served by parietal, occipital, and occipito-temporal regions.3 Although PCA has been recognised for more than two decades, the condition remains difficult to diagnose. In one recent series from a dementia clinic, only 5% (24 of 523) presented with predominant visual symptoms consistent with PCA.4 Coexisting mood disorders may contribute to misdiagnosis as a “functional” or non-organic disorder. The vague nature of the complaint may lead to premature closure of the differential diagnosis and can lead to multiple pairs of ineffective reading spectacle or cataract surgery.5–7
Several aetiologies should be considered in patients with PCA, including AD, CJD, corticobasal degeneration (CBD), dementia with Lewy bodies (DLB), and subcortical gliosis. In another large series that reported pathological data in 21 patients with PCA, 16 (76%) had AD variant pathology (isolated AD pathology or in combination with Lewy bodies and Parkinson disease), 3 (15%) had CBD pathology (isolated or in combination with subcortical gliosis), and 2 (10%) had prion disease.8 The high prevalence of AD pathology has been confirmed in multiple other series as well. Tang-Wai et al.9 reported that 78% of PCA patients had AD pathology and the remaining 22% had CBD. In our patient, the possibility of DLB and CBD were considered unlikely because of the clinical presentation. DLB is a progressive, degenerative dementia of unknown aetiology. Affected patients generally present with dementia preceding motor signs, particularly with visual hallucinations and episodes of reduced responsiveness. Clinical features that may help to distinguish DLB from AD include the fluctuations in cognitive function and level of alertness and attention, Parkinsonian motor features, visual and non-visual hallucinations, unexplained syncopal episodes, and rapid eye movement sleep disorder. The rate of progression in DLB is often more rapid than in typical AD. CBD is a sporadic neurodegenerative tauopathy that includes characteristic movement and cognitive dysfunction (corticobasal syndrome). The corticobasal syndrome is typically defined by progressive dementia, parkinsonism, and limb apraxia. In addition, in CBD (in contrast to AD), there are less “cognitive” abnormalities (e.g., right-left disorientation, naming difficulty, acalculia) and more “frontal-executive” deficits (e.g., distractibility, perseveration, loss of judgment, motor planning deficit even on the less motor-impaired side). Eye movement abnormalities, dystonia, rigidity, and myoclonus are also more frequently seen in patients affected by CBD.
PCA due to VVAD occurs usually in patients younger than those affected with typical AD,10–12 and these patients are often more aware of their deficit.13 As VVAD affects mostly the parieto-occipital cortex, involvement of the retrochiasmal visual pathway can occur. The occurrence of a homonymous hemianopic visual field defect in PCA/VVAD varies widely in the literature but may have a prevalence of up to 78%.14,15
The right hemisphere is often more severely affected than the left hemisphere in most patients with VVAD,4,11,16 as in our present case. The signs and symptoms of VVAD depend on whether the dorsal or ventral visual processing pathway is the primary site of disease.12,17 The dorsal (occipito-parietal) system is the most frequently involved (the “where” pathway) and can result in a complete or incomplete Balint syndrome (e.g., simultanagnosia, oculomotor apraxia, optic ataxia), Gerstmann syndrome (e.g., agraphia, acalculia, right-left disorientation, finger agnosia), dressing apraxia, and aphasia. In contrast, lesions of the ventral (occipito-temporal) system (the ”what” pathway) can produce alexia without agraphia, visual object agnosia, and prosopagnosia (inability to recognise faces).17 However, the complete Balint or Gerstmann syndrome is rare in PCA, and the most common associated features are simultanagnosia and acalculia.18 Our patient presented with typical complaints of PCA/VVAD disease characterised by progressive, painless, reading difficulty, a homonymous hemianopsia with no structural correlate on neuroimaging, and progressive visual processing difficulties.
Alzheimer disease is a clinical diagnosis. However, ancillary imaging studies and laboratory tests may help physicians to exclude other possible treatable causes for dementia and to make an early diagnosis. This is paramount in order to recognise prodromal AD patients and provide appropriate treatments in routine clinical practice.19
Notably, functional cerebral imaging, by PET or single-photon emission computed tomography (SPECT), will usually reveal parieto-occipital dysfunction, even in absence of anatomical cortical atrophy on CT or MRI.20
Newer PET technology (such as Florbetapir F18 PET) that detects β-amyloid plaque in the living brain has been validated and approved as an adjunctive biomarker in the diagnosis of AD.21 This technology may be useful because β-amyloid neuropathological changes occur decades before the first clinical symptoms appear.
Measurements of biochemical markers in cerebrospinal fluid (CSF) are increasingly used in the diagnostic process of dementia and other related disorders. Since CSF is an ideal source for developing viable biomarkers in AD, as it directly interacts with the extracellular space in the brain, CSF analysis may potentially reflect the associated biochemical/pathological changes.22 The sensitivity when using the combination of CSF Aβ 42 and total tau (t-tau)/phospho-tau (p-tau) for recognition of Alzheimer disease is high.23–25
CSF levels of tau and phosphorylated tau are often elevated in AD, whereas amyloid levels are usually low, perhaps because the amyloid is deposited in the brain rather than the CSF.19,26
In conclusion, because visual symptoms are prominent manifestations of VVAD and ophthalmologists are often the first to evaluate these patients,27 they should be aware of the possibility of neurodegenerative disease in patients presenting with such symptoms. Although many patients with VVAD have insight in to their deficits, the chief complaint of “brought in by spouse” is often the first clue that the problem is dementia related. Careful clinical examination, clinically oriented tests, and their accurate review are the three steps to reach a correct diagnosis of a dementing illness.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- [1].Brazis PW, Lee AG, Graff-Radford N, Desai NP.. Homonymous visual field defects in patients without corresponding structural lesions on neuroimaging. J Neuroophthalmol 2000;20:92–96. [DOI] [PubMed] [Google Scholar]
- [2].Benson DF, Davis RJ, Snyder BD.. Posterior cortical atrophy. Arch Neurol 1988;45:789–793. [DOI] [PubMed] [Google Scholar]
- [3].Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Nick CF.. Posterior cortical atrophy. Lancet Neurol 2012;11:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Snowden JS, Stopford CL, Julien CL, Thompson JC, Davidson Y, Gibbons L, Pritchard A, Lendon CL, Richardson AM, Varma A, Neary D, Mann D.. Cognitive phenotypes in Alzheimer’s disease and genetic risk. Cortex 2007;43:835–845. [DOI] [PubMed] [Google Scholar]
- [5].Zakzanis KK, Boulos MI.. Posterior cortical atrophy. Neurologist 2001;7:341–349. [DOI] [PubMed] [Google Scholar]
- [6].Trick GL, Trick LR, Morris P, Wolf M.. Visual field loss in senile dementia of the Alzheimer’s type. Neurology 1995;45:68–74. [DOI] [PubMed] [Google Scholar]
- [7].Renner JA, Burns JM, Hou CE, McKeel DW Jr, Storandt M, Morris JC. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology 2004;63:1175–1180. [DOI] [PubMed] [Google Scholar]
- [8].Tang-Wai DF, Graff-Radford NR, Boeve BF, Dickson DW, Parisi JE, Crook RJ, Knopman DS, Petersen RC.. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004;63:1168–1174. [DOI] [PubMed] [Google Scholar]
- [9].Tang-Wai DF, Graff-Radford NR, Boeve BF, Dickson DW, Parisi JE, Crook RJ, Knopman DS, Petersen RC.. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004;63:1168–1174. [DOI] [PubMed] [Google Scholar]
- [10].Charles RF, Hillis AE.. Posterior cortical atrophy. Behav Neurol 2005;16:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mendez MF, Ghajarania M, Perryman KM.. Posterior cortical atrophy: clinical characteristics and differences compared to Alzheimer’s disease. Dement Geriatr Cogn Disord 2002;14:33–40. [DOI] [PubMed] [Google Scholar]
- [12].Kas A, de Souza LC, Samri D, Bartolomeo P, Lacomblez L, Kalafat M, Migliaccio R, Thiebaut de Schotten M, Cohen L, Dubois B, Habert MO, Sarazin M. Neural correlates of cognitive impairment in posterior cortical atrophy. Brain 2011;134(Pt 5):1464–1478. [DOI] [PubMed] [Google Scholar]
- [13].Vighetto A. Towards an earlier diagnosis of Alzheimer’s disease presenting with visuospatial disorders (posterior cortical atrophy). Rev Neurol (Paris) 2013;169:687–694. [DOI] [PubMed] [Google Scholar]
- [14].Pelak VS, Smyth SF, Boyer PJ, Filley CM.. Computerized visual field defects in posterior cortical atrophy. Neurology 2011;77:2119–2122. [DOI] [PubMed] [Google Scholar]
- [15].Formaglio M, Krolak-Salmon P, Tilikete C, Bernard M, Croisile B, Vighetto A.. Homonymous hemianopia and posterior cortical atrophy. Rev Neurol (Paris) 2009;165:256–262. [DOI] [PubMed] [Google Scholar]
- [16].Harrison AR, Lee MS.. The woman who needed a pet. Surv Ophthalmol 2006;51:592–955. [DOI] [PubMed] [Google Scholar]
- [17].Kaeser PF, Ghika J, Borruat FX.. Visual signs and symptoms in patients with the visual variant of Alzheimer disease. BMC Ophthalmol 2015;15:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fletcher WA. Ophthalmological aspects of Alzheimer’s disease. Curr Opin Ophthalmol 1994;5:38–44. [PubMed] [Google Scholar]
- [19].Blennow K, Dubois B, Fagan AM, Lewczuk P, de Leon MJ, Hampel H.. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement 2015;11:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schmidtke K, Hull M, Talazko J.. Posterior cortical atrophy: variant of Alzheimer’s disease? A case series with PET findings. J Neurol 2005;252:27–35. [DOI] [PubMed] [Google Scholar]
- [21].Degenhardt EK, Witte MM, Case MG, Yu P, Henley DB, Hochstetler HM, D’Souza DN, Trzepacz PT.. Florbetapir F18 PET amyloid neuroimaging and characteristics in patients with mild and moderate alzheimer dementia. Psychosomatics 2016;57:208–216. [DOI] [PubMed] [Google Scholar]
- [22].Hampel H, Lista S, Khachaturian ZS.. Development of biomarkers to chart all Alzheimer’s disease stages: the royal road to cutting the therapeutic Gordian Knot. Alzheimers Dement 2012;8:312–336. [DOI] [PubMed] [Google Scholar]
- [23].Fagan AM, Shaw LM, Xiong C, Vanderstichele H, Mintun MA, Trojanowski JQ, Coart E, Morris JC, Holtzman DM. Comparison of analytical platforms for cerebrospinal fluid measures of beta-amyloid 1–42, total tau, and ptau181 for identifying Alzheimer disease amyloid plaque pathology. Arch Neurol 2011;68:1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Andreasen N, Vanmechelen E, Vanderstichele H, Davidsson P, Blennow K.. Cerebrospinal fluid levels of total-tau, phospho-tau and A beta 42 predicts development of Alzheimer’s disease in patients with mild cognitive impairment. Acta Neurol Scand Suppl 2003;179:47–51. [DOI] [PubMed] [Google Scholar]
- [25].Mulder C, Verwey NA, van der Flier WM, Bouwman FH, Kok A, van Elk EJ, Scheltens P, Blankenstein MA Amyloid-beta(1–42), total tau, and phosphorylated tau as cerebrospinal fluid biomarkers for the diagnosis of Alzheimer disease. Clin Chem 2010;56:248–253. [DOI] [PubMed] [Google Scholar]
- [26].Blennow K, Hampel H.. CSF markers for incipient Alzheimer’s disease. Lancet Neurol 2003;2:605–613. [DOI] [PubMed] [Google Scholar]
- [27].Lee AG, Martin CO.. Neuro-ophthalmic findings in the visual variant of Alzheimer’s disease. Ophthalmology 2004;111:376–381. [DOI] [PubMed] [Google Scholar]