

Abstract
Vasopressin (AVP) and CRH synergistically regulate adrenocorticotropin and insulin release at the level of the pituitary and pancreas, respectively. Here, we first extended these AVP and CRH coregulation processes to the adrenal medulla. We demonstrate that costimulation of chromaffin cells by AVP and CRH simultaneously induces a catecholamine secretion exceeding the one induced by each hormone alone, thus demonstrating a net potentiation. To further elucidate the molecular mechanisms underlying this synergism, we coexpressed human V1b and CRH receptor (CRHR)1 receptor in HEK293 cells. In this heterologous system, AVP also potentiated CRH-stimulated cAMP accumulation in a dose-dependent and saturable manner. This effect was only partially mimicked by phorbol ester or inhibited by a phospholipase C inhibitor respectively. This finding suggests the existence of an new molecular mechanism, independent from second messenger cross talk. Similarly, CRH potentiated the AVP-induced inositol phosphates production. Using bioluminescence resonance energy transfer, coimmunoprecipitation, and receptor rescue experiments, we demonstrate that V1b and CRHR1 receptors assemble as heterodimers. Moreover, new pharmacological properties emerged upon receptors cotransfection. Taken together, these data strongly suggest that direct molecular interactions between V1b and CRHR1 receptors play an important role in mediating the synergistic interactions between these two receptors.
Vasopressin (AVP) is a nine-amino acid neurohormone synthesized by parvocellular and magnocellular hypothalamic neurons. This hormone exhibits three main physiological roles: regulation of water homeostasis, vascular constriction, and control of ACTH secretion. More recently, AVP has also been demonstrated to be involved in modulating social behavior in human and rodents (for review see Ref. 1). The physiological effects of AVP are mediated by three specific receptor subtypes belonging to the G protein-coupled receptor (GPCR) family A presenting a nanomolar affinity for the natural hormone. The V2 receptor is positively coupled to adenylyl cyclase (AC) and mediates water reabsorption in the kidneys, whereas the V1a isoform induces vascular constriction via phospholipase C (PLC) activation (2). The V1b receptor, the third subtype, which was discovered last, is also coupled to the PLC pathway and has a well-known effect on ACTH secretion (3). In addition, it appears to be implicated in cognitive functions, and recent pharmacological advances have enlightened its anxiogenic role. In particular, the first V1b-selective antagonist clearly showed antidepressive and anxiolytic properties in rat and mouse (4). Indeed, V1b receptor knockout mice display a decrease in aggressive behavior (5) and secrete less ACTH than wild-type mice after a chronic stress (6).
CRH is a neuropeptide also involved in the control of stress-related behaviors (7). CRH exerts its physiological effects by acting on two well-characterized receptor subtypes belonging to the GPCR family B. The CRH receptor (CRHR)1 isoform has been proposed to mediate CRH-induced hypothalamo-pituitary-adrenal activation and anxiety-related behavior (8), whereas CRHR2 might be predominantly involved in the regulation of feeding behavior and cardiac functions (see for review Ref. 7). When stimulated by nanomolar concentrations of CRH, both CRHR subtypes activate the AC pathway (9).
In native tissues, AVP and CRH coregulate at least two secretory responses by acting on V1b and CRHR1 receptors. In the pituitary, AVP and CRH trigger ACTH liberation from corticotroph cells. This process is synergistic, because coapplication of low amounts of AVP and CRH, which cannot elicit ACTH release on their own, results in a significant secretion of ACTH (10). More recently, a similar mechanism has been described in the pancreas, where AVP and CRH synergistically stimulate insulin secretion (11).
The molecular mechanisms underlying the synergistic actions of AVP and CRH in vivo remain partially unexplained. A cross talk at the level of the second messenger mechanisms between the stimulation of diacylglycerol and inositol phosphate (IP) production by the V1b receptor on one hand and the accumulation of cAMP by the CRHR1 receptor on the other hand could account in part for this synergism. In particular, an activation of protein kinase C (PKC) has been previously reported to contribute to the potentiating effect of AVP on CRH-stimulated cAMP accumulation in rat pituitary cells (12) and on AVP potentiation of CRH-induced insulin release in rat pancreas (11). However, the AVP/CRH synergism may also involve a direct allosteric interaction between these two receptors, possibly involving receptor heterodimerization. Numerous studies have demonstrated that GPCR are able to physically associate to form functional homo- and heterodimers or higher order oligomers exhibiting new pharmacological and/or functional properties. This has been well demonstrated for γ-aminobutyric acid (GABA)B (GABAB1 and GABAB2) and taste 1 and 3 isoforms receptors. These GPCR are inactive as monomers but transduce signal efficiently as heterodimers. Many studies also demonstrate the ability of class A and B GPCR, the families of AVP and CRHR, respectively, to homo- and heterodimerize (see for review Ref. 13). Thus, for example, fluorescence resonance energy transfer (FRET) or bioluminescence resonance energy transfer (BRET) experiments indicate that, in heterologous models, AVP and oxytocin (OT) receptors (V1a, V1b, and V2, and OT isoforms) could homo- or heterodimerize with no significant alteration of their respective coupling properties. However, there was an important effect on their internalization profiles (14–17). In native tissues, Albizu et al. (18) also demonstrated the presence of functional homodimers of OT receptors. Young et al. (19) proposed that the rat V1b and CRHR1 receptors heterodimerize. However, the tagged V1b receptor used in this latter study was inactive, thus preventing any pharmacological studies. Mikhailova et al. (20, 21) also proposed that the avian isoform of the V1b receptor (the vasotocin VT2 receptor) heterodimerized with the CRHR1 receptor and modified the CRH-induced cAMP accumulation.
In this article, we characterize a new biological model, the adrenal chromaffin cells, in which AVP and CRH synergistically regulate catecholamine release. We further analyzed the pharmacological consequences of V1b and CRHR1 receptors coexpression by using HEK293 cells as a cellular model and attempted to decipher whether only second messenger cross talk is responsible for a synergistic mechanism between AVP and CRH.
Results
AVP and CRH synergistically regulate catecholamine release from bovine chromaffin cells in primary cultures
In mammals, adrenomedullary chromaffin cells secrete catecholamines in response to various hormonal and electrical stimuli (for review see Ref. 22). We previously demonstrated that AVP controls such a mechanism via V1b receptors at least in rat and human (23). In this study, we extended this observation to bovine chromaffin cells and studied the influence of both CRH and AVP on catecholamine release.
Using an immunocytochemical approach, we analyzed the expression of AVP and CRHR in bovine adrenomedullary cells in primary culture. First, the use of a specific antibody directed against tyrosine hydroxylase (TH) showed that the amount of positively stained cells reached 93 ± 5% (three distinct cultures) (data not shown), suggesting that our primary culture mainly consists in chromaffin cells. We then took advantage of our recently characterized V1b-specific fluorescent ligand, d[Leu4-(Lys-Alexa647)8]VP, to quantify the V1b-expressing cells (24). We observed that 58 ± 2% of TH immunostained cells were labeled with d[Leu4-(Lys-Alexa647)8]VP. This labeling was totally displaced by a saturating concentration of AVP. The V1b labeling specificity was assessed using d[Cha4]AVP, a selective bovine V1b agonist (25). A low dose of d[Cha4]AVP (50 nm) almost completely displaced d[Leu4-(Lys-Alexa647)8]VP labeling, which was entirely suppressed by 500 nm d[Cha4]AVP (Fig. 1A). In addition, 72 ± 1% of TH immunostained cells were labeled with a specific commercial CRHR1 antibody, whose specificity was previously verified (Fig. 1B and data not shown). Incubation with d[Leu4-(Lys-Alexa647)8]VP and the selective CRHR1 and TH antibodies showed that 52± 1% chromaffin cells (TH immunostained) cumulated V1b (yellow) and CRHR1 (red) labelings (Fig. 1B and data not shown). These results unambiguously indicate that a majority of bovine chromaffin cells coexpressed V1b and CRHR1 receptors.
Fig. 1.

V1b and CRHR1 receptors expression in bovine adrenomedullary cells in primary culture. Primary culture of bovine adrenomedullary cells were incubated with 150 nm of the fluorescent selective V1b agonist d[Leu4-(Lys-Alexa647)8]VP for 2 h at 12 C as described in Materials and Methods. After fixation and nuclei labeling with Hoechst (blue), they were submitted to a selective anti-CRHR1 antibody (secondary coupled with Cy3, in red) and to an anti-TH antibody (secondary coupled to Dylight488, in green) in a PBS/BSA/Triton buffer. Cells imaging was performed by confocal microscopy as described in Materials and Methods. A, Cells were incubated with d[Leu4-(Lys-Alexa647)8]VP alone (control), in the presence of 100 nm AVP (nonspecific labeling) or of increasing amounts of a selective bovine V1b agonist d[Cha4]AVP, and V1b labeling (in yellow) was imaged. B, Cells were incubated with d[Leu4-(Lys-Alexa647)8]VP as described above, fixed, permeabilized, and further incubated with both the anti-TH and anti-CRHR1 antibodies and the Hoechst dye (see Materials and Methods). The four labelings were analyzed under selective conditions to preserve a specific labeling of each marker (V1b, yellow; CRHR1, red; TH, green; and nuclei, blue). Single labeling and merge images are illustrated. Images illustrated in A and B are representative from three distinct primary cultures. To determine the percent of cells exhibiting single, double, or triple labeling, more than 120 cells originating from three distinct experiments and platted on five distinct coverslips were analyzed. Scale bar, 10 μm.
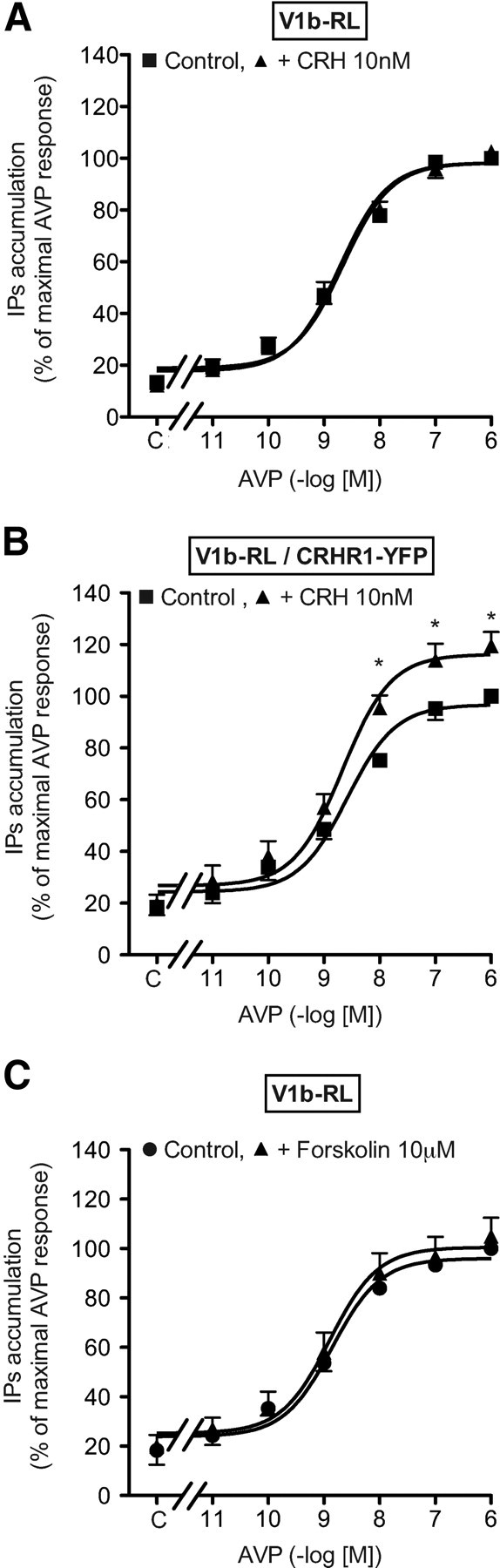
We then assessed the functionality of V1b and CRHR1 receptors in bovine chromaffin cells. As illustrated in Fig. 2A, AVP stimulated catecholamine release. This effect was dose dependent and saturable [concentration of analogue leading to half maximal effect (Kact) = 0.10 ± 0.01 nm, n = 3]. Similar effects were observed using d[Cha4]AVP confirming that the V1b receptor expressed in these cells is responsible for catecholamine secretion (Kact = 0.25 ± 0.03 nm, n = 3). As expected, AVP dose dependently activated IP accumulation in these same cells [Kact = 1.2 ± 0.06 nm; maximum velocity (Vmax) = 147 ± 1% of basal value, n = 3] and did not induce cAMP production, even when tested at 1 μm (Fig. 2, D and E). When similar experiments were performed using CRH, a similar dose-dependent release of catecholamines was observed (Kact = 27 ± 0.3 pm) (Fig. 2B). CRH-stimulated catecholamine release was fully inhibited by nanomolar concentrations of a selective CRHR1 antagonist, SSR125543 (26), demonstrating the involvement of the CRHR1 receptor (Fig. 2C). As expected, CRH dose dependently induced cAMP production (Kact = 30 ± 5 pm, n = 3) and did not induce IP accumulation, even when tested at 1 μm (Fig. 2, D and E).
Fig. 2.

Functional characterization of V1b and CRHR1 receptors expressed in bovine chromaffin cells in primary culture. A–C, Bovine chromaffin cells were loaded with [3H]norepinephrine, washed, and stimulated for 6 min at room temperature without (control) or with increasing concentrations of AVP or of the selective V1b agonist d[Cha4]AVP (A), CRH (B), or preincubated with increasing concentrations of the selective CRHR1 antagonist SSR125543 and further stimulated with 0.1 nm CRH (C). Catecholamines secreted in the medium were measured and expressed as percent of corresponding control values. D, Bovine chromaffin cells were loaded with [3H]myo-inositol. Cells were stimulated for 15 min at 37 C without (control) or with increasing concentrations of AVP or CRH. Total accumulated IP were measured and expressed as percent of basal response. E, Bovine chromaffin cells were loaded with [3H]adenine. Cells were stimulated for 15 min at 37 C without (control) or with increasing concentrations of AVP or CRH. Accumulated cAMP was measured and expressed as percent of control. Results are the mean ± sem of at least three distinct experiments, each performed in quadruplicates. C, Control.
Very low concentrations of AVP or CRH (10 pm each) did not significantly stimulate catecholamine release. However, when applied together, they induced a stimulatory effect, similar to the one obtained with maximal doses of either AVP or CRH (Figs. 3A and 2, A and B, respectively). This effect was more than additive, demonstrating a potentiating mechanism that could be reproduced by combining CRH with the selective V1b agonist, d[Cha4]AVP (Fig. 3A).
Fig. 3.

Synergistic action of AVP and CRH on catecholamine release and second messenger accumulation from primary bovine chromaffin cells. A, Bovine chromaffin cells were loaded with [3H]norepinephrine, washed, and stimulated for 6 min at room temperature without (control) or with 10 pm different combination of CRH, AVP, and d[Cha4]AVP. Catecholamines secreted in the medium were measured and expressed as percent of control. Results are the mean ± sem of three independent experiments, each performed in quadruplicates. *, P < 0.05 stands for comparison between catecholamines accumulation with or without each peptide. B, Bovine chromaffin cells loaded with [3H]myo-inositol were incubated without (C, Control) or with increasing amounts of AVP with or without 1 nm CRH. Total accumulated IP were measured and expressed as percent of values obtained on control cells incubated without CRH. *, P < 0.05 stands for comparison between corresponding IP accumulation with or without CRH. C, Bovine chromaffin cells loaded with [3H]adenine were incubated with or without (control) increasing amounts of CRH with or without 1 nm AVP. The accumulated cAMP was measured and expressed as percent of values obtained on control cells incubated without AVP. Results are the mean ± sem of three independent experiments, each performed in quadruplicates. When no errors bar is shown, it is comprised within the symbol. *, P < 0.05 stands for comparison between corresponding cAMP accumulation with or without AVP. D, Bovine chromaffin cells loaded with [3H]adenine were preincubated 15 min at 37 C with or without (C) increasing amounts of TPA. Then, CRH (10 nm) or vehicle were added in the incubation medium and the reaction allowed to proceed for another 30-min period. The accumulated cAMP was measured and expressed as percent of ATP converted in cAMP. Results are the mean ± sem of three independent experiments, each performed in quadruplicates. *, P < 0.05 as compared with corresponding control values without TPA. E, Bovine chromaffin cells loaded with [3H]adenine were preincubated 15 min at 37 C with or without (C) a combination of TPA and AVP. CRH (10 nm) or vehicle were then added in the incubation medium and the reaction allowed to proceed for another 30-min period. The accumulated cAMP was measured and expressed as percent of ATP converted in cAMP. Results are the mean ± sem of at least two independent experiments, each performed in pentaplicates. *, P < 0.05.
To analyze the molecular determinants of this synergistic mechanism, we studied the combined effects of AVP and CRH on second messenger accumulation. As illustrated in Fig. 3B, 1 nm CRH significantly shifted the AVP-stimulated dose-response curve of IP accumulation to the left by about 6-fold (Kact = 1.26 ± 0.06 vs. 0.11± 0.02 nm for experiments performed without and with CRH, respectively, n = 4). However CRH did not significantly modify the maximal level of AVP-stimulated IP accumulation. Corresponding experiments assessing the production of cAMP were performed using 1 nm AVP and increasing amounts of CRH. As illustrated in Fig. 3C, 1 nm AVP poorly affected the potency of CRH to stimulate cAMP (Kact of CRH = 36 ± 8 and 26 ± 10 pm for experiments performed without or with AVP, respectively, n = 4, nonsignificant) but significantly increased the maximal amount of cAMP accumulated upon CRH stimulation (45% of potentiation observed for 10 nm CRH).
Because PKC was previously shown to potentiate CRH-stimulated cAMP accumulation on rat hypophyseal cells in primary culture (12), we decided to verify whether a similar mechanism occurred in bovine chromaffin cells in primary culture. As illustrated in Fig. 3D, increasing amounts of the PKC activator 12-O-tetradecanoylphorbol 13-acetate (TPA) dose dependently stimulated the CRH-induced cAMP accumulation in bovine chromaffin cells, with a maximal effect at 1000 nm. To decipher whether the stimulatory effect of AVP on CRH-stimulated cAMP accumulation is only the consequence of PKC activation, we analyzed the combined effects of maximal doses of AVP and TPA. As illustrated (Fig. 3E), AVP and TPA alone potentiated CRH-stimulated cAMP accumulation. Used at maximal doses, AVP was even more efficient than TPA (maximal potentiation obtained for 100 nm AVP and 1 μm TPA, 108 and 40%, respectively). When AVP and TPA were added together, AVP still significantly increased the CRH-stimulated cAMP accumulation as compared with corresponding values obtained in the presence of TPA. The AVP-potentiating effect on cAMP production was yet reduced in the presence of TPA, suggesting that only part of this effect relies on PKC activation.
Altogether, these results indicate that V1b and CRHR1 receptors are coexpressed in bovine chromaffin cells and are involved in catecholamine secretion via the activation of IP and cAMP second messenger pathways, respectively. Interestingly, AVP and CRH act in synergism to release catecholamines via unknown molecular mechanisms that modify classical second messenger cascades. Two hypotheses can be envisaged: a cross talk between second messenger pathways and/or a modification of V1b and CRHR1 receptor coupling properties consecutive to their heterodimerization. To test these possibilities, we performed experiments in HEK293 cells cotransfected with tagged V1b and/or CRHR1 receptors to explore receptors dimerization. V1b and CRHR1 receptors carrying respectively myc and flag tags at their N termini were constructed, with addition of Renilla Luciferase (RL) for V1b and yellow fluorescent protein (YFP) for CRHR1 at the receptor C-terminal ends. This enables BRET and coimmunoprecipitation experiments to test for physical interaction between receptors, approaches which are not feasible in bovine native chromaffin cells due to the lack of specific V1b receptors antibodies.
V1b and CRHR1 receptors heterodimerize when coexpressed in HEK293 cells
Three complementary approaches were used to demonstrate receptor heterodimerization using RL and YFP tagged receptors.
Pharmacological and functional properties of V1b-RL and CRHR1-YFP receptors
First, we verified that the modified receptors were correctly expressed at the plasma membrane and exhibited functional properties similar to those of wild-type forms. Saturation binding experiments performed with [3H]AVP on crude plasma membrane derived from wild-type V1b or V1b-RL transfected cells showed a similar pharmacological pattern (Table 1). Both receptors bound [3H]AVP with nanomolar affinity. Similar results were obtained when comparing wild-type CRHR1 and CRHR1-YFP receptors with respect to [125I]CRH binding. Thus, addition of a tag at the N terminus and a bioluminescent agent at the C terminus of both V1b and CRHR1 receptors did not significantly modify their binding properties. Moreover, immunocytochemical experiments performed with antibodies directed against the myc epitope for V1b receptor combined with direct confocal microscopy to directly identify CRHR1-YFP confirmed that the modified receptors are appropriately addressed to the plasma membrane with some V1b and CRHR1 receptors also present in the cytoplasm (see Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org).
Table 1.
Pharmacological properties of wild-type and modified V1b and CRHR1 receptors expressed in HEK293 cells
| Expressed receptors | [3H]AVP binding parameters |
PLC activation |
||
|---|---|---|---|---|
| Kd (nm) | Bmax (fmol/mg prot) | Kact (nm) | Fold stimulation (% of basal) | |
| V1b WT | 0.24 ± 0.16 | 903 ± 342 | 0.26 ± 0.13 | 972 ± 141 |
| V1b-RL | 0.40 ± 0.12 | 381 ± 48 | 2.29 ± 0.49 | 688 ± 100 |
| V1b-RL/CRHR1-YFP | 0.41 ± 0.08 | 331 ± 57 | 2.80 ± 0.82 | 316 ± 41 |
| Expressed receptors | [125I]CRH binding parameters |
AC activation |
||
|---|---|---|---|---|
| Kd (nm) | Bmax (fmol/mg prot) | Kact (nm) | Fold stimulation (% of basal) | |
| CRHR1 WT | 0.14 ± 0.05 | 673 ± 136 | 0.26 ± 0.12 | 1460 ± 81 |
| CRHR1-YFP | 0.15 ± 0.03 | 1189 ± 255 | 2.01 ± 0.23 | 765 ± 225 |
| CRHR1-YFP/V1b-RL | 0.20 ± 0.05 | 1086 ± 284 | 3.80 ± 0.80 | 1087 ± 243 |
Saturation binding experiments using either [3H]AVP or [125I]CRH were performed on membranes derived from HEK293 cells transfected or cotransfected with wild-type (WT) or modified human V1b or CRHR1 receptors (see legends of Fig. 5, 6, and 10 for transfection conditions). Their Kd and Bmax were deduced from Scatchard representations (see Materials and Methods). The ability of AVP or CRH to stimulate IP or cAMP accumulation respectively, in HEK293 cells transfected or cotransfected with wild-type or modified receptors, was measured as described in Materials and Methods. Kact and the maximal second messenger stimulation observed (fold stimulation) were then determined. Results are the mean ± sem of at least three distinct experiments.
The functionality of V1b-RL and CRHR1-YFP receptors was also compared with that of their corresponding wild-type forms. As summarized in Table 1, in V1b-RL transfected cells, AVP still strongly stimulated IP accumulation (6.9-fold) with a nanomolar Kact. Similarly, in CRHR1-YFP transfected cells, CRH always strongly increased cAMP accumulation (7.6-fold). As found for the modified V1b receptor, the Kact for CRHR1-YFP receptor was increased by 10-fold, reflecting a moderate reduction in the potency of the YFP- and RL-fused receptors to couple to their respective signaling pathways, as compared with native receptors. Moreover, as summarized in Table 1, V1b-RL/CRHR1-YFP cotransfection did not significantly modify their binding properties. Coupling properties were however weakly affected. Only maximal IP accumulation upon AVP stimulation was significantly reduced by 2.1-fold.
Coimmunoprecipitation experiments
We performed immunoblotting experiments with an anti-RL antibody on cells expressing either V1b-RL, CRHR1-YFP, or both (Fig. 4A, left). Bands were detected at approximately 82 kDa, revealing the V1b-RL receptor (lanes 1 and 3). No band was detected in cells expressing CRHR1-YFP receptor (lane 2), demonstrating the specificity of the antibody used. We next performed a coimmunoprecipitation with the antigreen fluorescent protein (GFP) antibody followed by immunoblotting with the anti-RL antibody on cells coexpressing V1b and CRHR1 receptors (lane 4), cells only expressing V1b-RL receptor (lane 5), or a mix of membranes prepared from cells separately expressing one receptor or the other (lane 6). As shown in lane 5, no approximately 82-kDa band was observed with cells expressing only V1b-RL, thus validating the specificity of the immunoprecipitation. In lane 4, the broad band at approximately 82 kDa immunodetected by the anti-RL antibody represented the V1b-RL mature form that was immunoprecipitated by the anti-GFP antibody due to heterodimerization with the CRHR1-YFP receptor. We also observed a higher molecular weight band that might represent sodium dodecyl sulfate-resistant dimers or oligomers. The lower molecular weight band also observed probably represents immature receptor species consistent with the notion that dimerization occurs in the ER. Moreover, the absence of band in lane 6, corresponding to the mixture of cells expressing V1b-RL or CRHR1-YFP, clearly shows that heterodimerization occurred only in cotransfected cells and did not result from nonspecific receptors aggregation during the IP procedure. Figure 4, right, shows immunoblotting with the anti-GFP antibody on cells expressing either V1b-RL, CRHR1-YFP, or both receptors. Bands were detected at about 100 kDa, corresponding to the mature form of CRHR1-YFP along with other immature forms (∼80 and 70 kDa) (lanes 8 and 9). No band was detected in lane 7, attesting of the specificity of the antibody.
Fig. 4.

Heterodimerization between V1b and CRHR1 receptors. A, left, HEK293 cells transiently expressing either V1b-RL (lane 1), CRHR1-YFP (lane 2), or both (lane 3) were lysed and 1/20 of the lysates were submitted to SDS-PAGE. Bands were revealed, after immunoblotting (IB), using an anti-RL antibody. Middle, Lysate of HEK293 cells transiently coexpressing V1b-RL and CRHR1-YFP (lane 4), V1b-RL alone (lane 5), or a mixture of membranes from cells separately expressing V1b or CRHR1 (lane 6) were subjected to immunoprecipitation (IP) with an anti-GFP antibody followed by SDS-PAGE, Western blotting, and immunodetection with an anti-RL antibody. The absence of band in lane 5 demonstrates the specificity of the anti-GFP antibody used. Lane 6 shows that physical association did not occur when receptors are expressed separately and remixed. Right, After anti-RL immunodetection, the antibody was stripped off the nitrocellulose membrane (left panel) and reprobed with an anti-GFP antibody to reveal the mature CRHR1-YFP receptor. The results shown here are representative of three experiments (Mr, Molecular ratio). B, BRET saturation experiment was performed on intact HEK293 cells transfected with increasing amounts of CRHR1-YFP receptor plasmid (0–0.16 μg) along with a fixed amount of the V1b-RL receptor plasmid (0.04 μg). Gray dots corresponded to a 1:1 transfection ratio (0.4 μg of CRHR1-YFP/0.4 μg of V1b-RL). Net BRET is plotted as a function of total fluorescence emission from YFP over luminescence from RL. The Net BRET is presented as average from 10 consecutive measurements. The curves are obtained after collecting the results of three independent experiments.
BRET experiments
Heterodimerization of V1b-RL and CRHR1-YFP receptors was further explored using BRET, a biophysical technique relying on the occurrence of nonradiative energy transfer from the RL to the YFP moieties if both proteins are located within a 100-Å distance. Furthermore, this method is complementary to coimmunoprecipitation, because it allows the detection of protein-protein interactions in living cells. When HEK293 cells were cotransfected with V1b-RL and CRHR1-YFP, a BRET signal was observed, indicating that both receptors were in close proximity. The BRET signal observed was saturable, robust (maximal BRET net, 70 mU) and very reproducible between experiments. As shown in Fig. 4B, the net BRET signal increased, because the level of CRHR1-YFP expression was raised over the V1b-RL and reached a plateau. Saturation was observed for a 1:1 ratio, corresponding to 40 ng of the expression vector coding for V1b-RL and CRHR1-YFP (gray dots in the curve). This BRET saturation curve is a landmark of receptors interaction specificity, because nonspecific interactions are not saturable and therefore increase linearly with YFP bearing receptors at low YFP/RL ratios as observed when V1b-RL and GABAB2-YFP are coexpressed. We have also recorded the net BRET signal between V1b-RL and soluble YFP. For a transfection ratio leading to 75% of the saturating net BRET signal observed between V1b-RL and CRHR1-YFP receptors, no BRET signal was observed between these proteins (data not shown). Saturable BRET signal was also observed with the inverse constructions (i.e. myc-V1b-YFP and flag-CRHR1-RL receptors). In this condition, we observed a saturating net BRET signal as the level of V1b-YFP expression was raised over the CRHR1-RL, albeit at a lower maximal level (data not shown).
A CRHR1-specific pharmacological chaperone rescues V1b receptors from intracellular stores to the plasma membrane
Homo- and heterodimers of V1a, V1b, and V2 AVP receptors are expressed both on plasma and endoplasmic reticulum membranes (14, 17). Moreover, lipophilic antagonists of these receptors, like SSR49059 or SSR149415 (V1a and V1b antagonists, respectively), are known to rescue wild-type and mutated AVP receptors from internal stores to plasma membrane and to restore receptor functionality (27). We thus decided to verify whether SSR125543, a selective lipophilic antagonist of CRHR1 receptor that does not interact with the V1b receptor up to 1 μm (Table 2), would rescue the V1b receptor once coexpressed with the CRHR1 receptor (Fig. 5A).
Table 2.
Binding selectivity profiles of modified human V1b and CRHR1 receptors expressed in HEK 293 cells
| Analogues |
Ki (nm) |
||||
|---|---|---|---|---|---|
| V1b-RL |
V1b-RL/CRHR1-YFP |
CRHR1-YFP |
|||
| Abbreviations | Pharmacological properties | [3H]AVP | [3H]AVP | [125I]CRH | [125I]CRH |
| d[Cha4]AVP | Selective V1b agonist | 0.72 ± 0.12 | 0.75 ± 0.36 | n.d. | n.d. |
| SSR149415 | Selective V1b antagonist | 1.54 ± 0.41 | 1.54 ± 0.43 | 100 ± 10% | 109 ± 2% |
| Human CRH | Natural agonist | 97 ± 2%b | 86 ± 3%b | 4.6 ± 1.2 | 3.6 ± 1.4 |
| Astressin | Nonselective CRHR1antagonist | n.d. | n.d. | 15.9 ± 3.1a | 30.2 ± 4.1 |
| SSR125543 | Selective CRHR1 antagonist | 94 ± 2%b | 90 ± 2%b | 10.5 ± 2.3 | 11.9 ± 2.1 |
Binding experiments using either [3H]AVP and [125I]CRH were performed on membranes derived from V1b-RL or CRHR1-YFP mono-transfected HEK293 cells or from V1b-RL/CRHR1-YFP cotransfected HEK293 cells (see legend of Fig. 5 for transfection conditions). Dissociation inhibition constant (Ki) for unlabeled V1b or CRHR1 analogues were deduced from dose-dependent binding inhibition curves as illustrated Fig 5. The affinity (Kd) for both [3H]AVP and [125I]CRH used to calculate Ki were determined by saturation binding experiments performed on each plasma membrane preparations (see Table 1). Results are the mean ± sem of three to five distinct experiments each performed in triplicate. n.d., Not determined.
P < 0.025 compared with corresponding experiment performed on membranes derived from mono-transfected cells.
For analogues exhibiting a weak affinity, the specific binding determined in the presence of 1 μm was measured and expressed as percent of specific binding measured without unlabeled analogue.
Fig. 5.

A CRHR1 receptor antagonist rescues V1b receptor to the plasma membrane. HEK293 cells were transiently transfected with 2 μg of V1b-RL and 6 μg of CRHR1-YFP (B–D) or with 2 μg V1b-RL only (D). A, Schematic representation of V1b-RL receptor rescue by a CRHR1 antagonist. The lipophilic CRHR1 antagonist SSR125543 crosses the plasma membrane and interacts with CRHR1 receptors associated to the endoplasmic reticulum (step 1). Such binding induces the chaperoning and trafficking of CRHR1 receptors to the plasma membrane. If the CRHR1 is physically linked to the V1b receptor, both receptors will be driven to the plasma membrane (step 2). Thereafter, detection of V1b receptor at the plasma membrane using [3H]AVP, a nonpermeant peptide radioligand, will de facto reveal the rescue of the V1b/CRHR1 heterodimer (step 3). B, Scatchard representation of saturation binding experiments performed on HEK293 cells transiently expressing V1b-RL/CRHR1-YFP receptors. HEK293 cells were preincubated 24 h at 37 C with or without 1 μm SSR125543, extensively washed, and further incubated with increasing concentrations of [3H]AVP with (nonspecific binding) or without (total binding) 1 μm unlabeled AVP. Specific binding was calculated and Scatchard representation illustrated. C, Representation of Bmax and Kd values obtained from saturation binding experiments performed on control or SSR125543-stimulated HEK293 cells transiently expressing V1b-RL/CRHR1-YFP receptors. Values were obtained from three distinct experiments, each performed in triplicate. *, P < 0.05 compared with corresponding experiment performed on mono-transfected cells. 100% Bmax = 37,600 ± 4200 V1b-RL sites per cell, with 230,000 cells per well. D, V1b-RL receptor rescue by a CRHR1 antagonist. V1b-RL/CRHR1-YFP or V1b-RL mono-transfected HEK293 cells were preincubated 24 h at 37 C with or without (control) increasing amounts of SSR125543. Cells were further incubated with 3 nm [3H]AVP with (nonspecific binding) or without (total binding) 1 μm unlabeled AVP and specific binding calculated. Results, expressed as percent of control-specific binding are the mean ± sem of three distinct experiments each performed in triplicate. *, P < 0.05 for comparison between HEK293 cells incubated with or without SSR125543.
HEK 293 cells cotransfected with V1b-RL and CRHR1-YFP were preincubated for 18 h at 37 C with or without 1 μm SSR125543 and the pharmacological properties of the V1b receptor expressed at the plasma membranes determined. As shown in Fig. 5, B and C, SSR125543 pretreatment significantly increased the maximal binding capacity (Bmax) for [3H]AVP by approximately 40% without affecting the dissociation constant (Kd). As illustrated in Fig. 5D, this effect was dependent upon the SSR125543 concentration used for preincubation and saturable. This increase in V1b-RL receptor density was not observed unless CRHR1 receptors were expressed, suggesting a selective action of SSR125543 on CRHR1 receptors (Fig. 5D). V1b-RL rescue was also visualized in the same conditions by fluorescence microscopy (see Supplemental Fig. 2).
In summary, these experiments demonstrate that in HEK 293 cells, functionally active V1b-RL and CRHR1-YFP receptors physically interact. To determine whether these interactions induced modifications of receptor properties, we performed binding and functional experiments on single- and double-transfected HEK293 cells.
Functional consequences of V1b and CRHR1 receptor cotransfection in HEK 293 cells
Consequences on binding properties
Saturation binding experiments using [3H]AVP were performed on crude plasma membrane derived from HEK 293 cells transfected with V1b-RL and/or CRHR1-YFP receptors. As summarized in Table 1, V1b-RL affinity for [3H]AVP was similar whatever the membrane preparation used. Similarly, competition binding experiments were also performed using 1 nm [3H]AVP. As shown in Fig. 6, the V1b-RL pharmacological profile for d[Cha4]AVP and SSR149415 was similar to those earlier described for the wild-type V1b receptor (28). Moreover, inhibitory Kd (Ki) for these selective V1b analogs was unaffected upon CRHR1-YFP receptor cotransfection (Table 2). By contrast, a high concentration of CRH unable to displace [3H]AVP on plasma membrane expressing only V1b-RL receptors (1 μm) weakly but significantly inhibited [3H]AVP-specific binding on membranes coexpressing V1b-RL and CRHR1-YFP receptors (Fig. 6A).
Fig. 6.

Pharmacological profiles of V1b-RL or CRHR1-YFP receptors expressed alone or together in HEK293 cells. Crude membranes from HEK293 cells transiently transfected with 2 μg of V1b-RL only (A and C), 2 μg of CRHR1-YFP only (B and D), 2 μg of V1b-RL and 6 μg of CRHR1-YFP (A and E), or 6 μg of V1b-RL and 2 μg CRHR1-YFP (B and F) receptors were incubated either with 1 nm [3H]AVP (A, C, and E) or 0.1 nm [125I]CRH (B, D, and F) in the absence (C, Control) or in the presence of increasing amounts of the analogs to be tested. Specific binding was calculated in each experimental condition and expressed as percent of control-specific binding. 100% of [3H]AVP-specific binding corresponds to 1.16 ± 0.20 and to 1.14 ± 0.26 fmol of [3H]AVP specifically bound/μg protein for V1bRL and V1b-RL/CRHR1-YFP membranes preparations, respectively. 100% of [125I]CRH-specific binding corresponds to 0.37 ± 0.03 and to 0.28 ± 0.04 fmol of [125I]CRH specifically bound/μg protein for CRHR1-YFP and CRHR1-YFP/V1b-RL membranes preparations, respectively. Results are the mean ± sem of at least three distinct experiments, each performed in triplicate. *, P < 0.05 for comparison between corresponding values obtained on mono- or double-transfected cell membrane preparations.
The pharmacology of CRHR1-YFP receptor determined on single-transfected cells is similar to the one expected for human wild-type CRHR1 receptor (Table 2) (29). Upon V1b-RL and CRHR1-YFP cotransfection, saturation binding experiments using [125I]CRH did not exhibit significant modifications of Ki (Table 2). Similarly, the pharmacological profile of SSR125543 was not affected by V1b cotransfection. AVP, even tested at high doses, did not alter specific [125I]CRH binding on single- and double-transfected cells (Fig. 6B). By contrast, the affinity (Ki) of astressin, a nonselective CRHR1 antagonist, was slightly but significantly modified upon V1b-RL and CRHR1-YFP cotransfection (Fig. 6, D and F, and Table 2).
Consequences on cAMP accumulation
CRH dose dependently stimulated cAMP accumulation in HEK293 cells transfected with CRHR1-YFP receptors. Maximal stimulation was reached for 100 nm (Fig. 7A and Table 3). Neither the Kact nor the Vmax were affected by the presence of 10 nm AVP. In CRHR1-YFP/V1b-RL cotransfected cells, CRH stimulated cAMP accumulation with the same efficiency. However, in the presence of 10 nm AVP, a concentration allowing an almost complete saturation of V1b receptor, a strong potentiation of cAMP accumulation, was observed for saturating doses of CRH with a weak increase of Kact (Fig. 7B and Table 3).
Fig. 7.

Influence of AVP on CRH-stimulated cAMP accumulation in transiently transfected HEK293 cells. A and B, [3H]adenine-loaded HEK293 cells transiently transfected with 2 μg of CRHR1-YFP only (A) or 2 μg of CRHR1-YFP and 6 μg of V1b-RL receptors (B) were preincubated with or without (control cells) 10 nm AVP for 15 min at 37 C and further incubated 30 min with or without (C) increasing amounts of CRH. Accumulated cAMP was determined in each condition and expressed as percent of value obtained on control cells incubated with 1 μm CRH. C, [3H]adenine loaded HEK293 cells transiently transfected with 2 μg of CRHR1-YFP and 6 μg of V1b-RL receptors were incubated 5 min at 37 C with or without the V1b antagonist SSR149415 (1 μm). Then, increasing amounts of AVP, or vehicle (C, Control) were added in the incubation medium for 15 min and cells were further incubated 30 min with 10 nm CRH. Accumulated cAMP was measured and expressed as percent of values obtained on cells preincubated without AVP and SSR149415 but in the presence of 10 nm CRH (C). D, [3H]myo-inositol-loaded HEK293 cells transiently transfected with 2 μg of V1b-RL receptors were incubated for 15 min at 37 C with or without 1 μm SSR149415 and further incubated 15 min at the same temperature without (control) or with increasing concentrations of AVP. IP accumulated were measured and expressed as percent of AVP maximal response obtained on HEK293 cells incubated without SSR149415. E, HEK293 cells were transiently transfected with a constant amount of CRHR1-YFP plasmid (2 μg) and increasing amounts of V1b-RL plasmid (from 0 to 10 μg). The cells were [3H]adenine-loaded, preincubated with 10 nm AVP for 15 min at 37 C and further incubated for 30 min with 100 nm CRH. Accumulated cAMP was determined in each experimental condition and expressed as percent of corresponding value obtained on cells transfected only with CRHR1-YFP plasmid (ratio 2:0). Results were plotted over the CRHR1-YFP/V1b-RL transfection ratio. Results are the mean ± sem of at least three distinct experiments, each performed in triplicate. F, HEK293 cells were transiently transfected at the constant transfection ratio of 1:3 (CRHR1-YFP:V1b-RL) with increasing amounts of each of CRHR1-YFP and V1b-RL plasmids. The cells were [3H]adenine-loaded for 24 h and further incubated with or without 10 nm CRH for 30 min at 37 C left part). Accumulated cAMP was measured and expressed as percent of ATP converted in cAMP. Similar transfections and cAMP accumulation were performed but, in the presence of 10 nm AVP (F, right part), and the potentiation effect of AVP on CRH-stimulated cAMP accumulation was measured and expressed as indicated in E. Results are the mean ± sem of at least three distinct experiments, each performed in triplicate.
Table 3.
Influence of V1b and CRHR1 receptor co-transfection on cAMP and IP second messenger accumulation in HEK293 cells
| Expressed receptors | Conditions of stimulation | cAMP accumulation |
IP accumulation |
||
|---|---|---|---|---|---|
| Kact (nm) for CRH | CRH maximal stimulation (%) | Kact (nm) for AVP | AVP maximal stimulation (%) | ||
| V1b-RL | Control AVP | n.d. | n.d. | 2.3 ± 0.5 | 100% |
| Control AVP + 10 nm CRH | n.d. | n.d. | 2.4 ± 0.4 | 104 ± 1% | |
| V1b-RL/CRHR1-YFP | Control CRH | 3.8 ± 0.8 | 100% | n.d. | n.d. |
| Control CRH + 10 nm AVP | 7.9 ± 1.4 | 191 ± 11%a | n.d. | n.d. | |
| Control AVP | n.d. | n.d. | 2.8 ± 0.8 | 100% | |
| Control AVP + 10 nm CRH | n.d. | n.d. | 1.3 ± 0.3 | 119 ± 5%a | |
| CRHR1-YFP | Control CRH | 3.5 ± 0.9 | 100% | n.d. | n.d. |
| Control CRH + 10 nm AVP | 3.8 ± 0.6 | 97 ± 3% | n.d. | n.d. | |
HEK293 cells were transiently transfected with either V1b-RL and/or CRHR1-YFP receptors (see legends of Figs. 6 and 10 for transfection conditions) and further labeled with [3H]adenine (cAMP accumulation measurement) or [3H]myo-inositol (IP accumulation measurement). cAMP accumulation was measured in cells incubated with increasing amounts of CRH (designated as control CRH) with or without 10 nm AVP. IP accumulation was measured in cells incubated with increasing amount of AVP (designated as control AVP) with or without 10 nm CRH. Kact for each neuropeptide was deduced from dose-dependent stimulation curves as illustrated in Figs. 7 and 11. Maximal second messenger accumulation was also calculated and expressed as percent of maximal second messenger accumulation obtained in control HEK293 cells stimulated without hormonal coregulator in same experimental series. Results are the mean ± sem of at least three independent experiments each performed in triplicate. n.d., Not determined.
P < 0.05 compared with corresponding experiment performed on mono-transfected cells.
Because the functional properties (Kact) of V1b-RL and CRHR1-YFP receptors are slightly modified as compared with their corresponding wild-type forms (Table 1), we performed cAMP potentiation experiments with V1b and CRHR1 wild-type receptors and obtained the same effect (data not shown).
To test whether such potentiation required V1b-RL activation, CRHR1-YFP/V1b-RL cells were incubated with 10 nm CRH and increasing doses of AVP. As illustrated in Fig. 7C, AVP dose dependently potentiated CRH-stimulated cAMP accumulation. This effect was saturable, and the Kact obtained (0.14 ± 0.03 nm, n = 3) was found 16-fold lower as compared with the Kact of AVP measured in IP accumulation experiment (Table 3 and Fig. 7D). Moreover, 1 μm of the selective V1b antagonist SSR149415 shifted the AVP-potentiating effect on cAMP accumulation to the right. Interestingly, even in the presence of a large amount of AVP (10 μm), the inhibition by SSR149415 could not be fully reversed (Fig. 7C), suggesting a noncompetitive inhibitory process. In contrast, when similar experiments were performed in cells expressing only V1b-RL, SSR149415 competitively inhibited AVP-stimulated IP accumulation, because high concentrations of AVP completely reversed the inhibitory effect of 1 μm SSR149415 (Fig. 7D).
We further tested the influence of the receptor transfection ratio on the AVP-potentiating effect on CRH-stimulated cAMP accumulation. Increasing the amounts of V1b-RL receptor plasmid and keeping constant those of CRHR1-YFP led to an expected dose-dependent increase of the potentiating effect of AVP, which reached a plateau for a 1:1–1:3 CRHR1-YFP /V1b-RL transfection ratio. In these conditions, AVP maximally potentiated CRH-stimulated cAMP accumulation by around 40% (Fig. 7E). We then used an optimal ratio (1:3) and varied the amounts of receptors plasmid to explore a large scale of receptor expression. As seen in Fig. 7F, left, we first observed that the CRH-stimulated cAMP accumulation linearly increased with the amount of CRHR1 receptor plasmid used for transfection (r2 = 0.97) (data not shown). This indicates that the CRHR1 receptor expression was not functionally limiting in the range of plasmid used (between 0.03 and 2 μg). In the same experimental conditions, the potentiating effect of AVP on CRH-stimulated cAMP accumulation was not statistically affected by the amounts of CRHR1-YFP and V1b-RL receptors plasmids used (Fig. 7F, right). Altogether, these data indicate that the potentiating effect of AVP on CRH-stimulated cAMP accumulation was robust and that it could be observed even for low levels of receptor expression.
Potentiation of CRH-stimulated cAMP accumulation might be the consequence of AVP-stimulated PLC activation, inducing a downstream cross talk between V1b and CRHR1 second messenger cascades. As shown in Fig. 8A, the PLC inhibitor U73122 completely inhibited AVP-stimulated PLC activity with no significant effect on basal PLC activity. Maximal inhibition was obtained for 10 μm. Concerning the cAMP pathway, 20 μm U73122 modified neither basal nor CRH-stimulated levels of cAMP. However, it reduced the AVP-potentiating effect on CRH-stimulated cAMP accumulation by 70% (Fig. 8B). Increasing U73122 up to 30 μm did not further decrease this effect. These data suggested that the AVP-potentiating effect on CRH-stimulated cAMP accumulation was partially independent of the PLC activation (Fig. 8C). Because Abou-Samra et al. (12) showed in rat pituitary cells that the AVP-potentiating effect on CRH-stimulated cAMP may be due to PKC activation, we tested this possibility in our experimental model. First, we observed that 10 nm TPA potentiated the CRH-stimulated cAMP accumulation in HEK293 cells transfected with CRHR1-YFP receptor. As found for AVP potentiation, only the maximal cAMP response was affected (compare Figs. 7B and 9A). Moreover, this TPA effect was dose dependent, and saturable, maximal effect on CRH-induced cAMP production being reached for 10 nm TPA. Even in the presence of 100 nm, a dose that fully activates PKC, a significant potentiation of CRH-stimulated cAMP accumulation by 10 nm AVP could still be observed (Fig. 9C). However, as illustrated in Fig. 9D, TPA treatment led to a dose-dependent inhibition of the potentiation. For TPA concentrations higher than 30 nm, no further inhibition of the AVP-potentiating effect occurred. However, as previously observed in the presence of U73122, this effect was reduced by about 3-fold in the presence of TPA. These data suggest that the potentiating effect of AVP on cAMP accumulation induced by CRH is not just the consequence of PLC or PKC activation.
Fig. 8.

Influence of PLC activation on the AVP-potentiating effect on CRH-stimulated cAMP accumulation. A, [3H]myo-inositol-loaded HEK293 cells transiently transfected with 2 μg of V1b-RL receptor were preincubated for 15 min at 37 C with or without increasing concentrations of the PLC inhibitor U73122 and further incubated with or without 10 nm AVP for 30 min. Accumulated IP were measured. Results, expressed in disintegrations per minute per well, are the mean ± sem of at least three distinct experiments, each performed in triplicate. B, [3H]adenine-loaded HEK293 cells transiently transfected with CRHR1-YFP and V1b-RL plasmids (1:3 μg, respectively) were preincubated 5 min at 37 C with or without 20 μm U73122. AVP (10 nm) or vehicle were then added in the incubation medium for 10 min, and the cells further incubated 30 min at 37 C with or without 100 nm CRH. cAMP accumulated was measured and expressed as percent of ATP converted in cAMP. Results are representative of three distinct experiments each performed in quadruplicate. *, P < 0.05 for comparison between corresponding values measured with or without AVP. C, [3H]adenine-loaded HEK293 cells transiently transfected as described in B were preincubated with or without increasing amounts of U73122 and incubated with or without AVP and CRH as described in B. The potentiating effect of AVP over CRH stimulation was calculated from data illustrated in B as the difference between cAMP accumulation measured with or without AVP and expressed as percent of the same value measured in the absence of U73122. Results are the mean ± sem of at least three distinct experiments, each performed in quadruplicate. *, P < 0.05 for comparison between corresponding values measured with or without U73122.
Fig. 9.

Influence of PKC activation on the AVP-potentiating effect on CRH-stimulated cAMP accumulation. A, [3H]adenine-loaded HEK293 cells transiently transfected with the CRHR1-YFP receptor plasmid (2 μg) were incubated for 15 min at 37 C with or without (C, Control) 10 nm TPA and further incubated for 30 min with or without (C) increasing amounts of CRH. *, P < 0.05 for comparison between corresponding values obtained with or without TPA. B, [3H]adenine-loaded HEK293 cells transiently cotransfected with CRHR1-YFP and V1b-RL receptor plasmids (2:6 μg, respectively) were incubated for 15 min at 37 C with or without (C) increasing amounts of TPA and for 30 min with or without (control) 10 nm CRH. *, P < 0.05 as compared with corresponding control values without TPA. C, [3H]adenine-loaded HEK293 cells transiently transfected with CRHR1-YFP and V1b-RL receptor plasmids (2:6 μg, respectively) were preincubated for 15 min at 37 C with or without 10 nm AVP in the presence or absence of 100 nm TPA and for further 30 min with or without 10 nm CRH. *, P < 0.05. In each experimental condition (A–C), cAMP accumulated was measured and expressed as percent of ATP converted in cAMP. Results are the mean ± sem of at least three distinct experiments, each performed in triplicate. D, [3H]adenine-loaded HEK293 cells transiently transfected with CRHR1-YFP and V1b-RL receptor plasmids (2:6 μg, respectively) were preincubated and incubated as described in C with or without (control) increasing amounts of TPA. The potentiating effect of AVP over CRH stimulation was calculated from data illustrated in C as the difference between cAMP accumulation measured with or without AVP and expressed as percent of the same value measured in the absence of TPA. Results are the mean ± sem of at least three distinct experiments, each performed in quadruplicate. *, P < 0.05 for comparison between corresponding values obtained with or without TPA.
We also investigated a putative negative transregulation by a V1b-selective antagonist. On V1b-RL/CRHR1-YFP cotransfected cells, the selective V1b antagonist SSR149415 dose dependently inhibited CRH-stimulated cAMP accumulation without any significant effect on basal cAMP level (Fig. 10 and data not shown). The maximal effect was weak (15%) but specific because not observed in CRHR1-YFP mono-transfected cells. We also controlled that SSR149415 was unable to modify the cAMP accumulation measured in the presence of 10 nm CRH in V1b-RL mono-transfected cells (data not shown).
Fig. 10.

Influence of SSR149415 on CRH-stimulated cAMP accumulation in transiently transfected HEK293 cells. [3H]adenine prelabeled HEK293 cells transiently transfected with only CRHR1-YFP (2 μg) or CRHR1-YFP and V1b-RL receptor plasmids (2:6 μg) were preincubated 15 min at 37 C with or without (C, Control) increasing amounts of the V1b antagonist SSR149415; 10 nm CRH was further added to the incubation medium, and the reaction was allowed to proceed for an additional 30-min period. cAMP accumulated was measured and expressed as percent of control. Values are the mean ± sem of at least three distinct experiments, each performed in triplicate. *, P < 0.05 for comparison between mono- or double-transfected cells for similar SSR149415 concentrations.
Altogether, these data show that, once occupied by an agonist (or an antagonist), the V1b receptor positively (or negatively) regulates CRH-stimulated AC by combined actions of second messenger cross talk and of nonclassical mechanisms.
Consequences on IP accumulation
In V1b-RL transfected HEK293 cells, AVP dose dependently stimulated IP accumulation with a Kact = 2.3 ± 0.5 nm (n = 3). Maximal stimulation was reached for 0.1 μm (Fig. 11A and Table 3). In V1b-RL/CRHR1-YFP transfected cells, AVP stimulated PLC activity with the same efficiency (see Table 3). However, in the presence of 10 nm CRH, a concentration inducing an almost maximal activation of cAMP accumulation (Fig. 7A), a slight but significant increase of AVP-stimulated IP (19 ± 7%) was observed for high concentrations of AVP (Fig. 11B). In addition, a small leftward shift of the AVP-stimulated IP accumulation curve was observed (Table 3) as already observed on chromaffin cells (Fig. 3B). This CRH-potentiating effect was specific because not observed on V1b-RL transfected cells (Fig. 11A). To verify whether these effects could be due to cAMP produced by CRHR1 receptor activation, we performed control experiments using the classical AC activator, forskolin. Addition of 10 μm forskolin, a concentration leading to the same level of cAMP accumulation as that induced by 10 nm CRH (data not shown), did not modify the AVP-stimulated IP accumulation curve obtained on HEK293 cells transfected with V1b-RL receptor (Fig. 11C). This suggests that the CRH-potentiating effect observed on AVP-stimulated IP accumulation (Fig. 11B) does not result from protein kinase A activation.
Fig. 11.
Influence of CRH on AVP-stimulated IP accumulation in transiently transfected HEK293 cells. A and B, [3H]myo-inositol-loaded HEK293 cells transiently transfected with 2 μg of plasmid of V1b-RL receptor only or with V1b-RL and CRHR1-YFP receptors plasmids (2:6 μg, respectively) were preincubated 15 min at 37 C with or without 10 nm CRH (control cells) and further incubated 15 min with or without (C) increasing amounts of AVP. Accumulated IP was measured. C, [3H]myo-inositol-loaded HEK293 cells transiently transfected with 2 μg plasmid of V1b-RL receptor were preincubated for 15 min at 37 C with or without 10 μm forskolin (control cells) and further incubated for 15 min with or without (C) increasing amount of AVP. Accumulated IP was measured. Results, expressed as percent of maximal AVP response obtained on control cells, are the mean ± sem of at least three distinct experiments, each performed in triplicate. *, P < 0.05 for comparison between IP accumulation measured with or without CRH.
Because CRH significantly modulated AVP-stimulated IP accumulation, we tested the influence of the selective CRHR1 antagonist SSR125543 on AVP-stimulated IP production in V1b-RL/CRHR1-YFP cotransfected cells. No significant effect was observed even with 10 μm SSR125543 (data not shown).
The present results indicate that CRH, by acting on CRHR1 receptors, might regulate AVP-sensitive PLC activity. These potentiating effects are smaller than that described for the AVP-potentiating effect on cAMP accumulation, but they are independent of second messenger cross talk.
Discussion
AVP and CRH act in synergism both in pituitary and pancreas by poorly explored mechanisms involving second messenger cross talk and possibly receptor heterodimerization. In this study, we first demonstrate that such synergism also occurs in the adrenal medulla. We also provide evidence for a functional role of heterodimerization in the synergism between V1b and CRHR1 receptors.
In rodents, the adrenal medulla expresses V1b and CRHR1 receptors (23, 30). As illustrated here, primary bovine chromaffin cells also express V1b and CRHR1 receptors, each of them being involved in controlling catecholamine release. Furthermore, a majority of chromaffin cells coexpressed V1b and CRHR1 receptors (Fig. 1) and AVP and CRH synergistically stimulate this secretion. Such observations reinforce the paradigm of a concerted action of these two neuropeptides on several endocrine glands as shown in pituitary and pancreas (10, 11). Because CRHR1 and V1b receptors are also expressed in brain regions like hippocampus, hypothalamus, olfactory bulb, or cortex (31–33), and both engaged in regulating stress and anxiety (34), one may wonder whether these two neurohormones control cognitive functions in a concerted manner. Moreover, as the secretion of AVP and CRH is increased under stress situations (7), these phenomena may be of great physiological importance.
The molecular mechanisms underlying the synergism between AVP and CRH remain poorly understood. Previous works indicated a crucial role for the second messenger pathways. Indeed, in primary culture of rat pituitary cells or in HeLa cells, Abou-Samra et al. (12) and Mikhailova et al. (21) demonstrated that AVP or vasotocin strongly potentiated the CRH-stimulated ACTH release by modifying the cAMP response. AVP (or vasotocin) increased the maximal CRH-stimulated cAMP response without affecting its Kact (12, 21). More interestingly, previous studies on rat pituitary cells and Langerhans islets showed that the potentiating effect of AVP on CRH-induced ACTH and insulin secretion is PKC dependent. Thus, in these two models, TPA mimicked the effect of AVP (11, 12). Our results are in agreement with those published earlier. However, we showed in bovine chromaffin cells that phorbol ester did not entirely mimic the effect of AVP, which still induced potentiation even in the presence of saturating amounts of TPA in both native and heterologous models. Moreover, we demonstrate that a PLC inhibitor, which completely inhibited the AVP-stimulated IP production, did not completely abolish AVP-induced potentiation, suggesting the contribution of another mechanism in addition to a classical cross talk between V1b and CRHR1 second messenger pathways.
We also looked for a correlation between V1b and CRHR1 receptors heterodimerization and modifications of their functional properties. We took advantage of HEK 293 cells heterologous system to monitor receptor heterodimerization and to study second messenger cross talk. We could explore a large range of receptor expression levels, including low densities as found in native tissues.
Using such system, we confirmed that modified but still active V1b and CRHR1 receptors could physically interact. When coexpressed, V1b and CRHR1 receptors could be coimmunoprecipitated, and a specific BRET signal could be observed, indicating the formation of V1b/CRHR1 dimers. Moreover, the pharmacological chaperone SSR125543, specific for the CRHR1 receptor, could rescue V1b receptors from intracellular stores to the plasma membrane. This only happened when V1b and CRHR1 receptors were cotransfected. These data confirm previous studies indicating that receptors belonging to the AVP/OT family could form homo- and heterodimers (14, 17, 18). They also validate previous work showing that the rat V1b and the avian vasotocin VT2 receptors heterodimerized with the CRHR1 receptor (19). However, all these data were almost exclusively obtained on heterologous models. Only recently, Albizu et al. (18) brought also robust evidence for the presence of OT homodimers within plasma membrane from rat lactating mammary gland.
By comparing the signaling properties of HEK293 mono- or cotransfected cells, we found that V1b-RL and CRHR1-YFP receptors coexpression leads to specific coupling properties. Indeed, we showed that AVP dose dependently potentiated CRH-stimulated cAMP accumulation via a V1b receptor-mediated mechanism and that CRH, to a lesser extent, potentiated AVP-stimulated IP accumulation. Interestingly, we also found that some second messenger cross talk observed in HEK293 cells expressing both V1b and CRHR1 receptors are also observed in bovine chromaffin cells (compare Figs. 3C and 7B) that coexpressed both receptors (Fig. 1), suggesting a general mechanism of interaction. In heterologous models, we also provide experimental evidence suggesting that V1b and CRHR1 receptors coexpression promotes modifications of their signaling activity independently of second messenger cross talk. These evidences are the following: 1) The alteration of [3H]AVP-specific binding by micromolar concentrations of CRH was only observed on plasma membrane preparation from cotransfected cells in experimental conditions in which CRH was not able to generate cAMP due to the absence of ATP and GTP in the binding assay medium. Such allosteric regulations have already been observed for various GPCR oligomers (see for review Ref. 35), including the AVP receptor family (36). 2) On doubled-transfected cells, the specific trans-inhibitory effect induced by the full V1b antagonist SSR149415 (4) on CRH-stimulated cAMP accumulation could not be due to second messenger production. 3) The effect of AVP on CRH-stimulated cAMP accumulation observed for a large range of AVP concentrations (between 0.1 nm and 1 μm) could not be explained by a Michaelian interaction between AVP and V1b receptor but rather by allosteric mechanisms, as it has been previously observed (37). Similarly, SSR149415, which competitively inhibited AVP-stimulated PLC activity, noncompetitively inhibited CRH-stimulated cAMP accumulation by probably similar nonclassical mechanisms. 4) The synergism between AVP and CRH could not be only the consequence of second messengers cross talk. Indeed, the effect of AVP on CRH-stimulated cAMP accumulation was still present when PLC was fully inhibited by U73122 or PKC fully activated by TPA. Furthermore, CRH synergism on AVP-stimulated IP accumulation could not be mimicked by forskolin, an activator of AC. 5) One might exclude that the AVP-potentiating effects on cAMP accumulation arose from Gβγ-subunits release upon V1b receptor stimulation as described earlier (38). Indeed, in our experimental conditions, we showed that maximal IP accumulation was achieved when V1b receptors expressed in HEK293 cells were stimulated with 10 nm AVP, suggesting that Gβγ associated to Gq was maximally released. In these experimental conditions, we did not observe a significant rise in cAMP accumulation (103 ± 3%, over basal, seven distinct experiments) (data not shown).
All these data are consistent with a role of V1b-RL and CRHR1-YFP heterodimerization in the AVP/CRH synergism observed here.
However, the moderate magnitude of the effect might also be due to a partial level of coexpression (in our experimental conditions, only 30 ± 5% HEK293 cells coexpressed V1b-RL and CRHR1-YFP) (data not shown). It could also be the consequence of a partial level of heterodimerization, both receptors being also able to homodimerize (17, 39).
In the absence of selective pharmacological tools targeting heterodimers or of mutated receptors that could prevent heterodimerization, the presence of heterodimers in native tissues remains difficult to ascertain, particularly for class A or B GPCR. Pin et al. (40) suggested that heterodimerization could be indirectly revealed by the emergence of a specific functional property like synergism or by positive or negative allosteric regulations. For example, oligomerization of the metabotropic glutamate receptor 2 and serotonin 2A receptor has recently been shown to trigger specific cellular responses when targeted by hallucinogenic drugs in rat (41). Similar pharmacological modifications were also observed upon V1b-RL and CRHR1-YFP cotransfection in HEK293 cells (Fig. 7, B and C).
In conclusion, this paper reinforces the paradigm that V1b and CRHR1 receptors act in synergism in native tissues to potentiate many secretory processes. Although this synergism involves cross talk at the level of second messengers, we obtained several lines of evidence to suggest that this synergism also involves V1b and CRHR1 receptor heterodimerization. Additional studies will be necessary to validate this hypothesis, in particular to elucidate the presence and role of heterodimers in native tissues.
Materials and Methods
Chemicals
Most standard chemicals were purchased from Sigma (St. Louis, MO) or Merck (Darmstadt, Germany), unless otherwise indicated. AVP came from Bachem (Bubendorf, Switzerland); CRH, SSR149415, and SR126768 were kindly given by Serradeil-Le Gal from Sanofi-Aventis (Toulouse, France). Fetal calf serum came from Sigma (Saint Quentin Fallavier, France). [3H]AVP (44 Ci/mmol), myo-[2-3H] inositol (20 Ci/mmol), and [3H] adenine (27.2 Ci/mmol) came from PerkinElmer (Courtabœuf, France). [125I]CRH 1-41 amide (2000 Ci/mmol) and DL-[7,8-3H]norepinephrin came from Amersham Biosciences (Piscataway, NJ). DMEM and penicillin-streptomycin were purchased from Invitrogen (Cergy Pontoise, France). Inositol-free DMEM came from ICN Biochemicals (Orsay, France). Dowex AG1-X8 formate form 200–400 mesh was purchased from Bio-Rad (Munich, Germany). d[Cha4]AVP and d[Leu4-(Lys-Alexa647)8]VP were synthesized and kindly provided by M. Manning (College of Medicine, University of Toledo, Toledo, OH). Dopamine β-hydroxylase antibody was purchased from Chemicon (Euromedex, France), antirabbit IgG fluorescein isothiocyanate came from Sigma Chemical Co. (St. Louis, MO). Polyethyleneimine 25Kd linear came from Polysciences, Inc. (Warrington, PA). The rabbit CRHR1-specific antibody was from GeneTex (Interchim, Paris, France) and the Cy3-coupled donkey antirabbit from The Jackson Laboratory (Suffolk, UK). The anti-TH antibody was from Abcam (Cambridge, UK), and the secondary antisheep IgG was a Dylight488-conjugated Affipure Fab fragment (Thermo Fischer, Rockford, IL).
Isolation and culture of chromaffin cells
Animal manipulations were performed according to the recommendation of the French Ethical Committee. Fresh bovine adrenal glands were obtained from a local slaughter house, and the adrenal medulla was dissected. Chromaffin cells were isolated from adrenal medulla as described in Ref.42 with slight modifications. The cells were dissociated, isolated by collagenase digestion, and then purified through a Percoll gradient, carefully collected, and washed with Ca2+/Mg2+-free Locke's solution. Finally, cells were resuspended in DMEM/F-12 (Life Technologies, Inc., Carlsbad, CA) supplemented with 10% fetal calf serum, 5 μm cytosine arabinoside, and 10 μm 5-fluorodeoxiuridine. Cells were used for experiments 2 d after plating and purity (>95%) determined by monoamine selective staining with Neutral Red Dye (Sigma) (43).
Characterization of V1b and CRHR1 receptors in the bovine adrenal primary culture
Dispersed cells prepared from bovine adrenals were plated on coverslips previously coated with collagen and placed in 12-well plates. After 2 d of culture, the adherent cells were incubated with the fluorescent agonist d[Leu4-(Lys-Alexa647)8]VP at 150 nm in the presence of 100 nm the selective OT antagonist SR126768A for 2 h at 12 C as previously described (24). The incubations were realized in the presence or absence of an excess of AVP to determine nonspecific labeling. After three washes with cold PBS, they were fixed in 4% paraformaldehyde overnight at 4 C. The coverslips were incubated overnight at 4 C in PBS, 2 mg/ml BSA, and 0.1% triton supplemented with a first rabbit anti-CRHR1 antibody (1:4000; GeneTex). After three washes, they were treated with a secondary donkey antirabbit-Cy3 antibody (1:10,000; The Jackson Laboratory) for 1 h at room temperature, washed three times, and finally incubated in PBS/Hoechst for 15 min. Simultaneously, a primary anti-TH first antibody was used (1:2000; Abcam polyclonal). It was revealed by the use of a secondary antibody antisheep IgG (Dylight488, 1:5000). The coverslips were mounted on glass slides with 10 μl of mowiol and observed. Cell imaging was performed with a Zeiss LSM510 confocal microscope (Zeiss, Oberkochen, Germany) in multitracking mode to detect the different channels with 405-nm diode for Hoechst nuclei labeling (blue), 488-nm Argon laser for TH labeling (green), 543-nm Helium-Neon laser for CRF1-Cy3 labeling (red), and 633-nm Helium-neon laser for V1b-Al647 labeling (yellow).
Catecholamines release assay
Bovine chromaffin cells were loaded with 4 μCi /ml DL-[7,8-3H] norepinephrine in their culture medium supplemented with 5 mm ascorbic acid. After a 2- to 4-h loading period, cells were washed in a Hanks' buffered saline (HBS), further incubated for 6 min at 25 C with or without the analogs to be tested. The incubation medium was collected and [3H] norepinephrine release measured. Cells were lysed by addition of 5% trichloroacetic acid to determine cellular [3H] norepinephrine content.
Generation of DNA constructs
To express the V1b and CRHR1 receptors in HEK293 cells, DNA constructs were generated by PCR amplification using the full-length human wild-type V1b receptor (kindly provided by Maria-Angeles Ventura, Institut Cochin, Paris, France) and the human CRHR1 receptor (University of Missouri, Rolla cDNA Resource Center, University of Missouri, Rolla, MO) as templates. Receptors fused to YFP (enhanced YFP) at their C terminus were produced using the vector pEYFP-N1 (Clontech, Mountain View, CA). Receptors fused to RL at their C terminus were produced using the vector pRL-CMV (Promega, Madison, WI). All constructs were verified for proper directional in-frame insertion and for sequence integrity by DNA sequencing.
cDNA expression of AVP/CRHR in HEK293 cells
Wild-type, V1b-RL, and/or CRHR1-EYFP receptors were transiently transfected in HEK293 cells by electroporation. The cells (107/0.3 ml) were suspended in 50 mm K2HPO4, 20 mm C2H3KO2, 20 mm KOH (pH 7.4), and 40 mm MgSO4 containing 0.03–10 μg of the DNA constructs completed up to 20 μg with the empty pRK5 expression vector as carrier. They were then incubated for 20 min at room temperature before being pulsed (260 V, 1000 μF) with a Bio-Rad apparatus (Bio-Rad Laboratories, Hercules, CA). The cells were finally plated in 150 mm Petri dishes or 24-well plates depending upon the experiment to be conducted in high-glucose DMEM supplemented with 10% fetal calf serum (Sigma), penicillin (100 UI/ml), streptomycin (100 μg/ml), and maintained at 37 C in 5% CO2. For the BRET and coimmunoprecipitation experiments, transfections were performed using polyethyleimine (44).
Membrane preparation
HEK293 cells transiently transfected with wild-type or mutated receptors were washed twice in PBS without CaCl2 and MgCl2, harvested in lysis buffer [15 mm Tris-HCl (pH 7.4), 2 mm MgCl2, and 0.3 mm EDTA], polytron homogenized, and centrifuged at 44,000 × g for 20 min at 4 C. Pellets were washed in buffer A [50 mm Tris-HCl (pH 7.4) and 3 mm MgCl2] and centrifuged at 44,000 × g for 20 min at 4 C. Membranes were resuspended in a small volume of buffer A. Protein concentration was determined by the method of Bradford (Bio-Rad protein assay kit) using BSA as a standard. Membranes were stored in liquid nitrogen.
Binding assays
Membrane incubations with [3H]AVP or [125I]CRH were performed as described (45). Briefly, 10- to 20-μg membrane proteins were incubated for 60 min at 30 C in a medium containing 50 mm Tris-HCl (pH 7.4), 3 mm MgCl2, 1 mg/ml BSA, and 0.01 mg/ml leupeptine for [3H]-AVP, with an additional 0.5 mg/ml soybean trypsin inhibitor for [125I]-CRH. Radioligand concentrations from 0.01 to 15 nm were added to the incubation medium with (nonspecific binding) or without (total binding) 1 μm unlabeled AVP or CRH. For Ki determination, increasing amounts of the unlabeled analog to be tested were added to a fixed concentration of tritiated ligand. The membrane-associated radioactivity was collected by filtration through GF/C filters and counted.
For binding experiments on intact cells, HEK293 transfected cells were grown for 24 h and further incubated with or without receptor antagonists for another 24 h period in the presence of 5 mm sodium butyrate. Cells were then washed three times with 250 μl of a ice-cold incubation medium [PBS supplemented with 5 mm MgCl2, 1 mg/ml BSA, and 1 mm phenylalanine (pH 7.4)]. Then, 250 μl of incubation medium containing 1–15 nm [3H]AVP were added with (nonspecific binding) or without (total binding) 1 μm AVP. Cells were incubated for 3 h at 4 C, washed four times with ice-cold incubation medium, and 400 μl of NaOH 0.1 m were added. The lysates were then incubated for 15 min at 37 C, and 100 μl of AcOH 0.4 m were added. Lysates were then counted for radioactivity.
For both types of experiment, specific binding was calculated as the difference between total and nonspecific binding values and expressed as percent of the specific binding determined without unlabeled analog.
IP assay
IP accumulation was determined as described previously (45). HEK293 cells transiently transfected with V1b and/or CRHR1 receptors were plated at 100,000 cells per well. Cell were grown for 24 h in their respective culture medium and further incubated for another 24 h period in serum- and inositol-free medium supplemented with 2 μCi/ml myo-[2-3H] inositol and 5 mm sodium butyrate. Cells were then washed twice with HBS, incubated for 15 min in this medium supplemented with 20 mm LiCl with or without CRH, and further stimulated for 15 min with increasing concentrations of AVP with or without CRH. Reaction was stopped by perchloric acid (5% vol/vol). Total IP were extracted, purified on Dowex AG1-X8 anion exchange chromatography column and counted. Free myo-[2-3H]inositol was also measured in each condition. This value, proportional to the number of cells per well, was used to normalize the IP values to optimize experimental reproducibility.
AC assay
AC activity was assessed as described previously (46). HEK293 cells transiently transfected with V1b and/or CRHR1 receptors were plated at 400,000 cells per well. Cells were grown for 24 h in their culture medium and further incubated for another 24 h in a serum-free medium supplemented with 3μCi/ml [3H] adenine and 5 mm sodium butyrate. Cells were washed twice with HBS, incubated for 15 min in HBS supplemented with 1 mm isobutylmethylxanthine with or without AVP, and further stimulated for 30 min with increasing concentration of CRH with or without AVP. The reaction was stopped by trichloroacetic acid (5% vol/vol). For each point, labeled cAMP and ATP were extracted, purified on Dowex and alumin columns, counted, and expressed as percent of ATP converted into cAMP (percent of conversion). Such calculations allowed to normalize the results to the number of cells per well and thus were fine tuned to detect small changes in receptor function.
Coimmunoprecipitation
HEK293 transfected cells in 10-cm plates were washed with PBS and scraped off in 1 ml of the following ice-cold lysate buffer [25 mm HEPES/NaOH (pH 7.4), 5 mm EDTA, 50 mm NaCl, 10% glycerol, 1% Triton X-100, 1 μg/ml each of aprotinin, leupeptin, and pepstatin A, and 0.5 mm phenylmethylsulfonyl fluoride as protease inhibitors]. Receptors were solubilized for 30 min at 4 C with gentle mixing. The lysates were clarified by centrifugation at 17,000 × g for 10 min at 4 C and protein concentrations assayed by BCA (Pierce, Rockford, IL). An amount of 250 μg of protein in 250 μl of lysate buffer was subjected to preclearing using 20 μl of slurry protein G beads (Protein G sepharose 4 Fast Flow; GE Healthcare, Princeton, NJ) for 1 h at 4 C. The immunoprecipitation procedure per se started by the addition of 2 μg of anti-GFP antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) to the cleared lysates followed by incubation for 18 h at 4 C. Then, 20 μl of protein G slurry beads were added and left for another hour at 4 C with gentle mixing. At the end of the incubation, beads were centrifuged and washed three times with lysate buffer. The beads were resuspended in 20 μl of 2× SDS-PAGE Laemmli loading buffer [100 mm Tris (pH 6.8), 286 mm β-mercaptoethanol, 138 mm sodium dodecyl sulfate, 20% glycerol, and bromophenol blue] and heated to 65 C for 15 min. The denatured immunoprecipitation products were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad, Montreal, Canada). The RL-tagged receptors coimmunoprecipitated with the YFP-tagged receptors were detected using an antibody directed against the RL moiety (Chemicon, Billerica, MA) in conjunction with an antimouse HRP-linked secondary antibody (Sigma). Receptors were visualized using a chemiluminescence-based detection system (SuperSignal; Pierce). To reveal the YFP-tagged receptors, the antibody complex was first striped off the membrane by two 0.2 n NaOH washes, and the membrane was reprobed with the same anti-GFP antibody as used for the coimmunoprecipitation followed by an antirabbit secondary antibody (Cell Signaling, Pickering, Canada).
Bioluminescence resonance energy transfer
BRET was performed essentially as described previously (47). HEK-293 cells were grown in complete medium [DMEM supplemented with 10% (vol/vol) fetal calf serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Invitrogen)] in 96-well plates. Transient transfections were performed with the indicated plasmids using JetPEI (Polyplus Transfection, Illkirch, France) following the manufacturer's protocol. The expression of all constructs in HEK-293 cells was confirmed by BRET signals. For BRET experiments, 48 h after transfection, HEK-293 cells were washed twice with HBS (130 mm NaCl, 3.5 mm KCl, 2.3 mm CaCl2, 0.98 mm MgCl2, 5 mm HEPES, and 0.5 mm EGTA) complemented with glucose (5.55 mm), and coelenterazine h substrate (Promega) was added at a final concentration of 2.5 μm in the total volume of 50 μl/well at 30 C. BRET readings were then performed immediately after coelenterazine addition by the Mithras LB 940 plate reader (Berthold Biotechnologies, Oak Ridge, TN). BRET signals were expressed in milliBRET units of BRET ratio as previously described (48).
Calculations and statistics
The radioligand binding data were analyzed using the GraphPad (GraphPad Software, Inc., San Diego, CA). The Ki for unlabeled AVP/CRH analogs was calculated from binding competition experiments according to the Cheng and Prusoff equation: Ki = IC50 × (1+[L]/Kd), where IC50 is the concentration of unlabeled analog leading to half-maximal inhibition of specific binding, [L] the concentration of the radioligand present in the assay, and Kd its affinity for the receptor studied. Kact, concentration of analog leading to half-maximal stimulation of second messenger accumulation, was calculated from functional studies using GraphPad Prism. Results are expressed as the mean ± sem of the number of distinct experiments indicated (n). Statistical analysis of the data was performed using the one-way ANOVA test. Homogeneity of variance was assessed by Bartlett's test, and P values were obtained from Dunnett's tables.
Acknowledgments
We thank Dr. C. Serradeil-Le Gal (Sanofi-Aventis) and Dr. M. Manning (National Institutes of Health, Toledo, OH) for their generous gifts of CRH, OT, and AVP selective analogs and Dr. M. A. Ventura for giving us the human V1b plasmid. We also thank M. Gien-Asari for expert drawing assistance and Dr. M. Desarménien and Dr. M. Bouvier for fruitful discussions and for critical reading of the manuscript.
This work was supported by Institut National de la Santé et de la Recherche Médicale/Fonds de Recherche Santé Québec, Agence Nationale de la Recherche (Maladies neurologiques et psychiatriques 2009), and INSERM “Recherche Clinique translationnelle 2010” grants. B.M. was supported by a grant from the Fondation pour la Recherche Medicale.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AC
- Adenylyl cyclase
- AVP
- vasopressin
- Bmax
- maximal binding capacity
- BRET
- bioluminescence resonance energy transfer
- CRHR
- CRH receptor
- GABA
- γ-aminobutyric acid
- GFP
- green fluorescent protein
- GPCR
- G protein-coupled receptor
- HBS
- Hanks' buffered saline
- IP
- inositol phosphate
- Kact
- concentration of analogue leading to half maximal effect
- Kd
- dissociation constant
- Ki
- inhibitory Kd
- OT
- oxytocin
- PKC
- protein kinase C
- PLC
- phospholipase C
- RL
- Renilla Luciferase
- TH
- tyrosine hydroxylase
- TPA
- 12-O-tetradecanoylphorbol 13-acetate
- Vmax
- maximum velocity
- YFP
- yellow fluorescent protein.
References
- 1. Young LJ , Wang Z. 2004. The neurobiology of pair bonding. Nat Neurosci 7:1048–1054 [DOI] [PubMed] [Google Scholar]
- 2. Barberis C , Morin D , Durroux T , Mouillac B , Guillon G , Seyer R , Hibert M , Tribollet E , Manning M. 1999. Molecular pharmacology of AVP and OT receptors and therapeutic potential. Drug News Perspect 12:279–292 [Google Scholar]
- 3. Guillon G , Gaillard RC , Kehrer P , Schoenenberg P , Muller AF , Jard S. 1987. Vasopressin and angiotensin induce inositol lipid breakdown in rat adenohypophysial cells in primary culture. Regul Pept 18:119–129 [DOI] [PubMed] [Google Scholar]
- 4. Serradeil-Le Gal C , Wagnon J , Tonnerre B , Roux R , Garcia G , Griebel G , Aulombard A. 2005. An overview of SSR149415, a selective nonpeptide vasopressin V(1b) receptor antagonist for the treatment of stress-related disorders. CNS Drug Rev 11:53–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wersinger SR , Ginns EI , O'Carroll AM , Lolait SJ , Young WS. 2002. Vasopressin V1b receptor knockout reduces aggressive behavior in male mice. Mol Psychiatr 7:975–984 [DOI] [PubMed] [Google Scholar]
- 6. Lolait SJ , Stewart LQ , Jessop DS , Young WS , O'Carroll AM. 2007. The hypothalamic-pituitary-adrenal axis response to stress in mice lacking functional vasopressin V1b receptors. Endocrinology 148:849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keck ME. 2006. Corticotropin-releasing factor, vasopressin and receptor systems in depression and anxiety. Amino Acids 31:241–250 [DOI] [PubMed] [Google Scholar]
- 8. Liebsch G , Landgraf R , Engelmann M , Lörscher P , Holsboer F. 1999. Differential behavioural effects of chronic infusion of CRH 1 and CRH 2 receptor antisense oligonucleotides into the rat brain. J Psychiatr Res 33:153–163 [DOI] [PubMed] [Google Scholar]
- 9. Grigoriadis DE , Lovenberg TW , Chalmers DT , Liaw C , De Souze EB. 1996. Characterization of corticotropin-releasing factor receptor subtypes. Ann NY Acad Sci 780:60–80 [DOI] [PubMed] [Google Scholar]
- 10. Gillies GE , Linton EA , Lowry PJ. 1982. Corticotropin releasing activity of the new CRF is potentiated several times by vasopressin. Nature 299:355–357 [DOI] [PubMed] [Google Scholar]
- 11. O'Carroll AM , Howell GM , Roberts EM , Lolait SJ. 2008. Vasopressin potentiates corticotropin-releasing hormone-induced insulin release from mouse pancreatic β-cells. J Endocrinol 197:231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abou-Samra AB , Harwood JP , Manganiello VC , Catt KJ , Aguilera G. 1987. Phorbol 12-myristate 13-acetate and vasopressin potentiate the effect of corticotropin-releasing factor on cyclic AMP production in rat anterior pituitary cells. Mechanisms of action. J Biol Chem 262:1129–1136 [PubMed] [Google Scholar]
- 13. Milligan G , Canals M , Pediani JD , Ellis J , Lopez-Gimenez JF. 2006. The role of GPCR dimerisation/oligomerisation in receptor signalling. Ernst Schering Found 145–161 [DOI] [PubMed] [Google Scholar]
- 14. Terrillon S , Durroux T , Mouillac B , Breit A , Ayoub MA , Taulan M , Jockers R , Barberis C , Bouvier M. 2003. Oxytocin and vasopressin V1a and V2 receptors form constitutive homo- and heterodimers during biosynthesis. Mol Endocrinol 17:677–691 [DOI] [PubMed] [Google Scholar]
- 15. Terrillon S , Barberis C , Bouvier M. 2004. Heterodimerization of V1a and V2 vasopressin receptors determines the interaction with β-arrestin and their trafficking patterns. Proc Natl Acad Sci USA 101:1548–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Devost D , Zingg HH. 2004. Homo- and hetero-dimeric complex formations of the human oxytocin receptor. J Neuroendocrinol 16:372–377 [DOI] [PubMed] [Google Scholar]
- 17. Robert J , Auzan C , Ventura MA , Clauser E. 2005. Mechanisms of cell-surface rerouting of an endoplasmic reticulum-retained mutant of the vasopressin V1b/V3 receptor by a pharmacological chaperone. J Biol Chem 280:42198–42206 [DOI] [PubMed] [Google Scholar]
- 18. Albizu L , Cottet M , Kralikova M , Stoev S , Seyer R , Brabet I , Roux T , Bazin H , Bourrier E , Lamarque L , Breton C , Rives ML , Newman A , Javitch J , Trinquet E , Manning M , Pin JP , Mouillac B , Durroux T. 2010. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat Chem Biol 6:587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Young SF , Griffante C , Aguilera G. 2007. Dimerization between vasopressin V1b and corticotropin releasing hormone type 1 receptors. Cell Mol Neurobiol 27:439–461 [DOI] [PubMed] [Google Scholar]
- 20. Mikhailova MV , Blansett J , Jacobi S , Mayeux PR , Cornett LE. 2008. Transmembrane domain IV of the Gallus gallus VT2 vasotocin receptor is essential for forming a heterodimer with the corticotrophin releasing hormone receptor. J Biomed Opt 13:031208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mikhailova MV , Mayeux PR , Jurkevich A , Kuenzel WJ , Madison F , Periasamy A , Chen Y , Cornett LE. 2007. Heterooligomerization between vasotocin and corticotropin-releasing hormone (CRH) receptors augments CRH-stimulated 3′,5′-cyclic adenosine monophosphate production. Mol Endocrinol 21:2178–2188 [DOI] [PubMed] [Google Scholar]
- 22. de Diego AM , Gandia L , Garcia AG. 2008. A physiological view of the central and peripheral mechanisms that regulate the release of catecholamines at the adrenal medulla. Acta Physiol 192:287–301 [DOI] [PubMed] [Google Scholar]
- 23. Grazzini E , Lodboerer AM , Perez-Martin A , Joubert D , Guillon G. 1996. Molecular and functional characterization of V1b vasopressin receptor in rat adrenal medulla. Endocrinology 137:3906–3914 [DOI] [PubMed] [Google Scholar]
- 24. Corbani M , Trueba M , Stoev S , Murat B , Mion J , Boulay V , Guillon G , Manning M. 2011. Design, synthesis, and pharmacological characterization of fluorescent peptides for imaging human V1b vasopressin or oxytocin receptors. J Med Chem 54:2864–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andrés M , Peña A , Derick S , Raufaste D , Trojnar J , Wisniewski K , Trueba M , Serradeil-Le Gal C , Guillon G. 2004. Comparative pharmacology of bovine, human and rat vasopressin receptor isoforms. Eur J Pharmacol 501:59–69 [DOI] [PubMed] [Google Scholar]
- 26. Gully D , Geslin M , Serva L , Fontaine E , Roger P , Lair C , Darre V , Marcy C , Rouby PE , Simiand J , Guitard J , Gout G , Steinberg R , Rodier D , Griebel G , Soubrie P , Pascal M , Pruss R , Scatton B , Maffrand JP , Le Fur G. 2002. 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1,3-thiazol-2-amine hydrochloride (SSR125543A): a potent and selective corticotrophin-releasing factor(1) receptor antagonist. I. Biochemical and pharmacological characterization. J Pharmacol Exp Ther 301:322–332 [DOI] [PubMed] [Google Scholar]
- 27. Morello JP , Salahpour A , Laperrière A , Bernier V , Arthus MF , Lonergan M , Petäjä-Repo U , Angers S , Morin D , Bichet DG , Bouvier M. 2000. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest 105:887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Derick S , Cheng LL , Voirol MJ , Stoev S , Giacomini M , Wo NC , Szeto HH , Ben Mimoun M , Andres M , Gaillard RC , Guillon G , Manning M. 2002. [1-deamino-4-cyclohexylalanine] arginine vasopressin: a potent and specific agonist for vasopressin V1b receptors. Endocrinology 143:4655–4664 [DOI] [PubMed] [Google Scholar]
- 29. Hoare SR , Sullivan SK , Schwarz DA , Ling N , Vale WW , Crowe PD , Grigoriadis DE. 2004. Ligand affinity for amino-terminal and juxtamembrane domains of the corticotropin releasing factor type I receptor: regulation by G-protein and nonpeptide antagonists. Biochemistry 43:3996–4011 [DOI] [PubMed] [Google Scholar]
- 30. Müller MB , Preil J , Renner U , Zimmermann S , Kresse AE , Stalla GK , Keck ME , Holsboer F , Wurst W. 2001. Expression of CRHR1 and CRHR2 in mouse pituitary and adrenal gland: implications for HPA system regulation. Endocrinology 142:4150–4153 [DOI] [PubMed] [Google Scholar]
- 31. De Souza EB. 1987. Corticotropin-releasing factor receptors in the rat central nervous system: characterization and regional distribution. J Neurosci 7:88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Young WS , Li J , Wersinger SR , Palkovits M. 2006. The vasopressin 1b receptor is prominent in the hippocampal area CA2 where it is unaffected by restraint stress or adrenalectomy. Neuroscience 143:1031–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hernando F , Schoots O , Lolait SJ , Burbach JP. 2001. Immunohistochemical localization of the vasopressin V1b receptor in the rat brain and pituitary gland: anatomical support for its involvement in the central effects of vasopressin. Endocrinology 142:1659–1668 [DOI] [PubMed] [Google Scholar]
- 34. Aguilera G , Subburaju S , Young S , Chen J. 2008. The parvocellular vasopressinergic system and responsiveness of the hypothalamic pituitary adrenal axis during chronic stress. Prog Brain Res 170:29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Springael JY , Urizar E , Costagliola S , Vassart G , Parmentier M. 2007. Allosteric properties of G protein-coupled receptor oligomers. Pharmacol Therapeut 115:410–418 [DOI] [PubMed] [Google Scholar]
- 36. Albizu L , Balestre MN , Breton C , Pin JP , Manning M , Mouillac B , Barberis C , Durroux T. 2006. Probing the existence of G protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol 70:1783–1791 [DOI] [PubMed] [Google Scholar]
- 37. Bridges TM , Lindsley CW. 2008. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem Biol 3:530–541 [DOI] [PubMed] [Google Scholar]
- 38. Quitterer U , Lohse MJ. 1999. Crosstalk between Gα(i)- and Gα (q)-coupled receptors is mediated by Gβγ exchange. Proc Natl Acad Sci USA 96:10626–10631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kraetke O , Wiesner B , Eichhorst J , Furkert J , Bienert M , Beyermann M. 2005. Dimerization of corticotropin-releasing factor receptor type 1 is not coupled to ligand binding. J Recept Signal Trans Res 25:251–276 [DOI] [PubMed] [Google Scholar]
- 40. Pin JP , Neubig R , Bouvier M , Devi L , Filizola M , Javitch JA , Lohse MJ , Milligan G , Palczewski K , Parmentier M , Spedding M. 2007. International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol Rev 59:5–13 [DOI] [PubMed] [Google Scholar]
- 41. González-Maeso J , Ang RL , Yuen T , Chan P , Weisstaub NV , López-Giménez JF , Zhou M , Okawa Y , Callado LF , Milligan G , Gingrich JA , Filizola M , Meana JJ , Sealfon SC. 2008. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 452:93–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilson SP , Viveros OH. 1981. Primary culture of adrenal medullary chromaffin cells in a chemically defined medium. Exp Cell Res 133:159–169 [DOI] [PubMed] [Google Scholar]
- 43. Role LW , Perlman RL. 1980. Purification of adrenal medullary chromaffin cells by density gradient centrifugation. J Neurosci Methods 2:253–265 [DOI] [PubMed] [Google Scholar]
- 44. Robitaille M , Heroux I , Baragli A , Hebert TE. 2009. Novel tools for use in bioluminescence resonance energy transfer (BRET) assays. Meth Mol Biol 574:215–234 [DOI] [PubMed] [Google Scholar]
- 45. Pena A , Murat B , Trueba M , Ventura MA , Bertrand G , Cheng LL , Stoev S , Szeto HH , Wo N , Brossard G , Serradeil-Le Gal C , Manning M , Guillon G. 2007. Pharmacological and physiological characterization of d[Leu4, Lys8]vasopressin, the first V1b-selective agonist for rat vasopressin/oxytocin receptors. Endocrinology 148:4136–4146 [DOI] [PubMed] [Google Scholar]
- 46. Côté M , Payet MD , Rousseau E , Guillon G , Gallo-Payet N. 1999. Comparative involvement of cyclic nucleotide phosphodiesterases and adenylyl cyclase on adrenocorticotropin-induced increase of cyclic adenosine monophosphate in rat and human glomerulosa cells. Endocrinology 140:3594–3601 [DOI] [PubMed] [Google Scholar]
- 47. Devost D , Zingg HH. 2003. Identification of dimeric and oligomeric complexes of the human oxytocin receptor by co-immunoprecipitation and bioluminescence resonance energy transfer. J Mol Endocrinol 31:461–471 [DOI] [PubMed] [Google Scholar]
- 48. Ayoub MA , Couturier C , Lucas-Meunier E , Angers S , Fossier P , Bouvier M , Jockers R. 2002. Monitoring of ligand-independent dimerization and ligand-induced conformational changes of melatonin receptors in living cells by bioluminescence resonance energy transfer. J Biol Chem 277:21522–21528 [DOI] [PubMed] [Google Scholar]