

Abstract
Bile acids are synthesized from cholesterol and are major risk factors for Barrett adenocarcinoma (BAC) of the esophagus. Caveolin-1 (Cav1), a scaffold protein of membrane caveolae, is transcriptionally regulated by cholesterol via sterol-responsive element-binding protein-1 (SREBP1). Cav1 protects squamous epithelia by controlling cell growth and stabilizing cell junctions and matrix adhesion. Cav1 is frequently down-regulated in human cancers; however, the molecular mechanisms that lead to this event are unknown. We show that the basal layer of the nonneoplastic human esophageal squamous epithelium expressed Cav1 mainly at intercellular junctions. In contrast, Cav1 was lost in 95% of tissue specimens from BAC patients (n = 100). A strong cytoplasmic expression of Cav1 correlated with poor survival in a small subgroup (n = 5) of BAC patients, and stable expression of an oncogenic Cav1 variant (Cav1-P132L) in the human BAC cell line OE19 promoted proliferation. Cav1 was also detectable in immortalized human squamous epithelial, Barrett esophagus (CPC), and squamous cell carcinoma cells (OE21), but was low in BAC cell lines (OE19, OE33). Mechanistically, bile acids down-regulated Cav1 expression by inhibition of the proteolytic cleavage of 125-kDa pre-SREBP1 from the endoplasmic reticulum/Golgi apparatus and nuclear translocation of active 68-kDa SREBP1. This block in SREBP1's posttranslational processing impaired transcriptional activation of SREBP1 response elements in the proximal human Cav1 promoter. Cav1 was also down-regulated in esophagi from C57BL/6 mice on a diet enriched with 1% (wt/wt) chenodeoxycholic acid. Mice deficient for Cav1 or the nuclear bile acid receptor farnesoid X receptor showed hyperplasia and hyperkeratosis of the basal cell layer of esophageal epithelia, respectively. These data indicate that bile acid-mediated down-regulation of Cav1 marks early changes in the squamous epithelium, which may contribute to onset of Barrett esophagus metaplasia and progression to BAC.
Barrett esophageal adenocarcinoma (BAC) is a malignancy of increasing incidence and high mortality (1). The main predisposing factor for BAC is Barrett esophagus metaplasia (BE), a preneoplastic lesion characterized by intestinal metaplasia (2, 3). Gastroesophageal reflux disease, in which the esophageal mucosa is chronically exposed to bile salts and acids (4), is the major risk factor for BE (2). The most abundant bile acids in the patient's refluxate are deoxycholic acid (DCA) and chenodeoxycholic acid (CDCA) (5, 6). These bile acids cause chronic inflammation and injury of the epithelium. Chronic esophagitis induces transformation of the normal squamous epithelium to BE and progression to BAC after many years.
Caveolin-1 (Cav1) is a cytoplasmic 22-kDa scaffold protein enriched in lipid rafts, which contain cholesterol and glycosphingolipids (7). Caveolins oligomerize to form caveolae, flask-shaped invaginations of the plasma membrane that regulate endocytosis, vesicular traffic, and cholesterol efflux (8). As a scaffold protein, Cav1 interacts with diverse adhesion and receptor molecules to control cell-cell and cell-matrix interactions and signal transduction. Cav1 thereby acts as a tumor modulator in a tissue type- and stage-dependent manner (9, 10). In esophageal squamous cell carcinoma (SCC), Cav1 is overexpressed and correlates with lymph node metastasis and a poor clinical prognosis (11, 12). In contrast, the expression and/or function of Cav1 in BAC is unknown. We show here, that the majority of BAC patients suffer from a loss of Cav1 expression in the tumor as compared with normal esophageal squamous epithelium.
To clarify the link between bile acids and down-regulation of Cav1 in BAC, we postulated that bile acid signaling is translated by a candidate transcription factor to the Cav1 gene. The nuclear receptors farnesoid X receptor (FXR), liver X receptor and sterol responsive element binding proteins (SREBP) are pivotal transcription factors regulating sterol/lipid metabolism (13). SREBP1 controls target gene expression in response to alterations in cellular sterol contents (14). Consistent with its function as a deficiency sensor, SREBP1 is activated by low sterol but inhibited by high sterol concentrations (e.g. oxysterol, cholesterol) (15). Pre-SREBP1 is synthesized as a 125-kDa precursor, which is inserted into the endoplasmic reticulum (ER) and retained in the ER by SREBP cleavage-activating protein (SCAP)/Insig proteins in presence of high sterols. In contrast, at low sterol conditions, pre-SREBP1 is transported to the Golgi and cleaved by site-specific proteases to release a soluble active 68-kDa fragment which is translocated to the nucleus, where it acts as a transcription factor at sterol-regulatory elements (SRE) in promoters of target genes involved in lipid synthesis (16). The Cav1 gene promoter harbors three G/C-rich SRE (14). Cav1 gene transcription is regulated by SREBP1 in a cell type-specific manner through binding to these SRE (14, 17–19).
We therefore proposed that bile acids mimic high-sterol conditions to inhibit activation of SREBP1 and thereby reduce Cav1 expression. Our data show that bile acids down-regulate Cav1 expression both in vitro and in vivo via SREBP1 and the nuclear bile acid receptor FXR. These signaling events may contribute to early bile acid-induced changes in the esophageal squamous epithelium, which precede the development of BE metaplasia and BAC.
Materials and Methods
Subjects
Tissue specimens from patients with esophageal cancer (EC) were collected, classified into SCC and BAC by histological assessment, and stored as described previously (20). The study was approved by the Ethics Committee of the Technische Universität München.
Animals
Wild-type (WT) C57BL/6 and FXR-knockout (KO) mice (strain B6.129X1(FVB)-Nr1h4tm1Gonz/J; stock number 007214) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice (male, 8 wk) were randomized (n = 5 per group) and fed a chow diet or a chow diet enriched with 1% (wt/wt) CDCA or 0.2% DCA (with 5 ppm Zn2+) for 7 d or 8 wk as described previously (21). Homozygous Cav1 knockout (Cav1-KO) (strain Cav1tm1Mls/J; stock number 004585) and matched WT (strain B6129SF2/J; stock number 101045) mice were purchased from The Jackson Laboratory. In vivo labeling with bromodeoxyuridine (BrdU) was performed as published (21). Animal studies were conducted in agreement with ethical guidelines of the Technische Universität München and approved by the appropriate government authorities.
Reagents
Polyclonal antisera were Cav1 (N-20, sc-894; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), SREBP1 (PA1–46142, Thermofisher Scientific, Waltham, MA), SREBP1 (C-20, sc-366), SCAP (C-20, sc-9675), FXR (goat, Q-20, sc-1205, C terminus), and FXR (rabbit, H-130, sc-13063, N-terminus), Ki-67 (SP6, DCS GmbH, Hamburg, Germany), HSP90 (H-114, sc-7947), Furin (H-220, sc-20801). Mouse monoclonal antibodies were MEK1 (H-8, sc-6250), Cav1 (2297, BD/Transduction Laboratories, San Jose, CA), FXR (mouse, clone A9033A, N terminus; R&D Systems, Wiesbaden-Nordenstadt, Germany), and β-Actin (AC74, Sigma, Taufkirchen, Germany). Inhibitor for calpain I and proteasome acetyl-l-leucyl-l-leucyl-l-norleucinal (ALLN) was from Sigma.
Cell culture
Human embryonic kidney HEK293, human hepatoma HepG2, human gastric adenocarcinoma AGS and MKN45, parental human EC cell lines OE19, OE21, and OE33 (all from the American Type Culture Collection, Manassas, VA) and stably transfected clones generated thereof were maintained as described previously (10). CPC and EPC cell lines were a generous gift from O. Opitz (Department of Pathology, Universität Freiburg, Freiburg, Germany) and cultivated as described elsewhere (22).
DNA constructs
The approximately 800-bp fragment of the proximal human Cav1 promoter (accession no. AF019742, position 69 to 859) (14) was amplified by PCR from the genomic DNA of normal human liver and cloned into the KpnI/HindIII sites of pGL3-luc luciferase reporter plasmid (Promega GmbH, Mannheim, Germany). Transient transfection and luciferase assays were performed as described previously (20). Small interfering RNA (siRNA) oligonucleotides against human SCAP were from Dharmacon (Thermofisher).
Equilibrium density ultracentrifugation, coimmunoprecipitation (CoIP), and Western blot (WB)
Cold detergent-insoluble (Triton X-100) low-density membrane domains (lipid rafts) and their associated proteins were purified using sucrose gradients as published earlier (23). CoIP was performed as described before (23). Detection of proteins in WB by chemoluminescence or infrared-labeled secondary antibodies was performed according to the manufacturer's instructions (Odyssey, Licor Biosciences, Lincoln, NE).
Immunofluorescence
Staining was performed in triple-color mode visualizing 4′,6-diamidino-2-phenylindole and, Alexa-488 and -594 using a digital camera-connected (Axiovision, release 4.4) fluorescence microscope (Axiovert 200M; Carl Zeiss MicroImaging GmbH, Hallbergmoos, Germany) (10).
Immunohistochemistry (IHC)
Antibody and hematoxylin-eosin (H&E) stainings were performed on paraffin sections as described elsewhere (20). The frequency and intensity of Cav1 staining in tissue microarrays from BAC patient specimens were evaluated and scored by the expert pathologist A.W.
Methylation-specific, quantitative RT-PCR (RT-qPCR), Chromatin-immunoprecipitation (ChIP), and EMSA
The methods were performed as published previously (20, 21, 24) using oligonucleotides listed in Table 1.
Table 1.
Oligonucleotides
| Forward (5′-3′) | Reverse (5′-3′) | Comments | |
|---|---|---|---|
| Methylation specific PCR (MSP) | |||
| MSP2 | TATTCGTTTTTTGTTCGTTGC | CAAAAATTTATTCTACTCGCGA | Anneals to bisulfite-converted, methylated DNA |
| MSP3 | GATGTATTGCGAAAAATATTCGT | CGCAACGAACAAAAAACGAA | Anneals to bisulfite-converted, methylated DNA |
| EMSA | |||
| SRE | AAGCACCCCAGCGCGGGACAACGTTCT | AGAACGTTGTCCCGCGCTGGGGTGCTT | Biotinylated at 5- and 3-ends (BIO) |
| Mutated SRE | AATTAATTAATCGCGGGACAACGTTCT | AGAACGTTGTCCCGCGATTAATTAATT | Unlabeled competitor |
Statistics
Results are means ± se from at least five animals per genotype or three independent experiments from different cell passages. The software Graphpad Prism (version 4.0; GraphPad Software Inc., San Diego, CA) was used to analyze the data. P values (* <0.05) were calculated using Student's t test.
Results
Expression of Cav1 in EC patient tissue and cell lines
To detect Cav1 in normal human esophageal tissue, IHC on paraffin sections from routine biopsies was performed using a monoclonal antibody (20). The normal stratified squamous epithelium in the lower esophagus expressed Cav1 in the cytoplasm and at the membrane junctions (Fig. 1A, top). IHC on microarrays with resected tumor tissue from BAC patients (n = 100) revealed that Cav1 staining was lost in 95% of BAC specimens (Fig. 1A, middle). However, a small number of patients (n = 5) showed a strong cytoplasmic expression of Cav1(Fig. 1A, bottom), which was associated with a very poor prognosis of disease-free and overall survival (Fig. 1B), similar to observations in other cancer entities (11, 12, 25). These data indicated that Cav1 is down-regulated upon malignant transformation of the normal squamous epithelium. However, the underlying molecular mechanisms of Cav1 silencing are unknown.
Fig. 1.

Expression of Cav1 in human BAC patients. A, Cav1 is lost in the majority of BAC patients. IHC detection of Cav1 protein in human nonneoplastic esophageal tissue and BAC specimens (n = 100) using a monoclonal antibody; N, normal squamous epithelium (top panels) with Cav1 at cell junctions; T, BAC (lower panels). The staining scores for Cav1 in BAC tumor cells were correlated to disease-free and overall survival. Score: 0, negative; +1, positive (weak); +2, positive (moderate); +3, positive (strong). B, Strong cytoplasmic expression of Cav1 (score 3+) in a small subgroup of BAC patients (n = 5) is a negative prognostic factor for survival. Kaplan-Meier curves are presented for overall and disease-free survival.
RT-qPCR analyses and WB confirmed expression of Cav1 mRNA and protein in human esophageal cancer (EC) specimens (Fig. 2, A and B) and cell lines (Fig. 2, C and D). Nontumorous tissue from patients with SCC or BAC expressed Cav1 protein to similar levels. Cav1 protein expression was low in five of 12 (42%) BAC specimens and low in BAC cell lines (OE19, OE33), while high in SCC specimens (11) and the SCC cell line OE21. Down-regulation of Cav1 mRNA was confirmed in BAC tumor specimens as compared with matched normal tissue. To visualize the subcellular localization of Cav1, immunofluorescence microscopy was performed. OE19 cells grew in clusters and were sphere shaped, whereas OE21 cells were spread out showing an epithelial morphology. Cav1 was present in OE21 but not in OE19 cells and was found to be evenly distributed in vesicular structures within the cytosol and in dotted structures along the plasma membrane, presumably corresponding to lipid rafts (Supplemental Fig. 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Cav1 was also present in the immortalized esophageal squamous epithelial cell line (EPC) and in the Barrett esophagus (BE) cell line (CPC). In the present study, we further employed EPC and OE21 cells as in vitro model systems for mechanistic studies on Cav1 gene regulation by bile acids.
Fig. 2.

Expression of Cav1 in human EC cell lines. A and B, Expression of Cav1 mRNA (A) and protein (B) in human esophageal cancer (EC) detected by RT-qPCR and WB. Cav1 is reduced in BAC as compared with matched normal tissue and SCC tissues. CT values from RT-qPCR were normalized to β2-microglobulin (β2M) and presented as -fold ± se (n = 5 patients); *, P < 0.05; N (Normal) vs. T (Tumor). Total tissue lysates were normalized to protein content and compared with β-actin. Representative WB gels are shown. SCC, squamous cell carcinoma; BAC, Barrett adenocarcinoma. C and D, Detection of Cav1 mRNA (C) and protein (D) in human EC cell lines. CT values from RT-qPCRs were normalized to β2-microglobulin (β2M) and presented as -fold ± se (n = 3); *, P < 0.05 vs. OE19. Total cell lysates were normalized to protein content and compared with β-actin. Representative WB gels are shown. OE19 and OE33, BAC cell lines; OE21, SCC cell line; EPC, immortalized esophageal squamous epithelial cell line; CPC, Barrett esophagus (metaplasia) cell line; HET1A, control squamous epithelial cell line. Rel, Relative.
Bile acids down-regulate Cav1 in normal and transformed esophageal squamous epithelial cells
To test the effect of bile acids on Cav1 expression, OE21 cells were incubated for 30 h in serum-free medium with increasing concentrations of CDCA, in the presence and absence of 10 μm ALLN, an inhibitor of SREBP1 catabolism (14). ALLN is a cell-permeable inhibitor of calpain/cathepsin, neutral cysteine proteases, and the proteasome. ALLN prevents degradation of the active nuclear 68-kDa SREBP1 by the proteasome. Thus, ALLN is expected to stabilize active 68-kDa SREBP1 and counteract the inhibitory effect of CDCA on 68-kDa SREBP1 steady-state levels.
WB analysis of whole-cell lysates demonstrated significant down-regulation (50 ± 10% at 100 μm; *, P < 0.05) of Cav1 protein in CDCA-treated (IC50 = 66 μm) vs. vehicle-treated cells (Fig. 3A). As expected, ALLN prevented the CDCA-induced decrease of Cav1 protein, indicating that CDCA causes a deficiency in the amount of 68-kDa active SREBP1 available for positive regulation of Cav1 gene expression. RT-qPCR studies confirmed down-regulation of Cav1 mRNA in presence of CDCA (at 75 μm) in both OE21 (48 ± 12%; *, P < 0.05) and EPC (33 ± 9%; *, P < 0.05) cells under serum-free conditions (Fig. 3B). Both OE21 (data not shown) and EPC (Fig. 3C) cells reduced Cav1 protein expression in response to CDCA (at 75 μm) in a time-dependent manner, starting after 9 h and reaching a minimum after 30 h. Prolonged exposure (> 30 h) to CDCA (at 75–100 μm) in the absence of serum led to toxicity and cell death (data not shown). Consistent with the decrease in Cav1 protein, the amount of active 68-kDa SREBP1 protein (IC50 = 10–20 μm) was also diminished by CDCA in a concentration- and time-dependent manner, both in EPC (Fig. 3C) and OE21 (Fig. 3D) cells. Similar to its effect on the Cav1 protein, ALLN also prevented the CDCA-mediated decrease of 68-kDa SREBP1 protein. These data indicated that Cav1 expression is repressed by bile acids at the transcriptional level, and this process presumably involves SREBP1.
Fig. 3.

Bile acids down-regulate Cav1 and SREBP1 in normal human esophageal and SCC cells. A, Concentration-dependent reduction of Cav1 protein by CDCA. OE21 cells were treated for 30 h, in serum-free medium, with the concentration indicated of CDCA, in the presence and absence of the protease inhibitor 10 μm ALLN. O.D. values from bands in WB gels are expressed as -fold ± se (n = 3); *, P < 0.05 CDCA vs. DMSO. Quantitative analyses (left) are presented with representative WB (right). B, Down-regulation of Cav1 mRNA by CDCA. OE21 and EPC cells were treated as in panel A. CT values from RT-qPCRs were normalized to β2-microglobulin (β2M) and presented as -fold ± se (n = 3); *, P < 0.05 CDCA vs. DMSO. C, Time-dependent reduction of Cav1 and 68-kDa active SREBP1 proteins by CDCA. EPC cells were treated for the indicated times with 75 μm CDCA. Representative WB are shown. D, Concentration-dependent decrease of 68-kDa active SREBP1 protein. OE21 cells were incubated for 30 h with the indicated concentrations of CDCA, in the presence and absence of 10 μm ALLN, respectively. Quantitative analyses (left) and representative WB (right) are shown. Hsp, Heat shock protein.
Bile acids inhibit binding of SREBP1 to the human Cav1 promoter
To test the effect of bile acids on the transcriptional activity of the Cav1 promoter, reporter gene assays were performed. OE21 cells were transiently transfected with a luciferase reporter gene plasmid driven by the proximal human Cav1 promoter (−800/+1) (14) (Table 2). Twenty four hours upon transfection, cells were incubated for 30 h with CDCA (30 and 100 μm) and 50 μm DCA, respectively, in the presence or absence of 10 μm ALLN. Luciferase activity was significantly decreased (by 25–45%; *, P < 0.05) in bile acid-treated cells compared with vehicle-treated cells (Fig. 4A). As before, ALLN prevented the bile acid-mediated down-regulation of the Cav1 promoter activity. Similar results were obtained from HEK293 cells. These findings suggested that bile acids lower the amount of active SREBP1 that is available for transcriptional activation of the Cav1 promoter.
Table 2.
Homo sapiens caveolin-1a gene, promoter region, and partial CDS
| 1 | aaataattct | acaattataa | acatttttgt | gtattttgca | aaatatggct | aacctgttgg |
|---|---|---|---|---|---|---|
| 61 | ctaaaattcc | attgttccag | aaaatatcgg | taataaaatt | atagaaaagt | taaagatctt |
| 121 | catttcttat | ttcgaagcgt | ttgggagaca | tttcagaaac | ggatgggaaa | tgttaaattc |
| 181 | tgcatgcctg | cttaagtttc | catccacacc | gactagatgt | aaacgagtgt | caccaaaagt |
| SRE1 | ||||||
| 241 | acaccacagg | cacccacaca | gattccttcc | ataagggatc | cacaaagttt | agatgtaaat |
| 301 | gtacctaaag | ttcctagccg | tctttcatcc | ctccctctgt | gaaacaggga | acacatgtgt |
| 361 | tttaaggcag | agatggaact | tgggcatggg | agggggtggg | gaggtgggaa | gggacggctt |
| 421 | aggacagggc | aggattgtgg | attgtttctg | ccgccttggt | tcgccatact | gggcatctct |
| SRE2 | MSP3 | |||||
| 481 | gcaggcgcgt | cggctccctc | cacccctgct | gagatGATGC | ACTGCGAAAA | CATTCGCtct |
| 541 | ccccgggacg | cctctcggtg | gttcagagca | gggaaaattg | ttgcctcagg | taaaataatc |
| SRE3 | PPAR | MSP2/3 | ||||
| 601 | tgcccaagca | ccccagcgcg | ggacaacgtt | ctCACTCGCT | CTCTGCTCGC | TGCGcgctcc |
| 661 | ccgccctctg | ctgccagaac | cttggggatg | tgcctagacc | cggcgcagca | cacgtccggg |
| MSP2 | ||||||
| 721 | ccaacCGCGA | GCAGAACAAA | CCTTTGgcgg | gcggccagga | ggctccctcc | cagccaccgc |
| CAAT-Box | ||||||
| 781 | ccccctccag | cgcctttttt | tccccccata | caatacaaga | tcttccttcc | tcagttccct |
| Start CDS | ||||||
| 841 | taaagcacag | cccagggaaa | cctcctcaca | gttttcatcc | accacgggcc | agcatgtctg |
| 901 | ggggcaaata | cgtagactcg | gaggtaggca | tccgtggggg | ggcgccggct | cgggcgtgcg |
| 961 | gggg |
Accession number: AF019742.1. DNA-binding sites for SREBP (SRE) and peroxisome proliferator activated receptor (PPAR) are marked in bold. Legend for primer amplicons: capital letters (MSP2/3), underlined (EMSA).
Fig. 4.
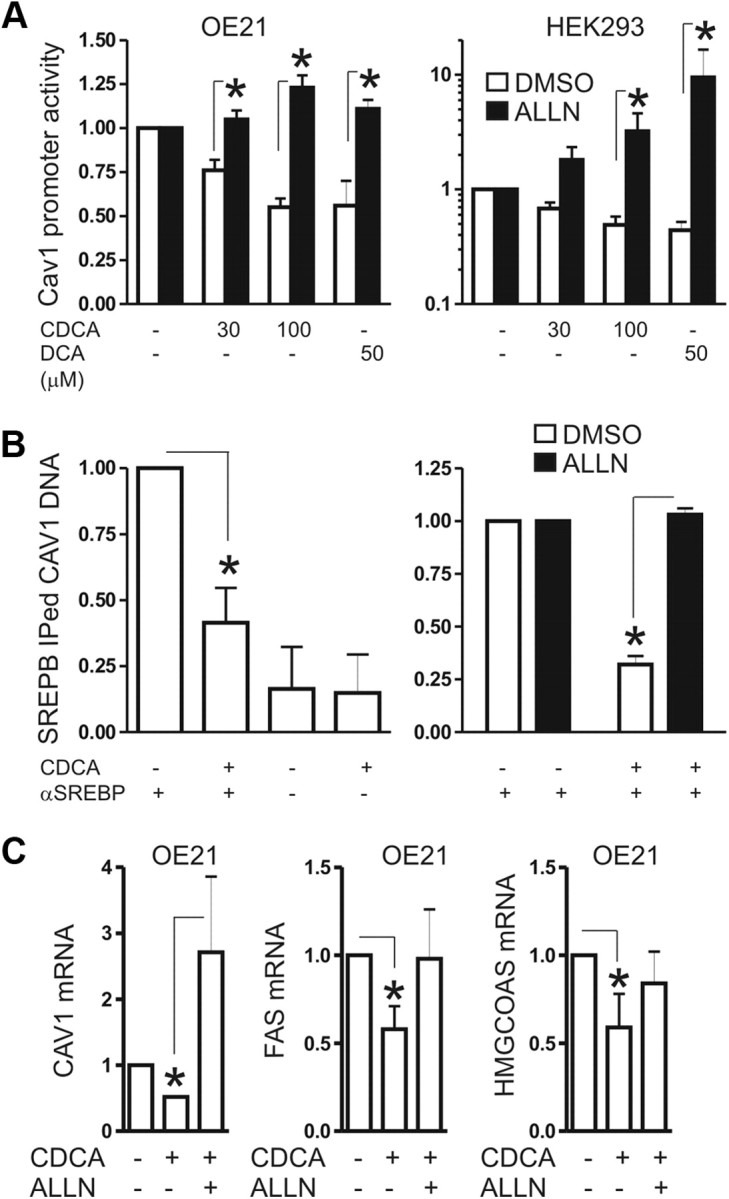
Bile acids inhibit activation of the human Cav1 promoter by active 68-kDa SREBP1. A, CDCA inhibits transactivation of the human Cav1 promoter. OE21 and HEK293 (control) cells were transiently transfected with a Cav1 promoter-driven luciferase reporter plasmid and treated for 30 h with CDCA (30 μm and 100 μm) and 50 μm DCA, with and without 10 μm ALLN, respectively. Luciferase activity was normalized to protein content and presented as -fold ± se (n = 3); *, P < 0.05 CDCA or DCA vs. DMSO. B, CDCA blocks binding of SREBP1 to sterol-response elements (SRE) in the human Cav1 promoter. Cells were treated as in panel A. IP was performed on cell lysates with rabbit polyclonal SREBP1 antiserum or control IgG. The CT values from genomic qPCR of IP-ed DNA were normalized to the CT values of input DNA and are expressed as -fold ± se. (n = 3) change of pulldown by CDCA compared with control; *, P < 0.05 CDCA vs. DMSO. C, Down-regulation of SREBP1 target genes by CDCA is reversed by ALLN, an inhibitor of active 68-kDa SREBP1's catabolism. OE21 cells were incubated for 30 h with 100 μm CDCA in the absence or presence of 10 μm ALLN. CT values from RT-qPCR were normalized to β2-microglobulin (β2M) and presented as -fold ± se (n = 3); *, P < 0.05 CDCA vs. DMSO or ALLN. FAS, Fatty acid synthase.
In the human Cav1 proximal promoter, three functional SREBP1-binding sites or sterol responsive elements (SRE) have been described (14) (Table 2). To measure SREBP1 binding to these sites, we designed ChIP primers flanking the first −500/+1 bp containing these regions until the transcriptional start site (Fig. 4B). OE21 were treated for 16 h with vehicle or 75 μm CDCA in the presence or absence of ALLN. Cell lysates were subjected to ChIP using polyclonal SREBP1 antiserum or control IgG. Data from genomic qPCR on IP-ed DNA fragments revealed that CDCA decreased (by 60 ± 13%; *, P < 0.05) the binding of SREBP1 to the SRE from the human Cav1-promoter (lane 2) compared with vehicle-treated control (lane 1). In contrast, ALLN abolished the negative effect of CDCA on the formation of the SREBP1/chromatin complex (lanes 3 and 4). These data indicated that CDCA prevented SREBP1 binding to SRE in the proximal promoter of the Cav1 gene.
To verify that inhibition of SREBP1 activation by bile acids causes down-regulation of Cav1, the expression of additional cognate SREPB1 target genes was investigated (16). Those included fatty acid synthase, low-density lipoprotein receptor (LDLR), and 3-hydroxy-3-methylglutaryl-coenzyme A synthase (HMGCoAS), which are all positively regulated by SREBP1. OE21 cells were incubated with CDCA (50 and 100 μm) in the absence or presence of 10 μm ALLN for 30 h. Analyses from RT-qPCR confirmed reduced (∼50%; *, P < 0.05) mRNA expression of SREBP1 target genes in CDCA-treated cells compared with vehicle controls (Fig. 4C). ALLN instead restored expression of the target genes in the presence of CDCA. These data confirmed that bile acids decrease the transactivation potential of SREBP1 on its target genes.
Bile acids inhibit nuclear translocation of active 68-kDa SREBP1 involving SCAP
To determine the subcellular localization of SREBP1, equilibrium density ultracentrifugation was performed (23). OE21 cells were treated for 30 h with either 100 μm CDCA or dimethylsulfoxide (DMSO) in serum-free medium, and cell lysates were loaded on a sucrose gradient followed by ultracentrifugation and WB analysis of individual fractions. The insoluble pellet (lane 3) consisted of proteins from the nuclear matrix, cytoskeleton, and membranes. In the presence of CDCA, the 125-kDa precursor and the 68-kDa SREBP1 fragment accumulated in the insoluble pellet (lane 3), which also contained proteins of the ER/Golgi membranes as evidenced by the expression of furin (Fig. 5A). As expected, the amount of membrane-bound Cav1 was considerably reduced by CDCA in this fraction (lane 3). These findings were consistent with SREPB1's reduced activity on Cav1 promoter-driven reporter gene activation in the presence of CDCA.
Fig. 5.

Bile acids inhibit nuclear accumulation of active 68-kDa SREBP1 involving SCAP. A, Sucrose gradient ultracentrifugation. OE21 cells were grown to confluency in 15-cm dishes and treated for 30 h with 100 μm CDCA or DMSO, respectively. Fractions were subjected to WB: 1,2, soluble ER and Golgi membrane proteins; 3, insoluble membrane, cytoskeleton, and matrix proteins; 4, input control. Note the reduced Cav1 protein and enhanced membrane retention (lane 3) of 125-kDa pre-SREBP1 and 68-kDa SREBP1 upon CDCA treatment. B, CDCA promotes interaction of pre-SREBP1 with SCAP. OE21 cells were grown and treated as in panel A, and CoIP was performed using SCAP and SREBP1 polyclonal antibodies. Representative WB are shown. C, SCAP is required for CDCA-mediated down-regulation of Cav1. OE21 cells were transiently transfected with siRNA oligonucleotides against SCAP or control siRNA, respectively, and treated with 75 μm CDCA for 30 h. RT-qPCR analyses (left) and representative WB (right) are shown. CT values from RT-qPCR were normalized to β2-microglobulin (β2M) and presented as -fold ± se (n = 3); *, P < 0.05 SCAP-siRNA vs. control-siRNA. Insert, Agarose gel from RT-PCR showing SCAP mRNA knockdown by siRNA. hsp90, Heat shock protein 90; IB, immunoblotting; IP, immunoprecipitation; MEK, MAPK kinase.
Immunofluorescence microscopy evinced that, in naive cells, the active 68-kDa SREBP1 was predominantly located in the nucleus (Supplemental Fig. 2). In contrast, in CDCA-treated cells, nuclear accumulation of the mature 68-kDa SREBP1 was reduced, and an enhanced perinuclear accumulation was evident that corresponded to the nuclear membrane and the ER, two characteristic locations of the immobilized 125-kDa SREBP1 precursor under high sterol conditions. This result confirmed our previous data that CDCA blocks maturation and nuclear translocation of active SREBP1.
To test whether CDCA promotes retention of SREBP1 in the ER/Golgi complex by SCAP, CoIP experiments were performed (Fig. 5B). OE21 cells were treated for 30 h with 75 μm CDCA, and whole-cell lysates were incubated with rabbit polyclonal SREBP1 and goat polyclonal SCAP antibodies, respectively. WB demonstrated that CDCA increased the interaction of 125-kDa pre-SREBP1 with SCAP protein. To further test the involvement of SCAP in the CDCA-mediated SREBP1 retention process, siRNA oligonucleotides directed against SCAP were transiently transfected into OE21 cells, which achieved approximately 75% knockdown compared with cells transfected with control RNAi (Fig. 5C, left). In cells with SCAP knockdown, CDCA failed to diminish Cav1 protein expression (lane 3) as compared with cells transfected with control siRNA (lane 4) (Fig. 5C, right). These data emphasized that SCAP is required for bile acid-mediated reduction of the active SREBP1 pool available for Cav1 gene expression.
Bile acids also down-regulate Cav1 in vivo and activate FXR
To assess whether bile acids inhibit Cav1 mRNA expression also in vivo, C57BL/6 WT mice were fed a chow diet containing 1% (wt/wt) CDCA (n = 5 per group) for 7 d. This diet has been shown to elicit a systemic bile acid/FXR response (21). IHC detecting SREBP1 using rabbit polyclonal antiserum confirmed its nuclear localization in esophageal tissue (Supplemental Fig. 3). The majority of positively stained nuclei were found in the basal layer and also in the differentiating cells of the squamous epithelium. RT-qPCR analyses revealed that the Cav1 mRNA was decreased in the esophageal tissue from mice under CDCA diet (by 75 ± 10%; *, P < 0.05) compared with mice on control diet (Fig. 6A). Bile acid also reduced the mRNA levels of the SREBP1-target genes Ldlr and Hmgcoas. Moreover, Icam1 mRNA was significantly reduced, indicative of alterations in cell-cell adhesion (26). Interestingly, the mRNA encoding for the nuclear bile acid (and CDCA) receptor FXR (Fxr) and its cognate target genes small heterodimer partner (Shp), Ibabp (13) and Keratin 13 (27) were concomitantly (∼2- to 3-fold) induced by CDCA (Fig. 6B). To examine the role of FXR in these gene regulations, FXR-KO and matched WT mice (n = 5 per group) were fed a chow diet enriched with 1% (wt/wt) CDCA or 0.2% (wt/wt) DCA for 8 wk. H&E stainings from paraffin sections of esophageal tissue did not reveal any macroscopic or histopathological signs of damage upon administration of CDCA or DCA (data not shown). Nevertheless, WT mice on CDCA or DCA suffered from hyperkeratosis (Fig. 6C), which was most pronounced in the squamous epithelium of the forestomach at the gastroesophageal junction. The keratinized sloughing tissue was thicker (∼ 8–10 μm vs. 4 ± 0,4 μm; * P < 0.05) and multilayered as compared with WT mice on control chow diet (Supplemental Fig. 4). FXR-KO mice exhibited enhanced hyperkeratosis (∼9–10 μm) already in the absence of bile acid diet, which was not further exacerbated by CDCA or DCA.
Fig. 6.
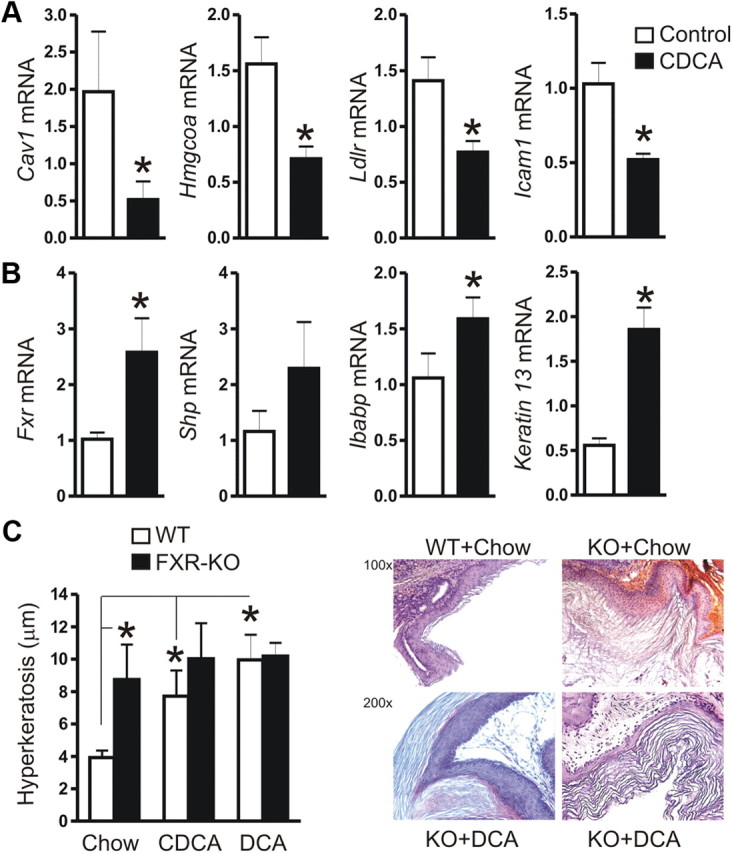
Bile acids down-regulate Cav1 also in vivo and activate FXR. A, Down-regulation of mRNAs encoding cell adhesion proteins (Cav1, Icam1) and SREBP1-target genes (Hmgcoas, Ldlr) in C57BL/6 mice on a 7-d chow diet enriched with 1% (wt/wt) CDCA as compared with littermates on control chow. CT values from RT-qPCR on total RNA extracted from resected esophagi were normalized to β2-microglobulin (β2M) and presented as means ± se (n = 5 per group); *. P < 0.05 CDCA vs. chow. B, Up-regulation of mRNA for FXR-target genes (Fxr, Shp, Ibabp, Keratin13). Data are presented as in panel A. C, FXR-KO mice show hyperkeratosis of the squamous epithelium. C57BL/6 WT and FXR-KO mice (n = 5 per group) were fed a chow diet with or without 1% (wt/wt) CDCA or 0.2% (wt/wt) DCA for 8 wk, respectively. H&E stainings from paraffin sections of esophageal and forestomach squamous epithelial tissue was evaluated for the thickness of the keratinizing sloughing tissue. The diameter of the keratin layer was measured (five random fields per mouse) and presented as μm ± se (n = 5 per group); *, P < 0.05 WT vs. FXR-KO; chow vs. CDCA/DCA. Quantitative analyses (left) are shown together with representative histology (right); magnifications ×100 and ×200.
These data indicated that bile acids down-regulate Cav1 also in vivo and activate FXR, resulting in an altered expression profile of downstream target genes, which may regulate cell adhesion (Cav1, Icam1) and differentiation (Keratin 13) (27) in the esophageal squamous epithelium.
WB (Supplemental Fig. 3) and RT-PCR (Supplemental Fig. 5) analyses confirmed expression of FXR mRNA (27) and protein in mouse esophageal tissue and in EPC cells, but not in other human EC cell lines. CDCA/FXR-induced SHP is a known repressor of Srebp1c gene transcription in mice (13). Thus, in FXR-positive EPC cells and tissues, bile acids may activate both the SCAP/SREBP1 and the FXR/SHP pathway to suppress postranslational processing of SREBP1 and Srebp1c gene transcription.
Cav1 deficiency promotes esophageal cell proliferation in vitro and in vivo
To finally assess the physiological consequences of bile acid-mediated loss of Cav1, we studied the BAC cell line OE19, which naturally lacks Cav1 expression. To explore the effect of ectopic Cav1 on the malignant phenotype of BAC cells, OE19 cells were stably transfected with an expression plasmid containing an oncogenic, dominant-negative mutant of Cav1 (P132L) (28) or an empty vector (EV) control plasmid, respectively. WB analyses confirmed expression of the transfectant compared with OE21-positive control cells (Fig. 7A). Clones expressing either the Cav1 mutant (OE19/P132L) or empty vector (OE19/EV) were subjected to MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) proliferation assays. In sum, the P132L clones exhibited an up to 2-fold (at d 6) faster growth rate than the EV clones (Fig. 7A). This result was in line with the oncogenic property described for this mutant in other tumor entities such as breast cancer (28, 29).
Fig. 7.
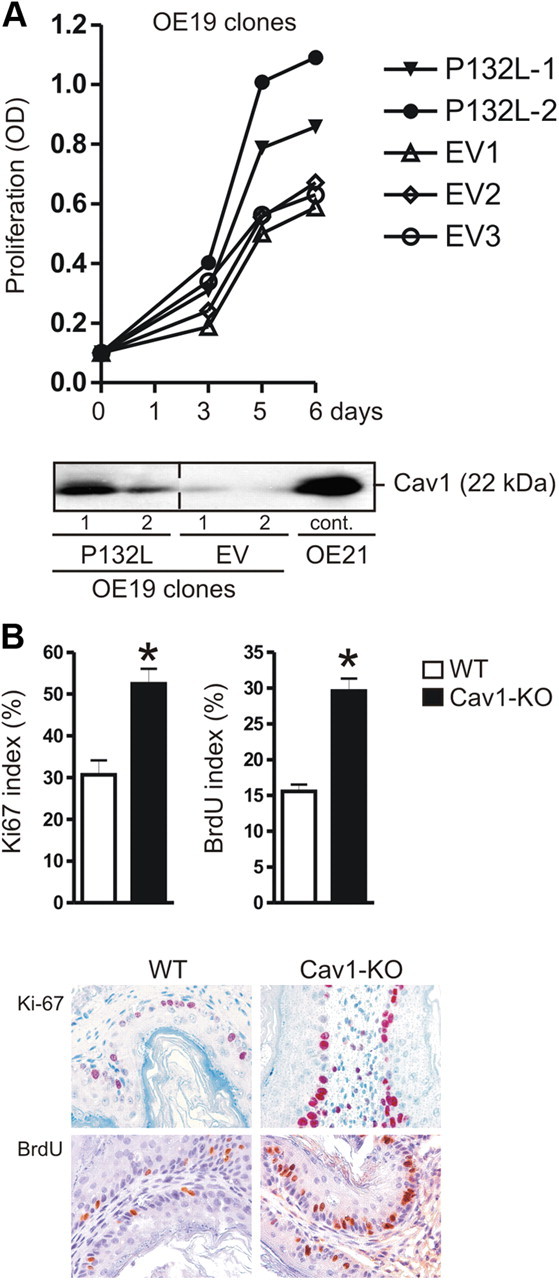
Cav1-deficiency increases proliferation in vitro and in vivo. A, The oncogenic dominant-negative Cav1 variant P132L promotes proliferation of OE19 cells. A (bottom panel), Cav1 protein expression in stably transfected OE19 cells. Representative WB are shown. A (top panel), Time course of proliferation of OE19 clones. OD values from colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays are presented as means ± se (n = 3) of Cav1-transfected (P132L) cells compared with EV-transfected cells; *, P < 0.05 P132L vs. EV. B, Cav1-KO mice display hyperplasia of the squamous epithelium. Quantitative analyses of IHC for Ki-67 and BrdU uptake; magnification, ×200. The ratio of positive to total nuclei in the basal layer of the squamous epithelium of forestomach and esophageal tissue was calculated as % ± se (n = 5 per genotype); *, P < 0.05 WT vs. Cav1-KO (both at an age of 4 months).
We then quantified proliferation in the esophageal and forestomach epithelia of 4-month old gender-matched Cav1-KO and WT mice. Consistent with previous results on the columnar epithelium of the gastric corpus (30), IHC against the proliferation marker Ki-67 (30 ± 2% KO vs. 16±1% WT; *, P < 0.05) and incorporated BrdU (52 ± 4% KO vs. 30±3% WT; *, P < 0.05) revealed that the cells from the basal layer of the squamous epithelia, both in esophagi and forestomach tissue adjacent to the glandular stomach (Fig. 7B), were significantly hyperplastic in Cav1-KO mice as compared with WT littermates.
These data indicated that loss of Cav1 expression potentiates the BAC cell's malignant behavior. Bile acid may initiate the silencing of the Cav1 gene through the SREBP/SCAP and FXR/SHP-mediated post/transcriptional events revealed in this study (Fig. 8).
Fig. 8.

Signaling model of bile acid-induced changes in esophageal squamous epithelia. CDCA or other bile acids (such as DCA in the refluxate) bind to the nuclear bile acid receptor FXR and activate expression of its cognate target gene SHP. SHP itself represses Srebp1c gene transcription. In parallel, CDCA mimics high-sterol conditions and inhibits proteolytic cleavage and nuclear accumulation of active 68-kDa SREBP1 at target gene promoters. Defective SREBP1 transactivation leads to reduced expression of target genes including Cav1. Other FXR-target genes such as Icam-1 and Keratin 13 may contribute to the observed hyperkeratosis phenotype of FXR-KO mice. Loss of Cav1 leads to squamous cell hyperplasia in Cav1-KO mice and may activate EGFR signaling and destabilize cell-cell and cell-matrix contacts. In sum, the SCAP/SREBP1 and the FXR/SHP pathways are expected to converge in vivo, to evoke early molecular changes in the esophageal squamous epithelium, which may ultimately facilitate BE metaplasia and transformation, preneoplastic lesions in the progression to BAC.
Discussion
The current study describes a novel molecular mechanism for bile acid-mediated silencing of Cav1 gene expression in human and murine esophageal squamous epithelial cells. We showed that Cav1, a scaffold protein of membrane caveolae and lipid rafts, is expressed at cell junctions in the basal layer of the healthy esophageal squamous epithelium, but is lost in the majority of specimens from BAC patients. Interestingly, Cav1 overexpression in a small subgroup of BAC patients, as already demonstrated for patients with esophageal SCC, was correlated with poor survival prognosis (11, 12). Inappropriate and high cytoplasmic expression of naturally membrane-associated Cav1 has been reported to alter its classical tumor-suppressive behavior into a survival-promoting role (31). For example, chronic phosphorylation of Cav1 at serine residues promotes its exocytosis from pancreatic tumor cells via the secretory pathway (32). Extracellular, soluble Cav1 acts as a survival and chemoattractant factor for prostate cancer cells during perineural invasion and metastasis (33, 34). Overexpression of Cav1 positively correlates with chemoresistance and aggressiveness of certain human tumors including esophageal SCC, rectal, renal, pancreatic, and lung cancer (as reviewed in Ref. 9). Our data are therefore consistent with the dual role of Cav1 as a tumor suppressor (upon loss of function) or promoter (upon gain of function) depending on its individual expression status in patients (9, 10). However, additional studies must decipher the role of cytoplasmic Cav1 in this specific subgroup of BAC patients.
In contrast, Cav1 down-regulation has been reported in many cancer entities (9, 10). However, the mechanisms that underlie this process are largely unknown and may comprise posttranscriptional and epigenetic changes, such as DNA methylation (35, 36). It has been suggested that the development of BE metaplasia occurs as a protective reaction of the mucosa against high concentrations of bile acids, and that specific transporters are synthesized for removal and detoxification of bile acids (37). Furthermore, bile acids may exert a direct role in carcinogenesis (38). Bile acids induce overexpression of the homeobox gene transcription factor CDX2 in BE mucosa (39), and CDX2 transgenic mice (40) represent a model to study the progression from squamous epithelium to columnar intestinal metaplasia. DCA causes membrane perturbations, for example, by affecting the membrane distribution of Cav1, in human HCT116 colorectal adenocarcinoma cells, and thereby triggers intracellular signaling cascades (41). Bile acids activate the epidermal growth factor (EGF) receptor (EGFR) in a non-ligand-dependent manner, via activation of src-kinase during the pathogenesis of BE (42, 43). Any alterations that lead to an overexpression or increased activation of EGF or EGFR may result in cancer (44, 45). Cav1 is a major inhibitor of EGFR activation (31) and an essential constituent of cell-cell junctions and cell-matrix adhesions (7, 8, 31). Thus, down-regulation of Cav1 by bile acids may directly contribute to carcinogenesis.
The results from the present study propose the existence of a bile acid-mediated retention mechanism for 125-kDa pre-SREBP1 through the ER-protein SCAP. As a consequence, nuclear translocation of the active 68-kDa SREBP1 fragment was impaired by CDCA treatment. This phenomenon may be caused by failure of proper ER-to-Golgi transport of pre-SREBP1 and/or consecutive proteolytic cleavage by Sp1 proteases in the Golgi apparatus. In contrast, SCAP deficiency abolished the repressive effect of CDCA on Cav1 expression, underlining that SCAP is required for the ability of CDCA to reduce the active pool of 68-kDa SREBP1 available for positive regulation of the Cav1 promoter. The Cav1 promoter contains three SRE (−646/−395/−287), and binding of SREBP1 to these sites has been demonstrated in human skin fibroblasts and endothelial cells (14, 46). In addition to Cav1, other bona fide SREBP1-target genes were down-regulated by CDCA in vitro and in vivo (16). The active nuclear 68-kDa SREBP1 is rapidly degraded by the ubiquitin-proteasome system (16). Consistently, the proteasome inhibitor ALLN prevented the bile acid-mediated decrease of Cav1 mRNA and restored proteolytic processing of 68-kDa SREBP1 together with SREBP-target gene transcription. The proximal SRE (−287) (14) of the human Cav1 promoter partially overlaps with a predicted PPARγ site (47), another important transcription factor involved in lipid synthesis. Thus, additional transcription factors that regulate the Cav1 promoter (such as PPARγ, p53, or SP1) may participate in bile acid-mediated inhibition of Cav1 gene transcription. An alternative mechanism of Cav1 silencing is DNA methylation (36, 48–50). The Cav1 promoter is located within an CpG island (36), which also harbors the SRE sites (14). The methylation inhibitor 5-azacytidine (Aza) restored Cav1 mRNA expression in AGS (51) and OE19 cells (data not shown). Methylation at two selected CpG-rich sites (methylation specific 2/3) overlapping with the CpG-rich SRE (Table 2) was detected in AGS cells. Thus, promoter methylation may complement defective transcription to silence Cav1 gene expression.
Our data suggest that, in addition to the SCAP/SREBP pathway, FXR signaling may contribute to repression of Srebp1c gene transcription. The FXR-target gene SHP is a known repressor of the Srebp1c gene (13). In contrast to all human EC lines tested, only EPC cells had detectable levels of 56-kDa FXR protein. However, all EPC and EC cell lines expressed a truncated mRNA of the FXRα variant (termed Δα1/2), as published previously for GC cell lines (27). In contrast to EPC cells, CDCA had no effect on Srebp1c mRNA levels in FXR-negative OE21 and HEK293 cells (data not shown). Thus, the decrease of Cav1 mRNA by CDCA in FXR-negative cells should be caused by reduced availability of active 68-kDa SREBP fragment through the SCAP/ER retention pathway and not through an FXR/SHP-dependent pathway. Instead, in FXR-positive EPC cells, both pathways may collaborate to inhibit Cav1 expression.
The in vivo data support this conclusion. In C57BL/6 mice, CDCA down-regulated Cav1 mRNA expression together with Icam1 and SREBP1-target genes, whereas FXR-target genes were up-regulated (Fxr, Shp, Ibabp, Keratin 13). Prolonged exposure of WT mice to CDCA and DCA resulted in hyperkeratosis of the squamous epithelia of the esophagus and the forestomach. FXR-null mice already suffered from spontaneous hyperkeratosis. Disruption of bile acid/cholesterol metabolism in mice (e.g. Insig, enzymes, apo-lipoproteins, transporters) (52–56) and humans (57–59) has been reported to promote hyperkeratosis, mainly in the skin epidermis. We demonstrated previously (27) that CDCA-activated FXR alters the expression of keratins in vitro, in human GC cell lines, and in vivo, in the mouse glandular stomach. An altered keratin profile characterizes premalignant lesions of squamous epithelia in humans and mice (60–63). Keratins are intermediary filaments of desmosomes (64), and Cav1 interacts with junction proteins (e.g. connexins) in keratinocytes (65, 66). Consistent with the previously observed hyperplasia of the gastric glandular epithelium (30), we confirmed hyperplasia of nuclei in the basal layer of the squamous epithelium from esophagus and forestomach in Cav1-deficient mice. These spontaneous and bile acid-induced phenotypes of Cav1- and FXR-KO mice point to a possible participation of both proteins, FXR and Cav1, in early molecular changes in the mucosa of the lower esophagus, where in humans BE metaplasia, BAC, and cancers of the cardia or gastro-esophageal junction arise.
Taken together, we propose a novel signaling model, how bile acids alter expression of esophageal genes important in regulation of cell proliferation, cell adhesion, and differentiation (Fig. 8). Bile acids activate SCAP/SREBP retention and FXR/SHP signaling pathways that converge to inhibit transcription of the Srebp1c gene and to block processing of pre-SREBP1, followed by an impaired transactivation of SREBP1-target genes, including Cav1. Inappropriate expression of other FXR-target genes (K13, Icam1, Ibabp, Fxr, Shp) may contribute to the phenotypical changes in the squamous epithelium.
What could be the clinical relevance of bile acid mimicry of cholesterol ? In addition to bile acid reflux, obesity is an established risk factor for the development of BAC (1, 67, 68). High blood cholesterol is associated with obesity and an age of 60 yr or older (69). This risk association of high sterol with age and BAC supports the concept that cholesterol and/or its mimicry by bile acids cause early alterations in the esophageal squamous epithelium before onset of BE. Thus, high-sterol conditions may facilitate posttranscriptional and/or epigenetic silencing of the tumor-suppressive and barrier-protective functions of SREBP1, Cav1, and other factors in the esophagus. These genes may constitute possible future biomarkers for early detection of preneoplastic lesions before histological manifestation of BE metaplasia. Presumably, a healthy and low-cholesterol diet may contribute to the reduction of blood cholesterol, and protect from the arising of BE/BAC. The clinical use of modified bile acids such as ECDCA and UDCA against cholestatic liver diseases may open novel perspectives in chemoprevention of BAC.
Acknowledgments
We thank Minhu Chen (Department of Gastroenterology, The First Affiliated Hospital of Sun Yat-sen University, 510275 Guangzhou, PR China) for support on collaborations and Hans-Peter Märki (F. Hoffmann-La Roche, CH-4070 Basel, Switzerland) for supply of ligands.
This study was supported by grants from the Deutsche Krebshilfe (108287) (to E.B. and M.E.) and DFG (BU-2285). M.E. is also supported by grants from the Deutsche Krebshilfe (107885), DFG (SFB 824, TP B1), Else Kröner Stiftung (Nr P14/07//A104/06), and BMBF (Mobimed 01EZ0802; KMU-innovativ no. 0315116B).
Tissue sample collection of human esophageal cancer (EC) specimens was approved by the Ethics Committees of the Technische Universität München. Animal housing and experiments were performed in agreement with the ethical guidelines of the Technische Universität München and approved by the appropriate government authorities.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ALLN
- Acetyl-l-leucyl-l-leucyl-l-norleucinal
- BAC
- Barrett adenocarcinoma
- BE
- Barrett esophagus metaplasia
- BrdU
- bromodeoxyuridine
- Cav1
- human/mouse caveolin-1
- CDCA
- chenodeoxycholic acid
- ChIP
- chromatin immunoprecipitation
- CoIP
- coimmunoprecipitation
- EC
- esophageal cancer
- EGF
- epidermal growth factor
- EGFR
- EGF receptor
- ER
- endoplasmic reticulum
- EV
- empty vector
- FXR
- farnesoid X receptor
- H&E
- hematoxylin and eosin
- HMGCoAS
- HMG-coenzyme A synthase
- IHC
- immunohistochemistry
- KO
- knockout
- P132L
- mutant Cav1
- RT-qPCR
- quantitative RT-PCR
- SCC
- squamous cell carcinoma
- SRE
- sterol-responsive element
- SREBP
- sterol-responsive element-binding protein
- SCAP
- SREBP cleavage-activating protein
- siRNA
- small interfering RNA
- WB
- Western blot
- WT
- wild type.
References
- 1. Lagergren J. 2005. Adenocarcinoma of oesophagus: what exactly is the size of the problem and who is at risk? Gut 54 Suppl 1:i1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shaheen NJ. 2005. Advances in Barrett's esophagus and esophageal adenocarcinoma. Gastroenterology 128:1554–1566 [DOI] [PubMed] [Google Scholar]
- 3. Anderson LA , Watson RG , Murphy SJ , Johnston BT , Comber H , Mc Guigan J , Reynolds JV , Murray LJ. 2007. Risk factors for Barrett's oesophagus and oesophageal adenocarcinoma: results from the FINBAR study. World J Gastroenterol 13:1585–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Flook NW , Wiklund I. 2007. Accounting for the effect of GERD symptoms on patients' health-related quality of life: supporting optimal disease management by primary care physicians. Int J Clin Pract 61:2071–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perez MJ , Briz O. 2009. Bile-acid-induced cell injury and protection. World J Gastroenterol 15:1677–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berr F , Pratschke E , Fischer S , Paumgartner G. 1992. Disorders of bile acid metabolism in cholesterol gallstone disease. J Clin Invest 90:859–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hansen CG , Nichols BJ. 2010. Exploring the caves: cavins, caveolins and caveolae. Trends Cell Biol 20:177–186 [DOI] [PubMed] [Google Scholar]
- 8. Williams TM , Lisanti MP. 2004. The caveolin genes: from cell biology to medicine. Ann Med 36:584–595 [DOI] [PubMed] [Google Scholar]
- 9. Goetz JG , Lajoie P , Wiseman SM , Nabi IR. 2008. Caveolin-1 in tumor progression: the good, the bad and the ugly. Cancer Metastasis Rev 27:715–735 [DOI] [PubMed] [Google Scholar]
- 10. Burgermeister E , Liscovitch M , Röcken C , Schmid RM , Ebert MP. 2008. Caveats of caveolin-1 in cancer progression. Cancer Lett 268:187–201 [DOI] [PubMed] [Google Scholar]
- 11. Ando T , Ishiguro H , Kimura M , Mitsui A , Mori Y , Sugito N , Tomoda K , Mori R , Harada K , Katada T , Ogawa R , Fujii Y , Kuwabara Y. 2007. The overexpression of caveolin-1 and caveolin-2 correlates with a poor prognosis and tumor progression in esophageal squamous cell carcinoma. Oncol Rep 18:601–609 [PubMed] [Google Scholar]
- 12. Kato K , Hida Y , Miyamoto M , Hashida H , Shinohara T , Itoh T , Okushiba S , Kondo S , Katoh H. 2002. Overexpression of caveolin-1 in esophageal squamous cell carcinoma correlates with lymph node metastasis and pathologic stage. Cancer 94:929–933 [PubMed] [Google Scholar]
- 13. Watanabe M , Houten SM , Wang L , Moschetta A , Mangelsdorf DJ , Heyman RA , Moore DD , Auwerx J. 2004. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113:1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bist A , Fielding PE , Fielding CJ. 1997. Two sterol regulatory element-like sequences mediate up-regulation of caveolin gene transcription in response to low density lipoprotein free cholesterol. Proc Natl Acad Sci USA 94:10693–10698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raghow R , Yellaturu C , Deng X , Park EA , Elam MB. 2008. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol Metab 19:65–73 [DOI] [PubMed] [Google Scholar]
- 16. Bengoechea-Alonso MT , Ericsson J. 2007. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol 19:215–222 [DOI] [PubMed] [Google Scholar]
- 17. Cao S , Fernandez-Zapico ME , Jin D , Puri V , Cook TA , Lerman LO , Zhu XY , Urrutia R , Shah V. 2005. KLF11-mediated repression antagonizes Sp1/sterol-responsive element-binding protein-induced transcriptional activation of caveolin-1 in response to cholesterol signaling. J Biol Chem 280:1901–1910 [DOI] [PubMed] [Google Scholar]
- 18. Yuan HY , Kuang SY , Zheng X , Ling HY , Yang YB , Yan PK , Li K , Liao DF. 2008. Curcumin inhibits cellular cholesterol accumulation by regulating SREBP-1/caveolin-1 signaling pathway in vascular smooth muscle cells. Acta Pharmacol Sin 29:555–563 [DOI] [PubMed] [Google Scholar]
- 19. Luo DX , Xia CL , Li JM , Xiong Y , Yuan HY , Tang ZW , Zeng Y , Liao DF. 2010. Static pressure accelerates ox-LDL-induced cholesterol accumulation via SREBP-1-mediated caveolin-1 downregulation in cultured vascular smooth muscle cells. Biochem Biophys Res Commun 403:52–58 [DOI] [PubMed] [Google Scholar]
- 20. Burgermeister E , Xing X , Röcken C , Juhasz M , Chen J , Hiber M , Mair K , Shatz M , Liscovitch M , Schmid RM , Ebert MP. 2007. Differential expression and function of caveolin-1 in human gastric cancer progression. Cancer Res 67:8519–8526 [DOI] [PubMed] [Google Scholar]
- 21. Xing X , Burgermeister E , Geisler F , Einwächter H , Fan L , Hiber M , Rauser S , Walch A , Röcken C , Ebeling M , Wright MB , Schmid RM , Ebert MP. 2009. Hematopoietically expressed homeobox is a target gene of farnesoid X receptor in chenodeoxycholic acid-induced liver hypertrophy. Hepatology 49:979–988 [DOI] [PubMed] [Google Scholar]
- 22. Fichter CD , Herz C , Münch C , Opitz OG , Werner M , Lassmann S. 2011. Occurrence of multipolar mitoses and association with Aurora-A/-B kinases and p53 mutations in aneuploid esophageal carcinoma cells. BMC Cell Biol 12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burgermeister E , Tencer L , Liscovitch M. 2003. Peroxisome proliferator-activated receptor-gamma upregulates caveolin-1 and caveolin-2 expression in human carcinoma cells. Oncogene 22:3888–3900 [DOI] [PubMed] [Google Scholar]
- 24. Yu J , Leung WK , Ebert MP , Leong RW , Tse PC , Chan MW , Bai AH , To KF , Malfertheiner P , Sung JJ. 2003. Absence of cyclin D2 expression is associated with promoter hypermethylation in gastric cancer. Br J Cancer 88:1560–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rödel F , Capalbo G , Rödel C , Weiss C. 2009. Caveolin-1 as a prognostic marker for local control after preoperative chemoradiation therapy in rectal cancer. Int J Radiat Oncol Biol Phys 73:846–852 [DOI] [PubMed] [Google Scholar]
- 26. Qin P , Borges-Marcucci LA , Evans MJ , Harnish DC. 2005. Bile acid signaling through FXR induces intracellular adhesion molecule-1 expression in mouse liver and human hepatocytes. Am J Physiol Gastrointest Liver Physiol 289:G267–G273 [DOI] [PubMed] [Google Scholar]
- 27. Lian F , Xing X , Yuan G , Schäfer C , Rauser S , Walch A , Röcken C , Ebeling M , Wright MB , Schmid RM , Ebert MP , Burgermeister E. 2011. Farnesoid X receptor protects human and murine gastric epithelial cells against inflammation-induced damage. Biochem J 438:315–323 [DOI] [PubMed] [Google Scholar]
- 28. Bonuccelli G , Casimiro MC , Sotgia F , Wang C , Liu M , Katiyar S , Zhou J , Dew E , Capozza F , Daumer KM , Minetti C , Milliman JN , Alpy F , Rio MC , Tomasetto C , Mercier I , Flomenberg N , Frank PG , Pestell RG , Lisanti MP. 2009. Caveolin-1 (P132L), a common breast cancer mutation, confers mammary cell invasiveness and defines a novel stem cell/metastasis-associated gene signature. Am J Pathol 174:1650–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayashi K , Matsuda S , Machida K , Yamamoto T , Fukuda Y , Nimura Y , Hayakawa T , Hamaguchi M. 2001. Invasion activating caveolin-1 mutation in human scirrhous breast cancers. Cancer Res 61:2361–2364 [PubMed] [Google Scholar]
- 30. Burgermeister E , Friedrich T , Hitkova I , Regel I , Einwächter H , Zimmermann W , Röcken C , Perren A , Wright MB , Schmid RM , Seger R , Ebert MP. 2011. The Ras inhibitors caveolin-1 and docking protein 1 activate peroxisome proliferator-activated receptor gamma through spatial relocalization at helix 7 of its ligand-binding domain. Mol Cell Biol 31:3497–3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williams TM , Lisanti MP. 2005. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol Cell Physiol 288:C494–C506 [DOI] [PubMed] [Google Scholar]
- 32. Schlegel A , Arvan P , Lisanti MP. 2001. Caveolin-1 binding to endoplasmic reticulum membranes and entry into the regulated secretory pathway are regulated by serine phosphorylation. Protein sorting at the level of the endoplasmic reticulum. J Biol Chem 276:4398–4408 [DOI] [PubMed] [Google Scholar]
- 33. Tahir SA , Yang G , Ebara S , Timme TL , Satoh T , Li L , Goltsov A , Ittmann M , Morrisett JD , Thompson TC. 2001. Secreted caveolin-1 stimulates cell survival/clonal growth and contributes to metastasis in androgen-insensitive prostate cancer. Cancer Res 61:3882–3885 [PubMed] [Google Scholar]
- 34. Ayala GE , Dai H , Tahir SA , Li R , Timme T , Ittmann M , Frolov A , Wheeler TM , Rowley D , Thompson TC. 2006. Stromal antiapoptotic paracrine loop in perineural invasion of prostatic carcinoma. Cancer Res 66:5159–5164 [DOI] [PubMed] [Google Scholar]
- 35. Lin SY , Yeh KT , Chen WT , Chen HC , Chen ST , Chang JG. 2004. Promoter CpG methylation of caveolin-1 in sporadic colorectal cancer. Anticancer Res 24:1645–1650 [PubMed] [Google Scholar]
- 36. Engelman JA , Zhang XL , Lisanti MP. 1999. Sequence and detailed organization of the human caveolin-1 and -2 genes located near the D7S522 locus (7q31.1). Methylation of a CpG island in the 5′ promoter region of the caveolin-1 gene in human breast cancer cell lines. FEBS Lett 448:221–230 [DOI] [PubMed] [Google Scholar]
- 37. Dvorak K , Watts GS , Ramsey L , Holubec H , Payne CM , Bernstein C , Jenkins GJ , Sampliner RE , Prasad A , Garewal HS , Bernstein H. 2009. Expression of bile acid transporting proteins in Barrett's esophagus and esophageal adenocarcinoma. Am J Gastroenterol 104:302–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hao Y , Sood S , Triadafilopoulos G , Kim JH , Wang Z , Sahbaie P , Omary MB , Lowe AW. 2007. Gene expression changes associated with Barrett's esophagus and Barrett's-associated adenocarcinoma cell lines after acid or bile salt exposure. BMC Gastroenterol 7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burnat G , Rau T , Elshimi E , Hahn EG , Konturek PC. 2007. Bile acids induce overexpression of homeobox gene CDX-2 and vascular endothelial growth factor (VEGF) in human Barrett's esophageal mucosa and adenocarcinoma cell line. Scand J Gastroenterol 42:1460–1465 [DOI] [PubMed] [Google Scholar]
- 40. Kong J , Crissey MA , Funakoshi S , Kreindler JL , Lynch JP. 2011. Ectopic Cdx2 expression in murine esophagus models an intermediate stage in the emergence of Barrett's esophagus. PLoS One 6:e18280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jean-Louis S , Akare S , Ali MA , Mash EA , Meuillet E , Martinez JD. 2006. Deoxycholic acid induces intracellular signaling through membrane perturbations. J Biol Chem 281:14948–14960 [DOI] [PubMed] [Google Scholar]
- 42. Hu Y , Williams VA , Gellersen O , Jones C , Watson TJ , Peters JH. 2007. The pathogenesis of Barrett's esophagus: secondary bile acids upregulate intestinal differentiation factor CDX2 expression in esophageal cells. J Gastrointest Surg 11:827–834 [DOI] [PubMed] [Google Scholar]
- 43. Avissar NE , Toia L , Hu Y , Watson TJ , Jones C , Raymond DP , Matousek A , Peters JH. 2009. Bile acid alone, or in combination with acid, induces CDX2 expression through activation of the epidermal growth factor receptor (EGFR). J Gastrointest Surg 13:212–222 [DOI] [PubMed] [Google Scholar]
- 44. Cronin J , McAdam E , Danikas A , Tselepis C , Griffiths P , Baxter J , Thomas L , Manson J , Jenkins G. 2011. Epidermal growth factor receptor (EGFR) is overexpressed in high-grade dysplasia and adenocarcinoma of the esophagus and may represent a biomarker of histological progression in Barrett's esophagus (BE). Am J Gastroenterol 106:46–56 [DOI] [PubMed] [Google Scholar]
- 45. Lanuti M , Liu G , Goodwin JM , Zhai R , Fuchs BC , Asomaning K , Su L , Nishioka NS , Tanabe KK , Christiani DC. 2008. A functional epidermal growth factor (EGF) polymorphism, EGF serum levels, and esophageal adenocarcinoma risk and outcome. Clin Cancer Res 14:3216–3222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yeh M , Cole AL , Choi J , Liu Y , Tulchinsky D , Qiao JH , Fishbein MC , Dooley AN , Hovnanian T , Mouilleseaux K , Vora DK , Yang WP , Gargalovic P , Kirchgessner T , Shyy JY , Berliner JA. 2004. Role for sterol regulatory element-binding protein in activation of endothelial cells by phospholipid oxidation products. Circ Res 95:780–788 [DOI] [PubMed] [Google Scholar]
- 47. Llaverias G , Vázquez-Carrera M , Sánchez RM , Noé V , Ciudad CJ , Laguna JC , Alegret M. 2004. Rosiglitazone upregulates caveolin-1 expression in THP-1 cells through a PPAR-dependent mechanism. J Lipid Res 45:2015–2024 [DOI] [PubMed] [Google Scholar]
- 48. Sunaga N , Miyajima K , Suzuki M , Sato M , White MA , Ramirez RD , Shay JW , Gazdar AF , Minna JD. 2004. Different roles for caveolin-1 in the development of non-small cell lung cancer versus small cell lung cancer. Cancer Res 64:4277–4285 [DOI] [PubMed] [Google Scholar]
- 49. Engelman JA , Zhang XL , Lisanti MP. 1998. Genes encoding human caveolin-1 and -2 are co-localized to the D7S522 locus (7q31.1), a known fragile site (FRA7G) that is frequently deleted in human cancers. FEBS Lett 436:403–410 [DOI] [PubMed] [Google Scholar]
- 50. Mori Y , Cai K , Cheng Y , Wang S , Paun B , Hamilton JP , Jin Z , Sato F , Berki AT , Kan T , Ito T , Mantzur C , Abraham JM , Meltzer SJ. 2006. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterology 131:797–808 [DOI] [PubMed] [Google Scholar]
- 51. Yamashita S , Tsujino Y , Moriguchi K , Tatematsu M , Ushijima T. 2006. Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2′-deoxycytidine treatment and oligonucleotide microarray. Cancer Sci 97:64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buxman MM , Goodkin PE , Fahrenbach WH , Dimond RL. 1979. Harlequin ichthyosis with epidermal lipid abnormality. Arch Dermatol 115:189–193 [PubMed] [Google Scholar]
- 53. Jong MC , Gijbels MJ , Dahlmans VE , Gorp PJ , Koopman SJ , Ponec M , Hofker MH , Havekes LM. 1998. Hyperlipidemia and cutaneous abnormalities in transgenic mice overexpressing human apolipoprotein C1. J Clin Invest 101:145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Evers BM , Farooqi MS , Shelton JM , Richardson JA , Goldstein JL , Brown MS , Liang G. 2010. Hair growth defects in Insig-deficient mice caused by cholesterol precursor accumulation and reversed by simvastatin. J Invest Dermatol 130:1237–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Smyth I , Hacking DF , Hilton AA , Mukhamedova N , Meikle PJ , Ellis S , Satterley K , Collinge JE , de Graaf CA , Bahlo M , Sviridov D , Kile BT , Hilton DJ. 2008. A mouse model of harlequin ichthyosis delineates a key role for Abca12 in lipid homeostasis. PLoS Genet 4:e1000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mirza R , Hayasaka S , Takagishi Y , Kambe F , Ohmori S , Maki K , Yamamoto M , Murakami K , Kaji T , Zadworny D , Murata Y , Seo H. 2006. DHCR24 gene knockout mice demonstrate lethal dermopathy with differentiation and maturation defects in the epidermis. J Invest Dermatol 126:638–647 [DOI] [PubMed] [Google Scholar]
- 57. Ishibashi S , Schwarz M , Frykman PK , Herz J , Russell DW. 1996. Disruption of cholesterol 7alpha-hydroxylase gene in mice. I. Postnatal lethality reversed by bile acid and vitamin supplementation. J Biol Chem 271:18017–18023 [DOI] [PubMed] [Google Scholar]
- 58. Hamanaka S , Ujihara M , Serizawa S , Nakazawa S , Otsuka F. 1997. A case of recessive X-linked ichthyosis: scale-specific abnormalities of lipid composition may explain the pathogenesis of the skin manifestation. J Dermatol 24:156–160 [DOI] [PubMed] [Google Scholar]
- 59. Mégarbané H , Mégarbané A. 2011. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome. Orphanet J Rare Dis 6:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takahashi K , Folmer J , Coulombe PA. 1994. Increased expression of keratin 16 causes anomalies in cytoarchitecture and keratinization in transgenic mouse skin. J Cell Biol 127:505–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schaaij-Visser TB , Bremmer JF , Braakhuis BJ , Heck AJ , Slijper M , van der Waal I , Brakenhoff RH. 2010. Evaluation of cornulin, keratin 4, keratin 13 expression and grade of dysplasia for predicting malignant progression of oral leukoplakia. Oral Oncol 46:123–127 [DOI] [PubMed] [Google Scholar]
- 62. Yao D , Alexander CL , Quinn JA , Porter MJ , Wu H , Greenhalgh DA. 2006. PTEN loss promotes rasHa-mediated papillomatogenesis via dual up-regulation of AKT activity and cell cycle deregulation but malignant conversion proceeds via PTEN-associated pathways. Cancer Res 66:1302–1312 [DOI] [PubMed] [Google Scholar]
- 63. van Baal JW , Bozikas A , Pronk R , Ten Kate FJ , Milano F , Rygiel AM , Rosmolen WD , Peppelenbosch MP , Bergman JJ , Krishnadath KK. 2008. Cytokeratin and CDX-2 expression in Barrett's esophagus. Scand J Gastroenterol 43:132–140 [DOI] [PubMed] [Google Scholar]
- 64. Magin TM , Vijayaraj P , Leube RE. 2007. Structural and regulatory functions of keratins. Exp Cell Res 313:2021–2032 [DOI] [PubMed] [Google Scholar]
- 65. Langlois S , Cowan KN , Shao Q , Cowan BJ , Laird DW. 2010. The tumor-suppressive function of Connexin43 in keratinocytes is mediated in part via interaction with caveolin-1. Cancer Res 70:4222–4232 [DOI] [PubMed] [Google Scholar]
- 66. Langlois S , Cowan KN , Shao Q , Cowan BJ , Laird DW. 2008. Caveolin-1 and -2 interact with connexin43 and regulate gap junctional intercellular communication in keratinocytes. Mol Biol Cell 19:912–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kant P , Hull MA. 2011. Excess body weight and obesity-the link with gastrointestinal and hepatobiliary cancer. Nat Rev Gastroenterol Hepatol 8:224–238 [DOI] [PubMed] [Google Scholar]
- 68. Abrams JA. 2009. Obesity and Barrett's oesophagus: more than just reflux. Gut 58:1437–1438 [DOI] [PubMed] [Google Scholar]
- 69. Flegal KM. 2000. Obesity, overweight, hypertension, and high blood cholesterol: the importance of age. Obes Res 8:676–677 [DOI] [PubMed] [Google Scholar]
- 70. Zhang Y , Kast-Woelbern HR , Edwards PA. 2003. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem 278:104–110 [DOI] [PubMed] [Google Scholar]