

Abstract
C-terminal binding protein (CtBP) is a transcriptional corepressor that plays an important role in mammalian development and tumorigenesis. We demonstrate that CtBP is expressed in adenohypophyseal cells and is expressed at high levels in human corticotroph, somatotroph, and lactotroph pituitary adenomas. CtBP interacts with Ikaros isoforms in GH4 and AtT20 pituitary tumor cells. Ikaros and CtBP1 expression is coordinately induced by hypoxia, and this response is abrogated by CtBP1 deficiency. Forced reduction of CtBP1 leads to reduced cell growth, up-regulation of Sprouty 2, and down-regulation of ectonucleotide pyrophosphatase phosphodiesterase 2 (Enpp2). Consistent with diminished Enpp2 activity, CtBP1-deficient pituitary cells are more susceptible to hypoxia-induced apoptosis, which is rescued by Enpp2-derived lysophosphatidic acid treatment. These results identify putative oncogenic properties of CtBP1 and provide new insights into the overlapping functions of two members of the chromatin remodeling network in the response to hypoxic pituitary tumor cell drive.
Tumors of the anterior pituitary exhibit a spectrum of hormonal and proliferative activities. Studies of cytodifferentiation and pituitary cell proliferation have provided valuable insight into the pathophysiology of tumorigenesis in this gland. The transcription factor Ikaros was identified as a necessary factor for corticotroph and somatotroph population expansion and cell-specific hormone expression during pituitary development (1, 2). Ikaros has also been implicated in pituitary tumorigenesis. The dominant-negative Ikaros isoform Ik6 was detected in a significant proportion of human pituitary tumors (3). Forced expression of Ik6 in mouse AtT20 corticotroph and rat GH4 mammosomatotroph tumor cells enhanced cell proliferation, evidenced by increased S-phase entry, colony formation in soft agar, and growth of xenografts in vivo, and increased protection against apoptosis by up-regulating Bcl-XL through selective acetylation of the Bcl-XL promoter (4).
In addition to transcriptional and epigenetic regulation of pituitary hormone genes (1, 2, 5), Ikaros has also been shown to regulate adenohypophyseal cell metabolism through cholesterol uptake (6). To gain further insight into Ikaros action and to identify associated regulators in pituitary cells, we considered members of the Ikaros nuclear complex in the lymphohematopoietic system. Two independent repression domains at the N and C termini of the Ikaros protein have been shown to recruit members of the nucleosome remodeling and histone deacetylation complex including Mi-2β, Sin3A, Sin3B, and class I histone deacetylases 1 and 2 (7, 8). Ikaros can also interact with the nucleosome remodeling and histone deacetylation complex member C-terminal binding protein (CtBP) through the PEDLS consensus sequence at its N terminus (9). CtBP interacts with endogenous Ikaros in both thymic nuclear extracts and reticulocyte lysates. CtBP was also purified from T-cell nuclear extracts, confirming the presence of the interaction both in vitro and in vivo. It was demonstrated that mutations in the Ikaros protein that abolish its interaction with CtBP weaken its ability to repress transcription of thymidine kinase and adenovirus major late promoters; however, there is little knowledge of common endogenous targets of the Ikaros and CtBP proteins (9). Because CtBP displays a broad range of oncogenic properties in many cell types (10–12) and there have been limited efforts to elucidate its role in the pituitary, we set out to characterize the role of CtBP in the pituitary and examine potential interactions between this corepressor and Ikaros isoforms in this gland.
Results
CtBP1 expression in the normal and neoplastic pituitary
Pituitary expression of CtBP has not been previously described. We examined CtBP expression in normal mouse pituitary at various stages of gestation and in the various hormone-producing cell types. CtBP expression has its onset at embryonic d 7.5 (13), and we identified expression at the earliest stages of pituitary development. In the mature gland, CtBP was localized to the nucleus of cells in the pars anterior, pars intermedia, and pars nervosa (Fig. 1A). Immunoreactivity was relatively homogeneous throughout the gland in the nuclei of almost all parenchymal cells and in vascular endothelium (Fig. 1B). Double staining for CtBP and the various pituitary hormones demonstrated colocalization of nuclear CtBP and cytoplasmic hormone in all cell types (Fig. 1C).
Fig. 1.

CtBP1 expression in the mouse and human pituitary. A, CtBP is localized in nuclei of cells throughout the pars anterior (PA), pars intermedia (PI), and pars nervosa (PN) of the normal mouse pituitary. B, Immunoreactivity is relatively homogeneous throughout the nuclei of almost all adenohypophyseal cells in the pars anterior and is present but weaker in the elongated nuclei of vascular endothelium (arrows). C, Double staining for CtBP (brown) and GH (red) demonstrates colocalization of nuclear CtBP and cytoplasmic GH as well as nuclear reactivity in cells that are negative for GH. The same pattern was seen in colocalization studies with other hormones. D, All but one of eight somatotroph adenomas exhibited very strong nuclear positivity in tumor cells. E, Corticotroph and lactotroph adenomas had moderate nuclear reactivity similar to that seen in the nontumorous adenohypophysis and in vascular endothelium (arrows); some lactotroph adenomas (illustrated) had focal loss of staining in scattered nuclei. F, All but one of seven null-cell adenomas were either negative or weakly positive. Vascular endothelium (arrow) served as an internal positive control.
To evaluate whether Ikaros impacts CtBP expression, we examined in a semiquantitative fashion CtBP expression in of the pituitaries of wild-type, Ik+/−, and Ik−/− mice. No detectable difference was identified. To verify this specifically in adenohypophyseal cells that are known to be impacted by Ikaros deficiency (1, 2), pituitary sections were costained for GH and CtBP or ACTH and CtBP and annotated, and the average intensity of CtBP staining was quantified in 75 cells per population. CtBP expression was comparable in GH- and ACTH-producing cells of all genotypes, further confirming that loss of Ikaros expression has little effect on CtBP stability in the primary mouse pituitary.
We next examined CtBP1 localization by immunohistochemistry in the human pituitary. This included five normal human pituitaries obtained at autopsy and 31 pituitary adenomas. All tumors were fully characterized according to currently accepted diagnostic criteria (14). They consisted of six corticotroph adenomas, eight somatotroph adenomas, four lactotroph adenomas, six gonadotroph adenomas, and seven null-cell adenomas. The staining pattern in the normal human pituitary was similar to that in the normal mouse gland. There was strong reactivity in vascular endothelium; other stromal cells were largely negative. All but one somatotroph adenoma exhibited very strong positivity (Fig. 1D). All corticotroph adenomas and lactotroph adenomas showed moderate reactivity, the latter with focal loss of staining (Fig. 1E). In contrast, four of the six gonadotroph adenomas and all but one of the null-cell adenomas were either negative or weakly positive (Fig. 1F).
Endogenous CtBP interacts with Ikaros isoforms in pituitary tumor cells
Ikaros has been shown to interact with CtBP in lymphocytes; however, this relationship has not been examined in neuroendocrine cells. Given our identification of prominent CtBP1 reactivity in somatotroph, lactotroph, and corticotroph adenomas, we selected pituitary GH4 and AtT20 cells to examine CtBP1 interaction with Ikaros in these cells that are known to express Ikaros (1, 2). Using Ik-V5 immunoprecipitation, we identified interaction between endogenous CtBP and the predominant Ikaros isoform Ik1 or the dominant-negative isoform Ik6 in transfected AtT20 cells (Fig. 2A). In complementary experiments, we show the reverse in GH4 pituitary tumor cells where CtBP1 immunoprecipitates showed interaction of CtBP with Ik1 or with Ik6 (Fig. 2B). Negative controls included immunoprecipitations of empty-vector-transfected cells. Immunoprecipitation with normal mouse IgG served as an additional negative control.
Fig. 2.

CtBP Interacts with Ikaros isoforms in pituitary tumor cells. Coimmunoprecipitation experiments were performed using mouse AtT20 corticotroph (A) and rat GH4 mammosomatotroph (B) pituitary tumor cell lysates. Individual lanes represent lysates from cells transfected with either the pcDNA empty vector (control), pcDNA-Ik1-V5, or pcDNA-Ik6-V5. Immunoprecipitation (IP) with anti-V5 antibody was used to detect V5-tagged Ik1 and Ik6 expression in AtT20 cells. In rat GH4 cells, immunoprecipitation was performed with anti-CtBP1 followed by immunoblotting with anti-V5 to detect Ikaros in Ik1- and in Ik6-transfected GH4 cells. Immunoblotting with anti-CtBP detects comparable protein levels. In all experiments, immunoprecipitation with normal mouse IgG was performed as an additional negative control.
CtBP1 deficiency impairs pituitary cell growth
To determine the functional impact of CtBP1 and based on its up-regulated expression in somatotroph and lactotroph adenomas, we generated CtBP1 stable knockdown in GH4 cells. GH4 cells stably expressing either a scramble short hairpin RNA (shRNA) sequence (control) or a CtBP1-specific shRNA sequence were screened for CtBP1 expression before each experiment. Two control clones (2 and 3) and two CtBP1-knockdown clones (9 and 16) with more than 70% protein reduction were selected for subsequent experiments (Fig. 3, A and B). Cell counting identified a significant reduction in CtBP1-deficient GH4 cell number compared with their scrambled control cells (Fig. 3, C and D).
Fig. 3.

Loss of CtBP1 impairs pituitary cell growth. A, Western blotting of CtBP expression in GH4 cells stably transfected with scramble shRNA (clones 2 and 3) or CtBP1-specific shRNA sequences (clones 9 and 16). CtBP2 expression is unaffected by CtBP1 knockdown. B, Densitometric quantification of CtBP1 protein reduction normalized to actin. The sd of three independent experiments are depicted by error bars. Significance using a paired, two-tailed Student's t test is shown. C, GH4 cell growth was monitored by counting cell numbers on d 3 and 6 after seeding 1 × 106 cells. The growth curves for control and CtBP1-knockdown cells represent cell counts performed in triplicate. D, Bar graph indicating the number of cells on d 6 of the assay where the mean of three independent experiments, each performed in triplicate, is shown. Cell viability was determined by trypan blue staining. Error bars depict sd; significance according to the Student's paired t test is indicated.
Induction of Ikaros expression by hypoxia is diminished in CtBP-deficient pituitary cells
Given the known protective effect of CtBP1 on the response to hypoxia (10) and our finding of CtBP/Ikaros interaction, we asked whether Ikaros can respond coordinately to this stimulus. As anticipated, CtBP1 levels rise in response to hypoxia in GH4 cells (Fig. 4A) and in AtT20 cells (data not shown). We also noted that endogenous Ikaros expression increases significantly (∼6-fold) in response to this hypoxic drive (Fig. 4A). Moreover, the Ikaros response to hypoxia appears at least partially dependent on the presence of CtBP1, because CtBP1-deficient GH4 cells show attenuated Ikaros induction to the same stimulus (Fig. 4B).
Fig. 4.

Hypoxia enhances the expression of Ikaros in pituitary tumor cells, and CtBP1 deficiency diminishes this effect. A, Western blotting depicting CtBP1 levels and Ikaros in wild-type GH4 cells before and after the indicated 16 h of hypoxia. Neither actin nor GAPDH loading controls are affected by hypoxia in these cells. B, Western blotting depicting Ikaros levels in control and CtBP1-deficient GH4 cells before and after 16 h of hypoxia. GAPDH was detected as a loading control. Densitometric quantification of Ikaros expression is normalized to GAPDH. Bar graphs indicate the mean values of two independent experiments, and sd are depicted by error bars. *, Significance according to the Student's paired t test: P < 0.01.
CtBP1 deficiency promotes hypoxia-induced apoptosis in pituitary tumor cells
CtBP activity is enhanced by the hypoxic environment that characterizes many tumors (10). Hypoxia is common in pituitary tumors as evidenced by reduced microvascular density of a portal vascular supply with potentially lower oxygen levels even in the normal gland (15). Thus, we examined the impact of graded hypoxia on the apoptotic response in CtBP1-deficient cells. In preliminary experiments, caspase cleavage and poly ADP ribose polymerase (PARP) degradation were identified in GH4 cells at the severely hypoxic level of 0.2% O2, consistent with hypoxia resistance. Control and CtBP1-deficient GH4 cells were then synchronized for 24 h and subsequently subjected to 16 h of 0.2% O2. CtBP1-deficient cells were measurably more susceptible to hypoxia-induced apoptosis as evidenced by enhanced PARP and caspase 3 cleavage (Fig. 5, A–C) compared with controls. Additionally, flow cytometry with annexin V staining confirmed increased apoptosis in response to CtBP1 down-regulation (Fig. 6, A and D). Moreover, CtBP1-deficient cells were more sensitive to hypoxia- mediated apoptosis compared with scrambled control cells (Fig. 6, B and E).
Fig. 5.
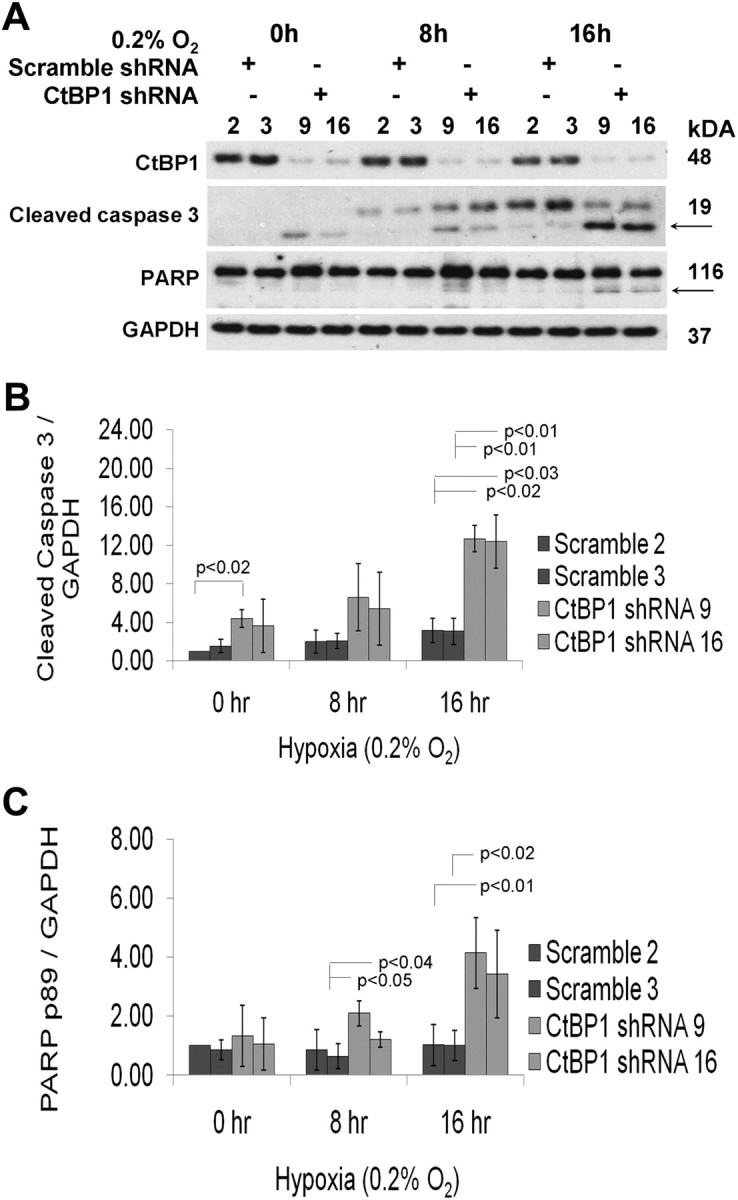
CtBP1 mediates hypoxia-induced apoptosis. A, Protein was extracted from synchronized GH4 cells after 0, 8, and 16 h of exposure to 0.2% O2. PARP cleavage and 17-kDa cleaved caspase 3 were used to quantify apoptosis and indicated by an arrow. GAPDH was detected as an endogenous control that was not affected by hypoxia. For every time point, individual lanes represent an independent clone expressing either scramble (clones 2 and 3) or CtBP1-specific (clones 9 and 16) shRNA. Densitometric quantification of 17-kDa cleaved caspase 3 (B) and 89-kDa cleaved PARP (C) expression are depicted by bar graphs. The sd (three experiments) are indicated by error bars. P values were calculated using paired Student's t test.
Fig. 6.

Impact of CtBP1 knockdown and LPA treatment on CtBP1-mediated hypoxic apoptosis. Pooled clones (n = 3) of GH4 pituitary cells with scrambled control (A) or shRNA targeting CtBP1 (D) were examined by flow cytometry after Annexin V staining. The proportion of cells undergoing early apoptosis is shown in the Q3 window (red circles). To examine the impact of hypoxia, cells were also examined under 0.2% O2 conditions. CtBP1 knockdown cells (E) show greater susceptibility to hypoxia-induced apoptosis compared with controls (B). Concomitant treatment with LPA 5 μm (C and F) rescues hypoxia-induced apoptosis in CtBP1 knockdown cells. Data shown are representative of two independent experiments.
Loss of CtBP1 leads to deregulation of genes involved in pituitary cell growth and survival
To identify novel targets potentially involved in mediating CtBP1 control of pituitary cell growth and apoptosis, we performed gene expression profiling of CtBP1-deficient GH4 cells. Genes differentially expressed by greater than 1.5-fold (P < 0.05) in CtBP1-knockdown vs. control cells were distinguished using an ANOVA test (Supplemental Tables 1 and 2, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Consistent with the observed phenotype of reduced growth by CtBP1-deficient vs. controls cells, several of the genes identified are implicated in growth factor signaling, which represented our focus. Real-time PCR confirmed up-regulation of sprouty 2 (Spry2) and somatostatin receptor 1 (Sstr1) and down-regulation of ectonucleotide pyrophosphatase phosphodiesterase 2 (Enpp2)/autotaxin in CtBP1-deficient cells (Fig. 7). These microarray-derived data and real-time PCR-validated results highlighted deregulation of genes that are common to proliferative and survival signaling pathways.
Fig. 7.
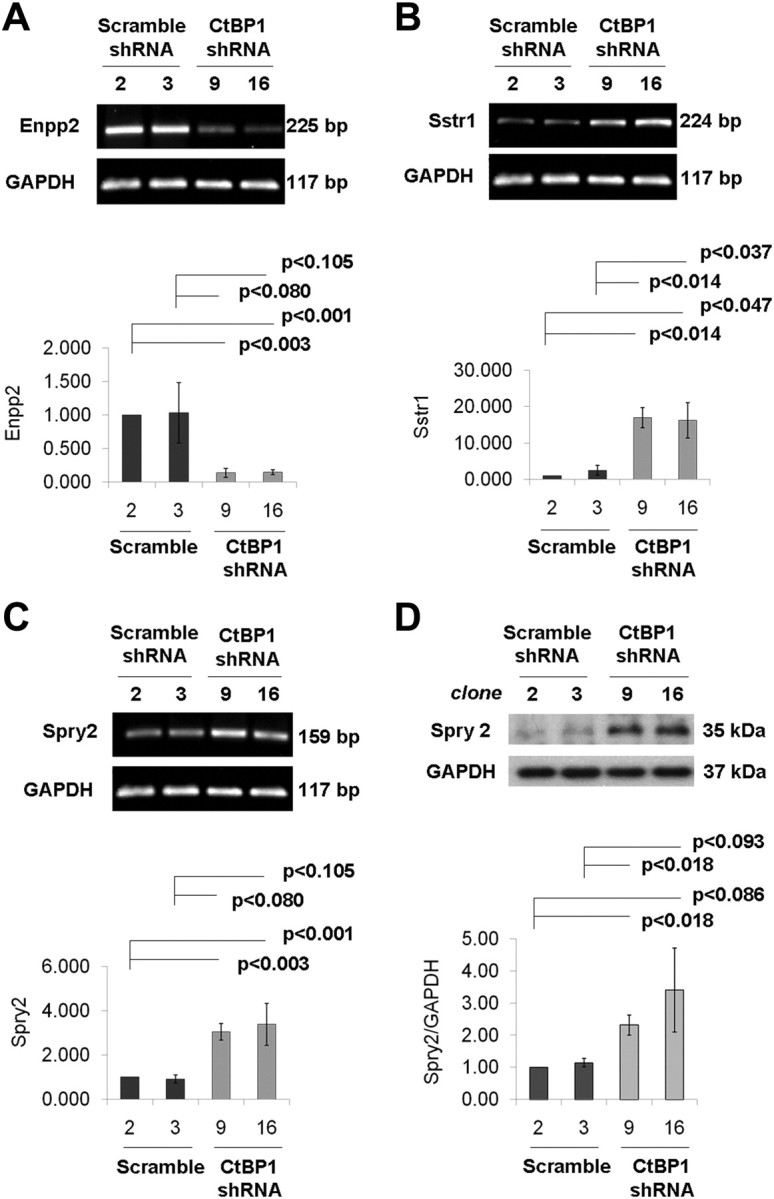
CtBP1 deficiency deregulates multiple genes involved in pituitary cell growth. Changes in gene expression at the mRNA and/or protein levels for Enpp2 (A), Sstr1 (B), and Spry2 (C, mRNA; D, protein). Relative mRNA levels were calculated using the 2−ΔΔCt method, with GAPDH, PGK1, and cyclophilin A as endogenous controls. Densitometric quantification of Spry2 are normalized to GAPDH expression. Bar graphs depict mean values for three experiments. Significance was calculated according to the Student's paired t test.
Functional validation of CtBP1 targets in the pituitary
The role of Sstr1 in the regulation of pituitary cell growth is well known; however, Enpp2 and Spry2 have not been previously examined in pituitary cells.
If Enpp2/autotaxin activity is functional in mediating CtBP1 action, we reasoned that intracellular generation of lysophosphatidic acid (LPA) would be diminished in CtBP1-deficient cells; this feature is potentially amenable to pharmacological rescue. We therefore treated hypoxic CtBP1-deficient and scrambled control GH4 cells with LPA. Figure 6, C and F, depicts the protective effect of LPA treatment on hypoxia-mediated apoptosis. Importantly, LPA rescued the effect of hypoxic apoptosis more strikingly in CtBP1-deficient than in control GH4 cells (Fig. 6, C and F). To further validate the involvement of Enpp2, we generated cells with stable knockdown of this enzyme. Figure 8 demonstrates the diminished cell growth of GH4 cells resulting from Enpp2 silencing. Moreover, given the putative effects of this phosphodiesterase on intracellular cAMP, Enpp2 knockdown cells exhibited a striking reduction of GH gene expression (Fig. 8).
Fig. 8.

Stable knockdown of Enpp2 diminishes GH4 cell growth and hormone expression. A, Pooled clones (n = 3) of GH4 pituitary cells with scrambled shRNA (control) or shRNA targeting Enpp2 were examined by Western blotting. B, Cell counting demonstrates reduced cell growth rate of Enpp2 knockdown cells compared with controls. Data shown are representative of three independent experiments.
Spry2 inhibits proliferation through several growth factor signaling pathways (16, 17). Consistent with this prediction, fibroblast growth factor (FGF) stimulation of CtBP1-deficient cells revealed blunted FRS2α (fibroblast growth factor receptor substrate 2 α) responses (Supplemental Fig. 1).
Discussion
We present here evidence that the transcription factor Ikaros interacts with the CtBP corepressor in the pituitary to modulate cell growth and survival. We show that loss of CtBP impairs pituitary cell growth and sensitizes these cells to apoptosis, likely through multiple deregulated signaling cascades.
The CtBP family of transcriptional corepressors, collectively referred to as CtBP, includes two highly homologous proteins, CtBP1 and CtBP2, which exhibit both unique and redundant functions. CtBP interacts with transcription factors through the E1A CtBP-binding motif (PXDLS) and is recruited to target promoters by various DNA-binding proteins and nuclear corepressor complexes (18). In the pituitary, CtBP is recruited to several gene promoters by the ZEB-corepressor for the repressor element-1 silencing transcription factor (CoREST)-lysine-specific demethylase-1 (LSD1) corepressor complex, which is known to repress GH transcription during lactotroph differentiation (19). In the present study, we demonstrate that endogenous CtBP interacts with Ikaros and with the pituitary tumor-derived dominant-negative isoform Ik6 in mouse and rat pituitary tumor cells. We also show for the first time that CtBP is expressed in human pituitary tumors, especially in corticotroph, somatotroph, and lactotroph types.
It is well appreciated that transcriptional regulators act in a cell- and context-specific manner. For example, the histone demethylase LSD1 plays a role in pituitary gene activation as part of the MLL-1 (mixed lineage leukemia-1) coactivator complex but also represses gene expression at a later stage in development through the ZEB1-CoREST-LSD1-CtBP complex (19). Temporal patterns of expression of specific LSD1-interacting proteins have been shown to modulate gene activation and repression programs during pituitary development. Pit1, a critical transcription factor for GH expression, is present on the promoter regulatory region of the GH gene as early as embryonic d 13.5–14.5 in the fetal mouse pituitary; however, LSD1 was not detected at this region of the GH promoter until embryonic d 16.5–17.5, at the time of GH gene activation (19). In this context, LSD1 is recruited to the GH promoter by a ZEB1-CoREST-CtBP repression complex that also contains the histone deacetylases, HDAC1 and HDAC2. Interestingly, Ikaros also regulates GH and PRL gene expression in mammosomatotroph cells through histone modifications that alter Pit1 binding to the proximal GH and PRL gene promoters (5). It is possible that Ikaros requires CtBP for context-specific gene regulation programs, as is the case for transcriptional regulation by ZEB and LSD1.
The CtBP proteins have been implicated in several aspects of tumorigenesis including promotion of epithelial-mesenchymal transition (10), repression of tumor suppressor genes (11), and antiapoptotic activity (20). Given CtBP's broad range of critical cellular functions, we found it pertinent to characterize CtBP function in the pituitary. We were interested in CtBP1specifically because homozygous CtBP1-mutant mice are one third smaller than their wild-type and heterozygous littermates, which is comparable to the dwarf phenotype of Ikaros-null mice (2). Here, we show that loss of CtBP1 impairs GH4 pituitary cell growth. Through microarray gene expression profiling, we identified changes in the expression of several genes in CtBP1-deficient cells, which may be implicated in the impaired pituitary proliferative phenotype. Down-regulation of autotaxin/Enpp2 and up-regulation of Sstr1 and Spry2 were confirmed by Western blotting and/or real-time quantitative PCR. Enpp2 was originally described for its ability to stimulate tumor cell motility (21) and has because been found to be overexpressed by a variety of tumors including breast cancer, glioblastoma, hepatocellular carcinoma, neuroblastoma, non-small-cell lung cancer, prostate cancer, renal cell carcinoma, and thyroid carcinoma (22). Of relevance to pituitary tumors, Enpp2 catalyzes the conversion of lysophosphatidylcholine to LPA. LPA enhances proliferation through G protein-coupled receptor signaling (23) and activation of the transcription factor cAMP response element-binding (CREB) (24). Overexpression of a transcriptionally inactive CREB mutant in pituitary somatotrophs leads to dwarfism and somatotroph hypoplasia in these mice, consistent with the prominent role of CREB in somatotroph growth (25). Our observed reduction of Enpp2 in CtBP1-deficient somatotrophs is therefore consistent with the impaired ability of these cells to grow. Furthermore, LPA treatment rescues CtBP1-deficient apoptotic cells, consistent with functional involvement of Enpp2 in mediating CtBP1 action. Independent knockdown of Enpp2 recapitulated the effect of CtBP1 by reducing cell growth through enhanced apoptosis. Taken together, these findings emphasize the highly conserved role of the G protein/adenylate cyclase pathway in pituitary somatotroph tumorigenesis (26).
We also show that the slower-growing CtBP1-deficient pituitary cells express higher levels of Sstr mRNAs. Sstrs mediate antiproliferative effects and induce G1 cell cycle arrest (27). Sstr1 expression is regulated in pituitary GH3 cells by the transcription factor Pit1, through two binding sites on the Sstr1 promoter (28). The Pit1 binding sites exhibit opposing activity, with negative regulatory activity by the distal site and positive regulatory activity at the proximal site. Given that CtBP interacts with Ikaros and ZEB1, both transcription factors that regulate Pit1 target genes, it is possible that CtBP also plays a role in transcriptional repression of the Sstr1 gene promoter. However, additional studies are required to specifically address this possibility.
We also identified a relationship between loss of CtBP1 in GH4 and increased Spry2 expression. Several groups have reported Spry2-mediated inhibition of cell migration and proliferation in response to growth factor stimulation. In HeLa cells, stable transfection of Spry2 inhibits cell growth in response to serum and impedes cell migration in response to epidermal growth factor (EGF), FGF, and platelet-derived growth factor (29). Forced expression of Spry2 in a human leiomyosarcoma cell line (SK-LMS) inhibits cell proliferation, anchorage-independent cell growth, and migration in wound-healing and in vitro invasion assays (30). Here we show that increased Spry2 expression in CtBP1 deficiency impairs phosphorylation of the FGF/EGF immediate substrate fibroblast growth factor receptor substrate 2. These studies demonstrate the tumor suppressive potential of Spry2 and are consistent with our finding that the slower-growing CtBP1-deficient GH4 cells exhibit elevated levels of Spry2.
Hypoxia describes a state of reduced oxygen supply to the body's cells or tissues. Neoplastic tissues are frequently exposed to hypoxia due to their high metabolic activity, which outstrips their blood supply (31). In particular, pituitary adenomas exhibit reduced microvascular density and lower oxygen supply than the normal gland (15); apoptosis occurs with low frequency in a subset of pituitary adenomas, in carcinomas, and in pituitary hyperplasia and is induced by some antitumor therapies such as dopamine agonists and somatostatin analogs (32–34). Hypoxia elevates the level of free intracellular reduced nicotinamide adenine dinucleotide, which enhances recruitment of CtBP to target promoters (10). The effect of nicotinamide adenine dinucleotide on CtBP function was first examined based on the observation that the CtBP proteins show marked structural and functional similarities to d-isomer-specific 2-hydroxy acid dehydrogenase (35). A key difference between the CtBP1 and CtBP2 proteins is the presence of a unique nuclear localization sequence within the N terminus of CtBP2, which contributes to its nuclear location. Thus, CtBP1 relies on increased hypoxic drives to undergo dimerization and enhanced affinity for PLDLS-containing binding partners to facilitate recruitment to target promoters (36). In the present study, we demonstrate that CtBP1-deficient GH4 cells are more susceptible to apoptosis and that this effect is significantly enhanced with hypoxia. This observation is consistent with previous reports that CtBP protects against apoptosis through the repression of proapoptotic genes including PERP, Bax, and Noxa (12). Our finding that CtBP1 deficiency impairs cell growth and induces apoptosis in GH4 cells implicates CtBP as having a potential oncogenic role in pituitary tumor cells. Indeed, CtBP1 deficiency rendered cells more apoptotic and even more sensitive to hypoxia-mediated apoptosis. We also observed that hypoxia is a potent stimulus serving to also induce Ikaros expression, consistent with the importance of its interaction with CtBP. The coordinated response of these two factors to hypoxia supports their putative functions in mediating pituitary tumor cell survival. Additional studies are now required to characterize the common transcriptional targets of these two corepressors in pituitary developmental and neoplastic transitions.
Materials and Methods
Cells and tissues
Rat pituitary mammosomatotroph GH4 cells were cultured in Ham's F10 medium (100 IU/ml penicillin and 100 IU/ml streptomycin) supplemented with 12.5% horse serum (Sigma-Aldrich Canada Ltd., Ontario, Canada) and 2.5% fetal bovine serum (Sigma-Aldrich Canada). Mouse pituitary corticotroph AtT20 cells were cultured in DMEM H21 (1.5 g/liter bicarbonate, 4.5 g/liter glucose, 100 IU/ml penicillin, and 100 IU/ml streptomycin) supplemented with 10% fetal bovine serum (Sigma-Aldrich Canada). All cells were incubated at 37 C in 95% humidity and 5% CO2 unless otherwise indicated.
Mouse pituitaries were collected from Ikaros-deficient mice and their control littermates as previously described (1, 2). Human pituitaries were collected at autopsy and human pituitary tumors were retrieved from the files of the University Health Network with institutional ethics approval.
Plasmids and transfections
Expression vectors encoding Ik1 (CDM8-1) or Ik6 (CDM8-6) were provided by Dr. K. Georgopoulos (Boston, MA). The Ik1 sequence was cloned into a pcDNA3.1D V5-His-TOPO vector. The Ik6 sequence was amplified using primers containing the V5 sequence and the resulting Ik6-V5 fragment was cloned into a pcDNA3.1 vector. Wild-type AtT20 and GH4 cells were transfected with pcDNA3.1 empty vector, pcDNA3.1D/V5-His-TOPO-Ik1, or pcDNA3.1-Ik6-V5 using GeneJuice transfection reagent (Merck KGaA, Darmstadt, Germany). To down-regulate CtBP1, GH4 cells were transfected with scrambled sequence, rat CtBP1 SureSilencing shRNA plasmids (SA Biosciences, Frederick, MD), or Enpp2 shRNA (GenePharma Co., Ltd., Shanghai, China) using GeneJuice Transfection Reagent (Merck). CtBP1 or Enpp2 levels were examined by Western blotting, and clones displaying greater than 75% knockdown were selected for experiments.
Growth factor stimulation
Cells were synchronized by serum starvation for 24 h followed by 10 min treatment with one of the following: 13 nm IGF-I (Sigma), 25 nm EGF (Sigma), 25 ng/ml FGF-1 (R&D Systems, Minneapolis, MN) with 10 U/ml heparin (Sigma) that promotes dimerization of FGF receptor, or 5 μm synthetic LPA (1-oleoyl-LPA 18:1; Avanti Polar Lipids, Alabaster, AL). Protein was extracted immediately after treatment according to the protocol described below.
Cell count assay
On d 0, one million GH4 cells were seeded in a 10-cm culture dish in growth medium and counted daily for 6 d. Viable adherent and floating cells were counted by collecting media and trypsinized adherent cells in a single tube, followed by centrifugation and addition of 10 ml PBS to the cell pellet. Each experiment included three independent counts per clone, and the mean number of cells at each time point was used to compare the proliferative rates of the different clones.
Coimmunoprecipitation
Coimmunoprecipitation experiments were performed using protein G and protein A Sepharose beads for immunoprecipitation with monoclonal and polyclonal antibodies, respectively. The beads were washed three times in washing buffer [50 mm Tris-HCl, 150 mm NaCl (pH 7.5), 0.3% Triton-XL] and incubated at 4 C with 1 μg antibody for 3 h. Antibody concentrations are detailed in Supplemental Table 3. The beads were then washed three times, antibody-bound beads were incubated overnight with 800 μg protein per sample, and unbound protein was removed in a final wash step. The beads were mixed with sample buffer in a 1:1 ratio, and Western blotting was performed.
Western blotting
Protein was extracted from adherent cells by 1 h incubation in buffer composed of 50 mm Tris, 150 mm NaCl (pH 7.5), 1% Nonidet P-40, 1 mm EDTA, 1 mm EGTA, and protease inhibitor cocktail. The cells were lysed for 1 h at 4 C, and protein was isolated after 20 min centrifugation at 12,000 rpm. Protein concentration was determined for samples and a BSA standard using the Bio-Rad Protein Assay Kit II (Bio-Rad Laboratories, Inc., Ontario, Canada) by measurement of absorbance at 595 nm. Equal amounts of protein (25 or 50 μg) were combined with sample buffer [0.5 m Tris-HCl (pH 6.8), 25% glycerol, 2% sodium dodecyl sulfate, 0.001% bromothymol blue, 5% B-mercaptoethanol] in a 1:1 ratio and heated in boiling water for 4 min. The samples were separated on a 12% polyacrylamide gel and electrophoretically transferred to polyvinylidene fluoride membranes. Membranes were blocked for 2 h in Tris-buffered saline containing 1% Tween 20 (TBS-T) and 5% nonfat dried milk powder and were incubated with primary antibody at 4 C overnight. Primary antibody dilutions are included in Supplemental Table 3. Membranes were washed in TBS-T, incubated for 1 h with antimouse or antirabbit IgG horseradish peroxidase-linked antibodies, and washed again in TBS-T. Proteins were detected using chemiluminescent horseradish peroxidase detection reagent (Denville Scientific, Inc., Metuchen, NJ). Densitometry was performed using Image J software (National Institutes of Health, Bethesda, MD). Protein levels were normalized to either β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) loading controls.
Immunohistochemical staining
Mouse and human pituitary tissue samples were fixed in formalin, embedded in paraffin, and sectioned at 3–4 μm for routine histology. Immunohistochemistry was performed to identify pituitary hormones as described previously (1–3). CtBP staining was performed using a mouse monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA; sc-11390) at a dilution of 1:100. Staining was quantified using Aperio Image Scope V10.0.36.1805. Stains were analyzed in the anterior, intermediate, and posterior lobes of the pituitary using a custom color deconvolution algorithm. GH or ACTH staining was used to identify somatotroph or corticotroph cells, and CtBP expression was analyzed in these cell populations based on the annotation of 75 positive cells per population per pituitary.
RNA examination
RNA was isolated using an RNeasy Mini Kit and RNase-Free DNase Set (QIAGEN, Ontario, Canada). The integrity of RNA was monitored by amplification of GAPDH mRNA transcripts and separation on 1% agarose gels. cDNA was prepared using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Life Technologies Corp., Foster City, CA) and 2 μg total RNA. The thermal cycling conditions were as follows: 25 C for 10 min, 37 C for 2 h, and 85 C for 5 min. Real-time PCR were prepared using the RT2 SYBR Green with ROX Master Mix (SA Biosciences). Primer sequences are detailed in Supplemental Table 4. To confirm that real-time PCR primers yielded a single product, dissociation curves with a single peak were confirmed, and real-time PCR products were run on 1% agarose gels.
Hypoxia treatment of cells
Hypoxia experiments were performed using an Invivo2 400 Hypoxia Workstation and Ruskinn Hypoxia Gas Mixer (Ruskin, Bridgend, UK). All experiments were carried out at 37 C in graded levels of hypoxia. Based on the responses of caspase cleavage and PARP degradation, response was optimized at 9% CO2 and 0.2% O2 for additional experiments. Cells were synchronized by 24 h serum starvation before hypoxia treatment, followed by the addition of growth medium to prevent cell death due to multiple stressors. Protein was extracted after 0, 8, and/or 16 h of hypoxia treatment.
Microarray analysis of gene expression
Total RNA was extracted from two independent stably transfected clones expressing scramble sequence or CtBP1-specific shRNA. Hybridization to the Affymetrix Rat GeneChip 1.0 ST Array was performed at The Centre for Applied Genomics (Hospital for Sick Children, Toronto, Ontario, Canada). Sample RNA in vitro transcription, labeling, and hybridization followed standard Affymetrix protocols. Hybridized chips were washed and scanned on an Affymetrix GeneChip 3000 scanner. Data was analyzed by ANOVA using Partek Genomics Suite 6.5β software (Partek, Inc., St. Louis, MO). Differentially expressed genes were identified based on a minimum of P < 0.05 and greater than 1.5-fold change.
Cell cycle analysis
After trypsinization, 1–3 × 106 cells were washed with PBS and fixed with cold 80% ethanol for 1 h on ice. Fixed cells were washed with staining buffer [0.2% Triton X-100 and 1 mm EDTA (pH 8.0) in PBS] and resuspended in the staining buffer containing 50 μg/ml ribonuclease A (Sigma Chemical Co., St. Louis, MO) and 50 μg/ml propidium iodide for 1 h. Cell cycle analysis was performed using flow cytometry (Becton Dickinson, San Jose, CA) after annexin V staining using Cellquest analysis. Data were analyzed using the Modfit DNA Analysis program (Verity Software House, Topsham, ME).
Statistical analysis
All data are presented as mean ± sd. Differences were assessed by Student's paired t test. Significance was assigned based on a value of P ≤ 0.05.
Acknowledgments
We thank Quyen Tran at The Centre for Applied Genomics for technical assistance. We also like to thank Dr. Richard Hill's laboratory for use of their Hypoxia Workstation and Gas Mixer.
This work was supported by Canadian Institutes of Health Research (MOP 79340) and funded in part by the Ontario Ministry of Health and Long Term Care (OMOHLTC). The views expressed do not necessarily reflect those of the OMOHLTC.
Disclosure Summary: The authors declare no conflict of interest.
Footnotes
- CoREST
- Corepressor for the repressor element-1 silencing transcription factor
- CREB
- cAMP response element-binding
- CtBP
- C-terminal binding protein
- EGF
- epidermal growth factor
- Enpp2
- ectonucleotide pyrophosphatase/phosphodiesterase 2
- FGF
- fibroblast growth factor
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- LPA
- lysophosphatidic acid
- LSD1
- lysine-specific demethylase-1
- PARP
- poly ADP ribose polymerase
- shRNA
- short hairpin RNA
- Spry2
- sprouty 2
- Sstr1
- somatostatin receptor 1
- TBS-T
- Tris-buffered saline containing 1% Tween 20.
References
- 1. Ezzat S , Mader R , Yu S , Ning T , Poussier P , Asa SL. 2005. Ikaros integrates endocrine and immune system development. J Clin Invest 115:1021–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ezzat S , Mader R , Fischer S , Yu S , Ackerley C , Asa SL. 2006. An essential role for the hematopoietic transcription factor Ikaros in hypothalamic-pituitary-mediated somatic growth. Proc Natl Acad Sci USA 103:2214–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ezzat S , Yu S , Asa SL. 2003. Ikaros isoforms in human pituitary tumors: distinct localization, histone acetylation, and activation of the 5′ fibroblast growth factor receptor-4 promoter. Am J Pathol 163:1177–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ezzat S , Zhu X , Loeper S , Fischer S , Asa SL. 2006. Tumor-derived Ikaros 6 acetylates the Bcl-XL promoter to up-regulate a survival signal in pituitary cells. Mol Endocrinol 20:2976–2986 [DOI] [PubMed] [Google Scholar]
- 5. Ezzat S , Yu S , Asa SL. 2005. The zinc finger Ikaros transcription factor regulates pituitary growth hormone and prolactin gene expression through distinct effects on chromatin accessibility. Mol Endocrinol 19:1004–1011 [DOI] [PubMed] [Google Scholar]
- 6. Loeper S , Asa SL , Ezzat S. 2008. Ikaros modulates cholesterol uptake: a link between tumor suppression and differentiation. Cancer Res 68:3715–3723 [DOI] [PubMed] [Google Scholar]
- 7. Koipally J , Renold A , Kim J , Georgopoulos K. 1999. Repression by Ikaros and Aiolos is mediated through histone deacetylase complexes. EMBO J 18:3090–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim J , Sif S , Jones B , Jackson A , Koipally J , Heller E , Winandy S , Viel A , Sawyer A , Ikeda T , Kingston R , Georgopoulos K. 1999. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 10:345–355 [DOI] [PubMed] [Google Scholar]
- 9. Koipally J , Georgopoulos K. 2000. Ikaros interactions with CtBP reveal a repression mechanism that is independent of histone deacetylase activity. J Biol Chem 275:19594–19602 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Q , Wang SY , Nottke AC , Rocheleau JV , Piston DW , Goodman RH. 2006. Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc Natl Acad Sci USA 103:9029–9033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mroz EA , Baird AH , Michaud WA , Rocco JW. 2008. COOH-terminal binding protein regulates expression of the p16INK4A tumor suppressor and senescence in primary human cells. Cancer Res 68:6049–6053 [DOI] [PubMed] [Google Scholar]
- 12. Grooteclaes M , Deveraux Q , Hildebrand J , Zhang Q , Goodman RH , Frisch SM. 2003. C-terminal-binding protein corepresses epithelial and proapoptotic gene expression programs. Proc Natl Acad Sci USA 100:4568–4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hildebrand JD , Soriano P. 2002. Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol 22:5296–5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Asa SL. 2011. Tumors of the pituitary gland. AFIP atlas to tumor pathology, series 4, fascicle 15. Washington, DC: American Registry of Pathology [Google Scholar]
- 15. Viacava P , Gasperi M , Acerbi G , Manetti L , Cecconi E , Bonadio AG , Naccarato AG , Acerbi F , Parenti G , Lupi I , Genovesi M , Martino E. 2003. Microvascular density and vascular endothelial growth factor expression in normal pituitary tissue and pituitary adenomas. J Endocrinol Invest 26:23–28 [DOI] [PubMed] [Google Scholar]
- 16. Hanafusa H , Torii S , Yasunaga T , Nishida E. 2002. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol 4:850–858 [DOI] [PubMed] [Google Scholar]
- 17. Edwin F , Singh R , Endersby R , Baker SJ , Patel TB. 2006. The tumor suppressor PTEN is necessary for human Sprouty 2-mediated inhibition of cell proliferation. J Biol Chem 281:4816–4822 [DOI] [PubMed] [Google Scholar]
- 18. Quinlan KG , Verger A , Kwok A , Lee SH , Perdomo J , Nardini M , Bolognesi M , Crossley M. 2006. Role of the C-terminal binding protein PXDLS motif binding cleft in protein interactions and transcriptional repression. Mol Cell Biol 26:8202–8213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J , Scully K , Zhu X , Cai L , Zhang J , Prefontaine GG , Krones A , Ohgi KA , Zhu P , Garcia-Bassets I , Liu F , Taylor H , Lozach J , Jayes FL , Korach KS , Glass CK , Fu XD , Rosenfeld MG. 2007. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446:882–887 [DOI] [PubMed] [Google Scholar]
- 20. Paliwal S , Kovi RC , Nath B , Chen YW , Lewis BC , Grossman SR. 2007. The alternative reading frame tumor suppressor antagonizes hypoxia-induced cancer cell migration via interaction with the COOH-terminal binding protein corepressor. Cancer Res 67:9322–9329 [DOI] [PubMed] [Google Scholar]
- 21. Stracke ML , Krutzsch HC , Unsworth EJ , Arestad A , Cioce V , Schiffmann E , Liotta LA. 1992. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J Biol Chem 267:2524–2529 [PubMed] [Google Scholar]
- 22. Yuelling LM , Fuss B. 2008. Autotaxin (ATX): a multi-functional and multi-modular protein possessing enzymatic lysoPLD activity and matricellular properties. Biochim Biophys Acta 1781:525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moolenaar WH , Kranenburg O , Postma FR , Zondag GC. 1997. Lysophosphatidic acid: G-protein signalling and cellular responses. Curr Opin Cell Biol 9:168–173 [DOI] [PubMed] [Google Scholar]
- 24. Kwon YJ , Sun Y , Kim NH , Huh SO. 2009. Phosphorylation of CREB, a cyclic AMP responsive element binding protein, contributes partially to lysophosphatidic acid-induced fibroblast cell proliferation. Biochem Biophys Res Commun 380:655–659 [DOI] [PubMed] [Google Scholar]
- 25. Struthers RS , Vale WW , Arias C , Sawchenko PE , Montminy MR. 1991. Somatotroph hypoplasia and dwarfism in transgenic mice expressing a non-phosphorylatable CREB mutant. Nature 350:622–624 [DOI] [PubMed] [Google Scholar]
- 26. Asa SL , Ezzat S. 2009. The pathogenesis of pituitary tumors. Annu Rev Pathol 4:97–126 [DOI] [PubMed] [Google Scholar]
- 27. Srikant CB. 1997. Human somatostatin receptor mediated antiproliferative action evokes subtype selective cytotoxic and cytostatic signaling. Yale J Biol Med 70:541–548 [PMC free article] [PubMed] [Google Scholar]
- 28. Baumeister H , Wegner M , Richter D , Meyerhof W. 2000. Dual regulation of somatostatin receptor subtype 1 gene expression by pit-1 in anterior pituitary GH3 cells. Mol Endocrinol 14:255–271 [DOI] [PubMed] [Google Scholar]
- 29. Yigzaw Y , Cartin L , Pierre S , Scholich K , Patel TB. 2001. The C terminus of sprouty is important for modulation of cellular migration and proliferation. J Biol Chem 276:22742–22747 [DOI] [PubMed] [Google Scholar]
- 30. Lee CC , Putnam AJ , Miranti CK , Gustafson M , Wang LM , Vande Woude GF , Gao CF. 2004. Overexpression of sprouty 2 inhibits HGF/SF-mediated cell growth, invasion, migration, and cytokinesis. Oncogene 23:5193–5202 [DOI] [PubMed] [Google Scholar]
- 31. Höckel M , Vaupel P. 2001. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 93:266–276 [DOI] [PubMed] [Google Scholar]
- 32. Kontogeorgos G , Sambaziotis D , Piaditis G , Karameris A. 1997. Apoptosis in human pituitary adenomas: a morphologic and in situ end-labeling study. Mod Pathol 10:921–926 [PubMed] [Google Scholar]
- 33. Kulig E , Jin L , Qian X , Horvath E , Kovacs K , Stefaneanu L , Scheithauer BW , Lloyd RV. 1999. Apoptosis in nontumorous and neoplastic human pituitaries: expression of the Bcl-2 family of proteins. Am J Pathol 154:767–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kapranos N , Kontogeorgos G , Horvath E , Kovacs K. 2004. Morphology, molecular regulation and significance of apoptosis in pituitary adenomas. Front Horm Res 32:217–234 [DOI] [PubMed] [Google Scholar]
- 35. Schaeper U , Boyd JM , Verma S , Uhlmann E , Subramanian T , Chinnadurai G. 1995. Molecular cloning and characterization of a cellular phosphoprotein that interacts with a conserved C-terminal domain of adenovirus E1A involved in negative modulation of oncogenic transformation. Proc Natl Acad Sci USA 92:10467–10471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumar V , Carlson JE , Ohgi KA , Edwards TA , Rose DW , Escalante CR , Rosenfeld MG , Aggarwal AK. 2002. Transcription corepressor CtBP is an NAD+-regulated dehydrogenase. Mol Cell 10:857–869 [DOI] [PubMed] [Google Scholar]