

Abstract
Despite the existence of effective antiandrogen therapy for prostate cancer, the disease often progresses to castration-resistant states. Elucidation of the molecular mechanisms underlying the resistance for androgen deprivation in terms of the androgen receptor (AR)-regulated pathways is a requisite to manage castration-resistant prostate cancer (CRPC). Using a ChIP-cloning strategy, we identified functional AR binding sites (ARBS) in the genome of prostate cancer cells. We discovered that a centrosome- and microtubule-interacting gene, transforming acidic coiled-coil protein 2 (TACC2), is a novel androgen-regulated gene. We identified a functional AR-binding site (ARBS) including two canonical androgen response elements in the vicinity of TACC2 gene, in which activated hallmarks of histone modification were observed. Androgen-dependent TACC2 induction is regulated by AR, as confirmed by AR knockdown or its pharmacological inhibitor bicalutamide. Using long-term androgen-deprived cells as cellular models of CRPC, we demonstrated that TACC2 is highly expressed and contributes to hormone-refractory proliferation, as small interfering RNA-mediated knockdown of TACC2 reduced cell growth and cell cycle progression. By contrast, in TACC2-overexpressing cells, an acceleration of the cell cycle was observed. In vivo tumor formation study of prostate cancer in castrated immunocompromised mice revealed that TACC2 is a tumor-promoting factor. Notably, the clinical significance of TACC2 was demonstrated by a correlation between high TACC2 expression and poor survival rates. Taken together with the critical roles of TACC2 in the cell cycle and the biology of prostate cancer, we infer that the molecule is a potential therapeutic target in CRPC as well as hormone-sensitive prostate cancer.
Androgen receptor (AR) is a nuclear hormone receptor that regulates expression of its target gene in a ligand-dependent manner (1) and functions as a key factor in determining the biology of prostate cancer (2, 3). Androgen deprivation therapy is initially effective for inhibiting the growth of prostate cancer by suppressing AR activity (2), although the disease often recurs as castration-resistant prostate cancer (CRPC). Accumulated evidence reveals that CRPC is often associated with enhanced AR expression and transcriptional activity (4–8). Nevertheless, the precise mechanisms underlying acquired resistance to androgen deprivation in the context of the AR-regulated gene network remain to be studied.
Recent advances in chromatin immunoprecipitation (ChIP) studies have demonstrated that the AR-regulated gene network involves cooperation of AR with other transcriptional factors and coregulators (9, 10). Global ChIP analyses using tiled oligonucleotide microarrays (ChIP-chip) and deep sequencing (ChIP-Seq) have defined various androgen-regulated genes in the vicinity of AR-binding sites (ARBS) and revealed the status of histone modification status around the ARBS (9, 11–15). In particular, AR cistrome identified the M-phase check point UBE2C as a direct AR target in castration-resistant prostate cancer cells in comparison with hormone-naïve cells (9). Thus, ChIP-based approaches are useful tools for elucidating the AR-regulated gene network in studies on androgen-dependent and castration-resistant prostate cancer.
Our previous approach, which used the ChIP-cloning technique, identified forkhead box protein P1 as a direct AR target in androgen-dependent LNCaP cells (16). In the present study, we describe transforming acidic coiled-coil protein 2 (TACC2) as a critical AR target. TACC2 is a centrosomal protein that interacts with microtubules (17–19). Although the association of TACC2 with cancer biology is controversial (20–22), the molecule may play a role in tumorigenesis with regard to its interaction with nuclear histone acetyltransferases (23). Our functional study and clinical data suggest that TACC2 is a prognostic factor of prostate cancer and a potential therapeutic target for CRPC.
Results
Identification of a strong ARBS in the TACC2 gene locus
For identification of direct AR target genes, we explored androgen-dependent ARBS in the genome by employing a modified strategy involving ChIP-cloning combined with PCR-based subtractive hybridization between ligand (R1881, 10 nm for 24 h)-treated ChIP DNA and vehicle-treated ChIP DNA derived from human prostate cancer LNCaP cells (16). A total of 100 sequences were obtained by ChIP-cloning and substantial ligand-dependent AR recruitment to the top five ARBS (ARBS nos. 1–5, Supplemental Table 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) was validated in a conventional ChIP assay (Fig. 1A). Surprisingly, AR occupancy at ARBS 4 was even higher than the occupancy at the known distal enhancer region of prostate-specific antigen (PSA) (>100-fold). To investigate the function of these ARBS, we constructed luciferase reporter vectors containing the ARBS sequences. The luciferase assay revealed that ligand-dependent enhancer activity exists for all five of the ARBS (Fig. 1B). Among these, ARBS 4 was found to have the highest enhancer activity to confer transcriptional activity on heterologous promoters in response to R1881. Using a BLAST search, we found that ARBS 4 is located in the vicinity of the TACC2 gene (Fig. 1C). The TACC2 ARBS was found to be located in a transcriptionally active chromatin region. The ChIP assay revealed activated histone modification marks of H3K4 monomethylation and H3/H4 acetylation, and ligand-dependent recruitment of RNA polymerase (Pol) II or phosphorylated Pol II (Fig. 1D).
Fig. 1.

Characterization of the ARBS in the genomic region of the TACC2 gene. A, AR binding ability of the top five (ARBSs 1–5) of 100 ARBS determined by the ChIP-cloning strategy. LNCaP cells were treated with R1881 (10 nm) or vehicle for 24 h. The known distal ARE in the enhancer region of PSA was used as a positive control. Fold enrichment of AR binding was quantified by real-time PCR. The data represent the mean ± sd, n = 3. B, Androgen-dependent promoter activity of the top five ARBS evaluated by the luciferase reporter assay. LNCaP cells were transfected with pGL promoter vectors containing ARBS sequences 1–5. Empty pGL3 promoter and mouse mammary tumor virus (MMTV) luciferase vectors were used as negative and positive controls, respectively. Cells were treated with R1881 (10 nm) or vehicle for 24 h. The data represent the mean ± sd, n = 3. C, Location of ARBS 4 (arrow) in the TACC2 gene mapped by the UCSC Genome Browser (human genome NCBI Build 35). D, Active histone modification and RNA Pol II-dependent transcription at TACC2 ARBS. LNCaP cells were treated with R1881 (10 nm) or vehicle for 24 h. ChIP analysis was performed using nonspecific IgG or specific antibodies targeting histone H3 monomethylated K4 (K4me1), acetylated histone H3 (AcH3), acetylated histone H4 (AcH4), Pol II, or phosphorylated Pol II (P-pol II). Fold enrichment was quantified by real-time PCR. The data represent the mean ± sd, n = 2. Chr, Chromosome; SV40, simian virus 40.
TACC2 is a novel androgen-regulated gene
To determine whether the ligand-dependent transcriptional activity of the TACC2 ARBS is directly regulated by AR, we evaluated the function of androgen response elements (ARE) involved in the TACC2 ARBS. Two canonical ARE sequences were identified in the TACC2 ARBS by TRANSFAC. We constructed luciferase vectors containing mutations of these ARE sequences (MutARE1 and MutARE2) and compared their transcriptional activities with that of the luciferase vector including the intact TACC2 ARBS (Fig. 2A). Substantial repression of luciferase activation was observed in each of the mutated constructs (Fig. 2B).
Fig. 2.

AR- and ligand-dependent transcription of TACC2. A, Schematic view of luciferase reporter constructs of TACC2 ARBS 4 (TACC2 ARBS-LUC: containing intact ARE, mutARE1-LUC: mutated ARE1 with intact ARE2, and mutARE2-LUC: intact ARE1 with mutated ARE2). Mutated bases in ARE are underlined. B, Luciferase activity of TACC2 ARBS luciferase vectors transfected into LNCaP cells. The pGL3 promoter and mouse mammary tumor virus (MMTV)-LUC vectors were used as negative and positive controls, respectively. Cells were treated with R1881 (10 nm) or vehicle for 24 h. The data represent the mean ± sd, n = 3. C, Androgen-dependent induction of TACC2 mRNA in LNCaP cells. Cells were stimulated with R1881 (10 nm) or vehicle and subjected to total RNA extraction for 6–72 h. The TACC2 mRNA level was analyzed by real-time PCR. The data represent the mean ± sd, n = 2. D, Isoform-specific regulation of TACC2 by androgen in prostate cancer cell lines. LNCaP cells or DU145 cells were stimulated with R1881 (10 nm) or vehicle and subjected to total RNA extraction for 24 h. TACC2 short or long isoform mRNA levels were analyzed by real-time PCR using specific primers. The data represent the mean ± sd, n = 2. E, Androgen-dependent TACC2 protein induction in LNCaP cells. Cells were treated with DHT (10 nm) or vehicle for 24 and 48 h. Whole-cell lysates were immunoblotted by anti-TACC2 and anti-β-actin. 293T cell lysates transfected with the expression vector for a short form of TACC2 (NM_206860) or empty vector were used as positive or negative controls, respectively. F, Modulation of TACC2 mRNA levels by DHT and antiandrogen bicalutamide. LNCaP cells were treated with vehicle, DHT (10 nm), DHT (10 nm) plus bicalutamide (1 or 10 μm), or bicalutamide (1 or 10 μm) alone for 24 h, and subjected to total RNA extraction. The TACC2 mRNA level was analyzed by real-time PCR. The data represent the mean ± sd, n = 3. *, P < 0.05 from DHT alone. G, Knockdown of AR represses TACC2 transcription. LNCaP cells were transfected with siControl (5 nm) or siAR (1 nm or 5 nm). G (left panel), Whole-cell lysates were immunoblotted with anti-AR 48 h after transfection. Anti-β-actin was used as a loading control. G (right panel), LNCaP cells were transfected with siControl (5 nm) or siAR (5 nm). The TACC2 mRNA level was analyzed by real-time PCR. The data represent the mean ± sd, n = 3. **, P < 0.01 from siControl. IB, Immunoblotting; mut, mutant; qRT-PCR, quantitative RT-PCR..
Androgen responsiveness of TACC2 expression was examined in LNCaP cells stimulated by R1881 and dihydrotestosterone (DHT) (10 nm each) (Fig. 2, C–F). The TACC2 mRNA was induced in a time-dependent manner, and an approximately 2-fold elevation was detected 24 h after R1881 treatment (Fig. 2C). With regard to the size of TACC2 isoforms, the mRNA expressed in LNCaP cells was found to be predominantly derived from the short isoform (NM_206860) as defined by the design of PCR primers (Fig. 2D). In addition, we examined androgen responsiveness of TACC2 protein expression in LNCaP cells using TACC2-specific antibody recognizing both isoforms (Fig. 2E). Androgen-dependent up-regulation of TACC2 protein was shown after stimulation of the LNCaP cells, the molecular size of which corresponds to the short isoform transiently expressed in 293T cells. DHT was also found to cause elevation of the TACC2 mRNA level by more than 2.5-fold 24 h after treatment, and the induction was significantly inhibited by the antiandrogen bicalutamide (10 μm) (Fig. 2F). To further investigate the direct contribution of AR to TACC2 expression, we performed knockdown of AR by small interfering RNA (siRNA) transfection. When the AR siRNA was transfected at a concentration sufficient for AR protein knockdown (Fig. 2G), the TACC2 mRNA level was reduced by about 40%.
We also found that a remarkable extent of AR recruitment at the TACC2 ARBS is detected in VCaP cells, another AR-positive prostate cancer cell line (30) and metastatic tissue of advanced prostate cancer in recent published ChIP-Seq data (15) (Supplemental Fig. 1). We also validated TACC2 induction in VCaP cells. Taken together, these results indicate that AR regulates TACC2 gene expression in a ligand-dependent manner in prostate cancer.
TACC2 promotes cell cycle progression for androgen-dependent prostate cancer cell proliferation
To investigate the function of TACC2 function in prostate cancer, we reduced the endogenous TACC2 expression by siRNA treatment. We used two siRNA sequences targeting different regions of TACC2 and confirmed that each siRNA transfection effectively represses the TACC2 mRNA level (Fig. 3A) as well as the TACC2 protein level in LNCaP cells (Fig. 3B). TACC2 knockdown was found to reduce the proliferation of cultured LNCaP cells in both basal and hormone-activated stages (Fig. 3, C and D). Next to determine the function of TACC2 in regulating the cell cycle, we performed cell cycle analysis using fluorescence-activated cell sorting (FACS). After a 96-h siRNA treatment, FACS analysis revealed that TACC2 silencing leads to G2/M accumulation (Fig. 3E). Immunofluorescence analysis showed that TACC2 expression is primarily observed in the cytoplasm during interphase in LNCaP cells (Supplemental Fig. 2A). During mitosis, TACC2 expression is compacted in centrosomes or colocalizes with microtubules (Supplemental Fig. 2, A and B). In addition, siRNA-mediated silencing of TACC2 increases the proportion of cells at prometaphase during the M-phase (Supplemental Fig. 2C). These data suggest that TACC2 plays a modulatory role in the G2/M cell cycle regulation in prostate cancer cells. Taken together, these results indicate that TACC2 is a critical factor for cell proliferation and cell cycle progression in androgen-dependent prostate cancer cells.
Fig. 3.

TACC2 is associated with hormone-dependent prostate cancer cell proliferation by accelerating the M-phase transition. A, Silencing of TACC2 mRNA by siRNA. LNCaP cells were transfected with siControl 1 and 2 or siTACC2 1 and 2 (5 nm each). TACC2 mRNA levels were analyzed by real-time PCR. The data represent the mean ± sd, n = 2. B, siRNA-mediated knockdown of TACC2 protein. LNCaP cells were transfected with siControl 1 or siTACC2 1 and treated with R1881 (10 nm) for 48 h. Whole-cell lysates were immunoblotted with anti-TACC2 and anti-β-actin. C, TACC2 knockdown retards LNCaP cell proliferation. LNCaP cells were transfected with siControl or siTACC2 (5 nm each). The MTS assay was performed 1–3 d after transfection. The data represent the mean ± sd, n = 4. *, P < 0.05 from siControl 1. D, TACC2 knockdown suppresses androgen-dependent growth of LNCaP cells. The MTS assay was performed 1–5 d after treatment with vehicle or DHT (10 nm) (siRNA transfection was performed 48 h before ligand treatment). The data represent the mean ± sd., n = 4. *, P < 0.01 from siControl transfection with DHT treatment. E, TACC2 knockdown increases G2/M phase cells. LNCaP cells were transfected with siControl or siTACC2 and subjected to cell cycle analysis by FACS, 96 h after transfection. Each percentage of G2/M phase cells is described in the graph. Representative result of repeated three experiments is shown.
TACC2 is overexpressed in a castrate-resistant prostate cancer cell model
It has been previously reported that LNCaP cells will change to acquire the ability to mimic CRPC under long-term androgen-deprived (LTAD) conditions (25). Previous studies have shown that prostate cancer cells under LTAD conditions often acquire the ability to proliferate during periods of hormone deprivation and also gain insensitivity to bicalutamide (24–26). To analyze the molecular mechanism of developing castration-resistant prostate cancer, we generated two independent LTAD cell lines derived from parental LNCaP cells. We confirmed that LTAD cells proliferate during periods of hormone depletion as fast as LNCaP cells treated with androgen by performing an 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl-2H-tetrazolium) (MTS) assay (Supplemental Fig. 3A). In addition, AR protein was found to be remarkably up-regulated in these LTAD cells, (Fig. 4A) although PSA induction by androgen is repressed in LTAD cells as reported previously (Supplemental Fig. 3B) (11, 26). However, several androgen-responsive genes such as APP (13) or ARFGAP3 (31) are more sensitive to DHT at low concentrations (Supplemental Fig. 3C).
Fig. 4.

Androgen hypersensitivity increases TACC2 transcription during long-term androgen deprivation. A, Up-regulation of TACC2 and AR proteins in LNCaP-derived LTAD cells. Whole-cell lysates from parental LNCaP cells incubated under hormone-depleted conditions for 72 h and LTAD 1 and 2 cells were immunoblotted with anti-TACC2, anti-AR, and anti-β-actin antibodies. B, Up-regulation of TACC2 mRNA in LTAD cells compared with LNCaP cells under hormone-depleted conditions. TACC2 mRNA levels were evaluated by real-time RT-PCR. The data represent the mean ± sd, n = 3. C, siRNA-mediated knockdown of AR protein in LTAD 2 cells 48 h after transfection. D, AR knockdown represses TACC2 mRNA expression and induction 24 h after DHT 10 nm treatment in LTAD cells. LTAD 1 and 2 was treated 48 h after siAR 10 nm transfection with vehicle and DHT 10 nm. The data represent the mean ± sd, n = 3. *, P < 0.05; **, P < 0.01 from siControl transfection, respectively. E and F, Enhanced AR recruitment at TACC2 ARBS in LTAD cells. After hormone depletion, parental LNCaP and LTAD 1and 2 cells were treated with vehicle or with 10 nm DHT for 24 h. Cells were subjected to ChIP analysis, and AR binding activity was evaluated and quantified by real-time PCR. E, Significant AR binding at TACC2 ARBS in LTAD cells treated with vehicle. F, Robust AR binding at TACC2 ARBS in LTAD cells treated with DHT (10 nm, 24 h). The data represent the mean ± sd, n = 2. G, Up-regulation of TACC2 proteins in LTAD cells treated with vehicle and DHT. Whole-cell lysates from parental LNCaP cells and LTAD 2 cells treated with vehicle and 10 nm DHT for 48 h after incubation under hormone-depleted conditions for 72 h were immunoblotted with anti-TACC2 and anti-β-actin antibodies. H, AR hypersensitivity was observed with TACC2 mRNA induction. LNCaP cells or LTAD cells were treated with DHT (0.1, 1, 10 nm) for 24 h. TACC2 mRNA levels were evaluated by real-time RT-PCR. Fold induction upon treatment with DHT over vehicle control is shown. The data represent the mean ± sd, n = 3. *, P < 0.05; **, P < 0.01, respectively, from LTAD cells. IB, Immunoblotting.
Interestingly, the expression of TACC2 mRNA and protein was found to be up-regulated in these LTAD cells as compared with parental cells under hormone-depleted conditions (Fig. 4, A and B). To investigate the contribution of AR to TACC2 overexpression, we performed AR knockdown (Fig. 4C). We observed that AR knockdown in LTAD cells reduces TACC2 mRNA levels and androgen-mediated up-regulation of TACC2 (Fig. 4D). This suggests that up-regulation of TACC2 in LTAD cells will be derived, in part, from the overexpression of AR. AR overexpression will exert greater occupancy of AR binding at the TACC2 ARBS in LTAD cells relative to parental LNCaP cells compared with other ARBS, either with or without DHT treatment (Fig. 4, E and F, and Supplemental Fig. 4). Furthermore, TACC2 up-regulation compared with LNCaP cells could be observed in the presence of DHT (Fig. 4G). We also examined the androgen sensitivity of TACC2 induction in LTAD cells (Fig. 4H). Compared with LNCaP cells, TACC2 induction at a low level of DHT concentration was observed.
TACC2 regulates castration-resistant cell proliferation by promoting the cell cycle under hormone-deprived conditions
We further investigated whether TACC2 is involved in cell cycle regulation under long-term androgen-deprived conditions. We showed that the siRNA targeting TACC2 effectively reduces the TACC2 expression level in LTAD cells (Fig. 5A). By cell proliferation assay, we demonstrated that TACC2 silencing itself can be an inhibitory factor for proliferation of LTAD cells under hormone-depleted conditions (Fig. 5B). Furthermore, we also analyzed AR and TACC2 cell proliferative function in the absence and presence of androgen using LTAD cells (Fig. 5C). Reduction of AR inhibited LTAD cell proliferation in both hormone-depleted and DHT-treated condition, suggesting AR involvement in castration-resistant cell proliferation as described in the past publication (11). Interestingly, siTACC2 treatment inhibited LTAD cell proliferation more effectively than siAR. TACC2 growth inhibition was more evident in hormone-depleted condition. In addition, addition of siAR to siTACC2-treated cells could not significantly inhibit castration-resistant cell proliferation (Fig. 5D).
Fig. 5.

TACC2 silencing modulates the cell cycle profile and represses LTAD cell growth under hormone-depleted conditions. A, LTAD cells (LTAD #2) were transfected with siControl (50 nm) or siTACC2 (20 nm or 50 nm) for 48 h. Cell lysates were immunoblotted with anti-TACC2 and anti-β-actin antibodies. B, Retardation of LTAD cell proliferation by siTACC2 transfection. LTAD cells (3000 cells per well) were transfected with siRNA and subjected to the MTS assay 1–5 d after transfection. The data represent the mean ± sd, n = 4. *, P < 0.05; **, P < 0.01 from siControl. C, Inhibition of vehicle and DHT treated-LTAD cell proliferation by siAR (10 nm) and siTACC2 (50 nm) transfection. LTAD cells were transfected with siRNAs 48 h before vehicle and DHT (1 or 10 nm) treatment and subjected to the MTS assay 5 d after vehicle or DHT stimulation. The data represent the mean ± sd, n = 5. *, P < 0.05; **, P < 0.01; ***, P < 0.001 siAR: from siControl, siTACC2: from siAR. D, LTAD cells were transfected with siRNAs (30 nm siControl, 20 nm siTACC2 + 10 nm siControl or 20 nm siTACC2 + 10 nm siAR) 48 h before vehicle and DHT (10 nm) treatment and subjected to the MTS assay 3 d after vehicle or DHT stimulation. The data represent the mean ± sd, n = 4. *, P < 0.05; **, P < 0.01; ***, P < 0.001 siTACC2: from siControl, siTACC2 +siAR: from siTACC2. N.S, not significant. E and F, Alteration of the cell cycle profile in LTAD cells. E, LTAD cells were transfected with siControl or siTACC2 (50 nm each) for 72 h. After serum starvation for 24 h, cells were treated with 20% FCS for 24 h after which cell cycle analysis was performed. The data represent the mean ± sd, n = 2. *, P < 0.05 from siControl. F, LTAD cells were transfected with siControl or siTACC2 (50 nm each) for 72 h. LNCaP cells were transfected with siControl (50 nm) for 72 h. The cells were then treated with nocodazole (0.5 μg/ml) for 14 h. Plates were washed with PBS and cells were cultured in fresh hormone-depleted medium. Cell cycle analysis was performed after 24 h incubation. The data represent the mean ± sd, n = 2. *, P < 0.05; **, P < 0.001 from siControl. N.S, not significant. IB, Immunoblotting.
To analyze the role of TACC2 in cell cycle progression, we synchronized the cells at G0/G1 phase and at G2/M phase. We performed FACS analysis after release from cell cycle arrest. We observed significant G2/M accumulation and a decrease in the proportion of cells at S-phase in LTAD cells transfected with siTACC2, 24 h after the release from synchronization at G0/G1 phase. This suggests that cell cycle inhibition occurs at G2/M (Fig. 5E). We further demonstrated that a robust decrease of the proportion of cells in S-phase could be caused by TACC2 knockdown after G2/M synchronization (Fig. 5F). Moreover, we also demonstrated that TACC2 knockdown inhibits cell proliferation of DU145 cells, AR-independent prostate cancer cell line (Supplemental Fig. 5). Thus, TACC2 is required for cell proliferation in both AR-positive hormone-sensitive and AR-negative hormone refractory prostate cancer cell lines.
To further analyze the mitotic features of LTAD cells treated with siTACC2, we did immunofluorescence analysis of these cells using anti-TACC2 and anti-γ-tubulin, centrosome marker (Supplemental Fig. 6). TACC2 knockdown reduces γ-tubulin staining of centrosomes in most cells or cells in mitosis exhibited abnormal mitotic spindles with more than two centrosomes or single, whereas in control cells two clear centrosomes were detected in mitotic cells (Supplemental Fig. 6, A and B). Furthermore in LTAD cells incubated with siTACC2 for a long time (>5 d), we observed chromatin instability during mitosis (Supplemental Fig. 6, C and D).
Overexpression of TACC2 promotes the cell cycle at G2/M phase
Furthermore, we examined whether the overexpression of TACC2 plays an oncogenic role in prostate cancer. We generated LNCaP cells stably expressing TACC2 and confirmed exogenous TACC2 expression by Western blot analysis (Fig. 6A). These TACC2-overexpressing cells proliferated faster than parental LNCaP cells (Fig. 6B). We further observed androgen responsiveness of cell proliferation in TACC2 overexpressing LNCaP cells by MTS assay (Fig. 6C). This result implied that AR functions almost normally to enhance cell proliferation even in the TACC2-overexpressing LNCaP cells.
Fig. 6.

TACC2 overexpression accelerates the cell cycle at G2/M phase for prostate cancer cell proliferation. A, Generation of LNCaP cells stably expressing TACC2. Expression of exogenous FLAG-tagged TACC2 was confirmed by Western blotting using anti-FLAG and anti-TACC2 antibodies. β-Actin antibody was used as a loading control. Arrow indicates the bands of endogenous TACC2 protein. B, Ectopic TACC2 expression promotes cultured LNCaP cell growth. Cell viability was measured by the MTS assay. The data represent the mean ± sd, n = 4. **, P < 0.0001 from vector-expressing cells (Vector 1 and 2). C, Androgen-dependent cell proliferation in TACC2 overexpressing LNCaP cells. Vector-expressing and TACC2-expressing LNCaP cells were treated with 10 nm DHT or vehicle. Cell viability was measured 3 d after treatment by the MTS assay. The data represent the mean ± sd, n = 4. **, P < 0.001; ***, P < 0.0001 from vehicle-treated cells. D and E, TACC2 overexpression accelerates cell cycle progression after G2/M synchronization. D, After serum starvation for 24 h, vector-expressing or TACC2-overexpressing cells were treated with 20% FCS for 0, 6, 12, and 24 h, after which cell lysates were immunoblotted with anti-cyclin D1 and β-actin antibodies. E, After treatment with nocodazole (0.5 μg/ml) for 14 h, plates were washed and cells were incubated in fresh medium. Cell lysates after 0-, 6-, and 24-h incubation were immunoblotted with anticyclin D1 and anti-β-actin antibodies. F, TACC2 overexpression promotes the cell cycle of LNCaP cells after G2/M synchronization. After cells were treated with nocodazole (0.5 μg/ml) for 14 h, the medium was changed to fresh medium. Cell cycle analysis was performed after a 24-h incubation period. Representative result of repeated three experiments (two clones for each group) is shown. Percentage of G2/M phase cells for each clone is described in the graph. IB, Immunoblotting;
To investigate the effect of the increased level of TACC2 expression on the cell cycle, we synchronized the cells, and a cell cycle analysis was performed. No significant differences were observed in cyclin D1 induction, which is important for the G1/S transition after release from G0/G1 arrest by fetal bovine serum (FBS) treatment, between vector-expressing and TACC2-overexpressing cells (Fig. 6D). In addition, the FACS analysis did not reveal any significant alterations after synchronization at G0/G1 (data not shown). However, after G2/M synchronization we observed rapid induction of cyclin D1 in TACC2-overexpressing cells relative to vector-expressing cells (Fig. 6E). Moreover, by FACS analysis 24 h after release from G2/M arrest, we identified a significant decrease of cells at G2/M-phase and accumulation of cells at S-phase (Fig. 6F). These data suggest that TACC2 plays an important role in G2/M cell cycle progression.
TACC2 is a novel prostate cancer prognostic factor associated with castration-resistant tumor proliferation
We investigated whether TACC2 overexpression is critical for castration resistance of prostate cancer cells by using immunocompromised mice. We sc injected LNCaP cells stably expressing TACC2 and performed castration when the initial tumor volume reached approximately 100 mm3. Even under hormone-depleted conditions, TACC2-expressing cells were found to express a substantial amount of TACC2 protein relative to androgen-treated LNCaP cells (Fig. 7A). Tumor growth was found to be robustly repressed in castrated mice implanted with parental cells, whereas substantial tumor growth was demonstrated in castrated mice implanted with TACC2-overexpressing cells (Fig. 7B). Taken together, our results show that TACC2 is a growth-promoting factor in a castration-resistant phase of prostate cancer.
Fig. 7.
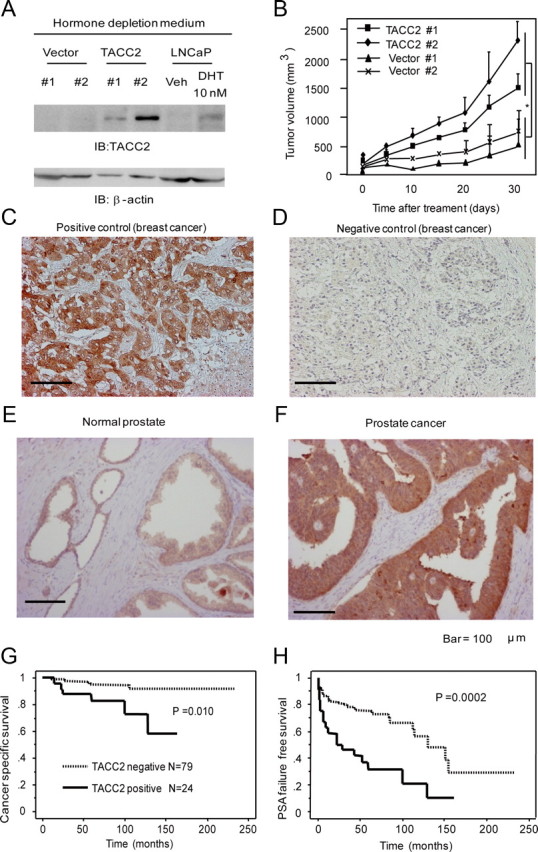
TACC2 overexpression is associated with castration-resistant tumor proliferation and can be a poor prognostic factor in prostate cancer cases. A, Expression levels of TACC2 in vector-expressing and TACC2-overexpressing LNCaP cells under hormone-depleted conditions. Cell lysates of vector-expressing and TACC2-overexpressing LNCaP cells and parental LNCaP cells treated with vehicle or 10 nm DHT for 48 h were harvested. Immunoblots were performed using anti-TACC2 and anti-β-actin antibodies. B, TACC2 overexpression substantially promotes in vivo tumor formation in male nude mice after castration. Data represent a 95% confidence interval of tumor volumes (n = 6 for each group; three per each clone). *, P < 0.01 from vector-expressing cells. C–F, Immunohistochemistry of TACC2 in prostate cancer tissues. TACC2 staining in breast cancer tissues (C) was used as positive control and staining with normal IgG (D) was used as negative control. Representative immunohistochemistry of TACC2 in normal prostate tissue with focal staining (E), and a prostate cancer tissue with intense and diffuse cytoplasmic staining (F). Scale bar, 100 μm. G and H, TACC2 immunoreactivity is positively correlated with a poor prognosis of postoperative prostate cancer patients. Kaplan-Meier analysis reveals lower cancer-specific survival (G) and PSA failure-free survival (H) in patients with positive TACC2 staining compared with those with negative staining. IB, Immunoblotting; Veh, vehicle.
We further evaluated TACC2 immunoreactivity in clinical samples. Immunohistochemical analysis was performed using breast cancer tissue as positive control (Fig. 7C), whereas we observed no staining when normal IgG was used as primary antibody (Fig. 7D). TACC2 immunoreactivity in normal prostate tissues was found to be usually weak and focused in the cytoplasm of prostate epithelial cells (Fig. 7E). In contrast, intense and diffuse TACC2 immunoreactivity was detected in the cytoplasm of prostate carcinoma cells (Fig. 7F). Positive status of TACC2 immunoreactivity was defined as complete cytoplasmic staining in more than 10% of the tumor cell population. Of 103 prostate cancer specimens, 24 samples were classified as TACC2-positive prostate carcinomas. Kaplan-Meier analysis revealed that TACC2-positive status is significantly correlated with poor cancer-specific survival (P = 0.01) (Fig. 7G) and PSA failure-free survival (P = 0.0002) (Fig. 7H) in this tumor population. In terms of associations between TACC2 immunoreactivity and clinicopathological parameters in 103 prostate carcinomas, the Gleason score was found to be positively correlated with the positive status of TACC2 (Supplemental Table 2; P = 0.047). Multivariate analysis revealed that the positive status of TACC2 is an independent prognostic factor for PSA failure (Supplemental Table 3; P = 0.01; relative risk, 2.2; 95% confidence intervals, 1.2–4.1).
Discussion
The present study showed that TACC2 is a critical androgen-regulated gene that promotes cell proliferation as well as in vivo tumor progression in prostate cancer by regulating cell cycle at mitosis. The ChIP-cloning strategy revealed functional ARBS capable of robust ligand-dependent AR recruitment in the vicinity of TACC2, which is associated with the androgen-dependent activated hallmarks of histone modification. Androgen-dependent TACC2 induction is directly regulated by AR, as confirmed by siRNA-mediated AR knockdown or the antiandrogen bicalutamide. In LTAD cells generated as a CRPC cell model, we demonstrated that TACC2 is abundantly expressed and contributes to hormone-refractory proliferation with siRNA-mediated knockdown of TACC2-reduced cell growth and suppressed cell cycle progression. An in vivo tumor formation study of prostate cancer in castrated immunocompromised mice revealed that TACC2 is a primary tumor-promoting factor. A clinicopathological study revealed a significant correlation between high TACC2 immunoreactivity and poor patient prognosis. Taken together, our data show that TACC2 is a primary AR downstream gene that modulates prostate cancer progression.
The members of the TACC family of proteins contain C-terminal coiled-coil domains (18) and are implicated in cancers via their function in the formation of the mitotic spindle during mitosis. With regard to the TACC functions, TACC1 and TACC3 have been shown to exert oncogenic roles in tumors: TACC1 promotes transformation and mammary tumorigenesis in a mouse model (32), and TACC3 is up-regulated in various cancer cells (33). TACC3 aberrations have also been described in patients with ovarian and non-small-cell lung cancer (34, 35). TACC2 has been initially identified as a potential tumor suppressor in breast cancer cells (20, 36), although its biological role in tumor formation remains controversial, because a recent report revealed that TACC2 correlates with poor prognosis in patients with breast cancer (22). Our findings revealed that TACC2 plays an oncogenic role in clinical prostate cancer tissues. The molecule is an independent prognostic factor in the prostate cancer population that we examined in the present study.
Resistance to hormonal therapies continues to be a major clinical problem in prostate cancer. Growing evidence suggests that AR plays an essential role in the transition to castration-resistant proliferation (3). For example, AR amplification (37), hypersensitivity to low levels of androgen in prostate tissues (8, 38), bidirectional signal cross talk between growth factor receptor pathway and AR (39), and AR variant (40) are major mechanisms that have been proposed previously. In the present study, we established a castration-resistant prostate cancer model by long-term androgen deprivation. In LTAD cells, we confirmed that AR expression is significantly up-regulated. Although the DHT-dependent up-regulation of TMPRSS2 mRNA was not substantial in LTAD cells compared with parental LNCaP cells, DHT up-regulated the mRNA expressions of other androgen target genes even at a low concentration, as exemplified by APP (13) and ARFGAP3 (31), which we previously identified as androgen-responsive growth-promoting factors. These data will suggest that the androgen sensitivity is enhanced at least in the transcriptional regulation of several gene promoters in LTAD cells compared with parental LNCaP cells. Therefore, we assume that our LTAD cell model is a suitable system for investigating the molecular mechanisms underlying the development of CRPC. We showed increased TACC2 expression in both the absence and presence of DHT and hypersensitivity of TACC2 induction in LTAD cells. Because the AR occupancy at the TACC2 ARBS was found to be increased in LTAD cells at both basal and hormone-stimulated states, AR regulation of TACC2 in LTAD cells was shown by AR-depleted LTAD cells. In addition, siTACC2 treatment inhibited cell proliferation more evidently than siAR, and addition of siAR to siTACC2-treated cells did not change cell proliferation significantly. These data suggests that TACC2 may play an important role in the amplified AR signaling for castration resistance. Published ChIP-Seq data support this proposal because the enrichment of AR binding at the TACC2 ARBS is also shown in metastatic prostate cancer tissues as well as in androgen-sensitive prostate cancer cell lines. Acceleration of tumor formation by TACC2 overexpression in castrated nude mice provides further evidence that the molecule will directly contribute to the development of CRPC.
We further characterized the involvement of TACC2 in cell cycle regulation. In unsynchronized LNCaP cells, we observed TACC2 knockdown leads to G2/M accumulation. This suggests that TACC2 plays a role in G2/M progression. On the contrary, overexpression of TACC2 increases S-phase entry after release from G2/M synchronization. Therefore, our results indicate that TACC2 functions in cell cycle progression at G2/M. We further investigated the role of TACC2 in cell cycle progression for castration-resistant cell proliferation. In our FACS analysis, we demonstrated that TACC2 silencing leads to G2/M accumulation, whereas there is a decrease in the cell population at S-phase in LTAD cells after the release from G0/G1 synchronization. Moreover, we observed that there is a more evident decrease of cell population in the S-phase after the G2/M synchronization rather than G0/G1 synchronization. In addition to the role in hormone-dependent cell cycle regulation, TACC2 plays an important role in castration-resistant cell proliferation by controlling G2/M progression. The cell cycle-regulatory function of TACC2 in LTAD cells will be important to investigate, because cell cycle progression at G2/M phase is a key regulatory signal of AR in CRPC (11).
To further investigate the molecular basis for TACC2 involvement in cell cycle regulation necessary for castration-resistant proliferation, we observed centrosomes by immunofluorescence analysis in mitotic LTAD cells. In our analysis, TACC2-depleted cells exhibited abnormal cell mitosis with multipolar, monopolar, or no staining of centrosomes. Moreover, chromosome instability with no centrosome in mitosis was observed. Taken together, these data indicated that TACC2 is necessary to promote cell division by forming centrosomes and maintain chromatin stability in mitosis.
According to the recent review (41), several markers such as AMACR, EZH2, or fusion genes have been tested for assigning patients to prognostic groups. However, no immunochemical marker is currently used for this purpose. In this study, we found TACC2 overexpression is an independent prognostic marker of PSA-free survival, suggesting that TACC2 may be a prognostic biomarker to predict aggressive tumor. Moreover, because TACC2 expression is significantly correlated with Gleason score and TACC2 functions for the progression of G2/M, we assume that TACC2 expression can be a novel marker of tumor proliferation rate.
Our findings indicate that TACC2-dependent mitotic spindle assembly and chromosomal stability are required for proliferation of hormone-naive prostate cancer cells as well as CRPC cells with the amplification of AR signaling. The cell cycle-promoting effect of TACC2 will be aberrantly modulated by exogenous factors such as viral large T antigen, because the interaction of T antigen with TACC2 induces microtubule dysfunction, leading to disorganized mitotic spindles, slow progression of mitosis, and chromosome missegregation (17). Furthermore, the cell cycle-related nature of the TACC family has also been reported in TACC3, which is highly expressed during the G2/M phase where it localizes to the centrosome and spindle apparatus (43). Because TACC3 silencing in NIH3T3 cells causes a p53-dependent induction of p21WAF and subsequent cell cycle arrest and leads to the sensitization of cells for paclitaxel treatment (44), TACC2 knockdown will potentiate the chemoresistance of CRPC cells by modulating the functions of cell cycle checkpoints. Because we observed nuclear staining of TACC2 during interphase and the TACC protein family is known to interact with the histone acetylation transferase family (23), we consider that TACC2 will also be involved in the activation of transcriptional machinery and chromatin remodeling in prostate cancer at both the androgen-dependent and hormone-refractory stages.
In summary, we here propose that TACC2 is a critical factor for the progression of androgen-dependent and -independent prostate cancers in the AR-regulated gene network. Considering its modulating functions of transcription activation and cell cycle progression, TACC2 will be a potential therapeutic target in prostate cancer, especially in CRPC.
Materials and Methods
Cell lines, plasmid constructs, and reagents
LNCaP and DU145 cells, which are human prostate cancer cells, were grown in RPMI 1640 supplemented with 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin. Before androgen treatment, the cells were cultured in phenol red-free medium containing 5% charcoal-stripped FBS for 48–72 h. VCaP and 293T cells were maintained in DMEM supplemented with 10% FBS. Subline LTAD cells were established by maintaining LNCaP cells in phenol-red free RPMI 1640 supplemented with 10% charcoal-stripped FBS for more than 9 months as described elsewhere (24–26). For construction of ARBS-containing luciferase vectors, ARBS sequences were amplified from Bluescript vectors containing AR ChIP DNA prepared for sequence analysis (16). Amplified PCR products were inserted into pGL3-promoter vectors (Mlu Ι–Xho Ι). The full-length TACC2 gene was amplified by PCR from TACC2 (NM_206860) cDNA (IMAGE: 5295870) purchased from Invitrogen (Carlsbad, CA). The PCR product was inserted into pcDNA3-Flag tagged at N-terminus (Not Ι–Xho Ι) in frame. LNCaP cells were transfected with expression vectors using FUGEN 6 reagent (Roche Applied Science, Indianapolis, IN). For generating stable cell lines of pcDNA3-Flag-TACC2 or empty vectors, cells were selected in 0.5 mg/ml G418 (Wako, Saitama, Japan). Antibodies for α-tubulin, Flag, AR, AcH3, AcH4, phospho-RNA Pol II, and RNA Pol II were used as described previously (12, 16, 29). Antibodies for H3K4mono-methyl (antibody 8895) and γ-tubulin (antibody 11316) were purchased from Abcam (Cambridge, UK). Antibody for TACC2 (07-228) was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Antibody for CyclinD1 (HD11) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). R1881 was purchased from PerkinElmer (Waltham, MA). Dihydrotestosterone (DHT) was purchased from Wako. Bicalutamide and Nocodazole were purchased from Sigma (St. Louis, MO).
ChIP and ChIP cloning
ChIP was performed as described previously (12–14). LNCaP cells or LTAD cells were fixed with 1% formaldehyde for 5 min at room temperature. Chromatin was sheared to an average size of 500 bp by sonication using Bioruptor ultrasonicator (Cosmo-bio, Tokyo, Japan). Lysates were rotated at 4 C overnight with anti-AR, AcH3, AcH4, AcH4, H3K4mono-methyl, phospho-RNA Pol II, and RNA Pol II antibody. Protein A-agarose was added and rotated for 1 h. Washing and reversal of cross-linking at 65 C were performed. DNA fragments were obtained by ethanol precipitation.
For ChIP-cloning, representational difference subtractive hybridization between ChIP DNA from R1881-treated LNCaP cells and ChIP DNA from vehicle-treated LNCaP cells was performed using GeneFisher PCR Subtraction System (TaKaRa Bio, Kyoto, Japan) with some modification (16). In brief, ChIP DNA from vehicle-treated LNCaP cells was blunted, ligated to linkers with BamHΙ site, and amplified by PCR followed by treatment with BamHΙ for removing linkers. ChIP DNA from R1881-treated LNCaP cells was also blunted, ligated to another set of linkers with BamHΙ site, and mixed with amplified ChIP DNA from vehicle-treated cells for subtractive hybridization. After hybridization, not hybridized DNA was amplified by PCR. The linkers were removed by BamHΙ digestion. This procedure was repeated twice for enhancing the specificity of AR binding ChIP DNA. DNA fragments were cloned to Bluescript vector and sequenced. We performed sequence analysis of inserted DNA in more than 100 clones. By determining their genomic location by BLAST searching, we identified 69 fragments that are distributed within 50 kb apart from transcriptional start sites of Refseq genes or within Refseq genes. Sequences for human RefSeq transcripts (hg 15) were retrieved from the UCSC Genome Browser (28). Enrichment analysis of androgen response elements (ARE) was performed based on a position-weighted matrix method, using position-weighted matrices retrieved from TRANSFAC (12). The threshold score of the ARE motif was 1e-4 against the background. Among them, we set 30 ChIP primers targeting genomic regions of putative ARBS (including ARE-like sequences) for validation by performing conventional ChIP analysis. We observed ligand-dependent increase of AR binding at least on 12 AR-binding sites (>2-fold). We selected the top five ARBS for further study.
Quantitative RT-PCR
Total RNA was isolated using ISOGEN reagent (NIPPON Gene, Tokyo, Japan). First-strand cDNA was synthesized using the Primescript RT reagent kit (TaKaRa Bio) according to manufacturer's protocol. The primer sequences for AR and TACC2 are listed in Supplemental Table 4. mRNA was quantified by real-time-PCR using SYBR green PCR mix (Applied Biosystems, Foster City, CA) and ABI Prism 7000 system (Applied Biosystems) based on SYBR Green fluorescence. The evaluation of relative differences of PCR product amounts among the treatment groups was carried out by the comparative cycle threshold (Ct) method, using glyceraldehyde-3-phosphate dehydrogenase as an internal control (12).
Quantitative ChIP-PCR
Fold enrichments relative to IgG or vehicle control were quantified by real-time PCR. Relative differences in the amounts of PCR products among the treatment groups were evaluated by the comparative cycle threshold (Ct) method, using glyceraldehyde-3-phosphate dehydrogenase as an internal control (12). The primer sequences for ARBS 1–5 are listed in Supplemental Table 4. The primer sequences for ARBS 3 (UGT1A) and positive control PSA enhancer were described previously (12).
Patients and tissue samples
Surgical samples (103) of prostate cancer were obtained from Tokyo University Hospital (Tokyo, Japan) after informed consent was obtained from the patients. The study was approved by the Tokyo University Ethics Committee. Clinicopathological data of the patients are listed in Supplemental Table 2.
Immunohistochemistry
Formalin-fixed tissues were embedded in paraffin and sectioned. A Histofine Kit (Nichirei, Tokyo, Japan), which employs the streptavidin-biotin amplification method, was used for immunohistochemical analysis of TACC2. Antigen retrieval was performed by heating the slides in an autoclave at 120 C for 5 min in citric acid buffer [2 mm citric acid and 9 mm trisodium citrate dehydrate (pH 6.0)]. The antigen-antibody complex was visualized with 3,3′-diaminobenzidine solution [1 mmol/liter 3,3′-diaminobenzidine, 50 mmol/liter Tris-HCl buffer (pH 7.6), and 0.006% H2O2] and counterstained with hematoxylin (29). As a negative control, normal rabbit IgG was used instead of the primary antibodies.
TACC2 immunoreactivity was detected in the cytoplasm, and the immunoreactivity was evaluated in more than 1000 carcinoma cells for each case. Subsequently, the percentage of immunoreactivity was determined by two pathologists. Cases with more than 10% of TACC2 immunoreactivity were considered TACC2-positive prostate cancers in this study. An association between TACC2 immunoreactivity and clinicopathological factors was evaluated using a Student's t test, a cross-table using the χ2 test, or a correlation coefficient (r) and regression equation. Cancer-specific survival and PSA failure-free survival curves were generated according to the Kaplan-Meier method, and the statistical significance was calculated using the log-rank test. Uni- and multivariate analyses were evaluated using Cox's proportional hazard model with PROC PHREG in SAS software (version 9.2; SAS Institute, Inc.).
Immunocytochemistry
For synchronizing LNCaP cells, cells were stimulated by 10% serum after serum starvation for 24 h. After 24-h incubation, cells were washed three times with PBS and fixed in 4% formaldehyde in PBS for 15 min at room temperature. After another washing step with PBS and blocking in 5% normal goat serum/PBS for 30 min, cells were first incubated with mouse antitubulin monoclonal and rabbit anti-TACC2 polyclonal antibodies in 5% normal goat serum/PBS overnight, washed three times with PBS, and then incubated with fluorescein isothiocyanate-conjugated antirabbit IgG and rhodamine-conjugated antimouse IgG antibodies for 1 h. Nuclei were stained with 4′,6-diamidino-2-phenylindole. Cover glasses were mounted in glycerol and visualized. We compared the proportion of mitosis (premetaphase and postmetaphase) in control siRNA-transfected LNCaP cells with siTACC2-transfected cells using the two-sided χ2 test. We randomly surveyed five fields and determined whether cells in mitosis are in premetaphase or postmetaphase.
Cell cycle analysis
For unsynchronized cell cycle analysis, cells were harvested after 96-h incubation after transfection of siControl and siTACC2. For synchronization at G0/G1, serum starvation for 24 h was performed using phenol-red free RPMI 1640 after which cells were treated with 20% fetal calf serum (FCS). For synchronization at G2/M, we added Nocodazole (0.5 μg/ml) for 14 h after which medium was changed. After 24-h incubation, cells were centrifuged, washed in PBS, and then fixed by slow addition of 3 ml of ice-cold 70% ethanol with mild shaking. They were stored at 4 C until use. On the day of cycle analysis, the cells were centrifuged, washed in PBS, resuspended in 1 ml of PBS containing 100 μg/ml ribonuclease (TaKaRa Bio) per 106 cells, and incubated at 37 C for 30 min. To determine DNA content, 30,000 cells were analyzed by FACS Calibur flow cytometry using Cell Quest software (BD Biosciences, San Diego, CA).
Western blot analysis
Whole-cell lysates were prepared using lysis buffer [50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 0.1% TritonX-100, 1.5 mm MgCl2, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mm phenylmethylsulfonylfluoride]. Lysates were resolved by 8% SDS-PAGE and electroblotted onto Immobilon-P Transfer Membrane (Millipore Corp., Billerica, MA). Membranes were first incubated with primary antibody [anti-TACC2 (1:1000), anti-Flag (1:1000), anti-CyclinD1 (1:500), anti-β-actin (1:5000)] and then with peroxidase-conjugated antirabbit or antimouse IgG antibody for 1 h. After extensive washing, the antibody-antigens were detected using enhanced chemiluminescence plus Western blotting detector system (GE Healthcare, Buckinghamshire, UK).
Cell proliferation assay
Cells were plated at 3000 cells per well on 96-well plates. For stable cell lines of pcDNA3-Flag-TACC2, cells were seeded in RPMI 40 containing 1% FBS. For RNA interference (RNAi) experiments, cells were transfected with siRNA 24 h after cell plating. MTS assay was carried out using cell titer reagent (Promega Corp., Madison, WI) according to the manufacturer's protocol. The experiments were performed in quintuplicate.
RNAi
Stealth RNAi (used in Fig. 5 and Supplemental Fig. 4) targeting TACC2 (HSS116289) and negative control RNAi were purchased from Invitrogen (Carlsbad, CA). Silencer Select siRNA targeting AR (s1539), TACC2 (s20765, s20767: used in Fig. 3), and negative controls 1 and 2 were purchased from Ambion (Austin, TX). Cells were transfected with RNA using Lipofectamine RNAi Max reagent (Invitrogen) for stealth RNAi or siPort NeoFX transfection reagent (Ambion) for Silencer Select siRNA 48–72 h before experiments.
Prostate cancer xenograft model
Five million cells (two clones of LNCaP cells expressing TACC2 or two vector control clones) in 100 μl medium with 100 μl of Matrigel (BD Biosciences, San Jose, CA) were injected sc into each side of 5-wk-old male nude mice (Nihon Crea, Tokyo, Japan). These mice were untreated until tumors could be observed. Tumors were measured with calipers two times per wk. When the volume of tumors reached approximately 100 mm3 (i.e. in 3–4 wk after injection) castration was performed (n = 6 for each group; three per each clone). Tumor volume was determined using the formula 0.5 × r1 × r2 × r3 (r1 < r2 < r3: three axes of tumor). Mice were killed by cervical dislocation 30 d after castration. Animal care was maintained in accordance with institutional guidelines.
Luciferase assay
LNCaP cells were plated at a density of 30,000 cells per well in a 24-well culture plate and cultured for 48 h in phenol red-free RPMI 1640 containing 5% charcoal-stripped FBS. Cells were transfected with plasmids using the transfection reagent FuGENE 6 and, 24 h later, were treated with R1881 (10 nm) or vehicle (0.1% ethanol) for 24 h. Luciferase activity of cell lysates was determined by the Dual Luciferase Assay Kit (Promega Corp.) according to standard procedures. A Renilla Luciferase Reporter Tk-pRL was cotransfected as a control for evaluating transfection efficiency.
Statistical analyses
For examining statistical analysis, each experiment was analyzed in duplicate or in triplicate. For the cell proliferation assay, we analyzed four wells. For the growth assay in vitro and in vivo of TACC2 stable cell lines, we performed two-way ANOVA at each time point. For other cell line experiments, statistical differences (P values) among groups were obtained using a two-sided Student's t test. All experiments were performed at least twice and similar results were obtained. P < 0.05 was considered to be statistically significant. Statistical procedures were performed using GraphPad Prism 5 software (GraphPad Software, San Diego, CA) or Excel.
Acknowledgments
This work was supported by Grants of the Cell Innovation Program (to S.I.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; by Grants (to S.I., T.U., and S.T) from the Japan Society for the Promotion of Science, Japan; by Grants-in-Aid (to S. I.) from the Ministry of Health, Labour, and Welfare, Japan; by the Program for Promotion of Fundamental Studies in Health Sciences (to S.I.), National Institute of Biomedical Innovation, Japan.
Disclosure summary: The authors have nothing to disclose.
NURSA Molecule Pages†:
Nuclear Receptors: AR;
Ligands: R1881.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- AR
- Androgen receptor
- ARE
- androgen response element
- ARBS
- AR-binding site
- ChIP
- chromatin immunoprecipitation
- ChIP-chip
- ChIP analyses using tiled oligonucleotide microarrays
- ChIP-Seq
- ChIP analyses using deep sequencing
- CRPC
- castration-resistant prostate cancer
- DHT
- dihydrotestosterone
- FACS
- fluorescence-activated cell sorting
- FBS
- fetal bovine serum
- FCS
- fetal calf serum
- LTAD
- long-term androgen-deprived
- MTS
- 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl-2H-tetrazolium
- Pol
- polymerase
- PSA
- protstate-specific antigen
- RNAi
- RNA interference
- siRNA
- short-interfering RNA
- TACC2
- transforming acidic coiled-coil protein 2.
References
- 1. Dehm SM , Tindall DJ. 2006. Molecular regulation of androgen action in prostate cancer. J Cell Biochem 99:333–344 [DOI] [PubMed] [Google Scholar]
- 2. Debes JD , Tindall DJ. 2004. Mechanism of androgen-refractory prostate cancer. N Engl J Med 351:1488–1490. [DOI] [PubMed] [Google Scholar]
- 3. Suzuki H , Ueda T , Ichikawa T , Ito H. 2003. Androgen receptor involvement in the progression of prostate cancer. Endocr Relat Cancer 10:209–216 [DOI] [PubMed] [Google Scholar]
- 4. Balk SP , Knudsen KE. 2008. AR, the cell cycle, and prostate cancer. Nucl Recept Signal 6:e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen CD , Welsbie DS , Tran C , Baek SH , Chen R , Vessella R , Rosenfeld MG , Sawyers CL. 2004. Molecular determinants of resistance to antiandrogen therapy. Nat Med 10:33–39 [DOI] [PubMed] [Google Scholar]
- 6. Feldman BJ , Feldman D. 2001. The development of androgen-independent prostate cancer. Nat Rev Cancer 1:34–45 [DOI] [PubMed] [Google Scholar]
- 7. Gregory CW , He B , Johnson RT , Ford OH , Mohler JL , French FS , Wilson EM. 2001. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res 61:4315–4319 [PubMed] [Google Scholar]
- 8. Waltering KK , Helenius MA , Sahu B , Manni V , Linja MJ , Jänne OA , Visakorpi T. 2009. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res 69:8141–8149 [DOI] [PubMed] [Google Scholar]
- 9. Wang Q , Li W , Liu XS , Carroll JS , Jänne OA , Keeton EK , Chinnaiyan AM , Pienta KJ , Brown M. 2007. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 27:380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shang Y , Myers M , Brown M. 2002. Formation of the androgen receptor transcription complex. Mol Cell 9:601–610 [DOI] [PubMed] [Google Scholar]
- 11. Wang Q , Li W , Zhang Y , Yuan X , Xu K , Yu J , Chen Z , Beroukhim R , Wang H , Lupien M , Wu T , Regan MM , Meyer CA , Carroll JS , Manrai AK , Jänne OA , Balk SP , Mehra R , Han B , Chinnaiyan AM , Rubin MA , True L , Fiorentino M , Fiore C , Loda M , Kantoff PW , Liu XS , Brown M. 2009. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takayama K , Kaneshiro K , Tsutsumi S , Horie-Inoue K , Ikeda K , Urano T , Ijichi N , Ouchi Y , Shirahige K , Aburatani H , Inoue S. 2007. Identification of novel androgen response genes in prostate cancer cells by coupling chromatin immunoprecipitation and genomic microarray analysis. Oncogene 26:4453–4463 [DOI] [PubMed] [Google Scholar]
- 13. Takayama K , Tsutsumi S , Suzuki T , Horie-Inoue K , Ikeda K , Kaneshiro K , Fujimura T , Kumagai J , Urano T , Sakaki Y , Shirahige K , Sasano H , Takahashi S , Kitamura T , Ouchi Y , Aburatani H , Inoue S. 2009. Amyloid precursor protein is a primary androgen target gene that promotes prostate cancer growth. Cancer Res 69:137–142 [DOI] [PubMed] [Google Scholar]
- 14. Takayama K , Tsutsumi S , Katayama S , Okayama T , Horie-Inoue K , Ikeda K , Urano T , Kawazu C , Hasegawa A , Ikeo K , Gojyobori T , Ouchi Y , Hayashizaki Y , Aburatani H , Inoue S. 2011. Integration of cap analysis of gene expression and chromatin immunoprecipitation analysis on array reveals genome-wide androgen receptor signaling in prostate cancer cells. Oncogene 30:619–630 [DOI] [PubMed] [Google Scholar]
- 15. Yu J , Yu J , Mani RS , Cao Q , Brenner CJ , Cao X , Wang X , Wu L , Li J , Hu M , Gong Y , Cheng H , Laxman B , Vallaichamy A , Shankar S , Li Y , Dhanasekaran SM , Morey R , Barrette Y , Lonigro RJ , Tomlins SA , Varambally S , Qin ZS , Chinnaiyan AM. 2010. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17:443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takayama K , Horie-Inoue K , Ikeda K , Urano T , Murakami K , Hayashizaki Y , Ouchi Y , Inoue S. 2008. FOXP1 is an androgen-responsive transcription factor that negatively regulates androgen receptor signaling in prostate cancer cells. Biochem Biophys Res Commun 374:388–393 [DOI] [PubMed] [Google Scholar]
- 17. Tei S , Saitoh N , Funahara T , Iida S , Nakatsu Y , Kinoshita K , Kinoshita Y , Saya H , Nakao M. 2009. Simian virus 40 large T antigen targets the microtubule-stabilizing protein TACC2. J Cell Sci 122:3190–3198 [DOI] [PubMed] [Google Scholar]
- 18. Gergely F , Karlsson C , Still I , Cowell J , Kilmartin J , Raff JW. 2000. The TACC domain identifies a family of centrosomal proteins that can interact with microtubules. Proc Natl Acad Sci USA 97:14352–14357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peset I , Vernos I. 2008. The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol 18:379–388 [DOI] [PubMed] [Google Scholar]
- 20. Chen HM , Schmeichel KL , Mian IS , Lelièvre S , Petersen OW , Bissell MJ. 2000. AZU-1: a candidate breast tumor suppressor and biomarker for tumor progression. Mol Biol Cell 11:1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lauffart B , Gangisetty O , Still IH. 2003. Molecular cloning, genomic structure and interactions of the putative breast tumor suppressor TACC2. Genomics 81:192–201 [DOI] [PubMed] [Google Scholar]
- 22. Cheng S , Douglas-Jones A , Yang X , Mansel RE , Jiang WG. 2010. Transforming acidic coiled-coil-containing protein 2 (TACC2) in human breast cancer, expression pattern and clinical/prognostic relevance. Cancer Genomics Proteomics 7:67–73 [PubMed] [Google Scholar]
- 23. Gangisetty O , Lauffart B , Sondarva GV , Chelsea DM , Still IH. 2004. The transforming acidic coiled coil proteins interact with nuclear histone acetyltransferases. Oncogene 23:2559–2563 [DOI] [PubMed] [Google Scholar]
- 24. Culig Z , Hoffmann J , Erdel M , Eder IE , Hobisch A , Hittmair A , Bartsch G , Utermann G , Schneider MR , Parczyk K , Klocker H. 1999. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer 81:242–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kokontis J , Takakura K , Hay N , Liao S. 1994. Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res 54:1566–1573 [PubMed] [Google Scholar]
- 26. Inoue T , Yoshida T , Shimizu Y , Kobayashi T , Yamasaki T , Toda Y , Segawa T , Kamoto T , Nakamura E , Ogawa O. 2006. Requirement of androgen-dependent activation of protein kinase Cζ for androgen-dependent cell proliferation in LNCaP Cells and its roles in transition to androgen-independent cells. Mol Endocrinol 20:3053–3069 [DOI] [PubMed] [Google Scholar]
- 27. Azuma K , Urano T , Horie-Inoue K , Hayashi S , Sakai R , Ouchi Y , Inoue S. 2009. Association of estrogen receptor alpha and histone deacetylase 6 causes rapid deacetylation of tubulin in breast cancer cells. Cancer Res 69:2935–2940 [DOI] [PubMed] [Google Scholar]
- 28. Kent WJ , Sugnet CW , Furey TS , Roskin KM , Pringle TH , Zahler AM , Haussler D. 2002. The human genome browser at UCSC. Genome Res 12:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suzuki T , Inoue A , Miki Y , Moriya T , Akahira J , Ishida T , Hirakawa H , Yamaguchi Y , Hayashi S , Sasano H. 2007. Early growth responsive gene 3 in human breast carcinoma: a regulator of estrogen-mediated invasion and potent prognostic factor. Endocr Relat Cancer 14:279–292 [DOI] [PubMed] [Google Scholar]
- 30. Makkonen H , Kauhanen M , Jääskeläinen T , Palvimo JJ. 2011. Androgen receptor amplification is reflected in the transcriptional responses of vertebral-cancer of the prostate cells. Mol Cell Endocrinol 331:57–65 [DOI] [PubMed] [Google Scholar]
- 31. Obinata D , Takayama K , Urano T , Ikeda K , Horie-Inoue K , Ouchi Y , Takahashi S , Inoue S. 6 June 2011. ARFGAP3, an androgen target gene, promotes prostate cancer cell proliferation and migration. Int J Cancer. 10.1002/ijc. 26224 [DOI] [PubMed] [Google Scholar]
- 32. Cully M , Shiu J , Piekorz RP , Muller WJ , Done SJ , Mak TW. 2005. Transforming acidic coiled coil 1 promotes transformation and mammary tumorigenesis. Cancer Res 65:10363–10370 [DOI] [PubMed] [Google Scholar]
- 33. Still IH , Vince P , Cowell JK. 1999. The third member of the transforming acidic coiled coil-containing gene family, TACC3, maps in 4p16, close to translocation breakpoints in multiple myeloma, and is upregulated in various cancer cell lines. Genomics 58:165–170 [DOI] [PubMed] [Google Scholar]
- 34. Lauffart B , Vaughan MM , Eddy R , Chervinsky D , DiCioccio RA , Black JD , Still IH. 2005. Aberrations of TACC1 and TACC3 are associated with ovarian cancer. BMC Women's Health 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jung CK , Jung JH , Park GS , Lee A , Kang CS , Lee KY. 2006. Expression of transforming acidic coiled-coil containing protein 3 is a novel independent prognostic marker in non-small cell lung cancer. Pathol Int 56:503–509 [DOI] [PubMed] [Google Scholar]
- 36. Conte N , Delaval B , Ginestier C , Ferrand A , Isnardon D , Larroque C , Prigent C , Séraphin B , Jacquemier J , Birnbaum D. 2003. TACC1-chTOG-Aurora A protein complex in breast cancer. Oncogene 22:8102–8116 [DOI] [PubMed] [Google Scholar]
- 37. Yuan X , Balk SP. 2009. Mechanisms mediating androgen receptor reactivation after castration. Urol Oncol 27:36–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Locke JA , Guns ES , Lubik AA , Adomat HH , Hendy SC , Wood CA , Ettinger SL , Gleave ME , Nelson CC. 2008. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res 68:6407–6415 [DOI] [PubMed] [Google Scholar]
- 39. Guo Z , Dai B , Jiang T , Xu K , Xie Y , Kim O , Nesheiwat I , Kong X , Melamed J , Handratta VD , Njar VC , Brodie AM , Yu LR , Veenstra TD , Chen H , Qiu Y. 2006. 2006 Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10:309–319 [DOI] [PubMed] [Google Scholar]
- 40. Sun S , Sprenger CC , Vessella RL , Haugk K , Soriano K , Mostaghel EA , Page ST , Colema IM , Nguyen HM , Sun H , Nelson PS , Plymate SR. 2010. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 120:2715–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bjartell A , Montironi R , Berney DM , Egevad L. 2011. Tumour markers in prostate cancer II: diagnostic and prognostic cellular biomarkers. Acta Oncol 50 (Suppl 1):76–84 [DOI] [PubMed] [Google Scholar]
- 42. Berney DM , Gopalan A , Kudahetti S , Fisher G , Ambroisine L , Foster CS. 2009. Ki-67 and outcome in clinically localized prostate cancer: Analysis of conservatively treated prostate cancer patients from the Trans-Atlantic Prostate Group study. Br J Cancer 100:888–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Piekorz RP , Hoffmeyer A , Duntsch CD , McKay C , Nakajima H , Sexl V , Snyder L , Rehg J , Ihle JN. 2002. The centrosomal protein TACC3 is essential for hematopoietic stem cell function and genetically interfaces with p53-regulated apoptosis. EMBO J 21:653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schneider L , Essmann F , Kletke A , Rio P , Hanenberg H , Schulze-Osthoff K , Nürnberg B , Piekorz RP. 2008. TACC3 depletion sensitizes to paclitaxel-induced cell death and overrides p21WAF-mediated cell cycle arrest. Oncogene 27:116–125 [DOI] [PubMed] [Google Scholar]